
Method Article
Transkription Start Site Mapping mit Super-low Input Carrier-CAGE
In diesem Artikel
Zusammenfassung
Cap Analysis of Gene Expression (CAGE) ist eine Methode zur genomweiten quantitativen Kartierung von mRNA 5'ends zur Erfassung von RNA-Polymerase-II-Transkriptionsstartstellen mit einer einzigen Nukleotid-Auflösung. Diese Arbeit beschreibt ein Low-Input-Protokoll (SLIC-CAGE) zur Erzeugung hochwertiger Bibliotheken mit Nanogramm-Mengen von Gesamt-RNA.
Zusammenfassung
Die Cap-Analyse der Genexpression (CAGE) ist eine Methode zum Nachweis der Single-Nukleotid-Auflösung von RNA-Polymerase-II-Transkriptionsstartstellen (TSS). Die genaue Erkennung von TSS verbessert die Identifizierung und Entdeckung von Kernpromotoren. Darüber hinaus können aktive Enhancer durch Signaturen der bidirektionalen Transkriptionsinitiierung erkannt werden. Hier wird ein Protokoll zur Durchführung von Super-Low Input Carrier-CAGE (SLIC-CAGE) beschrieben. Diese SLIC-Anpassung des CAGE-Protokolls minimiert die RNA-Verluste, indem die RNA-Menge durch den Einsatz eines in vitro transkribierten RNA-Trägermixes, der der betreffenden Probe zugesetzt wird, künstlich erhöht wird, wodurch die Bibliotheksvorbereitung aus Nanogramm-Mengen von RNA (d. h. Tausende von Zellen). Der Träger imitiert die erwartete DNA-Bibliotheksfragmentlängenverteilung und eliminiert so Verzerrungen, die durch die Fülle eines homogenen Trägers verursacht werden könnten. In den letzten Phasen des Protokolls wird der Träger durch Degradation mit homing endonucleases entfernt und die Zielbibliothek verstärkt. Die Ziel-Probenbibliothek ist vor Degradation geschützt, da die Homing-Endonuklease-Erkennungsstellen lang sind (zwischen 18 und 27 bp), was die Wahrscheinlichkeit ihrer Existenz in den eukaryotischen Genomen sehr gering macht. Das Endergebnis ist eine DNA-Bibliothek, die für die Sequenzierung der nächsten Generation bereit ist. Alle Schritte im Protokoll bis zur Sequenzierung können innerhalb von 6 Tagen abgeschlossen werden. Die Transportvorbereitung erfordert einen vollen Arbeitstag; Es kann jedoch in großen Mengen zubereitet und bei -80 °C gefroren gehalten werden. Nach der Sequenzkönnen können die Lesevorgänge verarbeitet werden, um genomweite Single-Nukleotid-Auflösungs-TSSs zu erhalten. TSS können für die Kernpromotor- oder Enhancer-Entdeckung verwendet werden, um Einblicke in die Genregulation zu geben. Nach der Aggregation zu Promotoren können die Daten auch für 5'-zentrierte Ausdrucksprofilierung verwendet werden.
Einleitung
Die Cap-Analyse der Genexpression (CAGE) ist eine Methode zur genomweiten Kartierung von RNA-Polymerase-II-Transkriptionsstartstellen (TSS)1. Seine quantitative Natur ermöglicht auch 5'-Ende zentrische Ausdruck Profiling. Regionen, die die TSS umgeben (ca. 40 bp vor und nachgelagert) sind Kernpromotoren und stellen den physikalischen Ort dar, an dem RNA-Polymerase II und allgemeine Transkriptionsfaktoren binden (zuvor untersucht2,3). Informationen über die genauen Standorte von TSS können für die Kern-Promoter-Entdeckung und für die Überwachung der Projektdynamik verwendet werden. Darüber hinaus können CAGE-Daten, da aktive Enhancer Signaturen der bidirektionalen Transkription aufweisen, auch zur Enhancer-Erkennung und Überwachung der Enhancer-Dynamik 4 verwendet werden. Die CAGE-Methodik hat in letzter Zeit aufgrund ihrer breiten Anwendung und Verwendung in hochkarätigen Forschungsprojekten wie ENCODE5, modENCODE6und FANTOM-Projekten7an Popularität gewonnen. Darüber hinaus erweisen sich TSS-Informationen auch als wichtig für die Unterscheidung von gesundem und krankem Gewebe, da krankheitsspezifische TSS für diagnostische Zwecke verwendet werden können8.
Obwohl mehrere Methoden für TSS-Mapping verfügbar sind (CAGE, RAMPAGE, STRT, nanoCAGE, nanoCAGE-XL, Oligo-Capping), haben wir und andere kürzlich gezeigt, dass CAGE die unvoreingenommenste Methode ist, um echte TSS mit der geringsten Anzahl falschpositiver Ergebnisse zu erfassen9 , 10. Das jüngste CAGE-Protokoll, nAnT-iCAGE11, ist das unvoreingenommeneste Protokoll für TSS-Profiling, da es verhindert, dass die Fragmente mit Restriktionsenzymen auf kurze Tags geschnitten werden und keine PCR-Verstärkung verwendet wird. Eine Einschränkung des nAnT-iCAGE-Protokolls ist die Anforderung an eine große Menge an Ausgangsmaterial (z.B. 5 g Gesamt-RNA für jede Probe). Um spezifische, biologisch relevante Fragen zu beantworten, ist es oft unmöglich, so hohe Mengen an Ausgangsmaterial zu erhalten (z.B. für FACS-sortierte Zellen oder frühe embryonale Stadien). Wenn nAnT-iCAGE erfolgreich ist, stehen von jeder Probe nur 1-2 ng DNA-Bibliotheksmaterial zur Verfügung, wodurch die erreichbare Sequenzierungstiefe begrenzt wird.
Um das TSS-Profiling mit nur Nanogramm der gesamten RNA zu ermöglichen, haben wir vor kurzem Super-low Input Carrier-CAGE10 entwickelt (SLIC-CAGE, Abbildung 1). SLIC-CAGE benötigt nur 10 ng der gesamten RNA, um Bibliotheken mit hoher Komplexität zu erhalten. Unser Protokoll stützt sich auf den sorgfältig entworfenen synthetischen RNA-Träger, der der RNA von Interesse hinzugefügt wird, um insgesamt 5 g RNA-Material zu erreichen. Der synthetische Träger imitiert die Ziel-DNA-Bibliothek in der Längenverteilung, um potenzielle Verzerrungen zu vermeiden, die durch homogene Moleküle im Überschuss verursacht werden könnten. Die Sequenz des Trägers basiert aus zwei Gründen auf der Sequenz des Escherichia coli leucyl-tRNA Synthetase-Gens (Tabelle 1). Erstens wird jedes Übrige des Trägers in der endgültigen Bibliothek, selbst wenn es sequenziert wird, nicht zu einem eukaryotischen Genom zugeordnet. Zweitens, da E. coli eine mesophile Spezies ist, sind seine Haushaltsgene für den für SLIC-CAGE geeigneten Temperaturbereich optimiert. Die Trägersequenz ist auch in homing Endonuklease-Erkennungsstellen eingebettet, um einen spezifischen Abbau der DNA zu ermöglichen, die aus den TRÄGER-RNA-Molekülen abgeleitet wurde. Die Zielbibliothek, die von Beispielen abgeleitet wurde, bleibt intakt, da die Homing-Endonuklease-Erkennungsstellen lang sind (I-CeuI = 27 bp; I-SceI = 18 bp) und statistisch unwahrscheinlich, in eukaryotischen Genomen gefunden zu werden. Nach der spezifischen Degradierung des Trägers und dem Entfernen von Fragmenten durch Größenausschluss ist die Zielbibliothek PCR verstärkt und bereit für die Sequenzierung der nächsten Generation. Je nach Start-RNA-Menge (1-100 ng) werden zwischen 13-18 PCR-Verstärkungszyklen erwartet. Die endgültige DNA-Menge pro Probe liegt zwischen 5-50 ng, was genügend Material für eine sehr tiefe Sequenzierung ergibt. Bei Verwendung von nur 1-2 ng der gesamten RNA können echte TSS s detektiert werden; Es wird jedoch erwartet, dass die Bibliotheken von geringerer Komplexität sein werden. Da SLIC-CAGE auf dem nAnT-iCAGE-Protokoll11basiert, ermöglicht es das Multiplexing von bis zu acht Samples vor der Sequenzierung.
Protokoll
1. Vorbereitung des Beförderers
-
Erstellung von DNA-Vorlagen für die In-vitro-Transkription
- Bereiten Sie den PCR-Mix für jede PCR-Vorlage vor, indem Sie 41 l Wasser, 20 l 5x HF-Puffer, 8 x L mit 2,5 mM dNTPs, 10 l mit 10 'M eindeutiger Vorwärtsprimer (PCR_GN5_f1, Tabelle2; Primer werden gelöst und in Wasser verdünnt) , 10 l mit 2 ng/l Schablonenplasmid, das das synthetische Trägergen enthält, und 1 l Phusion-Polymerase. Mischen Sie den PCR-Mix durch Pipettieren. Ein Master-Mix für alle 10 Vorlagen kann auf einmal vorbereitet werden (Vorbereitung auf 11 Reaktionen).
- Fügen Sie 90 l des PCR-Mixes zu 10 l von jedem 10-M-Reverse-Primer hinzu (PCR_N6_r1-r10, Tabelle 2). Mix durch Pipetten.
- PCR verstärken die Schablonen mit folgendem Programm: 98 °C für 60 s, (98 °C für 10 s, 50 °C für 30 s, 72 °C für 30 s) 35 Zyklen, 72 °C für 10 min, bei 4 °C halten.
-
Gelreinigung von PCR-verstärkten DNA-Vorlagen
- Bereiten Sie ein 1% Agarose-Gel vor (niedrigschmelzende Agarose wird empfohlen).
- Um das Volumen zu verringern, konzentrieren Sie die PCR-Reaktionsgemische mit dem Vakuumkonzentrator bei niedriger mittlerer Temperatur (30–40 °C) von 100 l bis 20 l Gesamtvolumen.
- Fügen Sie 6 l des 6x Ladefarbstoffs hinzu, mischen Sie sie gut und belasten Sie das Gel. Elektrophorese für 30 min im 1x TAE Puffer bei der Spannung laufen, die für den verwendeten Elektrophoresetank (5–10 V/cm) geeignet ist. Parallel laufen eine 100 bp oder 1.000 bp DNA-Leiter.
- Mit einem sauberen Skalpell, verbrauchen Sie die Gelscheiben, die das Ziel-PCR-Produkt enthalten. Vermeiden Sie überschüssiges Agarose-Gel. Reinigen Sie die PCR-Produkte mit einem Gel-Extraktionskit (gemäß den Anweisungen des Herstellers).
HINWEIS: A260/A230-Verhältnisse von DNA, die aus Agarose-Gelen isoliert wurden, sind in der Regel niedrig (0,1–0,3). Erwartete Zielprodukte und Nebenprodukte sind in Abbildung 2Adargestellt. Die erwarteten Erträge von 100 l PCR-Reaktionen liegen bei 1,2 bis 3 g. Die Reaktionen können skaliert werden, um eine höhere Ausbeute zu erzielen.
-
In-vitro-Transkription von Trägermolekülen
- Transkribieren Träger-RNA in vitro mit der T7-RNA-Polymerase gemäß den Anweisungen des Herstellers. Richten Sie 10–20 L-Reaktionen ein (das empfohlene Kit befindet sich in Tabelle der Materialien).
- Reinigen Sie die in vitro transkribierte RNA mit einem RNA-Reinigungskit. Richten Sie die DNA-Verdauung in Lösung unter Verwendung von DNase I gemäß den Standardanweisungen des Herstellers ein, und legen Sie die RNA in 50 l Wasser auf. Um die Elutionsausbeute zu erhöhen, lassen Sie das Wasser in der Säule 5 min vor der Zentrifugation.
HINWEIS: Achten Sie darauf, die maximale Bindungskapazität der Säulen nicht zu überschreiten (im In-Kit, das in der Tabelle der Materialienerwähnt wird, beträgt die Kapazität bis zu 100 g). Die erwartete Ausbeute aus PCR-Vorlagen 1–10 (1 kbp bis 200 bp Länge) beträgt 25–50 g von 10 l In-vitro-Transkriptionsreaktionen. Reaktionen können skaliert werden, um einen größeren Bestand an Trägermolekülen zu erhalten.
-
Verkappung von in vitro transkribierten TRÄGER-RNA-Molekülen
- Bereiten Sie die Verschlussmischung vor, indem Sie 2 l 10x Verschlusspuffer, 1 l mit 10 mM GTP, 1 l mit 2 mM SAM (frisch verdünnt) und 1 l Vaccinia-Verschlussenzym pro Träger-RNA kombinieren.
- Mischen Sie bis zu 10 g jedes Trägermolekülin in 15 l Gesamtvolumen und Denaturieren für 10 min bei 65 °C. Sofort auf Eis legen, um eine sekundäre Strukturbildung zu verhindern.
- Mischen Sie die denaturierte Träger-RNA mit 5 l der Verschlussmischung und brüten für 1 h bei 37 °C.
- Reinigen Sie gekappte RNA-Moleküle mit einem RNA-Reinigungskit – folgen Sie dem Reinigungsprotokoll des Herstellers. Elute-RNA in 30 l Wasser. Um die Elutionsausbeute zu erhöhen, lassen Sie das Wasser in der Säule 5 min vor der Zentrifugation.
HINWEIS: Messen Sie die Konzentration mit dem Mikrovolumenspektrophotometer. Erwartetes A260/A280-Verhältnis ist >2 und A260/A230 ist >2. Beachten Sie, dass bei einigen RNA-Proben A260/A230 zwischen 1,3 und 2 liegen kann. Die erwartete Ausbeute bei verwendung von 10 g ungekapselter RNA beträgt 9–10 g gedeckelte RNA.
- Bereiten Sie die Mischung des gedeckelten und des nicht gedeckelten Trägers vor, indem Sie die in Tabelle 3beschriebenen Beträge kombinieren. Mischen Sie gut, indem Sie das Rohr streichen und messen Sie die Konzentration mit dem Mikrovolumen-Spektrophotometer.
HINWEIS: Wenn eine höhere Konzentration des Trägers erforderlich ist, um in die Umgekehrte Transkriptionsreaktion zu passen (siehe unten), kann der Trägermix mit dem Vakuumkonzentrator bei niedriger mittlerer Temperatur (30-35 °C) bis zum Erreichen der gewünschten Endkonzentration konzentriert werden. Die Schritte 2–14 werden vom Standard-nAnT-iCAGE-Protokoll geändert, das von Murata et al.11 gemeldet wird.
2. Umgekehrte Transkription
- Kombinieren Sie 1 l des RT-Primers (2,5 mM TCT-N6 in Wasser gelöst, für Sequenz siehe Zusatztabelle 1), 10 ng der gesamten RNA von Interesse und 4.990 ng Trägermischung (Tabelle 3) in 10 l gesamtvolumen in einer niedrig bindenden PCR-Platte. Mischen Sie, indem Sie die Röhre.
HINWEIS: Wenn die Proben-RNA für eine umgekehrte Transkription zu verdünnt ist (siehe unten), kombinieren Sie sie mit der entsprechenden Menge des Trägers, konzentrieren Sie sich mit dem Vakuumkonzentrator auf das Gesamtvolumen von 9 l und fügen Sie 1 L des RT-Primers hinzu. Das Hinzufügen des Trägerearls, um insgesamt 5 g RNA zu erreichen, verhindert den Probenverlust. - Die Mischung von Schritt 2.1 bei 65 °C 5 min erhitzen und sofort auf Eis legen, um eine Renaturierung zu verhindern.
-
Vorbereiten des Reverse-Transkriptions-Mix (RT).
- Kombinieren Sie für jede Probe 6,1 l Wasser (RNase- und DNase-frei), 7,6 l 5x Erststrangpuffer, 1,9 l 0,1 m DTT, 1 l mit 10 mM dNTPs, 7,6 l Trehalose/Sorbitol-Mischung (siehe Rezept in Murata et al.11) und 3,8 l der empfohlenen Reverse-Transkriptase (siehe 6>Tabelle der Materialien). Mischen Sie gut, indem Sie die Röhre.
- Fügen Sie 28 L des RT-Mixes in die PCR-Röhre mit 10 l RNA, Träger und RT-Primer (Gesamtvolumen 38 l) ein. Durch Pipettieren gut mischen.
HINWEIS: Die Mischung ist aufgrund von Trehalose/Sorbitol hochviskos. Mischen, bis sie sichtbar homogen sind. - Inkubieren Sie in einem thermischen Cycler mit dem folgenden Programm: 25 °C für 30 s, 50 °C für 60 min und bei 4 °C halten.
-
Reinigung von cDNA:RNA-Hybriden mit SPRI-Magnetperlen
- Fügen Sie 68,4 l der empfohlenen RNAse- und DNase-freien SPRI-Perlen (siehe Materialtabelle)bis 38 L des RT-Mixes (Perlen zum Probenverhältnis 1,8:1) hinzu. Gut durch Pipetten mischen und 5 min bei Raumtemperatur (RT) brüten.
- Trennen Sie die Perlen auf einem magnetischen Ständer für 5 min. Entsorgen Sie den Überstand und waschen Sie die Perlen zweimal mit 200 l 70% Ethanol (frisch zubereitet).
HINWEIS: Das Ethanol wird den Perlen ohne Mischen und während der Röhre auf dem magnetischen Ständer zugesetzt. Das zugesetzte Ethanol wird sofort entfernt. Es sollte darauf geachtet werden, keine Perlen während der Wasse zu verlieren, da dies zu Probenverlust führen kann. - Während sich das Rohr noch auf dem magnetischen Ständer befindet, entfernen Sie alle Spuren von Ethanol. Ethanoltröpfchen können mit einer P10-Pipette aus dem Rohr geschoben werden. Lassen Sie die Perlen nicht trocknen.
- 42 L Wasser bei 37 °C vorgewärmt in die Perlen geben und die Probe durch Pipettieren 60x nach oben und unten elute.
HINWEIS: Achten Sie darauf, nicht durch Pipettieren zu schäumen, da es zu einem Verlust von Perlen (d. h. gebundene Probe) im Schaum führen kann. - Bei 37 °C für 5 min ohne Deckel inkubieren, um eine Verdunstung von Ethanolspuren zu ermöglichen.
- Trennen Sie die Perlen auf einem magnetischen Ständer für 5 min und übertragen Sie den Überstand auf eine neue Platte.
HINWEIS: Versuchen Sie, alle Überstand, um Probenverlust zu verhindern, während die Perlenübertragung zu vermeiden. Verwenden Sie die P10 Pipette, um die letzten Probentröpfchen zu erhalten.
3. Oxidation
- Fügen Sie der gereinigten RT-Reaktion 2 l von 1 M NaOAc (pH 4.5) hinzu. Durch Pipettieren mischen, 2 l von 250 mM NaIO4 hinzufügen und erneut mischen.
- 45 min auf Eis bebrüten. Bedecken Sie die Platte mit Aluminiumfolie, um Licht zu vermeiden.
- Fügen Sie 16 L Tris-HCl (pH 8.5) in den Oxidationsmix ein, um den pH-Wert zu neutralisieren.
- Reinigen Sie oxidierte cDNA:RNA-Hybriden mit SPRI-Magnetperlen. Fügen Sie 108 L SPRI-Perlen zu 60 l des Oxidationsmixes hinzu (1,8:1 Perlen zum Probenverhältnis). Wiederholen Sie die Reinigung wie in den Schritten 2.6.1–2.6.6 beschrieben. Elute mit 42 l Wasser vorgewärmt auf 37 °C.
HINWEIS: 250 mM NaIO4 frisch vorbereiten, indem Sie 18,7 l Wasser pro 1 mg NaIO4hinzufügen. NaIO4 ist lichtempfindlich; Halten Sie die Lösung daher in einem mit Aluminiumfolie überzogenen Rohr oder in einem lichtbeständigen Rohr auf.
4. Biotinylierung
- Fügen Sie 4 l von 1 M NaOAc (pH 6.0) in das Rohr, das die gereinigte oxidierte Probe enthält, und mischen Sie sie durch Pipettieren.
- Fügen Sie 4 l von 10 mM Biotinlösung hinzu, mischen Sie durch Pipettieren und brüten Sie 2 h bei 23 °C in einem thermischen Cycler, um Licht zu vermeiden.
HINWEIS: Bereiten Sie die Biotinlösung vor, indem Sie 50 mg Biotin mit 13,5 ml DMSO mischen. Machen Sie Einweg-Aliquots und frieren Sie bei -80 °C ein. - Reinigen Sie biotinylierte cDNA:RNA-Hybriden mit SPRI-Magnetperlen. Fügen Sie 12 l 2-Propanol und mischen durch Pipetten. Fügen Sie 108 L SPRI-Perlen (1,8:1 Perlen zum Probenverhältnis) hinzu und wiederholen Sie die Reinigung, wie in den Schritten 2.6.1–2.6.6 beschrieben. Elute mit 42 l Wasser vorgewärmt bei 37 °C.
HINWEIS: Das Protokoll kann hier angehalten und Proben bei -80 °C eingefroren werden.
5. RNase I Ddigestion
- Bereiten Sie die RNase I-Mischung vor, indem Sie 4,5 l 10x RNase I Puffer mit 0,5 l RNase I (10 U/L) pro Probe mischen. Mix durch Pipetten.
- Zu jeder gereinigten Probe 5 l der Mischung hinzufügen (insgesamt 45 l). Durch Pipettieren mischen und 30 min bei 37 °C inkubieren.
6. Vorbereitung der Streptavidin Perlen
- Für jede Probe 30 l der Streptavidin-Perlenschlämme mit 0,38 l von 20 mg/ml tRNA mischen. 30 min auf Eis bebrüten und alle 5 min durch Streichen der Röhre mischen.
HINWEIS: Setzen Sie die Streptavidin Perlen Gülle lange vor dem Pipettieren durch Streichen der Flasche. Die tRNA-Lösung sollte nach Murata et al.11 - Trennen Sie die Perlen auf dem magnetischen Ständer für 2-3 min. Entfernen Sie den Überstand.
- Waschen Sie die Perlen, indem Sie in 15 l Puffer A wieder aufhängen. Trennen Sie die Perlen auf dem magnetischen Ständer für 2-3 min und entfernen Sie den Überstand. Wiederholen Sie die Wäsche und entfernen Sie den Überstand.
- Setzen Sie die Perlen in 105 l Puffer A wieder auf und fügen Sie 0,19 l von 20 mg/ml tRNA hinzu. Durch Pipettieren gut mischen.
HINWEIS: Die Perlen sollten vor der Verwendung frisch zubereitet werden. Beginnen Sie mit der Vorbereitung der Perlen während der RNase I Verdauung. Für mehrere Proben bereiten Sie die Perlen zusammen in einem einzigen Rohr.
7. Cap-Trapping
-
Beispielbindung
- Fügen Sie der RNase I-behandelten Probe 105 l präparierte Streptavidinperlen hinzu. Durch Pipettieren gut mischen und bei 37 °C für 30 min inkubieren. Alle 10 min durch Pipettieren mischen.
- Trennen Sie die Perlen auf dem magnetischen Ständer für 2-3 min. Entfernen Sie den Überstand.
-
Waschperlen
- Fügen Sie 150 l Waschpuffer A hinzu und setzen Sie die Perlen durch Pipettieren wieder auf. Trennen Sie die Perlen auf dem magnetischen Ständer für 2-3 min und entfernen Sie den Überstand.
- Fügen Sie 150 l des Waschpuffers B hinzu und setzen Sie die Perlen durch Pipettieren wieder auf. Trennen Sie die Perlen auf dem magnetischen Ständer für 2-3 min und entfernen Sie den Überstand.
- Fügen Sie 150 l des Waschpuffers C hinzu und setzen Sie die Perlen durch Pipettieren wieder auf. Trennen Sie die Perlen auf dem magnetischen Ständer für 2-3 min und entfernen Sie den Überstand.
HINWEIS: Die Puffer B und C sollten auf 37 °C vorgewärmt werden. Rezepte für Waschpuffer A, B und C sind wie in Murata et al.11 beschrieben
-
cDNA-Freisetzung
- Bereiten Sie 1x RNase I Puffer durch Mischen von 58,5 l Wasser mit 6,5 l von 10x RNase I Puffer.
- Setzen Sie die Perlen in 35 l des 1x RNase I Puffers wieder auf. Bei 95 °C für 5 min inkubieren und 2 min direkt auf Eis übertragen, um eine Erneute Assoziation von cDNA zu verhindern. Halten Sie die Deckel während der Übertragung auf Eis, wie sie durch Druck aufbau fallen können.
- Trennen Sie die Perlen für 2-3 min auf einem magnetischen Ständer und übertragen Sie den Überstand auf eine neue Platte.
- Setzen Sie die Perlen in 30 l des 1x RNase I Puffers wieder auf. Trennen Sie die Perlen auf dem magnetischen Ständer für 2-3 min und übertragen Sie den Überstand auf den zuvor gesammelten Überstand (das Gesamtvolumen der eluierten cDNA sollte etwa 65 l betragen).
8. RNA-Entfernung durch RNase H und RNase I Verdauung
- Kombinieren Sie pro Probe 2,4 l Wasser, 0,5 l mit 10x RNase I Puffer, 0,1 l RNase H und 2 l RNase I.
- Fügen Sie 5 l der Mischung zu den 65 l der freigesetzten cDNA-Probe hinzu und mischen Sie sie durch Pipettieren. Bei 37 °C 15 min inkubieren und bei 4 °C halten.
- Reinigen Sie cDNA aus dem RNase-Verdauungsmix mit SPRI-Magnetperlen. Fügen Sie 126 L SPRI-Perlen zu 70 L Abbaureaktion hinzu und mischen Sie sie durch Pipettieren. Befolgen Sie die Reinigungsschritte, wie sie für die SPRI-Perlenreinigung in 2.6.1–2.6.6 beschrieben sind. Elute mit 42 l Wasser bei 37 °C vorgewärmt, wie beschrieben.
- Bereiten Sie rNase I-Mischung durch Die Kombination von 4,5 l 10x RNase I Puffer und 0,5 l RNase I.
- Fügen Sie 5 l der RNase-Mischung in die 40-L-L der gereinigten cDNA-Probe ein. Durch Pipettieren mischen und bei 37 °C 30 min inkubieren. Bei 4 °C halten.
- Reinigen Sie die Probe mit SPRI-Magnetperlen. Fügen Sie 81 L SPRI-Perlen zu 45 l Abbaureaktion hinzu und mischen Sie sie durch Pipettieren. Befolgen Sie die Reinigungsschritte, wie sie für die SPRI-Perlenreinigung in 2.6.1–2.6.6 beschrieben sind. Elute mit 42 l Wasser wie beschrieben.
9. Ligation von 5' Linker
- Konzentrieren Sie die gereinigte cDNA-Probe mit dem Vakuumkonzentrator auf 4 l. Halten Sie die Temperatur bei 30–35 °C. Testen Sie das Volumen mit einer Pipette. Wenn die Probe bis zur Vollständigkeit getrocknet ist, lösen Sie sie auf, indem Sie 4 l Wasser hinzufügen.
HINWEIS: Es ist besser, das Trocknen bis zur Vollständigkeit zu vermeiden, um Probenverlust zu verhindern. - Inkubieren Sie die konzentrierte Probe bei 95 °C für 5 min und legen Sie sofort auf Eis für 2 min, um Renaturierung zu verhindern. Halten Sie die Deckel beim Übertragen der Rohre, da die Deckel aufgrund von Druckaufbau abspringen können.
- Inkubieren Sie 4 l des 2,5-M-5-Glinkers bei 55 °C für 5 min und legen Sie es sofort für 2 min auf Eis, um eine Renaturierung zu verhindern.
- Mischen Sie 4 l des 2,5-M-5-G-Linkers mit 4 l der Probe.
HINWEIS: Die 5' Linker sollten gemäß Der Zusatztabelle 2, Der Zusatztabelle 3, der Zusatztabelle 4und der Zusatztabelle 5erstellt werden. Verdünnen Sie den 10 M 5' Linker mit 100 mM NaCl vor der Anwendung auf eine Konzentration von 2,5 m. - Fügen Sie dem gemischten 5'-Linker und der Probe 16 l des Ligations-Premixes (siehe Materialtabelle)hinzu und mischen Sie sie gut durch Pipettieren. Bei 16 °C für 16 h inkubieren.
- Reinigen Sie den Ligationsmix mit SPRI-Magnetperlen. Fügen Sie 43,2 L SPRI-Perlen hinzu und folgen Sie den Schritten 2.6.1–2.6.6. Elute, wie beschrieben mit 42 l Wasser vorgewärmt bei 37 °C.
- Wiederholen Sie die in Schritt 9.6 durchgeführte Reinigung, indem Sie dem übertragenen Überstand 72 L SPRI-Perlen (1,8:1 Perlen zum Probenverhältnis) hinzufügen.
HINWEIS: 5' Linker enthalten Barcodes, die das Poolen von bis zu acht Proben vor der Sequenzierung ermöglichen (acht Trinukleotid-Barcodes sind verfügbar, wie in Murata et al.11 und Supplementary Table 1beschrieben).
10. Ligation von 3' Linker
- Konzentrieren Sie die gereinigte Probe mit dem Vakuumkonzentrator wie in Schritt 9.1 beschrieben auf 4 l.
- Inkubieren Sie die konzentrierte Probe bei 95 °C für 5 min und legen Sie sofort auf Eis für 2 min, um Renaturierung zu verhindern. Halten Sie die Deckel beim Übertragen der Rohre, da die Deckel aufgrund von Druckaufbau abspringen können.
- Inkubieren Sie 4 l des 2,5-M-3-Linkers bei 65 °C für 5 min und legen Sie es sofort für 2 min auf Eis, um eine Renaturierung zu verhindern.
- Fügen Sie 4 l des 2,5-M-3-Linkers zu den 4 l der konzentrierten Probe hinzu.
- Fügen Sie 16 l des Ligations-Premixhinzuers hinzu und mischen Sie sie gut durch Pipettieren. Bei 16 °C für 16 h inkubieren.
- Reinigen Sie den Ligationsmix mit SPRI-Magnetperlen. Fügen Sie 43,2 L SPRI-Perlen hinzu und folgen Sie den Schritten 2.6.1–2.6.6. Elute, wie beschrieben mit 42 l Wasser vorgewärmt auf 37 °C.
HINWEIS: Die 3' Linker sollten gemäß den Zusatztabellen 6 und der Ergänzenden Tabelle 7erstellt werden. Verdünnen Sie den 10-M-3-Linker mit 100 mM NaCl auf eine Konzentration von 2,5 m.
11. Dephosphorylierung
- Bereiten Sie den SAP-Mix vor, indem Sie 4 l Wasser, 5 l 10x SAP-Puffer und 1 l SAP-Enzym kombinieren.
- Fügen Sie der gereinigten ligaierten Probe 10 l SAP-Mischung (Gesamtvolumen 50 l) hinzu und inkubieren Sie im Thermocycler mit folgendem Programm: 37 °C für 30 min, 65 °C für 15 min und halten Sie bei 4 °C.
12. Abbau von 3' Linker Upper Strand mit Uracil Specific Excision Enzyme
- Der dephosphorylierten Probe 2 L des uracilspezifischen Exzisionsenzyms (siehe Materialtabelle)hinzufügen, durch Pipettieren mischen und im Thermocycler mit folgendem Programm inkubieren: 37 °C für 30 min, 95 °C für 5 min, und sofort 2 min auf Eis legen, um verhindern, dass der fragmentierte obere Strang wieder annungstiniert wird.
- Reinigen Sie das Reaktionsgemisch, indem Sie der 52-L-Mischung 93,6 l SPRI-Magnetperlen hinzufügen und durch Pipettieren gut mischen. Wiederholen Sie die Reinigungsschritte 2.6.1–2.6.6. Elute mit 42 l Wasser, wie beschrieben bei 37 °C vorgewärmt.
13. Zweite Strangsynthese
- Bereiten Sie den zweiten Strangsynthesemix (Volumen werden pro Probe ausgedrückt) durch Kombination von 5 l 10x DNA-Polymerase-Reaktionspuffer, 2 l Wasser, 1 l von 10 mM dNTPs, 1 l von 50 nAnT-iCAGE zweiten Strangprimer (Sequenz ist in Supplementary Table 1) und 1 l von D NA exonuklease-defizidierende Polymerase (siehe empfohlene Polymerase in Tabelle der Materialien).
- Fügen Sie der gereinigten Probe 10 l der Mischung hinzu und mischen Sie sie gut durch Pipettieren (Gesamtvolumen beträgt 50 l). Inkubieren Sie im Thermischen Cycler mit folgendem Programm: 95 °C für 5 min, 55 °C für 5 min, 72 °C für 30 min und halten Sie bei 4 °C.
14. Abbau des zweiten Strangsynthese-Primers mit Exonuklease I
- Fügen Sie dem zweiten Strangsynthesegemisch 1 L Exonuklease I hinzu. Gut durch Pipettieren mischen und bei 37 °C 30 min lang inkubieren, gefolgt von 4 °C.
- Reinigen Sie die doppelsträngige DNA, indem Sie der mit Exonuclease I-behandelten Probe 91,8 L SPRI-Magnetperlen zu51 l hinzufügen. Wiederholen Sie die in 2.6.1–2.6.6 beschriebenen Löschschritte. und elute mit 42 l Wasser vorgewärmt auf 37 °C wie beschrieben.
- Konzentrieren Sie die Probe mit dem Vakuumkonzentrator auf 15 l, wie in Schritt 9.1 beschrieben.
15. Qualitäts- und Mengenkontrolle
- Verwenden Sie 1 L der konzentrierten Proben und führen Sie einen hochempfindlichen DNA-Chip auf einem DNA-Qualitätsanalysator aus. Das erwartete Profil/die erwartete Menge ist in Abbildung 3dargestellt.
16. Erste Runde der Deniedergangsrunde des Trägers
- Bereiten Sie den Abbaumix vor, indem Sie 2 l Wasser, 2 l 10x Restriktionsenzympuffer, 1 l I-SceI und 1 l I-CeuI kombinieren.
- Fügen Sie 6 l des Abbausmixes auf 14 l der konzentrierten Probe hinzu und mischen Sie sie durch Pipettieren. Bei 37 °C für 3 h inkubieren, gefolgt von 20 min Deaktivierung bei 65 °C und halten bei 4 °C.
- Reinigen Sie den Abbaumix mit SPRI-Magnetperlen. Fügen Sie 5 l Wasser hinzu, um das Volumen des Abbaumixes zu erhöhen, und fügen Sie 45 L SPRI-Perlen (1,8:1 Perlen zum Probenverhältnis) hinzu. Wiederholen Sie die Reinigung, wie in den Schritten 2.6.1–2.6.6 beschrieben. und mit 42 l Wasser vorgewärmt auf 37 °C.
- Konzentrieren Sie die eluierte Probe von 42 l auf 20 l des Gesamtvolumens, wie in Schritt 9.1 beschrieben.
17. Kontrolle des Abbauniveaus und Bestimmung der Anzahl der PCR-Amplifikationszyklen
- Bereiten Sie den qPCR-Mix zur Verstärkung ganzer Bibliotheken vor (Adaptor-Mix). Kombinieren Sie 3,8 l Wasser, 5 l qPCR-Vormischung (2x), 0,1 l mit 10 M-Adapter_f1-Primer (5'-AATGATACGGCGACCACCGA-3') und 0,1 l von 10 M-Adapter-R1-Primer (5'-CAAGCAGAGAAGACACACACGA-3') für jede Probe (siehe Tabelle der Materialien für den empfohlenen qPCR-Vormix).
- Kombinieren Sie 9 L qPCR-Adapter-Mix mit 1 l Probe aus Schritt 16.4 und mischen Sie gut durch Pipettieren.
- Bereiten Sie den qPCR-Mix zur Verstärkung der dna aus dem Träger (Trägermix) vor. Kombinieren Sie 3,8 l Wasser, 5 l qPCR-Vormischung (2x), 0,1 l mit 10 M Träger-F1-Primer (5'-GCGGCAGCGcGTTCGCTATAAC-3') und 0,1 l mit 10 M Adapter_r1-Primer für jede Probe
- Kombinieren Sie 9 l qPCR Trägermischung mit 1 l der Probe aus Schritt 16.4 und mischen Sie gut durch Pipettiern.
- Set qPCR-Programm: 95 °C für 3 min (95 °C für 20 s, 60 °C für 20 s, 72 °C für 2 min) 40x wiederholt, gefolgt von instrumentenspezifischer Denaturierungskurve (65–95 °C) und halten bei 4 °C.
HINWEIS: Bereiten Sie eine negative Kontrolle vor, indem Sie die Probe durch Wasser ersetzen.
18. PCR-Verstärkung der Zielbibliothek
- Bereiten Sie den PCR-Amplifikationsmix vor, indem Sie 6 l Wasser, 0,5 l mit 10 M Adapter_f1-Primer, 0,5 l mit 10 M Adapter_r1-Primer und 25 l PCR-Vormischung (2x) kombinieren. Mischen Sie durch Pipettierung (siehe Tabelle der Materialien für den empfohlenen PCR-Premix).
- Fügen Sie 32 L des PCR-Mixes ab Schritt 16.4 auf 18 l der Probe hinzu. Durch Pipettieren gründlich mischen.
- PcR-Verstärkung einstellen: 95 °C für 3 min, (98 °C für 20 s, 60 °C für 15 s, 72 °C für 2 min) 12-18 Zyklen, 72 °C für 2 min und halten bei 4 °C.
HINWEIS: Die genaue Anzahl der PCR-Zyklen wird durch qPCR-Ergebnisse bestimmt und entspricht dem Ct-Wert, der mit dem Adapterprimermix ermittelt wird (anzahl der PCR-Zyklen ist gleich dem Ct-Wert). - Reinigen Sie die verstärkte Probe, indem Sie der verstärkten Probe 90 L SPRI-Magnetperlen zu 50 l der verstärkten Probe hinzufügen und durch Pipettieren gründlich mischen. Wiederholen Sie die in den Schritten 2.6.1–2.6.6 beschriebenen Reinigungsschritte. und die Probe mit 42 L Wasser wie beschrieben zu löschen.
19. Zweite Runde der Deniedergangsrunde des Trägers
- Wiederholen Sie die Schritte 16.1–16.3.
- Reinigen Sie den Abbaumix mit SPRI-Magnetperlen. Fügen Sie der Probe 10 l Wasser hinzu, um das Volumen zu erhöhen, und mischen Sie sie mit 30 L SPRI-Perlen (1:1 Perlen zum Probenverhältnis). Wiederholen Sie die Reinigung, wie in den Schritten 2.6.1–2.6.6 beschrieben. und elute mit 42 l Wasser vorgewärmt bei 37 °C wie beschrieben.
- Konzentrieren Sie die eluierte Probe von 42 l auf 30 l des Gesamtvolumens.
20. Bibliotheksgrößenauswahl
- Mischen Sie 24 L SPRI-Magnetperlen mit 30 l der Probe ab Schritt 19.3. (0,8:1 Perlen zum Stichprobenverhältnis). Wiederholen Sie die Inden, wie in den Schritten 2.6.1–2.6.6 beschriebenen Reinigungsschritte. und die Probe wie beschrieben in 42 l Wasser zu löschen.
- Konzentrieren Sie die Probe auf ca. 14 l, wie in Schritt 9.1 beschrieben.
21. Qualitätskontrolle
-
Bewertung der Größenverteilung
- Führen Sie 1 l der Probe auf dem hochempfindlichen DNA-Chip aus. Die erwarteten Ergebnisse sind in Abbildung 4dargestellt.
HINWEIS: Wenn Fragmente mit weniger als 200 bp sichtbar sind (siehe Beispiel in Abbildung 4A, C), sollte die Größenauswahl (Schritte 20.1–20.2) wiederholt werden, bis die kurzen Fragmente entfernt werden ( Abbildung4B,D). In der Regel reicht eine zusätzliche Runde der Größenauswahl aus. Wenn die Anzahl der kurzen Fragmente stark ist (wie in Abbildung 4C), sollte das Verhältnis von Perlen zu Stichproben auf 0,6:1 verringert werden.
- Führen Sie 1 l der Probe auf dem hochempfindlichen DNA-Chip aus. Die erwarteten Ergebnisse sind in Abbildung 4dargestellt.
-
Qualitätskontrolle der Carrier-Degradation
- Wiederholen Sie die Schritte 17.1–17.5.
HINWEIS: Abhängig von der Konzentration der im HS-DNA-Chiplauf geschätzten Bibliotheken (Regionsanalyse) müssen die Proben vor qPCR verdünnt werden. Verwenden Sie 0,5 l der Probe, um Probenverlust zu vermeiden und 100–500x in Wasser zu verdünnen (auf 1–20 pg/L Endkonzentration verdünnen). Die erwartete Differenz zwischen den Ct-Werten, die mit Adapter- und Trägermischung ermittelt werden, beträgt 5–10.
- Wiederholen Sie die Schritte 17.1–17.5.
-
Bibliotheksquantifizierung
- Bereiten Sie die Arbeitsverdünnung des Lambda-DNA-Standards vor, indem Sie 20 l von 100 mg/ml Lambda-DNA-Standard mit 980 l 1x TE mischen (Vorbereitung durch Verdünnung von 20x TE im DNA-Quantifizierungskit). Die Verdünnung der Lambda-DNA kann bei -20 °C gespeichert werden.
- Bereiten Sie die Lambda DNA Standard serielle Verdünnungen durch Mischen der verdünnten Lambda-Standard und 1x TE nach Ergänzenden Tabelle 8.
HINWEIS: Für eine höhere Genauigkeit wird empfohlen, allen Rohren 100 L 1x TE-Puffer hinzuzufügen und das 1x TE-Volumen je nach Volumen des zu zu zu zu zu setzenden verdünnten Lambda zu entfernen. Verwenden Sie nicht mehr als 1 L der Bibliothek; Für diese Messung werden 384 Wellplatten empfohlen.
Ergebnisse
Dieser Bericht beschreibt das vollständige SLIC-CAGE-Protokoll zum Abrufen sequenzieller Bibliotheken aus Nanogrammen des beginnenden gesamten RNA-Materials (Abbildung 1). Um den synthetischen RNA-Trägermix zu erhalten, müssen zunächst PCR-Trägervorlagen vorbereitet und gelgereinigt werden, um PCR-Seitige Produkte zu eliminieren (Abbildung 2A). Jede PCR-Vorlage (insgesamt zehn) wird mit einem gemeinsamen Vorwärts-, aber einem anderen Reverse-Primer (Tabelle2) erstellt, was zu unterschiedlichen Längen der PCR-Vorlage führt, um größenvariabilität synthetischer RNA-Träger zu ermöglichen. Nach der Reinigung werden PCR-Vorlagen zur In-vitro-Transkription der Trägermoleküle verwendet. Ein einzelnes RNA-Trägerprodukt wird erwartet, wenn die Schablonen gelgereinigt sind (siehe repräsentative Gelanalyse in Abbildung 2B). Die Vorbereitung des Trägers kann je nach Bedarf und bei Derpräparation, Misch- und Tiefkühlung bei -80 °C für die zukünftige Verwendung hochskaliert werden.
Mit der empfohlenen minimalen Menge an Sample-Gesamt-RNA (10 ng) in Kombination mit 16-18 Zyklen PCR-Amplifikation können hochkomplexe SLIC-CAGE-Bibliotheken erreicht werden. Die Anzahl der PCR-Zyklen, die erforderlich sind, um die endgültige Bibliothek zu verstärken, hängt stark von der Menge der verwendeten gesamten Eingangs-RNA ab (die erwartete Anzahl von Zyklen ist in Tabelle 4dargestellt).
Nach der ersten Abbaurunde beträgt in qPCR-Ergebnissen (Schritt 17) die erwartete Differenz zwischen den Ct-Werten, die mit adaptor_f1 oder carrier_f1 Primer erzielt wurden, 1-2, wobei die Ct-Werte mit adaptor_f1 niedriger als mit carrier_f1 erhalten wurden.
Die Verteilung der Fragmentlängen in der endgültigen Bibliothek liegt zwischen 200-2.000 bp mit einer durchschnittlichen Fragmentgröße von 700-900 bp (basierend auf der Regionsanalyse mit Bioanalyzer-Software, Abbildung 4B,D). Kürzere Fragmente, wie in Abbildung 4A,Cdargestellt, müssen durch zusätzliche Größenausschlussrunden entfernt werden (Schritte 20-21). Diese kurzen Fragmente sind PCR-Verstärkungsartefakte und nicht die Zielbibliothek. Beachten Sie, dass kürzere Fragmente besser auf den Sequenzierungsflusszellen gruppiert werden und Sequenzierungsprobleme verursachen können.
Die erwartete Menge an Bibliotheksmaterial pro Probe liegt zwischen 5-50 ng. Deutlich geringere Beträge deuten auf einen Probenverlust während des Protokolls hin. Wenn die erhaltene geringe Menge für die Sequenzierung ausreicht (2-3 ng der gepoolten Bibliotheken werden benötigt), können die Bibliotheken von geringerer Komplexität sein (siehe unten).
Je nach Sequenzierungsmaschine muss die Menge der auf die Durchflusszelle geladenen Bibliothek möglicherweise optimiert werden. Mit einer Illumina HiSeq 2500 ergibt das Laden von 8-12 pM SLIC-CAGE-Bibliotheken durchschnittlich 150-200 Millionen Lesevorgänge, wobei >80% der Lesevorgänge den Qualitätswert Q30 als Schwellenwert übergeben.
Die erhaltenen Lesevorgänge werden dann dem Referenzgenom zugeordnet [für 50 bp liest, Bowtie212 kann mit Standardparametern verwendet werden, die null Nichtübereinstimmungen pro Seed-Sequenz (22 bp)] zulassen. Die erwarteten Mapping-Effizienzen hängen von der gesamten RNA-Eingangsmenge ab und sind in Tabelle 5dargestellt. Die eindeutig abgebildeten Lesevorgänge können dann in die grafische und statistische Computerumgebung13 geladen und mit CAGEr (Bioconductor-Paket14) verarbeitet werden. Die Paketvignette ist einfach zu befolgen und erklärt den Workflow und die Verarbeitung der zugeordneten Daten im Detail. Eine einfache visuelle Kontrolle der Bibliothekskomplexität ist die Verteilung der Promotorbreite, da Bibliotheken mit geringer Komplexität künstlich enge Promotoren haben werden (Abbildung5A, SLIC-CAGE-Bibliothek abgeleitet aus 1 ng gesamtRNA, für Details siehe vorherige Veröffentlichung10). Doch selbst die sLIC-CAGE-Bibliotheken mit geringer Komplexität ermöglichen die Identifizierung echter CTSS, mit einer höheren Genauigkeit als alternative Methoden für TSS-Mapping mit niedrigem und mittlerem Input (Abbildung 5B, C).

Abbildung 1: Schritte im SLIC-CAGE-Protokoll. Die Proben-RNA wird mit dem RNA-Trägermix gemischt, um insgesamt 5 g RNA-Material zu erreichen. cDNA wird durch Reverse-Transkription synthetisiert und die Kappe wird mit Natriumperiodat oxidiert. Oxidation ermöglicht die Befestigung von Biotin an der Kappe mit Biotinhydrazid. Biotin wird am 3-Zoll-Ende der mRNA befestigt, da es auch mit Natriumperiodat oxidiert wird. Um Biotin aus mRNA:cDNA-Hybriden mit unvollständig synthetisierter cDNA und von den 3-Zoll-Enden der mRNA zu eliminieren, werden die Proben mit RNase I behandelt. cDNA, die das 5-Zoll-Ende der mRNA erreicht hat, wird dann durch Affinitätsreinigung auf Streptavidin-Magnetperlen ausgewählt ( Cap-Trapping). Nach der Freisetzung von cDNA werden 5-Linker und 3-Linker ligiert. Die Bibliotheksmoleküle, die aus dem Träger stammen, werden mit I-SceI und I-CeuI Homing Endonukleasen abgebaut und die Fragmente werden mit SPRI-Magnetperlen entfernt. Die Bibliothek wird dann PCR verstärkt. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
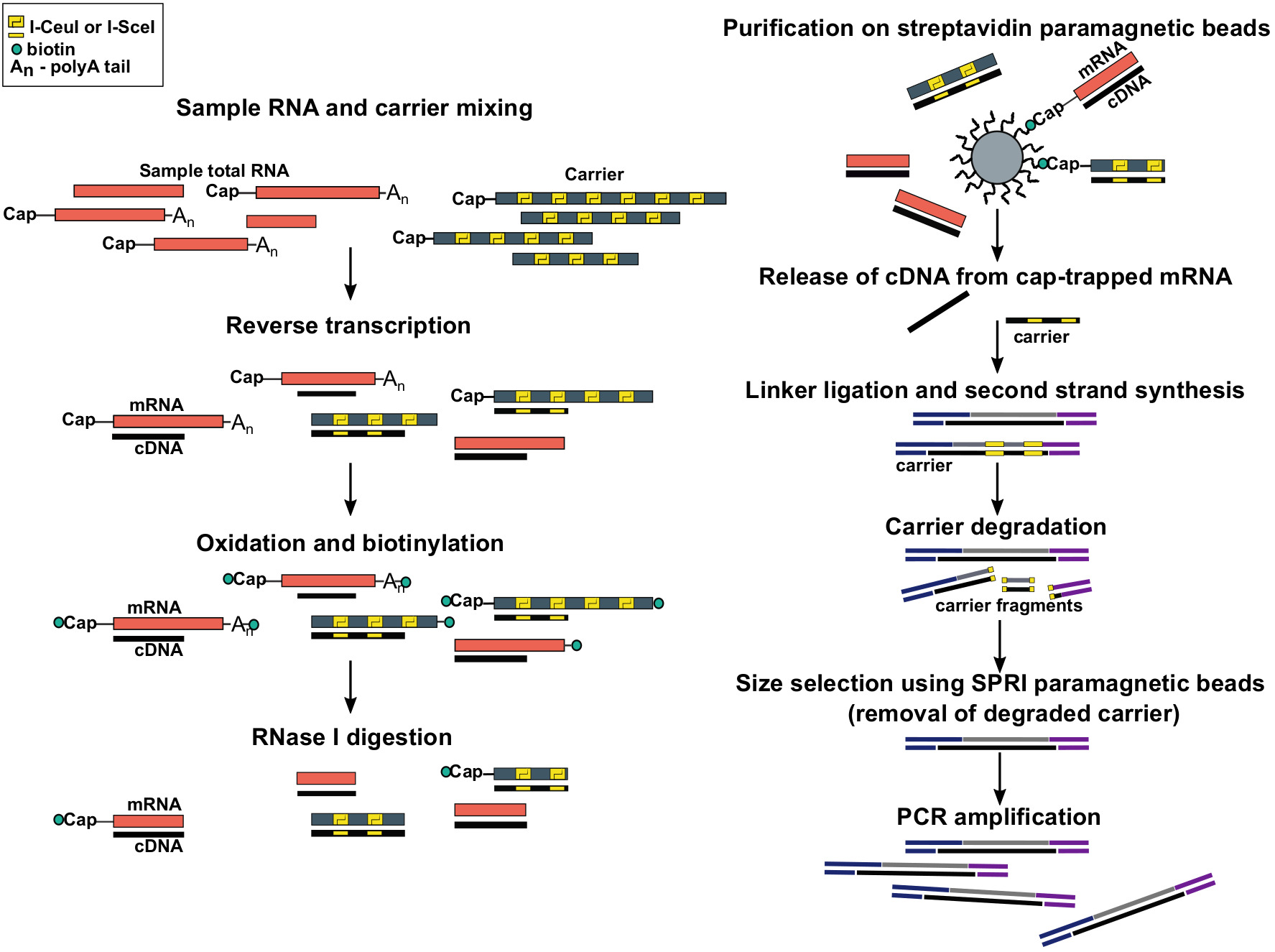{kind=link}

Abbildung 2: Repräsentative Gelanalyse von Träger-PCR-Vorlagen und Träger-In-vitro-Transkripten. (A) Carrier PCR-Vorlagen vor der Gelreinigung: Der erste Brunnen enthält den 1 kbp Marker, gefolgt von Carrier PCR-Vorlagen 1, 1-10. (B) Carrier In-vitro-Transkripte: Der erste Brunnen enthält den 1 kbp-Marker, gefolgt von Trägertranskripten 1-10. Die Trägertranskripte wurden vor dem Beladen für 5 min bei 95 °C erhitzt. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 3: Repräsentative DNA-Qualität (Hochempfindlichkeits-DNA-Chip) Spur von SLIC-CAGE vor der ersten Runde des Trägerabbaus. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 4: Repräsentative DNA-Qualität (Hochempfindlichkeits-DNA-Chip) Spuren von SLIC-CAGE-Bibliotheken nach PCR-Verstärkung. (A) SLIC-CAGE-Bibliothek, die eine zusätzliche Größenauswahl zum Entfernen kurzer Fragmente erfordert. (B) SLIC-CAGE-Bibliothek nach Größenauswahl mit 0,6x SPRI-Perlen zum Stichprobenverhältnis. (C) SLIC-CAGE-Bibliothek mit geringerer Ausgabemenge, die Größenauswahl für die Entfernung von kurzen Fragmenten erfordert. (D) SLIC-CAGE-Bibliothek mit geringerem Ausgangsbetrag nach Größenauswahl mit 0,6:1 SPRI-Perlen zum Stichprobenverhältnis. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
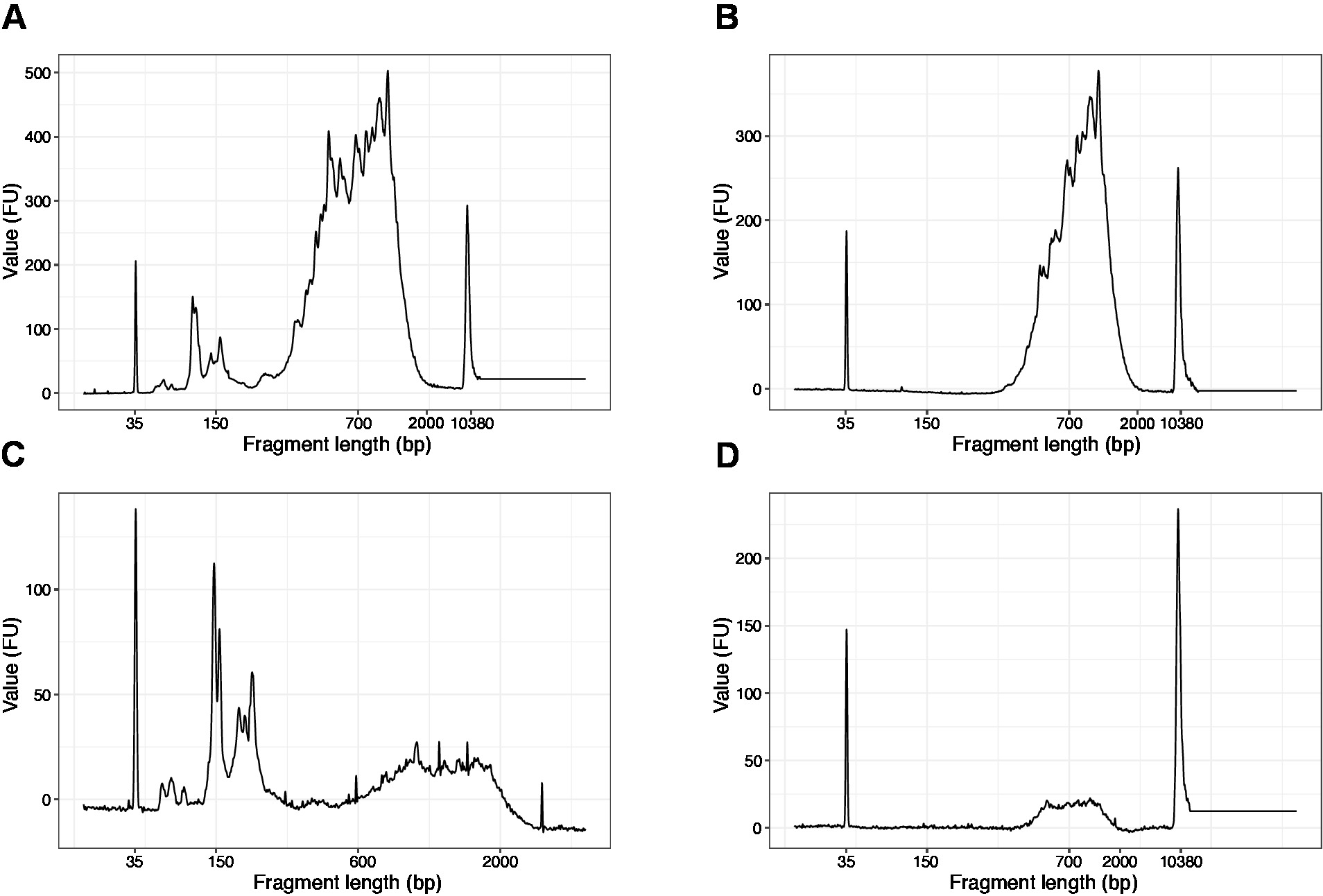{kind=link}

Abbildung 5: Validierung von SLIC-CAGE-Bibliotheken. (A) Verteilung der interquantilen Breiten des Tag-Clusters in SLIC-CAGE-Bibliotheken, die aus 1, 5 oder 10 ng der S. cerevisiae-Gesamt-RNA hergestellt werden, und in der nAnT-iCAGE-Bibliothek, die aus 5 g S. cerevisiae gesamter RNA hergestellt wird. Eine hohe Anzahl von schmalen Tag-Clustern in der 1 ng SLIC-CAGE-Bibliothek weist auf ihre geringe Komplexität hin. (B) ROC-Kurven zur CTSS-Identifikation in S. cerevisiae SLIC-CAGE-Bibliotheken. Alle S. cerevisiae nAnT-iCAGE CTSSs wurden als echtes Set verwendet. (C) ROC-Kurven zur CTSS-Identifikation in S. cerevisiae nanoCAGE-Bibliotheken. Alle S. cerevisiae nAnT-iCAGE CTSSs wurden als echtes Set verwendet. Der Vergleich von ROC-Kurven zeigt, dass SLIC-CAGE bei der CTSS-Identifikation deutlich übertrifft. Es wurden Daten von ArrayExpress E-MTAB-6519 verwendet. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
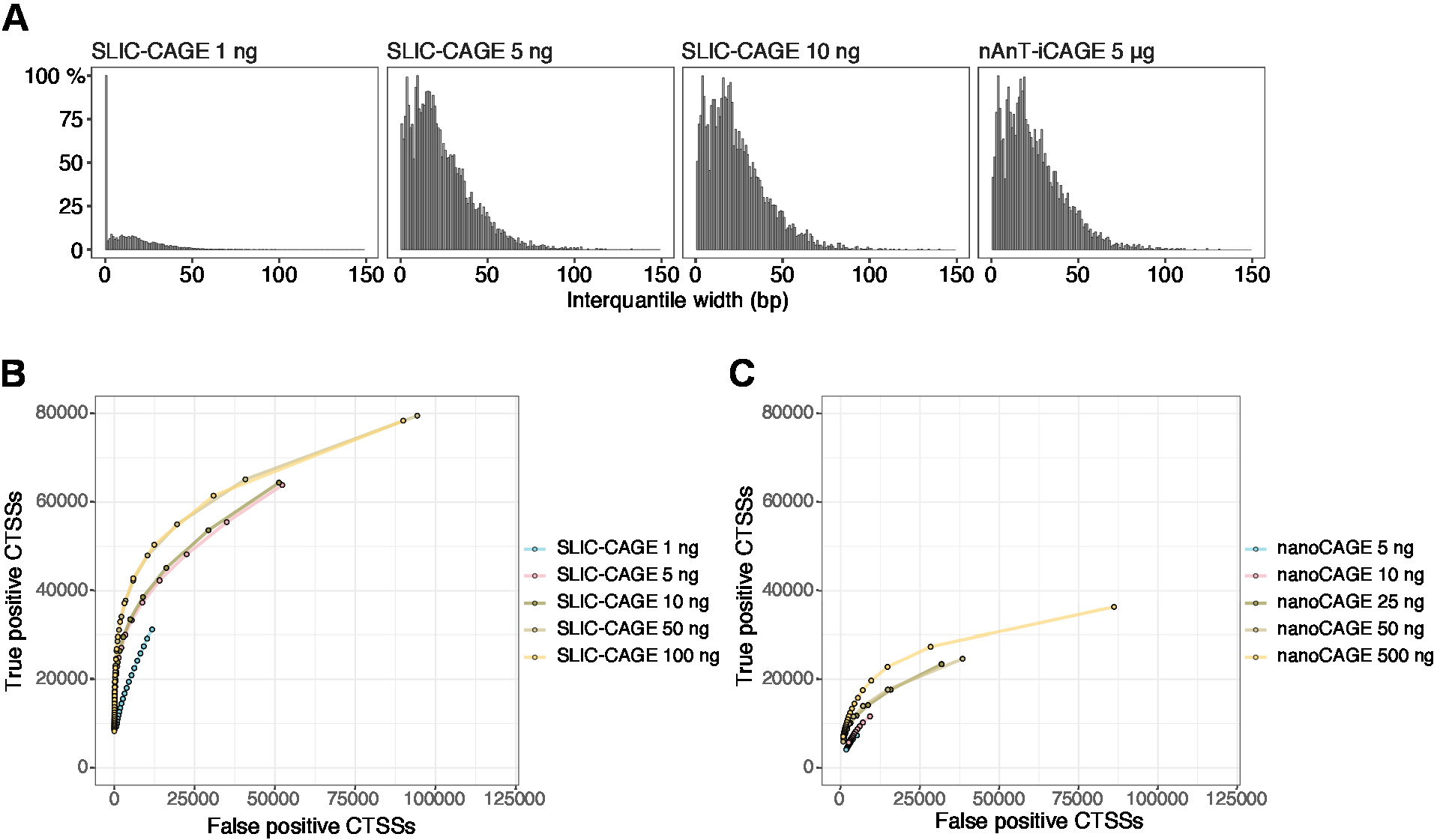{kind=link}
Tabelle 1: Sequenz des synthetischen Trägergens. I-SceI-Sites sind fett und kursiv in Violett, und I-CeuI-Erkennungsseiten sind grün. Bitte klicken Sie hier, um diese Datei herunterzuladen.
| spediteur | Reverse Primer 5'-3' | PCR Produktlänge / bp | |
| 1 | PCR_N6_r1: NNNNNNCTACGTGTCGCAGACGAATT | 1034 | |
| 2 | PCR_N6_r2: NNNNNNTATCCAGATCGTTGAGCTGC | 966 | |
| 3 | PCR_N6_r3: NNNNNNCACTGCGGGATCTCTTTACG | 889 | |
| 4 | PCR_N6_r4: NNNNNNGCCGTCGATAACTTGTTCGT | 821 | |
| 5 | PCR_N6_r5: NNNNNNAGTTGACCGCAGAAGTCTTC | 744 | |
| 6 | PCR_N6_r6: NNNNNNGTGAAGAATTTCTGTTCCCA | 676 | |
| 7 | PCR_N6_r7: NNNNNNCTCGCGGCTCCAGTCATAAC | 599 | |
| 8 | PCR_N6_r8: NNNNNNTATACGATGTTGTCGTAC | 531 | |
| 9 | PCR_N6_r9: NNNNNNCGCCGCGCCCCCGCAGG | 454 | |
| 10 | PCR_N6_r10: NNNNNNCAGGACGTTTTGCCCAGCA | 386 | |
| * Vorwärts-Primer ist für alle Trägervorlagen gleich. Unterstrichen ist die T7-Promoter-Sequenz. PCR_GN5_f1: TAATACGACTCACTATAGNNNNNCAGCGTTCGCTA | |||
Tabelle 2: Primer für Trägervorlagenverstärkung. Vorwärts-Primer ist für alle Carrier-Vorlagen gleich. Unterstrichen ist die T7-Promoter-Sequenz. PCR_GN5_f1: TAATACGACTCACTATAGNNNNNCAGCGTTCGCTA. Mit unterschiedlichen Reverse Primern werden PCR-Vorlagen und damit Träger-RNAs unterschiedlicher Länge hergestellt.
| spediteur | länge | ungekapert/g | gekappt/g |
| 1 | 1034 | 3,96 | 0,45 |
| 2 | 966 | 8,36 | 0,95 |
| 3 | 889 | 4.4 | 0,5 |
| 4 | 821 | 6.6 | 0,75 |
| 5 | 744 | 4.4 | 0,5 |
| 6 | 676 | 3,08 | 0,35 |
| 7 | 599 | 4.4 | 0,5 |
| 8 | 531 | 3,96 | 0,45 |
| 9 | 454 | 2,64 | 0,3 |
| 10 | 386 | 2.2 | 0,25 |
Tabelle 3: RNA-Trägermix. Insgesamt 49 g des Trägersmix 0,3-1 kbp: ungekapselt = 44 g, gedeckelt = 5 g.
| RNA-Eingang insgesamt /ng | PCR-Zyklen |
| 1 ng | 18 |
| 2 ng | 17 |
| 5 ng | 16 |
| 10 ng | 15-16 |
| 25 ng | 14-15 |
| 50 ng | 13-15 |
| 100 ng | 12-14 |
Tabelle 4: Erwartete Anzahl von PCR-Zyklen in Abhängigkeit von der gesamten RNA-Eingabe der Probe. Ungefähre Anzahl der Zyklen basiert auf Experimenten, die mit Saccharomyces cerevisiae, Drosophila melanogaster, und Mus musculus total RNA durchgeführt.
| RNA-Eingang insgesamt/ng | % insgesamt kartiert | % eindeutig kartiert | % Träger |
| 1 ng | 30 | 20-30 | 30 |
| 2 ng | 60 | 20-50 | 10 |
| 5 ng | 60-70 | 40-60 | 5-10 |
| 10 ng | 60-70 | 40-60 | 5-10 |
| 25 ng | 65-80 | 40-70 | 0-5 |
| 50 ng | 65-80 | 40-70 | 0-3 |
| 100 ng | 70-85 | 40-70 | 0-2 |
Tabelle 5: Erwartete Mapping-Effizienz und Abhängigkeit von der gesamten RNA-Eingangsmenge. Ungefähre Zahlen werden dargestellt und basieren auf Experimenten, die mit Saccharomyces cerevisiae und Mus musculus total RNA durchgeführt wurden.
Diskussion
Für erfolgreiche SLIC-CAGE-Bibliothekspräparate ist es wichtig, niedrigverbindliche Spitzen und Tuben zu verwenden, um Probenverluste durch Probenadsorption zu verhindern. In allen Schritten, die das Abrufen des Überstandes beinhalten, wird empfohlen, das gesamte Sample-Volume wiederherzustellen. Da das Protokoll mehrere Schritte hat, führt kontinuierlicher Beispielverlust zu erfolglosen Bibliotheken.
Wenn CAGE (nAnT-iCAGE) nicht routinemäßig durchgeführt wurde, ist es am besten, SLIC-CAGE mit unterschiedlichen Eingangsmengen (10 ng, 20 ng, 50 ng, 100 ng, 200 ng) der gleichen Gesamt-RNA-Probe zu testen und mit nAnT-iCAGE-Bibliotheken zu vergleichen, die mit 5 g Gesamt-RNA erstellt werden. Wenn die nAnT-iCAGE-Bibliothek nicht erfolgreich ist (weniger als 0,5-1 ng der DNA-Bibliothek pro Probe), ist es unwahrscheinlich, dass SLIC-CAGE funktioniert, und der Probenverlust muss minimiert werden.
Ein entscheidender Schritt, um qualitativ hochwertige Bibliotheken ohne ungekapselte, degradierte RNA oder rRNA zu gewährleisten, ist das in Abschnitt 7 beschriebene Cap-Trapping. Es ist sehr wichtig, dass die Streptavidinperlen gründlich in Waschpuffern resuspendiert werden und dass die Waschpuffer vor dem nächsten Waschschritt oder der Elution von cDNA entfernt werden.
Wenn die Ergebnisse der qPCR nach der ersten Runde der Trägerdegradation keinen Unterschied zwischen der Verwendung von adaptor_f1 und carrier_f1 Primern aufweisen, wird die Fortsetzung des Protokolls weiterhin empfohlen. Wenn nach der zweiten Runde des Trägerabbaus der Unterschied in den Ct-Werten weniger als fünf beträgt, wird eine dritte Runde der Trägerdegradation empfohlen. Wir haben nie eine dritte Abbaurunde für notwendig befunden, und wenn sie eintritt, wird empfohlen, die homing endonuklease-Bestände zu ersetzen.
Zusätzliche Runden der PCR-Verstärkung können dem Protokoll hinzugefügt werden, wenn der endgültige Betrag der erhaltenen Bibliothek nicht für die Sequenzierung ausreicht. Die PCR-Verstärkung kann dann mit einer minimalen Anzahl von Amplifikationszyklen eingestellt werden, die benötigt werden, um genügend Material für die Sequenzierung zu liefern, unter Berücksichtigung von Probenverlusten, die bei der Größenauswahl nicht vermieden werden können. Die Reinigung oder Größenauswahl mit SPRI-Magnetperlen sollte dann durchgeführt werden, bis alle kleinen (<200 bp) Fragmente entfernt werden (falls erforderlich, 0,6:1 Perlen zum Stichprobenverhältnis verwenden), und die Bibliothek sollte mit Picogreen quantifiziert werden.
Bibliotheken können im Single-End- oder Paired-End-Modus sequenziert werden. Mithilfe der paired-end-Sequenzierung können Informationen über Transkription-Isoformen abgerufen werden. Darüber hinaus können Informationen aus dem sequenzierten 3'-Ende als eindeutige molekulare Identifikatoren (UMI) verwendet werden, um PCR-Duplikate zu reduzieren ( TCT-N6, N6 ist ein zufälliger Hexamer). Da eine moderate Anzahl von PCR-Verstärkungszyklen verwendet wird (bis zu 18), wurde die Verwendung von UMIs bisher als unnötig befunden.
Da der Kern des Protokolls auf nAnT-iCAGE11basiert, verwendet SLIC-CAGE acht Barcodes. Daher wird das Multiplexing von mehr als acht Samples derzeit nicht unterstützt. Darüber hinaus eignen sich sowohl SLIC-CAGE als auch nAnT-iCAGE nicht für die Erfassung von RNAs kleiner als 200 bp, da die Protokolle entwickelt wurden, um Linker und PCR-Artefakte durch Größenausschluss mit AMPure XP Perlen zu entfernen.
SLIC-CAGE ist die einzige unvoreingenommene Methode zur Auflösung der Single-Nukleotid-Auflösung mit niedrigem Eindräuen zur Kartierung von Transkriptionsinittierungsstartstellen mit Nanogrammen von rna-Gesamtmaterial. Alternative Methoden basieren auf der Vorlagen-Switching-Aktivität der Reverse-Transkriptase zu Barcode-capped RNA anstelle von Cap-Trapping (z.B. NanoCAGE15 und NanoPARE16). Aufgrund des Template-Switchings weisen diese Methoden sequenzspezifische Verzerrungen bei der TSS-Erkennung auf, was zu einer erhöhten Anzahl falsch positiver TSS und einer verringerten Anzahl echter TSSs9,10führt.
Offenlegungen
Ein Patent für abbaubare Träger-RNA/DNA wurde abgefüllt.
Danksagungen
Diese Arbeit wurde durch das Wellcome Trust-Stipendium (106954) unterstützt, das B. L. und Medical Research Council (MRC) Core Funding (MC-A652-5QA10) gewährt wurde. N. C. wurde von EMBO Long-Term Fellowship (EMBO ALTF 1279-2016) unterstützt; E. P. wurde vom Medical Research Council UK unterstützt; B. L. wurde vom Medical Research Council UK unterstützt (MC UP 1102/1).
Materialien
| Name | Company | Catalog Number | Comments |
| 2-propanol, Bioultra, for molecular biology, ≥99.5% | Sigma-Aldrich | 59304-100ML-F | Used in RNAclean XP purification. |
| 3' linkers | Sequences are described in Murata et al. 2014 and Supplementary Table 1 of this manuscript. Annealing of strands to produce 3'linkers is described in the supplementary of this protocol. | ||
| 5' linkers | Sequences are described in Murata et al. 2014 and Supplementary Table 1 of this manuscript. Annealing of strands to produce 5'linkers is described in the supplementary of this protocol. | ||
| Agencourt AMPure XP, 60 mL | Beckman Coulter | A63881 | Purification of DNA |
| Agencourt RNAClean XP Kit | Beckman Coulter | A63987 | Purification of RNA and RNA:cDNA hybrids in CAGE steps. |
| Axygen 0.2 mL Polypropylene PCR Tube Strips and Domed Cap Strips | Axygen (available through Corning) | PCR-0208-CP-C | Or any 8-tube PCR strips (used only for water and mixes). |
| Axygen 1 x 8 strip domed PCR caps | Axygen (available through Corning) | PCR-02CP-C | Caps for PCR plates. |
| Axygen 1.5 mL Maxymum Recovery Snaplock Microcentrifuge Tube | Axygen (available through Corning) | MCT-150-L-C | Low-binding 1.5 mL tubes, used for enzyme mixes or sample concentration. |
| Axygen 96 well no skirt PCR microplate | Axygen (available through Corning) | PCR-96-C | Low-binding PCR plates - have to be used for all steps in the protocol. Note that plates should be cut to contain 2 x 8 wells for easier visibility of the samples |
| Bioanalyzer (or Tapestation): RNA nano and HS DNA kits | Agilent | To determine quality of RNA, efficient size selection and final quality of the library (Tapestation can also be used) | |
| Biotin (Long Arm) Hydrazide | Vector laboratories | SP-1100 | Biotinylation/tagging |
| Cutsmart buffer | NEB | Restriction enzyme buffer | |
| Deep Vent (exo-) DNA Polymerase | NEB | M0259S | Second strand synthesis |
| DNA Ligation Kit, Mighty Mix | Takara | 6023 | Used for 5' and 3'-linker ligation |
| dNTP mix (10 mM each) | ThermoFisher Scientific | 18427013 | dNTP mix for production of carrier templates (or any dNTPs suitable for PCR) |
| Dynabeads M-270 Streptavidin | Invitrogen | 65305 | Cap-trapping. Do not use other beads as these are optimised with the buffers used. |
| DynaMag-2 Magnet | ThermoFisher Scientific | 12321D | Magnetic stand for 1.5 mL tubes - used to prepare Streptavidin beads. |
| DynaMag-96 Side Skirted Magnet | ThermoFisher Scientific | 12027 | Magnetic stand for PCR plates (96 well-plates) - used with cut plates to contain 2 x 8 wells. |
| Ethanol, BioUltra, for molecular biology, ≥99.8% | Sigma-Aldrich | 51976-500ML-F | Used in AMPure washes. Any molecular biology suitable ethanol can be used. |
| Exonuclease I (E. coli) | NEB | M0293S | Leftover primer degradation |
| Gel Loading Dye, Purple (6x), no SDS | NEB | B7025S | agarose gel loading dye |
| HiScribe T7 High Yield RNA Synthesis Kit | New England Biolabs | E2040S | Kit for carrier in vitro transcription |
| Horizontal electrophoresis apparatus | purification of carrier DNA templates from agarose gels | ||
| I-Ceu | NEB | R0699S | Homing endonuclease used for carrier degradation. |
| I-SceI | NEB | R0694S | Homing endonuclease used for carrier degradation. |
| KAPA HiFi HS ReadyMix (2x) | Kapa Biosystems (Supplied by Roche) | KK2601 | PCR mix for target library amplification |
| KAPA SYBR FAST qPCR kit (Universal) 2x | Kapa Biosystems (Supplied by Roche) | KK4600 | qPCR mix to assess degradation efficiency and requiered number of PCR amplification cycles |
| Micropipettes and multichannel micropipettes (0.1-10 µL, 1-20 µL, 20-200 µL) | Gilson | Use of Gilson with the low-binding Sorenson tips is recommended. Other micropippetes might not be compatible. Different brand low-binding tips may not be of equal quality and may increase sample loss. | |
| Microplate reader | For Picogreen concentration measurement of the final library. Microplates are used to allow small volume measurement and reduce sample waste. | ||
| nuclease free water | ThermoFisher Scientific | AM9937 | Or any nuclease (DNase and RNase) free water |
| PCR thermal cycler | incubation steps and PCR amplficication | ||
| Phusion High-Fidelity DNA Polymerase | ThermoFisher Scientific | F530S | DNA polymerase for amplification of carrier templates (or any high fidelity polymerase) |
| QIAquick Gel Extraction Kit (50) | Qiagen | 28704 | Purification of carrier PCR templates from agarose gels. |
| qPCR machine | determining PCR amplification cyle number and degree of carrier degradation | ||
| Quant-iT PicoGreen dsDNA Reagent | ThermoFisher Scientific | P11495 | Used to measure final library concentration - recommended as, in our hands, it is more accurate and reproducible than Qubit. |
| Quick-Load Purple 100 bp DNA Ladder | NEB | N0551S | DNA ladder |
| Quick-Load Purple 1 kb Plus DNA Ladder | NEB | N0550S | DNA ladder |
| Ribonuclease H | Takara | 2150A | Digestion of RNA after cap-trapping. |
| RNase ONE Ribonuclease | Promega | M4261 | Degradation of single stranded RNA not protected by cDNA. |
| RNase-Free DNase Set | Qiagen | 79254 | Removal of carrier DNA templates after in vitro transcription. |
| RNeasy Mini Kit | Qiagen | 74104 | For cleanup of carrier RNA from in vitro transcription or capping |
| Sodium acetate, 1 M, aq.soln, pH 4.5 RNAse free | VWR | AAJ63669-AK | Or any nuclease (DNase and RNase) free solution |
| Sodium acetate, 1 M, aq.soln, pH 6.0 RNAse free | Or any nuclease (DNase and RNase) free solution | ||
| Sodium periodate | Sigma-Aldrich | 311448-100G | Oxidation of vicinal diols |
| Sorenson low binding aerosol barrier tips, MicroReach Guard, volume range 10 μL, Graduated | Sorenson (available through SIGMA-ALDRICH) | Z719390-960EA | Low-binding tips - recommended use throughout the protocol to minimise sample loss. |
| Sorenson low binding aerosol barrier tips, MultiGuard, volume range 1,000 μL , Graduated | Sorenson (available through SIGMA-ALDRICH) | Z719463-1000EA | Low-binding tips - recommended use throughout the protocol to minimise sample loss. |
| Sorenson low binding aerosol barrier tips, MultiGuard, volume range 20 μL , Graduated | Sorenson (available through SIGMA-ALDRICH) | Z719412-960EA | Low-binding tips - recommended use throughout the protocol to minimise sample loss. |
| Sorenson low binding aerosol barrier tips, MultiGuard, volume range 200 μL , Graduated | Sorenson (available through SIGMA-ALDRICH) | Z719447-960EA | Low-binding tips - recommended use throughout the protocol to minimise sample loss. |
| SpeedVac Vacuum Concentrator | concentrating samples in various steps to lower volume | ||
| SuperScript III Reverse Transcriptase | ThermoFisher Scientific | 18080044 | Used for reverse transcription (1st CAGE step) |
| Trehalose/sorbitol solution | Preparation is described in Murata et al. 2014. | ||
| Tris-HCl, 1 M aq.soln, pH 8.5 | 1 M solution, DNase and RNase free | ||
| tRNA (20 mg/mL) | tRNA solution. Preparation is described in Murata et al. 2014. | ||
| UltraPure Low Melting Point Agarose | ThermoFisher Scientific | 16520050 | Or any suitable pure low-melt agarose. |
| USB Shrimp Alkaline Phosphatase (SAP) | Applied Biosystems (Provided by ThermoFisher Scientific) | 78390500UN | |
| USER Enzyme | NEB | M5505S | Degradation of 3'linker's upper strand, Uracil Specific Excision Reagent/Enzyme |
| Vaccinia Capping System | NEB | M2080S | Enzymatic kit for in vitro capping of carrier molecules |
| Wash buffer A | Cap trapping washes. Preparation is described in Murata et al. 2014. | ||
| Wash buffer B | Cap trapping washes. Preparation is described in Murata et al. 2014. | ||
| Wash buffer C | Cap trapping washes. Preparation is described in Murata et al. 2014. |
Referenzen
- Shiraki, T., et al. Cap analysis gene expression for high-throughput analysis of transcriptional starting point and identification of promoter usage. Proceedings of the National Academy of Sciences of the United States of America. 100 (26), 15776-15781 (2003).
- Haberle, V., Lenhard, B. Promoter architectures and developmental gene regulation. Seminars in Cell and Developmental Biology. 57, 11-23 (2016).
- Haberle, V., Stark, A. Eukaryotic core promoters and the functional basis of transcription initiation. Nature Reviews Molecular Cell Biology. 19 (10), 621-637 (2018).
- Andersson, R., et al. An atlas of active enhancers across human cell types and tissues. Nature. 507 (7493), 455-461 (2014).
- Consortium, E. P. An integrated encyclopedia of DNA elements in the human genome. Nature. 489 (7414), 57-74 (2012).
- Celniker, S. E., et al. Unlocking the secrets of the genome. Nature. 459 (7249), 927-930 (2009).
- Consortium, F., et al. A promoter-level mammalian expression atlas. Nature. 507 (7493), 462-470 (2014).
- Boyd, M., et al. Characterization of the enhancer and promoter landscape of inflammatory bowel disease from human colon biopsies. Nature Communications. 9 (1), 1661(2018).
- Adiconis, X., et al. Comprehensive comparative analysis of 5'-end RNA-sequencing methods. Nature Methods. , (2018).
- Cvetesic, N., et al. SLIC-CAGE: high-resolution transcription start site mapping using nanogram-levels of total RNA. Genome Research. 28 (12), 1943-1956 (2018).
- Murata, M., et al. Detecting expressed genes using CAGE. Methods in Molecular Biology. 1164, 67-85 (2014).
- Langmead, B., Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nature Methods. 9, 357(2012).
- R Core Team. A language and environment for statistical computing. , Available from: https://www.R-project.org/ (2017).
- Haberle, V., Forrest, A. R., Hayashizaki, Y., Carninci, P., Lenhard, B. CAGEr: precise TSS data retrieval and high-resolution promoterome mining for integrative analyses. Nucleic Acids Research. 43 (8), e51(2015).
- Poulain, S., et al. NanoCAGE: A Method for the Analysis of Coding and Noncoding 5'-Capped Transcriptomes. Methods in Molecular Biology. 1543, 57-109 (2017).
- Schon, M. A., Kellner, M. J., Plotnikova, A., Hofmann, F., Nodine, M. D. NanoPARE: parallel analysis of RNA 5' ends from low-input RNA. Genome Research. 28 (12), 1931-1942 (2018).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten