
Method Article
Trascrizione Inizio mapping del sito utilizzando vettore-CAGE di input super-basso
In questo articolo
Riepilogo
Cap Analysis of Gene Expression (CAGE) è un metodo per la mappatura quantitativa a livello di genoma di mRNA 5'end per catturare i siti di inizio della trascrizione della polimerasi II dell'RNA a una risoluzione mononucleotide. Questo lavoro descrive un protocollo a basso input (SLIC-CAGE) per la generazione di librerie di alta qualità utilizzando quantità di nanogrammi di RNA totale.
Abstract
L'analisi del cap dell'espressione genica (CAGE) è un metodo utilizzato per il rilevamento della risoluzione mononucleotide dei siti di avvio della trascrizione della polimerasi II (TSS) di RNA. Il rilevamento accurato dei TSS migliora l'identificazione e la scoperta dei promotori di base. Inoltre, gli esaltatori attivi possono essere rilevati attraverso le firme dell'avvio della trascrizione bidirezionale. Di seguito è descritto un protocollo per l'esecuzione di un vettore di input super-basso-CAGE (SLIC-CAGE). Questo adattamento SLIC del protocollo CAGE riduce al minimo le perdite di RNA aumentando artificialmente la quantità di RNA attraverso l'uso di un mix di portatori di RNA trascritto in vitro che viene aggiunto al campione di interesse, consentendo così la preparazione della libreria da nanogrammi-quantità di totale RNA (cioè migliaia di cellule). Il vettore imita la distribuzione della lunghezza del frammento della libreria del DNA prevista, eliminando così i pregiudizi che potrebbero essere causati dall'abbondanza di un vettore omogeneo. Nelle ultime fasi del protocollo, il vettore viene rimosso attraverso la degradazione con endonucleases homing e la libreria di destinazione viene amplificata. La libreria di campioni di destinazione è protetta dalla degradazione, poiché i siti di riconoscimento dell'endonuclealo sono lunghi (tra i 18 e i 27 bp), rendendo molto bassa la probabilità di loro esistenza nei genomi eucarioti. Il risultato finale è una libreria di DNA pronta per il sequenziamento di nuova generazione. Tutti i passaggi del protocollo, fino al sequenziamento, possono essere completati entro 6 giorni. La preparazione del trasportatore richiede una giornata lavorativa completa; tuttavia, può essere preparato in grandi quantità e tenuto congelato a -80 gradi centigradi. Una volta sequenziate, le letture possono essere elaborate per ottenere TSS a risoluzione mononucleotide a livello di genoma può essere utilizzato per la scoperta del promotore o del potenziatore del nucleo, fornendo informazioni sulla regolazione genica. Una volta aggregati ai promotori, i dati possono essere utilizzati anche per la profilatura delle espressioni incentrate su 5'.Once aggregated to promoters, the data can also be used for 5'--centric expression profiling.
Introduzione
L'analisi del cap dell'espressione genica (CAGE) è un metodo utilizzato per la mappatura a livello di genoma a risoluzione mononucleotide dei siti di inizio della trascrizione della polimerasi II (TSS)1. La sua natura quantitativa consente anche una profilazione dell'espressione cintesa da 5'-end. Le regioni che circondano i TSS (circa 40 bp a monte e a valle) sono promotori fondamentali e rappresentano la posizione fisica in cui l'RNA polimerasi II e i fattori di trascrizione generali si legano (rivisti in precedenza2,3). Le informazioni sulle posizioni esatte dei TSS possono essere utilizzate per la scoperta del promotore del core e per il monitoraggio delle dinamiche promotori. Inoltre, poiché gli stimolatori attivi esibiscono firme di trascrizione bidirezionale, i dati CAGE possono essere utilizzati anche per la scoperta e il monitoraggio dell'esaltatore delle dinamiche potenziatori4. La metodologia CAGE è recentemente aumentata in popolarità grazie alla sua ampia applicazione e utilizzo in progetti di ricerca di alto profilo come ENCODE5, modENCODE6e FANTOM progetti7. Inoltre, le informazioni tSS si stanno rivelando importanti per distinguere i tessuti sani e malati, in quanto gli TSS specifici della malattia possono essere utilizzati per scopi diagnostici8.
Anche se sono disponibili diversi metodi per la mappatura TSS (CAGE, RAMPAGE, STRT, nanoCAGE, nanoCAGE-XL, oligo-capping), noi e altri abbiamo recentemente dimostrato che CAGE è il metodo più imparziale per catturare i Veri TSS con il minor numero di falsi positivi9 , 10.Il recente protocollo CAGE, nAnT-iCAGE11, è il protocollo più imparziale per la profilazione TSS, in quanto evita di tagliare i frammenti a tag brevi utilizzando enzimi di restrizione e non utilizza l'amplificazione PCR. Una limitazione del protocollo nAnT-iCAGE è la necessità di una grande quantità di materiale di partenza (ad es., 5 g di RNA totale per ogni campione). Per rispondere a domande specifiche e biologicamente rilevanti, è spesso impossibile ottenere tali elevate quantità di materiale di partenza (ad esempio, per le cellule in ordine FACS o per le fasi embrionali precoci). Infine, se nAnT-iCAGE ha successo, solo 1-2 ng di materiale della libreria del DNA è disponibile da ogni campione, limitando così la profondità di sequenziamento raggiungibile.
Per consentire la profilazione TSS utilizzando solo nanogrammi di RNA totale, abbiamo recentemente sviluppato Super-low Input Carrier-CAGE10 (SLIC-CAGE, Figura 1). SLIC-CAGE richiede solo 10 ng di RNA totale per ottenere librerie ad alta complessità. Il nostro protocollo si basa sul vettore di RNA sintetico attentamente progettato aggiunto all'RNA di interesse per ottenere un totale di 5 g di materiale di RNA. Il vettore sintetico imita la libreria del DNA bersaglio nella distribuzione della lunghezza per evitare potenziali pregiudizi che potrebbero essere causati da molecole omogenee in eccesso. La sequenza del portatore si basa sulla sequenza del gene sintetasi Escherichia coli leucyl-tRNA per due motivi. In primo luogo, qualsiasi resista del vettore nella biblioteca finale, anche se sequenziato, non verrà mappato a un genoma eucariotico. In secondo luogo, poiché E. coli è una specie mesofilica, i suoi geni delle pulizie sono ottimizzati per la gamma di temperatura appropriata per SLIC-CAGE. La sequenza portante è anche integrata con siti di riconoscimento endonucleasi homing per consentire una degrado specifica del DNA derivato dalle molecole di RNA portante. La libreria di destinazione, derivata dal campione, rimane intatta, poiché i siti di riconoscimento endonuclease sono lunghi (I-CeuI - 27 bp; I-SceI 18 pb) e statisticamente improbabile nei genomi eucarioti. Dopo la degradazione specifica del vettore e la rimozione dei frammenti per esclusione di dimensioni, la libreria di destinazione è PCR amplificata e pronta per la sequenza di prossima generazione. A seconda della quantità iniziale di RNA (1-100 ng), si prevede che siano necessari tra 13-18 cicli di amplificazione PCR. La quantità finale di DNA per ogni campione varia tra 5-50 ng, producendo materiale sufficiente per il sequenziamento molto profondo. Quando si utilizza solo 1-2 ng di RNA totale, vero TSS può essere rilevato; tuttavia, si prevede che le librerie siano di minore complessità. Infine, poiché SLIC-CAGE si basa sul protocollo nAnT-iCAGE11, consente il multiplexing fino a otto campioni prima del sequenziamento.
Protocollo
1. Preparazione del vettore
-
Preparazione di modelli di DNA per la trascrizione in vitro
- Preparare il mix PCR per ogni modello di PCR combinando 41 - L di acqua, 20 L di 5x buffer HF, 8 L da 2,5 mM dNTP, 10 l di 10M primer in avanti unici (PCR_GN5_f1, Tabella 2; primer dissolti e diluiti in acqua) , 10 l di 2 ng/L modello di plasmide contenente il gene del vettore sintetico e 1 -L Phusion polymerase. Mescolare il mix PCR pipettando. Un master mix per tutti i 10 modelli può essere preparato in una sola volta (prepararsi per 11 reazioni).
- Aggiungete 90 l della miscela PCR a 10 gradi di ogni primer inverso da 10 M (PCR_N6_r1-r10, tabella2). Mescolare con il pipettaggio.
- La PCR amplifica i modelli utilizzando il seguente programma: 98 gradi centigradi per 60 s, 98 gradi centigradi per 10 s, 50 gradi centigradi per 30 s, 72 gradi centigradi per 30 s) 35 cicli, 72 gradi centigradi per 10 min, tenere a 4 gradi centigradi.
-
Purificazione del gel dei modelli di DNA amplificato da PCR
- Preparare un gel di agarose dell'1% (si raccomanda di fondersi agasina).
- Per diminuire il volume, concentrate le miscele di reazione PCR da 100 a 20 gradi di volume totale utilizzando il concentratore a vuoto a una temperatura medio-bassa (30-40 gradi centigradi).
- Aggiungere 6 l del colorante di carico 6x, mescolare bene e caricare sul gel. Eseguire l'elettroforesi per 30 min in 1x TAE buffer alla tensione appropriata per il serbatoio di elettroforesi usato (5-10 V/cm). In parallelo eseguire una scala di DNA 100 bp o 1.000 bp.
- Utilizzando un bisturi pulito, accisare le fette di gel contenenti il prodotto PCR di destinazione. Evitare l'eccesso di gel di agarose. Purificare i prodotti PCR utilizzando un kit di estrazione gel (secondo le istruzioni del produttore).
NOT: I rapporti A260/A230 di DNA isolato dai gel di agarose sono in genere bassi (0,1–0,3). Il prodotto di destinazione previsto e i prodotti laterali sono illustrati nella Figura 2A. Le rese attese da 100 reazioni PCR l sono di 1,2-3 g. Le reazioni possono essere aumentate per ottenere un rendimento più elevato.
-
Trascrizione in vitro di molecole portatrici
- Trascrivere l'RNA portante in vitro utilizzando la polimerasi T7 RNA secondo le istruzioni del produttore. Impostare 10-20 reazioni l (il kit consigliato è in Table of Materials).
- Purificare l'RNA trascritto in vitro utilizzando un kit di purificazione dell'RNA. Impostare la digestione del DNA nella soluzione utilizzando DNase I seguendo le istruzioni standard del produttore, ed eluire l'RNA in 50 - L di acqua. Per aumentare la resa dell'eluizione, lasciare l'acqua nella colonna per 5 min prima della centrifugazione.
NOT: Fare attenzione a non superare la capacità di rilegatura massima delle colonne (nel kit indicato nella Tabella dei Materiali, la capacità è fino a 100 g). Il rendimento previsto dai modelli PCR da 1 a 10 (da 1 kbp a 200 bp di lunghezza) è di 25-50 g da 10 volte in vitro reazioni di trascrizione. Le reazioni possono essere scalate per ottenere uno stock più grande di molecole portatrici.
-
Tappatura di molecole di RNA traninate in vitro
- Preparare il mix di capping combinando 2 o L di 10x tampone di capping, 1 L di 10 mM GTP, 1 luna di 2 mM SAM (appena diluito) e 1 L di Vaccinoa enzima di tappatura per RNA portante.
- Mescolare fino a 10 g di ciascuna molecola portante in 15 , l' di volume totale e denaturare per 10 min a 65 gradi centigradi. Mettere sul ghiaccio immediatamente per evitare la formazione secondaria della struttura.
- Mescolare l'RNA del supporto denaturato con 5 L del colle di tappatura e incubare per 1 h a 37 gradi centigradi.
- Purificare le molecole di RNA con un kit di purificazione dell'RNA: seguire il protocollo di pulizia del produttore. Elute RNA in 30 litri d'acqua. Per aumentare la resa dell'eluizione, lasciare l'acqua nella colonna per 5 min prima della centrifugazione.
NOT: Misurare la concentrazione utilizzando lo spettrofotometro a microvolume. Previsto rapporto A260/A280 è >2 e A260/A230 è >2. Si noti che per alcuni campioni di RNA A260/A230 può essere compreso tra 1,3-2. La resa prevista quando si utilizzano 10 g di RNA non tappato è di 9-10 g di RNA con tappo.
- Preparare la combinazione del vettore con tappo e senza sbarramenti combinando gli importi descritti nella tabella 3. Mescolare bene facendo scorrere il tubo e misurare la concentrazione utilizzando lo spettrofotometro microvolume.
NOT: Se è necessaria una maggiore concentrazione del portatore per adattarsi alla reazione di trascrizione inversa (vedi sotto), il mix del supporto può essere concentrato utilizzando il concentratore di vuoto a bassa temperatura media (30-35 gradi centigradi) fino a raggiungere la concentrazione finale desiderata. I passaggi da 2 a 14 sono modificati dal protocollo standard nAnT-iCAGE riportato da Murata et al.
2. Trascrizione inversa
- Unire 1 l dell'inn: L dell'inn: Rt (2,5 mM TCT-N6 disciolto in acqua, per la sequenza vedere tabella supplementare 1), 10 ng di RNA totale di interesse e 4.990 ng di mix di portante (tabella 3) in 10 L di volume totale in una piastra PCR a bassa legatura. Mescolare facendo scorrere il tubo.
NOT: Se l'RNA campione è troppo diluito per la trascrizione inversa (vedi sotto), combinarlo con la quantità appropriata del supporto, concentrarsi utilizzando il concentratore di vuoto a 9 l volume totale e aggiungere 1 l dell'introduzione al primer RT. L'aggiunta del conte portante, per raggiungere 5 g di RNA in totale impedisce la perdita del campione. - Riscaldare il mix da passo 2.1 a 65 gradi centigradi per 5 minuti, e mettere immediatamente sul ghiaccio per prevenire la rinaturazione.
-
Preparare la miscela di trascrizione inversa (RT).
- Per ogni campione combinare 6,1 litri di acqua (senza RNase e DNase), 7,6 ll di 5 volte buffer di primo filamento, 1,9 L di 0,1 M DTT, 1 / L di 10 m dNTP, 7,6 - L di miscela trehalose/sorbitolo (vedi ricetta in Murata et al.11) e 3,8 L della trascrizione inversa raccomandata (vedi c 6>Tabella deimateriali). Mescolare bene facendo scorrere il tubo.
- Aggiungere 28 l della miscela RT nel tubo PCR con 10 litri di RNA, portello e primer RT (volume totale 38 . Mescolare bene pipettando.
NOT: Il mix è altamente viscoso a causa di trehalose/sorbitol. Mescolare fino a quando visibilmente omogeneo. - Incubare in un ciclore termico utilizzando il seguente programma: 25 gradi centigradi per 30 s, 50 gradi centigradi per 60 min, e tenere a 4 gradi centigradi.
-
Purificazione degli ibridi cDNA:RNA utilizzando perline magnetiche SPRI
- Aggiungete 68,4 l delle perline SPRI raccomandate senza RNAse e DNase (vedi Tabella deimateriali) a 38 l della miscela RT (perline a rapporto campione 1.8:1). Mescolare bene pipettando e incubare per 5 min a temperatura ambiente (RT).
- Separare le perline su un supporto magnetico per 5 min. Scartare il supernatante e lavare le perline due volte con 200 luna del 70% di etanolo (appena preparato).
NOT: L'etanolo viene aggiunto alle perline senza miscelazione e mentre il tubo è sul supporto magnetico. L'etanolo aggiunto viene immediatamente rimosso. Bisogna fare attenzione a non perdere perline durante i lavamenti in quanto può portare alla perdita del campione. - Mentre il tubo è ancora sul supporto magnetico, rimuovere tutte le tracce di etanolo. Le goccioline di etanolo possono essere rimosse e spinte fuori dal tubo utilizzando una pipetta P10. Non lasciare asciugare le perline.
- Aggiungere 42 l di acqua preriscaldata a 37 gradi centigradi alle perline ed eluire il campione pipetting su e giù 60x.
NOT: Fare attenzione a non causare schiuma da pipettaggio in quanto può causare la perdita di perline (cioècampione legato) nella schiuma. - Incubare a 37 gradi centigradi per 5 min senza il coperchio per consentire l'evaporazione di tracce di quantità di etanolo.
- Separare le perline su un supporto magnetico per 5 min e trasferire il supernatante in una nuova piastra.
NOT: Cerca di recuperare tutto il supernatante per prevenire la perdita di campioni evitando il riporto del tallone. Utilizzare la pipetta P10 per ottenere le ultime goccioline campione.
3. Ossidazione
- Aggiungere nella reazione RT purificata 2 -L (pH 4,5). Mescolare con la pipettatura, aggiungere 2 -L l di 250 mM NaIO4 e mescolare di nuovo.
- Incubare sul ghiaccio per 45 min. Coprire la piastra con un foglio di alluminio per evitare la luce.
- Aggiungere 16 L di Tris-HCl (pH 8,5) nel mix di ossidazione per neutralizzare il pH.
- Purificare gli ibridi cDNA:RNA ossidati utilizzando perline magnetiche SPRI. Aggiungete 108 l di perline SPRI a 60 gradi l del mix di ossidazione (1.8:1 perline a rapporto campione). Ripetere la purificazione come descritto nei passaggi 2.6.1–2.6.6. Condire con 42 gradi di acqua preriscaldata a 37 gradi centigradi.
NOT: Preparare appena 250 mM NaIO4 aggiungendo 18,7 l di acqua per 1 mg di NaIO4. NaIO4 è sensibile alla luce; quindi, mantenere la soluzione in un tubo coperto con lamina di alluminio o in un tubo resistente alla luce.
4. Biotinylation
- Aggiungere 4 -L l di 1 M NaOAc (pH 6.0) nel tubo contenente il campione ossidato purificato e mescolare con la pipistrazione.
- Aggiungere 4 -L di 10 mM di soluzione di biotina, mescolare con il pipettaggio e incubare per 2 h a 23 gradi centigradi in un ciclore termico per evitare la luce.
NOT: Preparare la soluzione di biotina mescolando 50 mg di biotina con 13,5 mL di DMSO. Fare aliquote monouso e congelare a -80 gradi centigradi. - Purificare gli ibridi cDNA:RNA biotinylati utilizzando perline magnetiche SPRI. Aggiungere 12 -L di 2-propanol e mescolare con il pipettaggio. Aggiungere 108 l di perline SPRI (1.8:1 perline a rapporto campione) e ripetere la purificazione come descritto nei passaggi 2.6.1–2.6.6. Conevitate con 42 gradi di acqua preriscaldata a 37 gradi centigradi.
NOT: Il protocollo può essere messo in pausa qui, e i campioni congelati a -80 gradi centigradi.
5. RNase I Ddigestione
- Preparare il mix RNase I mescolando 4,5 ll di 10x buffer RNase I con 0,5 - L di RNase I (10 U / L) per ogni campione. Mescolare con il pipettaggio.
- Aggiungere 5 l'l del mix ad ogni campione purificato (45 - L in totale). Mescolare con pipettaggio e incubare per 30 min a 37 gradi centigradi.
6. Preparazione di perline Streptavidin
- Per ogni campione, mescolare 30 l di liquami di perline di streptavidina con 0,38 luna di 20 mg/mL di tRNA. Incubare sul ghiaccio per 30 min e mescolare ogni 5 min facendo scorrere il tubo.
NOT: Risospendere il lilmo streptavidina ben prima di pipettare facendo scorrere la bottiglia. La soluzione tRNA deve essere preparata secondo Murata et al. - Separare le perline sul supporto magnetico per 2-3 min. Rimuovere il supernatante.
- Lavare le perline risuspendendo in 15 - L di tampone A. Separare le perline sul supporto magnetico per 2–3 min e rimuovere il supernatante. Ripetere il lavaggio e rimuovere il supernatante.
- Risospendere le perline in 105 gradi l del buffer A e aggiungere 0,19 - L di 20 mg/mL di tRNA. Mescolare bene pipettando.
NOT: Le perline devono essere preparate fresche prima dell'uso. Iniziare la preparazione delle perline durante la digestione di RNase I. Per più campioni preparare le perline insieme in un unico tubo.
7. Cap-inpping
-
Associazione di esempio
- Aggiungete 105 l di perline di streptavidina preparate a 45 -L del campione trattato con RNase I. Mescolare bene con il pipettaggio e incubare a 37 gradi centigradi per 30 min. Mescolare pipettando ogni 10 min.
- Separare le perline sul supporto magnetico per 2-3 min. Rimuovere il supernatante.
-
Lavare le perline
- Aggiungere 150 l di tampone di lavaggio A e sospendere nuovamente le perline con la pipa. Separare le perline sul supporto magnetico per 2-3 min e rimuovere il supernatante.
- Aggiungere 150 l del tampone di lavaggio B e sospendere nuovamente le perline con il pipettaggio. Separare le perline sul supporto magnetico per 2-3 min e rimuovere il supernatante.
- Aggiungere 150 l del tampone di lavaggio C e sospendere nuovamente le perline con il pipettaggio. Separare le perline sul supporto magnetico per 2-3 min e rimuovere il supernatante.
NOT: I tamponi B e C devono essere preriscaldati a 37 gradi centigradi. Le ricette per i tamponi di lavaggio A, B e C sono come descritto in Murata et al.
-
rilascio cDNA
- Preparare il buffer 1x RNase I mescolando 58,5 l' di acqua con 6,5 ldi di 10x buffer RNase I.
- Risospendere le perline in 35 - L di 1x RNase I buffer. Incubare a 95 gradi centigradi per 5 min e trasferire direttamente sul ghiaccio per 2 min per evitare la riassociazione del cDNA. Tenere i coperchi durante il trasferimento al ghiaccio in quanto possono pop-off a causa di accumulo di pressione.
- Separare le perline per 2-3 min su un supporto magnetico e trasferire il supernatante in una nuova piastra.
- Risospendere le perline in 30 - L di 1x RNase I buffer. Separare le perline sul supporto magnetico per 2-3 min e trasferire il supernatore al supernatante precedentemente raccolto (il volume totale di cDNA eluito dovrebbe essere di circa 65 gradi).
8. Rimozione dell'RNA da parte di RNase H e RNase I Digestion
- Per campione, unire 2,4 l' di acqua, 0,5 l di 10x Tampone RNase I, 0,1 L di RNase H e 2 -L di RNase I.
- Aggiungere 5 l dell'innle del mix al 65 o Più dell'on. Incubare a 37 gradi centigradi per 15 min e tenere a 4 gradi centigradi.
- Purificare il cDNA dal mix di digestione di RNase utilizzando perline magnetiche SPRI. Aggiungete 126 lofan di perline SPRI a 70 gradi di reazione alla degradazione e mescolate con il pipettaggio. Seguire i passaggi di purificazione come descritto per la purificazione delle perline SPRI in 2.6.1–2.6.6. Elute usando 42 l di acqua preriscaldata a 37 gradi centigradi come descritto.
- Preparare rNase I mix combinando 4,5 ll di 10x RNase I buffer e 0,5 L di RNase I.
- Aggiungere 5 -L della miscela RNase al 40 OL del campione di cDNA purificato. Mescolare con pipettaggio e incubare a 37 gradi centigradi per 30 min.
- Purificare il campione utilizzando perline magnetiche SPRI. Aggiungete 81 l di perline SPRI a 45 - L di reazione alla degradazione e mescolate con il pipettaggio. Seguire i passaggi di purificazione come descritto per la purificazione delle perline SPRI in 2.6.1–2.6.6. Elutare usando 42 - L di acqua come descritto.
9. Ligazione del Linker 5'
- Concentrare il campione di cDNA purificato a 4 gradi utilizzando il concentratore a vuoto. Mantenere la temperatura a 30-35 gradi centigradi. Testare il volume utilizzando una pipetta. Se il campione si è asciugato fino alla completezza, sciogliere aggiungendo 4 l'acqua.
NOT: È meglio evitare l'essiccazione alla completezza per prevenire la perdita del campione. - Incubare il campione concentrato a 95 gradi centigradi per 5 min e mettere immediatamente sul ghiaccio per 2 min per prevenire la rinaturazione. Tenere i coperchi durante il trasferimento dei tubi come i coperchi possono pop-off a causa di accumulo di pressione.
- Incubare 4 l. del linker da 2,5 m 5' a 55 gradi centigradi per 5 min e mettere immediatamente sul ghiaccio per 2 min per prevenire la rinaturazione.
- Mescolare 4 l del linker da 2,5 M da 5' con 4 L del campione.
NOT: Il linker 5' deve essere preparato in base alla tabella supplementare2, alla tabellasupplementare 3, alla tabella supplementare 4e alla tabella supplementare 5. Diluire il linker da 10 M da 5 m a una concentrazione di 2,5 m utilizzando 100 mM NaCl prima dell'uso. - Aggiungete 16 l della premiatrice di legatura (vedi Tabella deimateriali) al linker misto da 5' e al campione e mescolate bene pipettando. Incubare a 16 gradi centigradi per 16 ore.
- Purificare la miscela di legatura utilizzando perline magnetiche SPRI. Aggiungete 43,2 l di perline SPRI e seguite i passaggi 2.6.1–2.6.6. Elute, come descritto utilizzando 42 gradi di acqua preriscaldata a 37 gradi centigradi.
- Ripetere la purificazione effettuata nel passaggio 9.6 aggiungendo 72 l'l di perline SPRI al supernatant trasferito (1.8:1 perline al rapporto campione).
NOTA: i linker 5' contengono codici a barre che consentono il pooling di un massimo di otto campioni prima della sequenza (sono disponibili otto codici a barre trinucleotide, come descritto in Murata et al.11 e supplementari tabella 1).
10. Ligazione del linker 3'
- Concentrate il campione purificato a 4 - l utilizzando il concentratore di vuoto come descritto al punto 9.1.
- Incubare il campione concentrato a 95 gradi centigradi per 5 min e mettere immediatamente sul ghiaccio per 2 min per prevenire la rinaturazione. Tenere i coperchi durante il trasferimento dei tubi come i coperchi possono pop fuori a causa di accumulo di pressione.
- Incubare 4 l. del linker da 2,5 m 3' a 65 gradi centigradi per 5 min e mettere immediatamente sul ghiaccio per 2 min per prevenire la rinaturazione.
- Aggiungere 4 L del linker da 2,5 M 3' al 4 O L del campione concentrato.
- Aggiungere 16 l della premiatrice di legatura e mescolare bene pipettando. Incubare a 16 gradi centigradi per 16 ore.
- Purificare la miscela di legatura utilizzando perline magnetiche SPRI. Aggiungete 43,2 l di perline SPRI e seguite i passaggi 2.6.1–2.6.6. Elute come descritto utilizzando 42 l di acqua preriscaldata a 37 gradi centigradi.
NOT: Il linker 3' dovrebbe essere preparato secondo le tabelle supplementari 6 e la tabella supplementare 7. Diluire il linker da 10 M da 3 m a una concentrazione di 2,5 M utilizzando 100 mM NaCl.
11. Dephosphorylation
- Preparare il mix SAP combinando 4 luna di acqua, 5 luna di 10 volte buffer SAP e 1 L di enzima SAP.
- Aggiungete 10 l di miscela SAP al campione purificato (volume totale 50) e incubate nel termociclore utilizzando il seguente programma: 37 gradi centigradi per 30 min, 65 gradi centigradi per 15 min, e tenere premuto a 4 gradi centigradi.
12. Degradazione del 3' del linker Upper Strand utilizzando l'enzima di escissione specifica Uracil
- Aggiungere 2 - L di enzima di escissione specifico dell'uracil (vedi Tabella deimateriali) al campione dephosphorylated, mescolare con pipettaggio e incubare nel termociclore utilizzando il seguente programma: 37 gradi C per 30 min, 95 gradi centigradi per 5 min, e mettere immediatamente sul ghiaccio per 2 min per impedire la reincerazione del filamento superiore frammentato.
- Purificare la miscela di reazione aggiungendo 93,6 l'l di perline magnetiche SPRI alla miscela di 52 l e mescolare bene con la pipettatura. Ripetere i passaggi di purificazione 2.6.1 – 2.6.6. Elute con 42 litri d'acqua preriscaldata a 37 gradi centigradi come descritto.
13. Seconda sintesi dell'ancato
- Preparare il secondo mix di sintesi del filamento (i volumi sono espressi per campione) combinando 5 .L di 10x buffer di reazione al polimerasi del DNA, 2 L di acqua, 1 ll di 10 mM dNTP, 1 L di 50 nAnT-iCAGE secondo filamento primer (sequenza è in supplementare tabella 1) e 1 L di D Polimerasi NA carente di esonuclea (vedere la polimerasi raccomandata nella tabella dei materiali).
- Aggiungere 10 l del mix al campione purificato e mescolare bene pipettando (il volume totale è 50 .L). Incubare nel ciclore termico utilizzando il seguente programma: 95 gradi centigradi per 5 min, 55 gradi centigradi per 5 min, 72 gradi centigradi per 30 min, e tenere a 4 gradi centigradi.
14. Degradazione del Secondo Filamento Sintesi Primer Utilizzando Exonuclease I
- Aggiungere 1 -L di Exonuclease I alla seconda miscela di sintesi del filamento. Mescolare bene con pipettaggio e incubare a 37 gradi centigradi per 30 min seguiti da una tenuta a 4 gradi centigradi.
- Purificare il DNA a doppio filamento aggiungendo 91,8 luna di perline magnetiche SPRI a 51 -L del campione trattato con Eonuclease I. Ripetere i passaggi di purificazione descritti in 2.6.1–2.6.6. ed elute con 42 litri di acqua preriscaldata a 37 gradi centigradi come descritto.
- Concentrare il campione utilizzando il concentratore a vuoto a 15 gradi, come descritto al punto 9.1.
15. Controllo qualità e quantità
- Utilizzare 1 l dei campioni concentrati ed eseguire un chip di DNA ad alta sensibilità su un analizzatore di qualità del DNA. Profilo/quantità prevista è presentato nella Figura 3.
16. Primo round di degradazione del vettore
- Preparare la degradazione combinando 2 gradi di acqua, 2 luna di 10x tampone enzimatico di restrizione, 1 luna di I-SceI e 1 L di I-CeuI.
- Aggiungete 6- l della degradazione a 14 -L del campione concentrato e mescolate con il pipettaggio. Incubare a 37 oC per 3 h seguito da 20 min di disattivazione a 65 gradi centigradi e tenere a 4 gradi centigradi.
- Purificare il mix di degradazione utilizzando perline magnetiche SPRI. Aggiungere 5 - L di acqua per aumentare il volume della miscela di degradazione e aggiungere 45 l of SPRI perline (1.8:1 perline al rapporto campione). Ripetere la purificazione come descritto nei passaggi 2.6.1–2.6.6. ed elute con 42 gradi di acqua preriscaldata a 37 gradi centigradi.
- Concentrare il campione eluito da 42 a 20 gradi del volume totale, come descritto al punto 9.1.
17. Controllo del livello di degradazione e determinazione del numero di cicli di amplificazione PCR
- Preparare il mix qPCR per amplificare intere librerie (mix di adattatori). Unire 3,8 l'acqua, 5 - L di premix qPCR (2x), 0,1 di 10 -M adattatore_f1 primer (5'-AATGATACGGCGACCGA-3') e 0,1 - L di 10M adaptor_r1 primer (5'-CAAGCAGAAGACGGCATACGA-3') per ogni campione (vedere Tabella dei materiali per i premi qRPCx consigliati).
- Unire 9 ll l di mix di adattatori qPCR con 1 ll di campione dal passo 16.4 e mescolare bene con la pipettatura.
- Preparare il mix qPCR per amplificare il DNA derivato dal vettore (mix di vettori). Unire 3,8 ll di acqua, 5 ll l di qPCR premix (2x), 0,1 di 10 m carrier_f1 primer (5'-GCGGCAGCGTTCGCTATAC-3') e 0,1 L di 10 m adaptor_r1 primer per ogni campione
- Unire 9 lll del mix di supporti qPCR con 1 l del campione del punto 16,4 e mescolare bene con il pipettaggio.
- Programma qPCR set: 95 s per 3 min (95 gradi centigradi per 20 s, 60 gradi centigradi per 20 s, 72 gradi c per 2 min) ripetuti 40x, seguiti da una curva di denaturazione specifica dello strumento (65-95 ) e tenere a 4 gradi centigradi.
NOT: Preparare un controllo negativo sostituendo il campione con acqua.
18. Amplificazione PCR della libreria di destinazione
- Preparare il mix di amplificazione PCR combinando 6 litri d'acqua, 0,5 l una di 10 -M adaptor_f1 primer, 0,5 - L di 10 -M adaptor_r1 primer e 25 l. di premiscela PCR (2x). Miscela mediante pipettaggio (vedere Tabella dei materiali per la premix PCR consigliata).
- Aggiungere 32 l dell'inmiscelato PCR a 18 gradi del campione dal punto 16,4. Mescolare accuratamente con il pipettaggio.
- Impostare l'amplificazione PCR: 95 gradi centigradi per 3 min, (98 gradi centigradi per 20 s, 60 gradi centigradi per 15 s, 72 gradi centigradi per 2 min) 12-18 cicli, 72 gradi C per 2 min e tenere a 4 gradi centigradi.
NOT: Il numero esatto di cicli PCR è determinato dai risultati qPCR e corrisponde al valore Ct ottenuto con il mix di primer dell'adattatore (il numero di cicli PCR è uguale al valore Ct). - Purificare il campione amplificato aggiungendo 90 L di perline magnetiche SPRI a 50 - L del campione amplificato e mescolare accuratamente con la pipettatura. Ripetere i passaggi di purificazione descritti nei passaggi 2.6.1–2.6.6. ed eluire il campione usando 42 - L di acqua come descritto.
19. Secondo ciclo di degradazione del vettore
- Ripetere i passaggi da 16.1– 16.3.
- Purificare il mix di degradazione utilizzando perline magnetiche SPRI. Aggiungere al campione 10 l di acqua per aumentare il volume e mescolare con 30 gradi di perline SPRI (1:1 perline a rapporto campione). Ripetere la purificazione come descritto nei passaggi 2.6.1–2.6.6. ed elutare con 42 litri di acqua preriscaldata a 37 gradi centigradi come descritto.
- Concentrare il campione eluito da 42 a 30 l di volume totale.
20. Selezione delle dimensioni della libreria
- Mescolare 24 ll di perline magnetiche SPRI con 30 l del campione dal passo 19.3. (0.8:1 perline a rapporto campione). Ripetere i passaggi di purificazione come descritto nei passaggi 2.6.1–2.6.6. ed eluire il campione in 42 - L di acqua come descritto.
- Concentrare il campione a circa 14 - come descritto nel passaggio 9.1.
21. Controllo qualità
-
Valutazione della distribuzione delle dimensioni
- Eseguire 1 l del campione sul chip di DNA ad alta sensibilità. I risultati attesi sono presentati nella Figura 4.
NOT: Se sono visibili frammenti inferiori a 200 bp (vedere l'esempio nella figura 4A,C), la selezione delle dimensioni (passaggi da 20.1–20.2) deve essere ripetuta fino a quando i frammenti brevi non vengono rimossi (Figura 4B, D). Di solito è sufficiente un ulteriore giro di selezione delle dimensioni. Se la quantità di frammenti brevi è grave (come illustrato nella figura 4C), il rapporto tra perline e campioni deve essere ridotto a 0,6:1.
- Eseguire 1 l del campione sul chip di DNA ad alta sensibilità. I risultati attesi sono presentati nella Figura 4.
-
Controllo della qualità della degradazione del vettore
- Ripetere i passaggi 17.1–17.5.
NOT: A seconda della concentrazione delle librerie stimate nell'esecuzione del chip HS (analisi della regione), i campioni devono essere diluiti prima di qPCR. Utilizzare 0,5 l del campione per evitare la perdita del campione e diluire 100-500x in acqua (diluire a 1–20 pg/L concentrazione finale). La differenza prevista tra i valori Ct ottenuti con adattatore e mix di vettori è compresa tra 5 e 10.
- Ripetere i passaggi 17.1–17.5.
-
Quantificazione biblioteca
- Preparare la diluizione di lavoro dello standard del DNA lambda mescolando 20 - L di 100 mg/mL di standard di DNA lambda con 980 - L di 1x TE (preparare diluindo 20x TE fornito nel kit di quantificazione del DNA). La diluizione del DNA lambda può essere immagazzinata a -20 gradi centigradi.
- Preparare le diluizioni seriali standard del DNA lambda mescolando lo standard lambda diluito e 1x TE secondo la tabella supplementari 8.
NOT: Per una maggiore precisione, si consiglia di aggiungere 100 luna di 1x buffer TE a tutti i tubi e rimuovere 1x volume TE come richiesto per volume dell'espressione lambda diluita da aggiungere. Non utilizzare più di 1 L della libreria; si raccomanda l'uso di 384 piastre per questa misura.
Risultati
Questo rapporto descrive il protocollo SLIC-CAGE completo per ottenere librerie pronte per il sequenziamento da nanogrammi di materiale RNA totale iniziale (Figura 1). Per ottenere il mix di portatori di RNA sintetici, in primo luogo, i modelli di portante PCR devono essere preparati e purificati in gel per eliminare i prodotti laterali PCR (Figura 2A). Ogni modello PCR (dieci in totale) viene prodotto utilizzando un inoltro comune, ma un'inversione diversa (Tabella 2), che porta a lunghezze diverse del modello PCR per consentire la variabilità delle dimensioni dei portatori di RNA sintetici. Una volta purificati, i modelli PCR vengono utilizzati per la trascrizione in vitro delle molecole portatrici. Un singolo prodotto portarna è previsto se i modelli sono purificati in gel (vedi analisi rappresentativa del gel nella Figura 2B). La preparazione del supporto può essere scalata a seconda della necessità, e quando preparato, mescolato e congelato a -80 gradi centigradi per un uso futuro.
Utilizzando la quantità minima raccomandata di RNA totale campione (10 ng) combinato con 16-18 cicli di amplificazione PCR, è possibile ottenere librerie SLIC-CAGE ad alta complessità. Il numero di cicli PCR necessari per amplificare la libreria finale dipende molto dalla quantità totale di RNA di input utilizzato (il numero previsto di cicli è indicato nella tabella4).
Dopo il primo ciclo di degradazione, nei risultati qPCR (passaggio 17), la differenza prevista tra i valori Ct ottenuti utilizzando adaptor_f1 o carrier_f1 primer è 1-2, con i valori Ct ottenuti con adaptor_f1 inferiore rispetto a carrier_f1.
La distribuzione delle lunghezze dei frammenti nella libreria finale è compresa tra 200-2.000 bp con la dimensione media del frammento di 700-900 bp (basata sull'analisi della regione utilizzando il software Bioanalyzer, Figura 4B,D). Frammenti più brevi, come illustrato nella figura 4A,C, devono essere rimossi da ulteriori cicli di esclusione di dimensioni (passaggi 20-21). Questi brevi frammenti sono artefatti di amplificazione PCR e non la libreria di destinazione. Si noti che i frammenti più brevi si raggruppano meglio sulle celle di sequenziamento e possono causare problemi di sequenziamento.
La quantità prevista di materiale di libreria ottenuto per campione è compresa tra 5-50 ng. Importi significativamente inferiori sono indicativi di perdita di campione durante il protocollo. Se la quantità bassa ottenuta è sufficiente per la sequenza (è necessaria 2-3 ng delle librerie in pool), le librerie potrebbero essere di minore complessità (vedere di seguito).
A seconda della macchina di sequenziamento, potrebbe essere necessario ottimizzare la quantità della libreria caricata nella cella di flusso. Utilizzando un Illumina HiSeq 2500, il caricamento di 8-12 pM librerie SLIC-CAGE dà in media 150-200 milioni di letture, con >80% delle letture che superano il punteggio di qualità Q30 come soglia.
Le letture ottenute vengono quindi mappate al genoma di riferimento [per letture da 50 bp, Bowtie212 può essere utilizzato con parametri predefiniti che consentono zero mancate corrispondenze per sequenza di semi (22 bp)]. L'efficienza di mappatura prevista dipende dalla quantità totale di ingresso dell'RNA e sono presentate nella tabella 5. Le letture mappate in modo univoco possono quindi essere caricate nell'ambiente di elaborazione grafico e statistico R13 ed elaborate utilizzando CAGEr (pacchetto Bioconductor14). La vignetta pacchetto è facile da seguire e spiega il flusso di lavoro e l'elaborazione dei dati mappati in dettaglio. Un facile controllo visivo della complessità della libreria è la distribuzione della larghezza del promotore, in quanto le librerie a bassa complessità avranno promotori artificialmente stretti (Figura 5A, libreria SLIC-CAGE derivata da 1 ng di RNA totale, per i dettagli si vedano pubblicazione10). Tuttavia, anche le librerie SLIC-CAGE a bassa complessità consentono l'identificazione di CTSS reali, con maggiore precisione rispetto ai metodi alternativi per il mapping TSS a basso/medio input (Figura 5B,C).

Figura 1: Passaggi del protocollo SLIC-CAGE. L'RNA campione viene miscelato con la miscela portante dell'RNA per ottenere 5 g di materiale totale di RNA. cDNA è sintetizzato attraverso la trascrizione inversa e il cappuccio viene ossidato utilizzando periodato di sodio. L'ossidazione consente l'attaccamento della biotina al cappuccio utilizzando l'idrato di biotina. La biotina si attacca all'estremità di 3, in quanto viene anche ossidata usando il periodato di sodio. Per eliminare la biotina dagli ibridi mRNA:cDNA con cDNA in completamente sintetizzato e dalle estremità di 3o mRNA, i campioni vengono trattati con RNase I. cDNA che ha raggiunto l'estremità 5' dell'mRNA viene quindi selezionato per purificazione di affinità su perline magnetiche streptavidin ( cap-trappola). Dopo il rilascio del cDNA, i linker da 5 a 3 vengono legati. Le molecole della biblioteca che provengono dal vettore sono degradate usando le endonucleasi i-SceI e I-CeuI e i frammenti vengono rimossi utilizzando perline magnetiche SPRI. La libreria viene quindi amplificata PCR. Fare clic qui per visualizzare una versione più grande di questa figura.
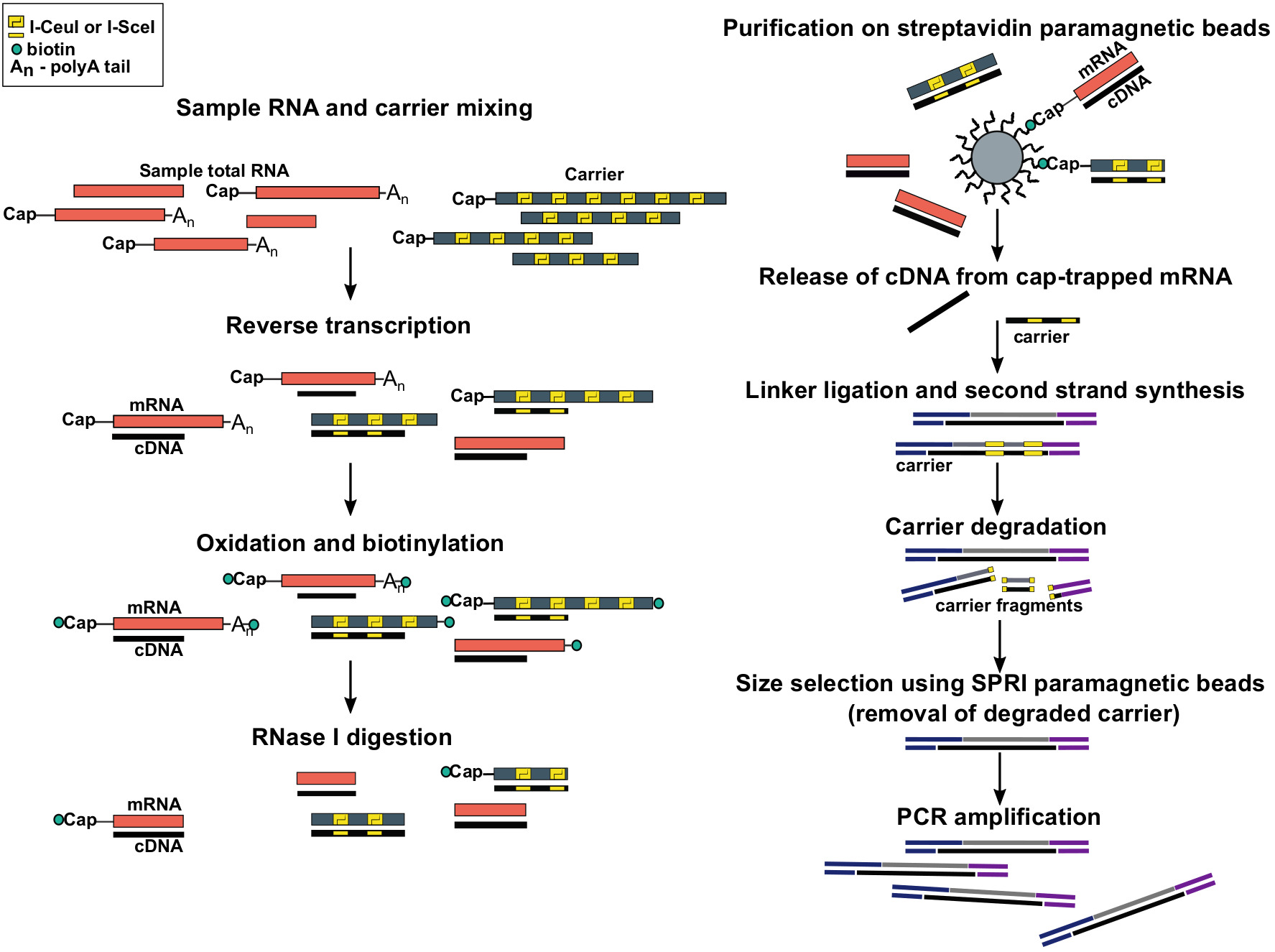{kind=link}

Figura 2: Analisi congiunta gel dei modelli PCR portante e trascrizioni carrier in vitro. (A) Modelli di portaggio PCR prima della purificazione del gel: il primo pozzo contiene il marcatore da 1 kbp, seguito dai modelli PCR portanti 1, 1-10. (B) Trascrizioni carrier in vitro: il primo pozzo contiene il marcatore da 1 kbp, seguito dalle trascrizioni del vettore 1-10. Le trascrizioni dei portante sono state denaturate riscaldando per 5 minuti a 95 gradi centigradi prima del caricamento. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 3: Traccia rappresentativa della qualità del DNA (chip di DNA ad alta sensibilità) di SLIC-CAGE prima del primo ciclo di degradazione del vettore. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 4: Tracce rappresentative di qualità del DNA (chip di DNA ad alta sensibilità) delle librerie SLIC-CAGE dopo l'amplificazione PCR. (A) libreria SLIC-CAGE che richiede un'ulteriore selezione delle dimensioni per la rimozione di brevi frammenti. (B) Libreria SLIC-CAGE dopo la selezione delle dimensioni utilizzando 0,6x perline SPRI al rapporto campione. (C) Libreria SLIC-CAGE di quantità di output inferiore che richiede la selezione delle dimensioni per la rimozione del frammento corto. (D) Libreria SLIC-CAGE di quantità di uscita inferiore dopo la selezione delle dimensioni utilizzando 0.6:1 Spri perline al rapporto campione. Fare clic qui per visualizzare una versione più grande di questa figura.
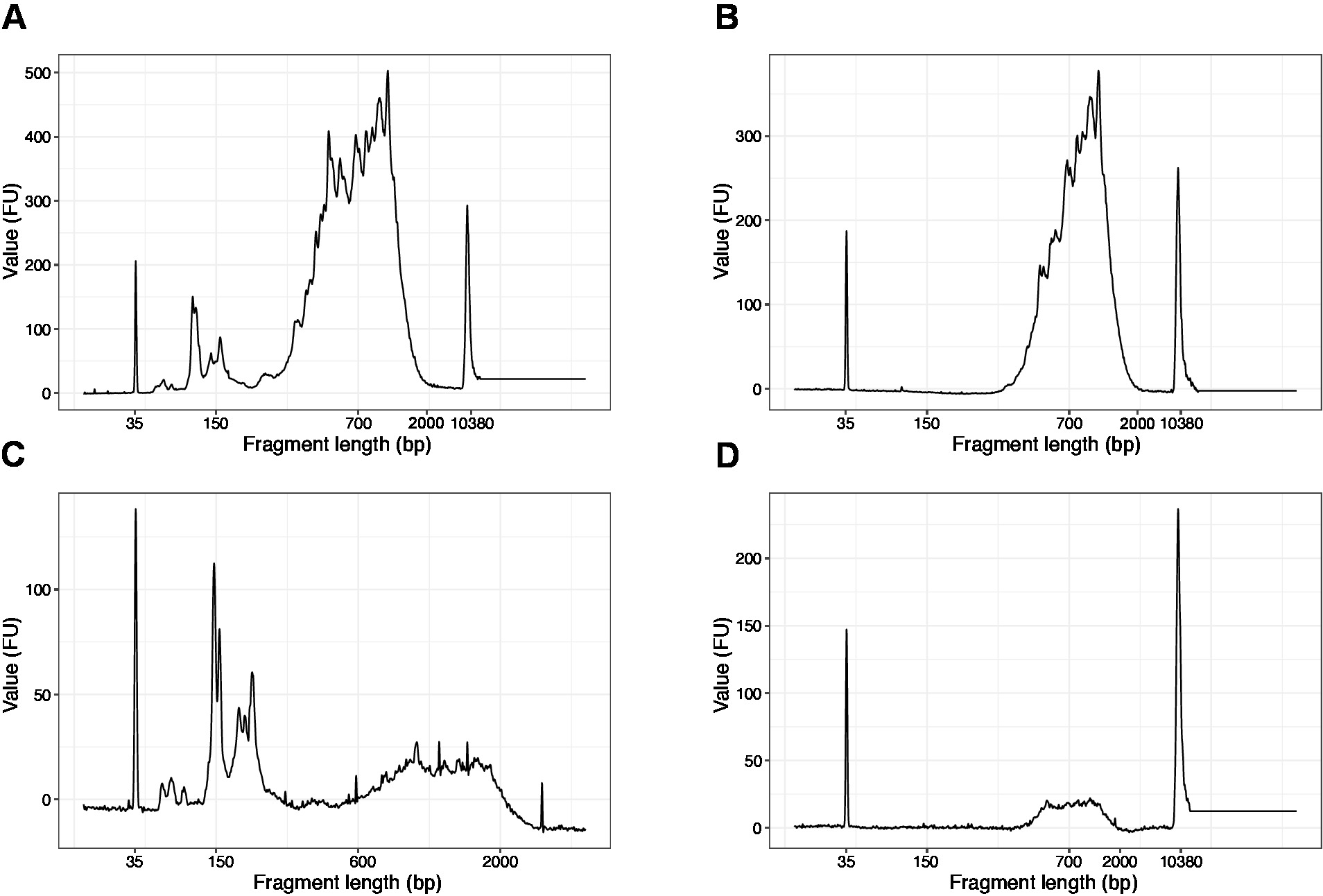{kind=link}

Figura 5: Convalida delle librerie SLIC-CAGE. (A) Distribuzione delle larghezze interquantili del tag nelle librerie SLIC-CAGE preparate a partire da 1, 5 o 10 ng di RNA totale S. cerevisiae, e nella libreria nAnT-iCAGE preparata da 5 g di S. cerevisiaeRNA totale. Una quantità elevata di cluster di tag stretti nella libreria SLIC-CAGE 1 ng ne indica la bassa complessità. (B) Curve ROC per l'identificazione CTSS nelle librerie S. cerevisiae SLIC-CAGE. Tutti i CTS S. cerevisiae nAnT-iCAGE sono stati utilizzati come un vero set. (C) Curve ROC per l'identificazione CTSS nelle librerie s. cerevisiae nanoCAGE. Tutti i CTS S. cerevisiae nAnT-iCAGE sono stati utilizzati come un vero set. Il confronto delle curve ROC mostra che SLIC-CAGE supera fortemente nanoCAGE nell'identificazione CTSS. Sono stati utilizzati i dati di ArrayExpress E-MTAB-6519. Fare clic qui per visualizzare una versione più grande di questa figura.
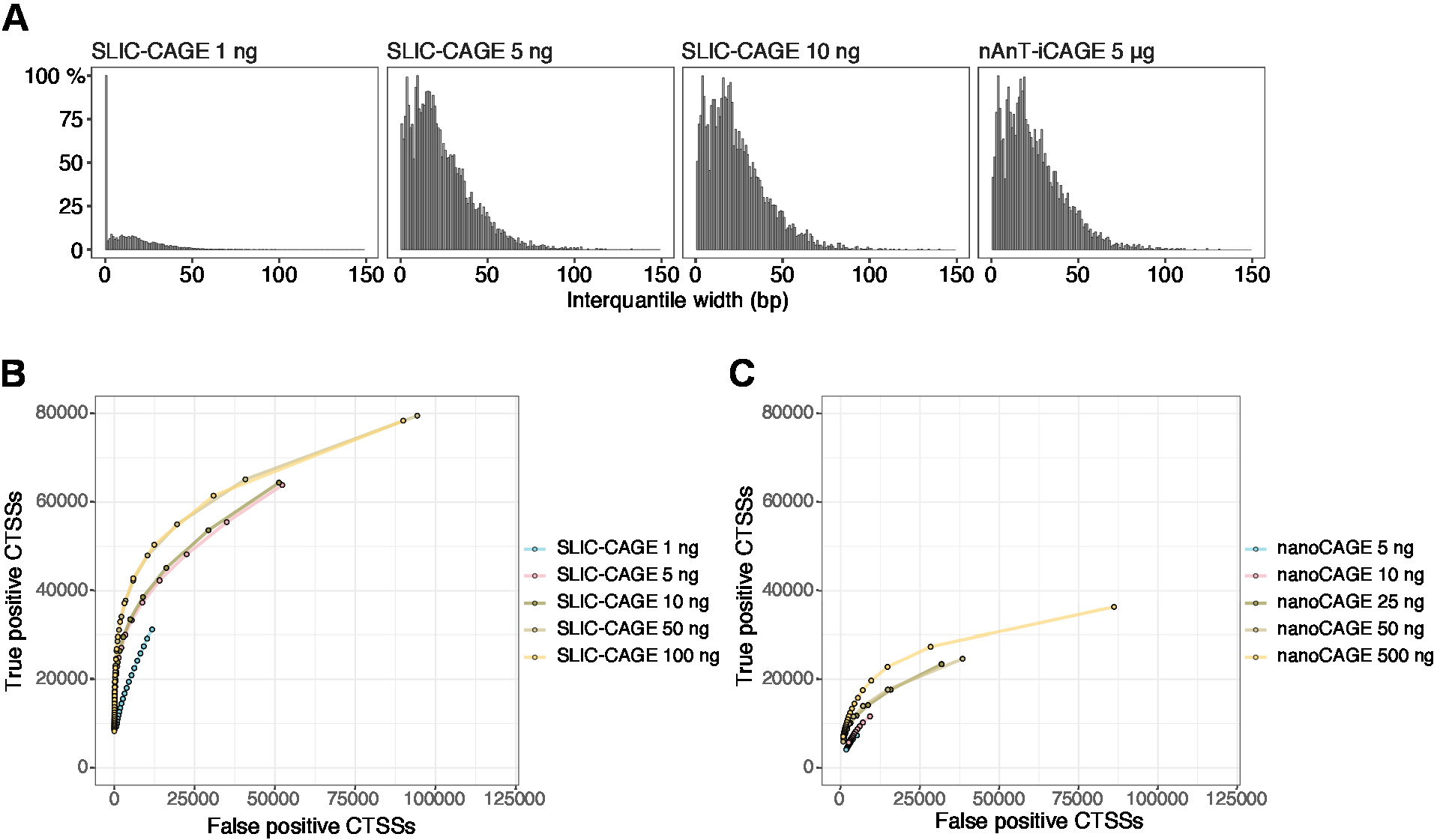{kind=link}
Tabella 1: Sequenza del gene sintetico portatore. I siti I-SceI sono in grassetto e corsivo in viola, e i siti di riconoscimenti I-CeuI sono verdi. Fare clic qui per scaricare questo file.
| portatore | primer inverso 5'-3' | Lunghezza del prodotto PCR / bp | |
| 1 : il nome del | PCR_N6_r1: NNNNNNCTACGTGTCGCAGAGAATT | 1034 | |
| 2 Il nome del sistema | PCR_N6_r2: NNNNNNTATCCAGATCGTTGAGCTGC | 966 | |
| 3 (COM del nome | PCR_N6_r3: NNNNNNCACTGCGGCTCTCTCT | 889 889 | |
| 4 DEL psu' | PCR_N6_r4: NNNNNNGCCGTCGATAACTTGTTGTTGT | 821 (in questo stato del | |
| 5 Del numero 3( | PCR_N6_r5: NNNNNNAGTTGACCGCAGAAGTCTCTC | 744 Del 144 | |
| 6 È possibile: | PCR_N6_r6: NNNNNNNNGTGAAGAATTCTTCTCTCCA | 676 del sistema | |
| 7 (in questo stato | PCR_N6_r7: NNNNNNCTCGCGGCTCTCTCATAAAC | 599 | |
| 8 (IN vio | PCR_N6_r8: NNNNNNTATACGCGATGTCGTCGTAC | 531 | |
| 9 (in vie | PCR_N6_r9: NNNNNNACCGGCGCCTCTCTGCAGG | 454 | |
| 10 del sistema | PCR_N6_r10: NNNNNNCAGGACGTTTTTGCCCAGCA | 386 mila | |
| Il primer di inoltro è lo stesso per tutti i modelli di vettori. Sottolineato è la sequenza promotore T7. PCR_GN5_f1: TAATACGACTCACTATAGNNNCAGCGTTCGCT | |||
Tabella 2: Primer per l'amplificazione del modello carrier. Forward primer è lo stesso per tutti i modelli di vettore. Sottolineato è la sequenza promotore T7. PCR_GN5_f1: TAATACGACTCACTATAGNNNNNCGCGTTCGCT. Utilizzando diverse primer inverse, vengono prodotti modelli PCR e quindi RNA portante di lunghezza diversa.
| portatore | lunghezza | non ancora | con tappo/g |
| 1 : il nome del | 1034 | 3.96 (in inglese) | 0,45 (in linguaggio da 1,05) |
| 2 Il nome del sistema | 966 | 8.36 (in inglese) | 0,95 (in questo da fwlinkin base al nome) |
| 3 (COM del nome | 889 889 | 4.4 (in questo stato del documento) | 0,5 0,5 |
| 4 DEL psu' | 821 (in questo stato del | 6.6 | 0,75 (in questo 0,05) |
| 5 Del numero 3( | 744 Del 144 | 4.4 (in questo stato del documento) | 0,5 0,5 |
| 6 È possibile: | 676 del sistema | Ore 3.08 | 0,35 (in questo da fwlinka che) |
| 7 (in questo stato | 599 | 4.4 (in questo stato del documento) | 0,5 0,5 |
| 8 (IN vio | 531 | 3.96 (in inglese) | 0,45 (in linguaggio da 1,05) |
| 9 (in vie | 454 | 2.64 | 0.3 0.3 |
| 10 del sistema | 386 mila | 2.2 2 Il suo | 0,25 |
Tabella 3: Mix di portatori di RNA. In totale 49 g del mix portante 0,3-1 kbp: non con tappo : 44 g, con tappo di 5 g.
| Ingresso TOTALE rna /ng | Cicli PCR |
| 1 ng | 18 mi lato |
| 2 ng | 17 mi lato |
| 5 ng | 16 |
| 10 ng | 15-16 anni |
| 25 ng | 14-15 anni |
| 50 ng | 13-15 anni |
| 100 ng | 12-14 anni |
Tabella 4: Numero previsto di cicli PCR in dipendenza dall'input totale di RNA campione. Il numero approssimativo di cicli si basa su esperimenti eseguiti utilizzando Saccharomyces cerevisiae, Drosophila melanogaster e Mus musculus total RNA.
| Ingresso/ng totale di RNA | % mappata complessivamente | % mappata in modo univoco | % vettore |
| 1 ng | 30 milio | 20-30 anni | 30 milio |
| 2 ng | 60 del sistema | 20-50 anni | 10 del sistema |
| 5 ng | 60-70 anni | 40-60 | 5-10 anni |
| 10 ng | 60-70 anni | 40-60 | 5-10 anni |
| 25 ng | 65-80 | 40-70 | Da 0 a 5 |
| 50 ng | 65-80 | 40-70 | 0-3 |
| 100 ng | 70-85 anni | 40-70 | 0-2 |
Tabella 5: Efficienza di mappatura prevista e dipendenza della quantità totale di ingresso dell'RNA. I numeri approssimativi sono presentati e basati su esperimenti eseguiti utilizzando Saccharomyces cerevisiae e Mus musculus total RNA.
Discussione
Per una corretta preparazione della libreria SLIC-CAGE, è fondamentale utilizzare suggerimenti e tubi a bassa legatura per evitare la perdita del campione a causa dell'adsorbimento del campione. In tutte le fasi che comportail recupero del super-natante, si raccomanda di recuperare l'intero volume del campione. Poiché il protocollo prevede più passaggi, la perdita continua del campione porterà a librerie non riuscite.
Se CAGE (nAnT-iCAGE) non è stato eseguito regolarmente, è meglio testare SLIC-CAGE con quantità di ingresso diverse (10 ng, 20 ng, 50 ng, 100 ng, 200 ng) dello stesso campione totale di RNA e confrontarlo con le librerie nAnT-iCAGE preparate utilizzando 5 g di RNA totale. Se la libreria nAnT-iCAGE non riesce (meno di 0,5-1 ng della libreria di DNA ottenuta per campione), è improbabile che SLIC-CAGE funzioni e che la perdita del campione sia ridotta al minimo.
Un passo fondamentale per garantire che le librerie di alta qualità prive di RNA o rRNA degradato non consorcoperto sia l'intrappolamento del tappo descritto nella sezione 7. È molto importante che le perline di streptavidina siano completamente risospese nei tamponi di lavaggio e che i tamponi di lavaggio vengano rimossi prima di continuare alla fase di lavaggio successiva o alla elusione del cDNA.
Se i risultati del qPCR dopo il primo ciclo di degradazione del vettore non mostrano alcuna differenza tra l'uso di primer adaptor_f1 e carrier_f1, è comunque consigliabile continuare il protocollo. Se dopo il secondo ciclo di degradazione del vettore, la differenza nei valori Ct è inferiore a cinque, si consiglia un terzo ciclo di degradazione del vettore. Non abbiamo mai trovato necessario un terzo ciclo di degradazione e, se si verifica, si raccomanda di sostituire gli stock di endonucleasi homing.
Ulteriori cicli di amplificazione PCR possono essere aggiunti al protocollo se la quantità finale della libreria ottenuta non è sufficiente per la sequenza. L'amplificazione PCR può quindi essere impostata con un numero minimo di cicli di amplificazione necessari per produrre materiale sufficiente per il sequenziamento, tenendo conto della perdita del campione che non può essere evitata nella selezione delle dimensioni. La purificazione o la selezione delle dimensioni mediante perline magnetiche SPRI devono quindi essere eseguite fino a quando tutti i piccoli frammenti (<200 bp) non vengono rimossi (se necessario, utilizzano 0.6:1 perline per il rapporto campione) e la libreria deve essere quantificata utilizzando Picogreen.
Le librerie possono essere sequenziate in modalità a estremità singola o accoppiata. Utilizzando il sequenziamento a fine accoppiato, è possibile ottenere informazioni sugli isoformi di trascrizione. Inoltre, poiché la trascrizione inversa viene eseguita utilizzando un primer casuale (TCT-N6, N6 essendo un esagono casuale), le informazioni dalla sequenza 3'-end possono essere utilizzate come identificatori molecolari univoci (UMI) per comprimere i duplicati PCR. Poiché viene utilizzato un numero moderato di cicli di amplificazione PCR (fino a 18), l'uso di UMI è stato precedentemente ritenuto non necessario.
Poiché il nucleo del protocollo si basa su nAnT-iCAGE11, SLIC-CAGE utilizza otto codici a barre. Pertanto, il multiplexing più di otto campioni non è attualmente supportato. Inoltre, sia SLIC-CAGE che nAnT-iCAGE non sono adatti per l'acquisizione di RNA più brevi di 200 bp, in quanto i protocolli sono progettati per rimuovere linker e artefatti PCR tramite l'esclusione delle dimensioni con perline AMPure XP.
SLIC-CAGE è l'unico metodo imparziale di risoluzione a basso nucleotide a basso input per la mappatura dei siti di avvio della trascrizione utilizzando nanogrammi di materiale RNA totale. I metodi alternativi si basano sull'attività di commutazione del modello della trascrizione inversa all'RNA con tappo a barre anziché all'abbondanza di tappo (ad esempio, NanoCAGE15 e NanoPARE16). A causa della commutazione del modello, questi metodi presentano distorsioni specifiche della sequenza nel rilevamento dei Contenuti, portando ad un aumento del numero di TSS falsi positivi e alla diminuzione del numero di TSSreali 9,10.
Divulgazioni
È stato riempito un brevetto per l'RNA/DNA portatore degradabile.
Riconoscimenti
Questo lavoro è stato sostenuto dalla sovvenzione Wellcome Trust (106954) assegnata a B. L. e Medical Research Council (MRC) Core Funding (MC-A652-5QA10). N. C. è stato supportato dalla EMBO Long-Term Fellowship (EMBO ALTF 1279-2016); E. P. è stato sostenuto dal Medical Research Council UK; B. L. è stato sostenuto dal Medical Research Council UK (MC UP 1102/1).
Materiali
| Name | Company | Catalog Number | Comments |
| 2-propanol, Bioultra, for molecular biology, ≥99.5% | Sigma-Aldrich | 59304-100ML-F | Used in RNAclean XP purification. |
| 3' linkers | Sequences are described in Murata et al. 2014 and Supplementary Table 1 of this manuscript. Annealing of strands to produce 3'linkers is described in the supplementary of this protocol. | ||
| 5' linkers | Sequences are described in Murata et al. 2014 and Supplementary Table 1 of this manuscript. Annealing of strands to produce 5'linkers is described in the supplementary of this protocol. | ||
| Agencourt AMPure XP, 60 mL | Beckman Coulter | A63881 | Purification of DNA |
| Agencourt RNAClean XP Kit | Beckman Coulter | A63987 | Purification of RNA and RNA:cDNA hybrids in CAGE steps. |
| Axygen 0.2 mL Polypropylene PCR Tube Strips and Domed Cap Strips | Axygen (available through Corning) | PCR-0208-CP-C | Or any 8-tube PCR strips (used only for water and mixes). |
| Axygen 1 x 8 strip domed PCR caps | Axygen (available through Corning) | PCR-02CP-C | Caps for PCR plates. |
| Axygen 1.5 mL Maxymum Recovery Snaplock Microcentrifuge Tube | Axygen (available through Corning) | MCT-150-L-C | Low-binding 1.5 mL tubes, used for enzyme mixes or sample concentration. |
| Axygen 96 well no skirt PCR microplate | Axygen (available through Corning) | PCR-96-C | Low-binding PCR plates - have to be used for all steps in the protocol. Note that plates should be cut to contain 2 x 8 wells for easier visibility of the samples |
| Bioanalyzer (or Tapestation): RNA nano and HS DNA kits | Agilent | To determine quality of RNA, efficient size selection and final quality of the library (Tapestation can also be used) | |
| Biotin (Long Arm) Hydrazide | Vector laboratories | SP-1100 | Biotinylation/tagging |
| Cutsmart buffer | NEB | Restriction enzyme buffer | |
| Deep Vent (exo-) DNA Polymerase | NEB | M0259S | Second strand synthesis |
| DNA Ligation Kit, Mighty Mix | Takara | 6023 | Used for 5' and 3'-linker ligation |
| dNTP mix (10 mM each) | ThermoFisher Scientific | 18427013 | dNTP mix for production of carrier templates (or any dNTPs suitable for PCR) |
| Dynabeads M-270 Streptavidin | Invitrogen | 65305 | Cap-trapping. Do not use other beads as these are optimised with the buffers used. |
| DynaMag-2 Magnet | ThermoFisher Scientific | 12321D | Magnetic stand for 1.5 mL tubes - used to prepare Streptavidin beads. |
| DynaMag-96 Side Skirted Magnet | ThermoFisher Scientific | 12027 | Magnetic stand for PCR plates (96 well-plates) - used with cut plates to contain 2 x 8 wells. |
| Ethanol, BioUltra, for molecular biology, ≥99.8% | Sigma-Aldrich | 51976-500ML-F | Used in AMPure washes. Any molecular biology suitable ethanol can be used. |
| Exonuclease I (E. coli) | NEB | M0293S | Leftover primer degradation |
| Gel Loading Dye, Purple (6x), no SDS | NEB | B7025S | agarose gel loading dye |
| HiScribe T7 High Yield RNA Synthesis Kit | New England Biolabs | E2040S | Kit for carrier in vitro transcription |
| Horizontal electrophoresis apparatus | purification of carrier DNA templates from agarose gels | ||
| I-Ceu | NEB | R0699S | Homing endonuclease used for carrier degradation. |
| I-SceI | NEB | R0694S | Homing endonuclease used for carrier degradation. |
| KAPA HiFi HS ReadyMix (2x) | Kapa Biosystems (Supplied by Roche) | KK2601 | PCR mix for target library amplification |
| KAPA SYBR FAST qPCR kit (Universal) 2x | Kapa Biosystems (Supplied by Roche) | KK4600 | qPCR mix to assess degradation efficiency and requiered number of PCR amplification cycles |
| Micropipettes and multichannel micropipettes (0.1-10 µL, 1-20 µL, 20-200 µL) | Gilson | Use of Gilson with the low-binding Sorenson tips is recommended. Other micropippetes might not be compatible. Different brand low-binding tips may not be of equal quality and may increase sample loss. | |
| Microplate reader | For Picogreen concentration measurement of the final library. Microplates are used to allow small volume measurement and reduce sample waste. | ||
| nuclease free water | ThermoFisher Scientific | AM9937 | Or any nuclease (DNase and RNase) free water |
| PCR thermal cycler | incubation steps and PCR amplficication | ||
| Phusion High-Fidelity DNA Polymerase | ThermoFisher Scientific | F530S | DNA polymerase for amplification of carrier templates (or any high fidelity polymerase) |
| QIAquick Gel Extraction Kit (50) | Qiagen | 28704 | Purification of carrier PCR templates from agarose gels. |
| qPCR machine | determining PCR amplification cyle number and degree of carrier degradation | ||
| Quant-iT PicoGreen dsDNA Reagent | ThermoFisher Scientific | P11495 | Used to measure final library concentration - recommended as, in our hands, it is more accurate and reproducible than Qubit. |
| Quick-Load Purple 100 bp DNA Ladder | NEB | N0551S | DNA ladder |
| Quick-Load Purple 1 kb Plus DNA Ladder | NEB | N0550S | DNA ladder |
| Ribonuclease H | Takara | 2150A | Digestion of RNA after cap-trapping. |
| RNase ONE Ribonuclease | Promega | M4261 | Degradation of single stranded RNA not protected by cDNA. |
| RNase-Free DNase Set | Qiagen | 79254 | Removal of carrier DNA templates after in vitro transcription. |
| RNeasy Mini Kit | Qiagen | 74104 | For cleanup of carrier RNA from in vitro transcription or capping |
| Sodium acetate, 1 M, aq.soln, pH 4.5 RNAse free | VWR | AAJ63669-AK | Or any nuclease (DNase and RNase) free solution |
| Sodium acetate, 1 M, aq.soln, pH 6.0 RNAse free | Or any nuclease (DNase and RNase) free solution | ||
| Sodium periodate | Sigma-Aldrich | 311448-100G | Oxidation of vicinal diols |
| Sorenson low binding aerosol barrier tips, MicroReach Guard, volume range 10 μL, Graduated | Sorenson (available through SIGMA-ALDRICH) | Z719390-960EA | Low-binding tips - recommended use throughout the protocol to minimise sample loss. |
| Sorenson low binding aerosol barrier tips, MultiGuard, volume range 1,000 μL , Graduated | Sorenson (available through SIGMA-ALDRICH) | Z719463-1000EA | Low-binding tips - recommended use throughout the protocol to minimise sample loss. |
| Sorenson low binding aerosol barrier tips, MultiGuard, volume range 20 μL , Graduated | Sorenson (available through SIGMA-ALDRICH) | Z719412-960EA | Low-binding tips - recommended use throughout the protocol to minimise sample loss. |
| Sorenson low binding aerosol barrier tips, MultiGuard, volume range 200 μL , Graduated | Sorenson (available through SIGMA-ALDRICH) | Z719447-960EA | Low-binding tips - recommended use throughout the protocol to minimise sample loss. |
| SpeedVac Vacuum Concentrator | concentrating samples in various steps to lower volume | ||
| SuperScript III Reverse Transcriptase | ThermoFisher Scientific | 18080044 | Used for reverse transcription (1st CAGE step) |
| Trehalose/sorbitol solution | Preparation is described in Murata et al. 2014. | ||
| Tris-HCl, 1 M aq.soln, pH 8.5 | 1 M solution, DNase and RNase free | ||
| tRNA (20 mg/mL) | tRNA solution. Preparation is described in Murata et al. 2014. | ||
| UltraPure Low Melting Point Agarose | ThermoFisher Scientific | 16520050 | Or any suitable pure low-melt agarose. |
| USB Shrimp Alkaline Phosphatase (SAP) | Applied Biosystems (Provided by ThermoFisher Scientific) | 78390500UN | |
| USER Enzyme | NEB | M5505S | Degradation of 3'linker's upper strand, Uracil Specific Excision Reagent/Enzyme |
| Vaccinia Capping System | NEB | M2080S | Enzymatic kit for in vitro capping of carrier molecules |
| Wash buffer A | Cap trapping washes. Preparation is described in Murata et al. 2014. | ||
| Wash buffer B | Cap trapping washes. Preparation is described in Murata et al. 2014. | ||
| Wash buffer C | Cap trapping washes. Preparation is described in Murata et al. 2014. |
Riferimenti
- Shiraki, T., et al. Cap analysis gene expression for high-throughput analysis of transcriptional starting point and identification of promoter usage. Proceedings of the National Academy of Sciences of the United States of America. 100 (26), 15776-15781 (2003).
- Haberle, V., Lenhard, B. Promoter architectures and developmental gene regulation. Seminars in Cell and Developmental Biology. 57, 11-23 (2016).
- Haberle, V., Stark, A. Eukaryotic core promoters and the functional basis of transcription initiation. Nature Reviews Molecular Cell Biology. 19 (10), 621-637 (2018).
- Andersson, R., et al. An atlas of active enhancers across human cell types and tissues. Nature. 507 (7493), 455-461 (2014).
- Consortium, E. P. An integrated encyclopedia of DNA elements in the human genome. Nature. 489 (7414), 57-74 (2012).
- Celniker, S. E., et al. Unlocking the secrets of the genome. Nature. 459 (7249), 927-930 (2009).
- Consortium, F., et al. A promoter-level mammalian expression atlas. Nature. 507 (7493), 462-470 (2014).
- Boyd, M., et al. Characterization of the enhancer and promoter landscape of inflammatory bowel disease from human colon biopsies. Nature Communications. 9 (1), 1661 (2018).
- Adiconis, X., et al. Comprehensive comparative analysis of 5'-end RNA-sequencing methods. Nature Methods. , (2018).
- Cvetesic, N., et al. SLIC-CAGE: high-resolution transcription start site mapping using nanogram-levels of total RNA. Genome Research. 28 (12), 1943-1956 (2018).
- Murata, M., et al. Detecting expressed genes using CAGE. Methods in Molecular Biology. 1164, 67-85 (2014).
- Langmead, B., Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nature Methods. 9, 357 (2012).
- A language and environment for statistical computing. Available from: https://www.R-project.org/ (2017)
- Haberle, V., Forrest, A. R., Hayashizaki, Y., Carninci, P., Lenhard, B. CAGEr: precise TSS data retrieval and high-resolution promoterome mining for integrative analyses. Nucleic Acids Research. 43 (8), e51 (2015).
- Poulain, S., et al. NanoCAGE: A Method for the Analysis of Coding and Noncoding 5'-Capped Transcriptomes. Methods in Molecular Biology. 1543, 57-109 (2017).
- Schon, M. A., Kellner, M. J., Plotnikova, A., Hofmann, F., Nodine, M. D. NanoPARE: parallel analysis of RNA 5' ends from low-input RNA. Genome Research. 28 (12), 1931-1942 (2018).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati