
Method Article
Perfiles de Complexome de Alta Resolución por Cryoslicing BN-MS Analysis
En este artículo
Resumen
Se presenta un protocolo BN-MS crioscrioso versátil que utiliza un microtome para perfiles de complexomo de alta resolución.
Resumen
Las proteínas generalmente ejercen funciones biológicas a través de interacciones con otras proteínas, ya sea en conjuntos dinámicos de proteínas o como parte de complejos establemente formados. Este último se puede resolver elegantemente según el tamaño molecular utilizando electroforesis de gel de poliacrilamida nativa (BN-PAGE). El acoplamiento de estas separaciones a la espectrometría de masas sensible (BN-MS) ha sido bien establecido y teóricamente permite una evaluación exhaustiva del complexomo extraíble en muestras biológicas. Sin embargo, este enfoque es bastante laborioso y proporciona una resolución y sensibilidad de tamaño complejo limitado. Además, su aplicación se ha restringido a abundantes proteínas mitocondriales y plastoides. Por lo tanto, para la mayoría de las proteínas, todavía falta información sobre la integración en complejos proteicos estables. Aquí se presenta un enfoque optimizado para el perfilado de complexome que comprende la separación BN-PAGE a escala preparatoria, el muestreo submilimétrico de carriles de gel anchos por corte criomicrotoya, y el análisis espectrométrico de masas con cuantificación de proteínas sin etiquetas. Los procedimientos y herramientas para los pasos críticos se describen en detalle. Como aplicación, el informe describe el análisis de complexoma de una fracción de membrana enriquecida con endosoma solubilizada de los riñones del ratón, con 2.545 proteínas perfiladas en total. Los resultados demuestran la identificación de proteínas de membrana uniformes y de baja abundancia, como los canales iónicos intracelulares, así como patrones complejos de ensamblaje de proteínas de alta resolución, incluidas las isoformas de glicosilación. Los resultados están de acuerdo con análisis bioquímicos independientes. En resumen, esta metodología permite la identificación integral e imparcial de los complejos proteicos (super)complejos y su composición de subunidades, proporcionando una base para investigar la estequiometría, el montaje y la dinámica de interacción de los complejos proteicos en cualquier sistema biológico.
Introducción
La separación BN-PAGE se acoplaba primero directamente al análisis LC-MS (BN-MS) por los grupos de investigación Majeran1 y Wessels2 utilizando la división manual de carriles de gel BN-PAGE. Sus análisis identificaron una serie de abundantes complejos proteicos de membrana con composición de subunidades conocidas a partir de plastoides vegetales y mitocondrias de células HEK, respectivamente. Sin embargo, estos análisis distan mucho de ser exhaustivos y no permitieron la identificación imparcial de conjuntos novedosos. El rendimiento de los espectrómetros de masas y los métodos de cuantificación sin etiquetas ha mejorado considerablemente desde entonces, lo que ha permitido análisis completos de BN-MS. Esto ha acuñado el término "perfilado complexome". Por ejemplo, Heide y sus compañeros de trabajo analizaron las mitocondrias del corazón de rata identificando y agrupando 464 proteínas mitocondriales, confirmando así muchos conjuntos conocidos. Además, encontraron que TMEM126B era una subunidad novedosa y crucial de un complejo de ensamblaje específico3. Se obtuvieron resultados comparables (con 437 perfiles de proteína mitocondrial) en un estudio paralelo de las mitocondrias de células HEK4.
A pesar de estas mejoras, han seguido varias cuestiones que restringen todo el potencial del BN-MS para la elaboración de perfiles de complexome. Una limitación importante es la resolución efectiva del tamaño de los complejos que está determinada por dos factores: la (i) calidad de la separación BN-PAGE, que depende de la uniformidad del gradiente de poros de la matriz de gel, así como de la estabilidad/solubilidad de los complejos de muestra, y (ii) tamaño de paso de muestreo de gel, que es en el mejor de los 1 mm cuando se utiliza el corte manual convencional5,6. La mala resolución de tamaño no sólo echa de menos las sutiles isoformas complejas y las heterogeneidades, sino que también afecta negativamente el rango dinámico y la confianza de la asignación y cuantificación de subunidades de novo imparciales.
Otros desafíos incluyen la precisión de la cuantificación de proteínas y la cobertura del rango dinámico real de abundancias de proteínas en la muestra mediante análisis espectrométricos de masas. Por lo tanto, la aplicación de perfiles de complexoma BN-MS ha permanecido restringida en gran medida a muestras biológicas con menor complejidad, alta expresión de complejos objetivo y propiedades favorables de solubilización (es decir, plastoides, mitocondrias y microorganismos)6,7,8,9,10.
Recientemente introdujimos BN-MS asistida por corte criomicrotome (csBN-MS), que combina un muestreo submilimétrico preciso de carriles de gel BN-PAGE con un análisis exhaustivo de la EM y un procesamiento de datos de EM elaborado para la determinación de perfiles proteicos con confianza11. La aplicación a una preparación de membrana mitocondrial a partir de cerebros de rata demostró una resolución efectiva de tamaño complejo no satisfecha y una cobertura máxima de subunidades de complejo de cadena respiratoria oxidativa (OXPHOS) (es decir, 90 de 90 MS accesibles). En este ejemplo también se identificaron varios conjuntos de proteínas novedosos.
Aquí se describen procedimientos optimizados para la separación BN-PAGE a escala preparatoria de los complejos proteicos (no restringido a una fuente biológica en particular), la fundición de grandes geles BN-PAGE preparativos, el corte de criomicrotome de carriles de gel amplios y los datos de EM Tratamiento. El rendimiento de la generación de perfiles de alta resolución está demostrado para una preparación compleja de proteínas a partir de membranas enriquecidas con endosomas renales de ratón. Por último, se analizan los beneficios del aumento de la resolución y la precisión de la cuantificación espectrométrica de masas.
Protocolo
1. Preparativo BN-PAGE
- Preparación de gel
- Utilice un sistema de electroforesis de gel vertical de formato medio a grande (>10 cm de distancia de separación del gel; 14 cm x 11 cm, espaciador de 1,5 mm) con un enfriamiento eficaz ajustado a 10 oC.
- Gel de gradiente de poro lineal o hiperbólico fundido (espaciadores de 1,5 a 3,0 mm) utilizando un mezclador de gradiente de dos cámaras de agitación accionado por una bomba (ver Tabla de Materiales y Reactivos). En el ejemplo presentado (gel de gradiente lineal 1%-13%):
- Preparar una solución de 13 ml para la cámara frontal (mezcla) que consta de: 13% de acrilamida (de 30% de solución en stock, 37,5:1,0 acrilamida:bisacrilamida), ácido aminocaproico de 0,75 M, 50 mM de Bis-Tris (pH a 7,0) y 10% glicerol.
- Preparar una solución de 10 ml para la cámara del depósito que consta de: 1% de acrilamida (de 30% de solución de stock, 37,5:1,0 acrilamida:bisacrilamida), ácido aminocaproico de 0,75 M, 50 mM de Bis-Tris (pH a 7,0) y detergente CL-47 al 0,2% .
- Inicie el agitador y agregue 30 microlitros de APS (peroxodisulfato de amonio, solución de stock del 10%) y 2,5 l de TEMED (N,N,N',N',N'-tetrametileeemetilediamina) y 2,5 microlitros de TEMED a la solución en la cámara frontal. Encienda la bomba y abra la válvula delantera (el flujo debe ajustarse para completar la fundición en 10 min). Después de 1 min añadir 90 s de APS y 5 l de TEMED a la solución en la cámara del depósito y abrir la conexión de la cámara.
- Deje que el gel polimerice lentamente pero a fondo durante al menos 24 horas a temperatura ambiente (RT) para generar un gradiente homogéneo de tamaño de poro. Cuando se mantiene húmedo, el gel polimerizado se puede almacenar en posición vertical a 4 oC durante un máximo de 1 semana.
NOTA: Intencionalmente, la parte superior del gel tendrá una consistencia suave / viscosa. Esto se eliminará más tarde, pero permite la entrada suave de proteínas en el gel, con un riesgo mínimo de precipitación de proteínas que de otro modo podría conducir a artefactos de migración (es decir, rayado o precipitación de proteínas).
- Preparación y carga de muestras
- Prepare las ranuras de carga insertando espaciadores apropiados (por ejemplo, tubos de silicio) entre las placas de vidrio para separar 0,5-2,0 mg de proteína. Las ranuras deben hacerse al menos 3 cm de ancho (o mejor, 5-6 cm de ancho).
- Solubiliza 2,5 mg de membrana (preparación enriquecida con endosoma de riñón de ratón) en 2 ml de tampón de solubilización que contiene 1% (p/v) detergente no desnaturalizante (ComplexioLyte CL-47) durante 30 minutos sobre hielo. Ultracentrífuga (corte de sedimentación a 200 S o menos; 130.000 x g/11 min se utiliza aquí).
- Concentrar el solubilisato en un corto 50%/20% (p/v, 0,3 ml cada uno) gradiente de paso de sacarosa por ultracentrifugación durante 1 h a 400.000 x g. El rendimiento final de la proteína debe ser de al menos 1 mg.
- Añadir 0.05% (p/v) Coomassie G-250 al solubilisato y cargar la muestra en el gel. Limite la carga proteica a una sección transversal de carril de gel de 10-15 g/mm2 para obtener una alta resolución y evitar artefactos resultantes de la precipitación proteica.
- Condiciones de funcionamiento de BN-PAGE
- Para tampones en ejecución, prepare un búfer de cátodo estándar que consista en tricine de 50 mM, 15 mM Bis-Tris y 0,01% coomassie G-250. Preparar un amortiguador de ánodo estándar compuesto por 50 mM Bis-Tris (pH a 7,0).
- Ejecuta un BN-PAGE preparativo a 10oC durante la noche utilizando un protocolo de voltaje de tres pasos13 que consta de: una fase de equilibrio durante 30 min a 100 V, luego una rampa lenta (3 h) a la tensión máxima (40-50 V/cm de longitud) que finalmente se mantiene durante al menos 6 h para enfoque final de las proteínas.
NOTA: Se recomienda pausar la electroforesis cuando el frente de migración ha alcanzado la mitad del gel e intercambiar el tampón de cátodo por un tampón fresco sin Coomassie G250. Esto ayuda a evitar los artefactos de precipitación en el gel resultante del colapso local de la estructura de los poros de la matriz.
2. Muestreo y digestión de gel
- Escisión de carriles de gel
- Después de la carrera, escanee los geles con fines de documentación mientras lo mantiene entre las placas de vidrio.
- Desmontar las placas e extirpar la(s) sección(s) de carril de interés.
- Tome una tira de muestra del carril para su análisis por 2D BN/SDS-PAGE y la tinción de proteínas o la hincha occidental (como se muestra en la Figura 1B)para determinar las regiones de interés, la resolución efectiva del tamaño complejo y la abundancia de proteínas.
- Arreglar los carriles de gel seleccionados dos veces durante al menos 30 minutos con 30% (v/v) de etanol y 15% (v/v) ácido acético.
- Transfiera la muestra al medio de incrustación y déjela empapar y equilibrar durante al menos 2 h a 4 oC, manteniendo la losa de gel en cámara lenta en un agitador orbital.
NOTA: La separación del gel debe inspeccionarse cuidadosamente para comprobar la calidad general de la separación y los artefactos de migración. Las bandas de gel que representan proteínas dominantes deben ser de intensidad libre de distorsión y homogéneas. Los artefactos locales en el gel deben ser extirpado o dejados fuera del análisis.
- Emincrustación y corte criomicrotome
NOTA: Esta es una versión mejorada del procedimiento de incrustación descrito y fotodocumentado previamente que permite la incrustación y corte de carriles de gel más amplios de hasta 8 cm11.- En primer lugar, cortar los carriles de gel fijos en secciones (aquí, 3 cm) exactamente paralelas al patrón de la parte frontal/banda de migración de proteínas. Para facilitar el manejo, coloque cada sección en un soporte de película de plástico con las mismas dimensiones.
- Transfiera los carriles en un tubo abierto con tapónes (cerrados en la parte inferior, perforados centralmente en la parte superior, ambos alineados con los extremos superior e inferior de la sección de gel).
- Sumerja el cilindro brevemente en nitrógeno líquido para iniciar rápidamente la solidificación. El medio de incrustación transparente se solidifica en cuestión de segundos y se vuelve de color blanco.
- Llene la cavidad con medio de incrustación, sumerja brevemente en nitrógeno líquido y congele el cilindro a -20 oC durante varias horas.
NOTA: Enfriar el cilindro rápidamente sumergiéndolo en nitrógeno líquido ayuda a evitar el desplazamiento de la losa de gel dentro del tubo. Debe evitarse la distorsión para garantizar una alta resolución en el siguiente análisis de la EP. - Después del desmontaje, retire la película de plástico y transfiera el bloque con la sección de gel incrustada a un cilindro de metal refrigerado, de mayor diámetro, colocado en un soporte plano (es decir, plato Petri) y sellado con medio de incrustación en el exterior del cilindro. Llene el cilindro con medio de incrustación y congele bien.
- Repita este procedimiento con el otro lado del cilindro para obtener un bloque sólido con superficies coplanares.
- Retire el bloque del cilindro, péguelo con un medio de incrustación en un soporte de metal preenfriado e inserte el soporte en la máquina de crioslice (criotome). La superficie del bloque debe estar cuidadosamente alineada con respecto al plano de corte. Permita que se equilibre a la temperatura óptima para el proceso de corte (aquí, -15 oC).
NOTA: Utilice un ciclo de corte manual que progresa lentamente de 0,1 mm de tamaño de paso hasta que golpee la superficie de la sección de gel incrustada para garantizar un posicionamiento correcto. - Cosecha las rodajas de gel una tras otra, con un espesor final deseado de 0,25 mm de tamaño de paso, y transfiéralas individualmente a tubos de reacción con propiedades de baja unión a proteínas.
NOTA: En esta configuración, las rodajas de gel uniformes se pueden obtener fácilmente tan delgadas como 0,1 mm y tan gruesas como 0,5 mm.
- Digestión tríptica
- Realizar la digestión tríptica en gel después de un lavado extensivo de las rodajas de gel (se recomiendan al menos tres rondas adicionales de lavado para eliminar los componentes poliméricos del medio de incrustación) siguiendo un procedimiento estándar11.
- Péptidos eluten secados al vacío y redissolve en ácido trifluoroacético del 0,5% (v/v) agitando a 37 oC (10 min) seguido de sonicación del baño (5 min) y una breve centrifugación.
3. Espectrometría de masas
- configuración de nanoHPLC y MS
- Cargue las muestras digeridas en una precolumna C18 (tamaño de partícula de 5 m; diámetro a 300 m) con ácido trifluoroacético del 0,05% (v/v) utilizando un nano-HPLC (libre de división) acoplado a un espectrómetro de masas con alta resolución.
- Elute capturó péptidos con un gradiente acuoso-orgánico (eluyente A): 5 min 3% B, 120 min de 3% B a 30% B, 20 min de 30% B a 99% B, 5 min 99% B, 5 min de 99% B a 3% B, 15 min 3% B (tasa de flujo a 300 nL/min).
NOTA: las rodajas de gel csBN-MS suelen dar como resultado muestras con abundancia de péptidos bajos a intermedios y un grado limitado de complejidad. Por lo tanto, el análisis nanoLC-MS/MS debe realizarse con una configuración que proporcione una sensibilidad y velocidad de secuenciación razonables, una alta resolución de masa (>100.000) y un rango dinámico máximo (efectivamente 3-4 órdenes de magnitud). Sin embargo, no requiere dimensiones de columna largas o gradientes de elución extendidos más allá de 3 h. - Separar los péptidos eludados en un emisor (i.d. 75 m; punta a 8 m) embalados manualmente aproximadamente 20 cm con material C18 (tamaño de partícula a 3 m). Electrospray las muestras a 2,3 kV (modo iónico positivo) en el capilar de transferencia calentado (250 oC) del espectrómetro de masas.
- Realizar análisis con los siguientes ajustes del instrumento11: tiempo máximo de inyección de MS/MS a 400 ms; duración de la exclusión: 60 s; umbral de señal mínimo: 5.000 recuentos, los 10 principales precursores fragmentados; anchura de aislamiento a 1,0 m/z).
NOTA: Para facilitar la calibración de la masa, el tiempo de retención y la asignación de señales de péptidos en un gran número de conjuntos de datos o mediciones, se recomienda realizar las respectivas series de medición de EM sin interrupciones ni cambios en los parámetros y el hardware ( es decir, en la misma columna/emisor C18).
- Identificación de proteínas (datos de MS evaluados como se describió anteriormente11)
- Extraiga listas de picos de espectros de iones de fragmentos utilizando la herramienta "msconvert.exe" (parte de ProteoWizard).
- Cambie todos los valores m/z precursores para cada conjunto de datos por el desplazamiento mediano m/z de todos los péptidos asignados a proteínas en una búsqueda preliminar de la base de datos con tolerancia de masa de péptidos de 50 ppm.
- Busque en las listas de picos corregidos con un motor de búsqueda adecuado (aquí, Mascot 2.6.2) contra todas las entradas del ratón de la base de datos UniProtKB/Swiss-Prot (versión 2018_11).
- Seleccione "Acetil (proteína N-término)", "Carbamidomethyl (C)", "Gln ? pyro-Glu (N-término Q), Glu piro-Glu (N-término E)", "Oxidación (M)" y "Propionamida (C)" como modificaciones variables.
- Ajuste la tolerancia de la masa de péptidos y fragmentos a 5 ppm y a 0,8 Da, respectivamente, y permita una escisión tríptica perdida. Establezca el límite de valor esperado para la identificación de péptidos en 0,5 o menos. Utilice una búsqueda de base de datos de señuelo para determinar la tasa de detección de falsos positivos (FDR). Establezca el FDR en 1% o aplique criterios de calidad adicionales para garantizar una identificación fiable.
NOTA: El experimento presentado identificó más de 3.500 proteínas, con un péptido medio FDR de 4,4 a 0,77% (n a 101 muestras de rodajas), o 3.000 proteínas cuando el péptido FDR se estableció en 1%. Es importante destacar que se utilizaron criterios más estrictos para la selección de proteínas perfiladas (2.568). Incluía todas las proteínas que se identificaron con al menos dos péptidos, al menos uno de ellos es específico de proteínas, en al menos una de las 101 muestras de rodajas.
- Cuantificación de proteínas
- Utilice intensidades de señal de péptidos (volúmenes máximos [PV]) para la cuantificación de proteínas que se obtienen de las exploraciones completas FT y corrigen el tiempo de retención y los cambios de masa utilizando el software adecuado (aquí, MaxQuant v1.6.3).
- Alinee los datasets de MS uno por uno para hacer referencia a los tiempos de elución de péptidos (promedio total) utilizando la regresión LOESS. Asigne PV a péptidos directamente (identificación basada en MS/MS) o indirectamente (es decir, en función de su m/z coincidente y tiempo de elución dentro de tolerancias muy estrechas).
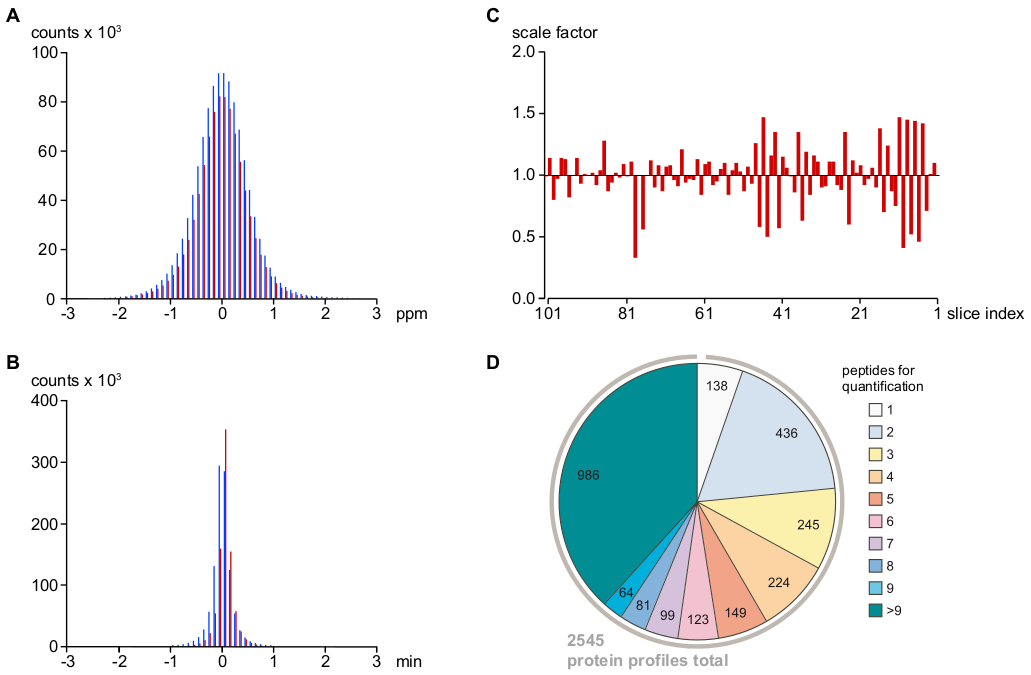
NOTA: Este protocolo utiliza software interno para la asignación de los péptidos "insertados". Los parámetros de conjunto dan como resultado tolerancias efectivas de coincidencia de tiempo de eliminación de tiempo de laución de 2 ppm y 1 min, respectivamente (véase la figura 2A,B). - Correcto para variaciones sistemáticas de la carga de péptidos y la eficiencia de la ionización mediante el reescalado de intensidad de péptidos calculado a partir de las diferencias medianas de intensidades relativas de péptidos entre muestras de rodajas vecinas(Figura 2C).
- Filtre los datos fotovoltaicos para los valores atípicos y las asignaciones de falsos positivos restantes identificadas por el análisis interno de coherencia fotovoltaica.
- Normalice los PV de cada péptido a sus valores máximos en todos los conjuntos de datos de sectores que producen perfiles de abundancia de péptidos relativos.
- Por último, calcule los perfiles de abundancia de proteínas relativas como promedios de al menos dos (y hasta seis o 50%, cualquier valor mayor) de los perfiles de péptidos mejor correlacionados sobre una ventana de tres rebanadas consecutivas. Esto permite puentear los valores fotovoltaicos que faltan y reducir el ruido.
NOTA: Esto finalmente dio lugar a 2.545 perfiles proteicos(Figura 2D).
- Caracterización de complejos proteicos
- Analice los perfiles de proteínas realizando primero la detección de picos utilizando el método de máxima local, y ajuste consecutivamente las distribuciones normales a estos picos, produciendo la posición (es decir, índice de rodajas o tamaño complejo aparente) de su máxima y FWHM (ancho completo a estos picos valores de intensidad media máxima) (recuadro de la Figura 4).
NOTA: En el conjunto de datos, los perfiles se analizan automáticamente mediante scripts personalizados. Los valores FWHM más pequeños son indicativos de la resolución de tamaño efectiva del enfoque (aquí, 6 x 0,25 x 1,5 mm). - Utilizar picos complejos proteicos de referencia con masa molecular definida (como se indica en la base de datos UniProtKB/Swiss-Prot) para el análisis de regresión lineal de los valores log10 (masa molecular predicha) para convertir índices de números de sectores en tamaños moleculares aparentes (es decir, tamaño complejo aparente en kDa).
NOTA: En este estudio se seleccionaron 23 complejos de marcadores en la muestra(Figura 4) basándose en (i) formas monodispersas de picos de perfil, (ii) soporte experimental de pesos moleculares y (iii) distribuciones a lo largo de las secciones de gel BN-PAGE investigadas.
- Analice los perfiles de proteínas realizando primero la detección de picos utilizando el método de máxima local, y ajuste consecutivamente las distribuciones normales a estos picos, produciendo la posición (es decir, índice de rodajas o tamaño complejo aparente) de su máxima y FWHM (ancho completo a estos picos valores de intensidad media máxima) (recuadro de la Figura 4).
Resultados
La gran mayoría de los estudios convencionales de BN-MS, así como el enfoque csBN-MS de alta resolución recientemente establecido se han aplicado a preparaciones mitocondriales y plastidas que son (i) fácilmente disponibles, (ii) tienen una complejidad limitada, y (iii) expresan complejos proteicos objetivo (membrana) a altas densidades. Este protocolo amplía la aplicación de perfiles de complexome de alta resolución a membranas no mitocondriales que expresan proteínas de bajo abundante, de las cuales se dispone de poca información sobre su integración en complejos. Para fines de demostración, elegimos una preparación de membrana enriquecida con endosomas a partir del riñón de ratón obtenida por centrifugación de gradiente de densidad.
La optimización de esta preparación se ha guiado por la proteína marcadora TPC1 que forma canales iónicos intracelulares predominantemente localizados a temprano y endosomas de reciclaje12. También se expresa altamente en células tubulares proximales renales, como se muestra en el análisis inmunohistoquímico de secciones de tejido renal(Figura 1A). Estas membranas fueron suavemente solubilizadas (ComplexioLyte 47 a una baja relación proteína:detergente de 1:8) y se concentraron en un cojín de sacarosa por ultracentrifugación. Este último resultó ser un paso importante para eliminar el exceso de componentes de menor peso molecular (es decir, detergentes, lípidos, sales, polímeros orgánicos y metabolitos) que tienden a afectar negativamente a la resolución de las separaciones preparativas de BN-PAGE.
La separación compleja en un gel de gradiente de poliacrilamida nativo de 1%-13% (p/v)(Figura 1B,panel medio) mostró bandas de proteínafuertemente teñidas con muy pocos artefactos de migración. La separación SDS-PAGE de una tira de gel BN-PAGE estrecha(Figura 1B, marco en caja en rojo) como segunda dimensión seguida de análisis de manchas occidentales mostró un patrón bien resuelto de poblaciones complejas asociadas a TPC1 distintas(Figura 1B , panel superior, marcado por flechas rojas), probablemente resultante de la asociación con subunidades de proteínas adicionales y/o modificaciones posttranslacionales (como la glicosilación12). Una sección de 3 cm de interés fue extirpada, fija y procesada para el corte de criomicrotome como se describe11. Los pasos individuales de este procedimiento (en particular, la alineación precisa de la sección de gel ancho), que es de importancia crítica para preservar la resolución durante el muestreo, se documentan en el vídeo adjunto. La sección de gel incrustada fue finalmente cortada en 101 rodajas de gel con un espesor uniforme de 0,25 mm(Figura 1B,panel inferior), que fueron digeridas y analizadas por separado mediante espectrometría de masas con acoplamiento LC de alto rendimiento.
Además de la resolución de tamaño, la calidad de la cuantificación de proteínas es clave para el perfilado de complexome exitoso. Con la configuración y los ajustes de La EM utilizados, el análisis de las muestras fue bastante completo, lo que resultó en una identificación media de más de 1.000 proteínas y 10.000 péptidos (8.200 de los cuales eran específicos de proteínas) por rodaja, y alrededor de 3.000 proteínas y 43.000 péptidos (38.500 de los cuales eran específicos de proteínas) en total. Sin embargo, debido a la naturaleza estocástica de la secuenciación de MS/MS dependiente de datos y sus limitaciones en el rango dinámico, la información de intensidad seguía siendo fragmentaria para proteínas menos abundantes. Por lo tanto, se realizó un elaborado procedimiento de procesamiento de datos de MS11que se basa en la asignación precisa de señales de péptidos (volúmenes máximos [PV] - intensidades de señal relacionadas con péptidos integradas a lo largo de m/z y tiempo) a lo largo de toda la serie de conjuntos de datos.
Como se muestra en la Figura 2A,B, las desviaciones de las señales de péptidos en masa y tiempo de retención que quedaron después de la calibración fueron idénticas para los PV secuenciados por EM y para los PV asignados indirectamente (con tolerancias muy estrechas de <1 ppm y <0,5 min para el 95% de los los PV), indicativo de una tasa muy baja para la asignación fotovoltaica falso-positiva. Los valores atípicos restantes se filtraron en función de su coherencia con otros PV relacionados. Puesto que todas las mediciones de MS se realizaron consecutivamente en la misma configuración de LC-MS sin cambios en los parámetros o componentes de hardware, variaciones de ejecución a ejecución (determinadas como la mediana de todas las intensidades fotovoltaicas en una muestra en relación con las de los sectores vecinos) fueron pequeños y fácilmente eliminados mediante el reescalado de los conjuntos de datos fotovoltaicos(Figura 2C). La información de intensidad del péptido resultante se utilizó entonces para reconstruir 2.545 perfiles de abundancia relativa de proteínas. Como se muestra en la Figura 2D,más del 75% de estos perfiles proteicos se basaron en al menos tres péptidos específicos de proteínas independientes.
A continuación, el protocolo evaluó la relevancia del tamaño escalonado del muestreo de gel BN-PAGE para la resolución de complejos proteicos. Para ello, los datasets de sectores se unieron sumando la información fotovoltaica de dos, tres o cuatro sectores consecutivos, simulando así el resultado para tamaños de paso de 0,5 mm, 0,75 mm y 1 mm (en comparación con el muestreo original de 0,25 mm). La Figura 3 ilustra los perfiles de abundancia resultantes para la proteína TPC1 como ejemplo (A-D). A 0,25 mm, las intensidades relativas y la separación del tamaño de las poblaciones complejas asociadas a TPC1(Figura 3A)estaban muy bien de acuerdo con los resultados del análisis de manchas occidentales(Figura 1B,panel superior); aunque, el perfil mostró algo de ruido, principalmente resultante de los valores faltantes ("huecos") en la matriz fotovoltaica utilizada para la cuantificación.
La unión de dos sectores correspondientes a 0,5 mm mantuvo las intensidades correctas y la separación de los complejos asociados al TPC1 y el ruido de cuantificación eliminado(Figura 3B). Por el contrario, los tamaños de paso más grandes de 0,75 mm y 1 mm(Figura 3C,D)llevaron a una pérdida de resolución de tamaño y abolió la discriminación de las subpoblaciones complejas de TPC1. Cabe señalar que la gran mayoría de los análisis convencionales publicados de BN-MS utilizan rebanadas de 2 mm cortadas manualmente (alrededor de 60 para cubrir todo el carril de gel)7,8,9,10.
La conversión de la distancia de migración o el índice de rodajas al tamaño molecular se basa generalmente en marcadores, ya sean proteínas estándar nativas disponibles comercialmente o complejos de proteínas endógenos bien caracterizados con composición de subunidades conocidas (principalmente [super] complejos de la cadena respiratoria oxidativa mitocondrial [OXPHOS])13. Sin embargo, dado que la separación BN-PAGE se basa en la sección transversal molecular eficaz que está determinada no sólo por la masa molecular, sino también por la estructura 3D y el número de lípidos asociados, detergente y moléculas de coomassie, las proteínas individuales pueden mostrar desviaciones más grandes. Por lo tanto, fue elegido para utilizar conjuntos más grandes de complejos proteicos como marcadores11. La gráfica de la Figura 4 muestra 23 marcadores seleccionados con una subunidad representativa que se muestra como un círculo negro, que indica los valores log10 de su masa molecular predicha (según la base de datos UniProtKB/Swiss-Prot) vs. el índice de sector del máximo máximo máximo de perfil correspondiente. Estos últimos se obtuvieron de los ajustes gaussianos automatizados a los datos de abundancia relativa, como se muestra en el recuadro de la Figura 4 que muestra el ejemplo con chaperona BCS1. La regresión lineal (línea roja) proporcionó una función para convertir los valores del índice de la rebanada a tamaños moleculares aparentes, que oscilaban entre 160-630 kDa, a lo largo de la sección de gel investigada.
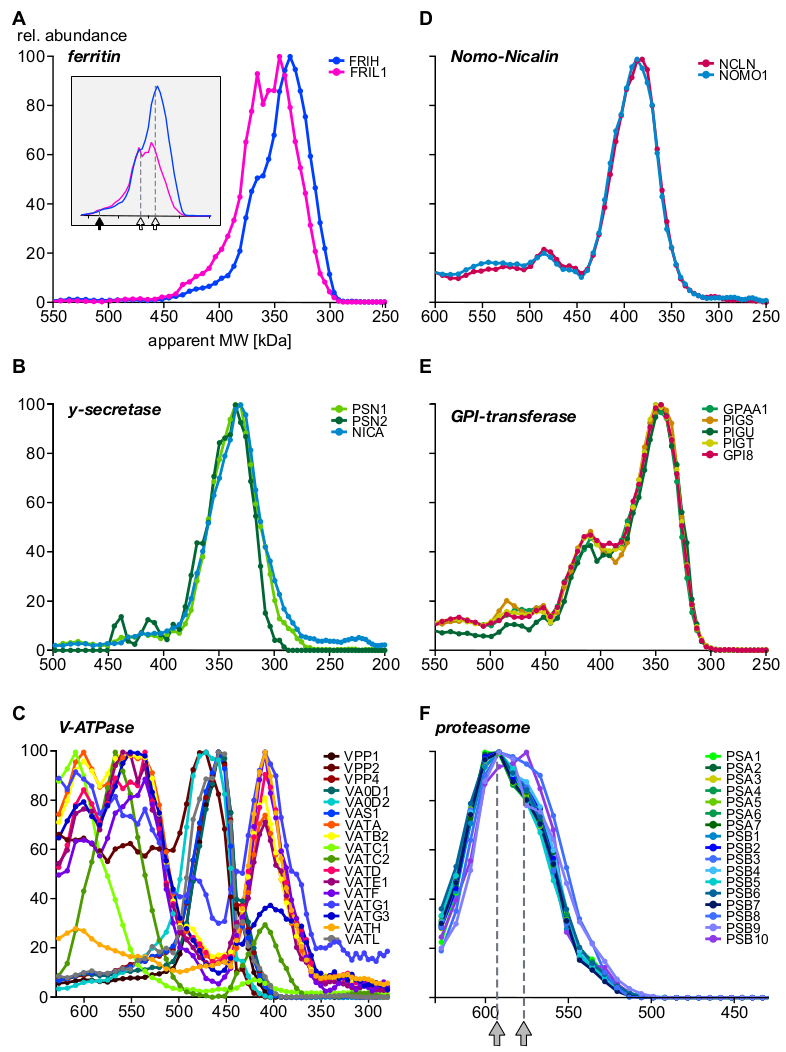
Por último, el análisis proporcionó información sobre complejos bien caracterizados y demostró la existencia de nuevas subunidades y ensamblajes complejos. En la Figura 5 se muestran ejemplos que destacan diferentes aspectos del complexomo (A-C: proteínas expresadas o preferiblemente ubicadas en compartimentos endosomales; D-F: complejos de otras localizaciones subcelulares). Se sabe que la proteína transportadora de hierro ferritina forma complejos a partir de 24 subunidades ligeras (FRIL1) y/o pesadas (FRIH), con un peso molecular total de 440 kDa14 (Figura 5A,flecha llena). Los perfiles de subunidades(Figura 5A)sugieren la existencia de al menos dos formas más pequeñas del complejo (con una masa aparente de 360 kDa y 340 kDa; flechas abiertas) con estequiometrías distintas de cadena pesada/ligera (mejor visible después de reescalar abundancias, enelconjunto de la Figura 5A) que están abundantemente presentes en los endosomas.
Por el contrario, los complejos de nicalin-nomo115 (Figura 5D), el complejo de núcleo gamma-secretasa16 (Figura 5B)y la maquinaria GPI-transamidasa17 (Figura 5E) muestran fijos proporciones de abundancia de sus subunidades principales en toda la gama de tamaños e independientes de la asociación con proteínas adicionales. Esto indica que sus subunidades son exclusivas entre sí. Vacuolar H+-ATPases son complejos multiproteicos ensamblados a partir de un conjunto de más de 20 subunidades de forma modular con un peso molecular total de alrededor de 900 kDa. La Figura 5C revela subcomplejos con composiciones distintas de al menos 17 subunidades, ya sea representando productos intermedios biológicos (dis) de ensamblaje o subcomplejos generados por las condiciones experimentales, algunos de los cuales también han sido observado en un reciente estudio de BN-MS18. Otro ejemplo complejo multiproteico es el proteosoma19 (Figura 5F). Una inspección minuciosa de los perfiles de abundancia de las subunidades alfa y beta que forman el núcleo de proteosoma 20S sugiere dos grandes poblaciones complejas con sutiles diferencias de tamaño (590 kDa y 575 kDa, indicadas por flechas grises) e integración de tres subunidades beta.
En resumen, el perfil de complexoma csBN-MS de membranas renales enriquecidas con endosomas proporciona resultados completos y detallados con respecto a la (i) integración de proteínas diana uniformes y de bajo abundante en complejos, ii) composición compleja general de subunidades y estoquiometría, y iii) heterogeneidades complejas, subestructuras y (dis)intermedios de ensamblaje.

Figura 1: Separación BN-PAGE preparativa de membranas enriquecidas con endosomaso solubilizado del riñón del ratón utilizando el canal intracelular TPC1 como marcador. (A) Localización inmunohistoquímica de la proteína TPC1 en túbulos proximales renales mediante microscopía confocal. Verde: anti-TPC1 anticuerpo12 tinción visualizada con Secundaria Cy3-biotinylated cabra anti-conejo IgG; rojo: tetragonolobus de loto biotintilado lectina (LTL, 10 g/ml, conjugado FITC) que marca la superficie luminal de las células del túbulo proximal. El recuadro muestra la tinción de una sección correspondiente de un riñón TPC1-KO como un control negativo. Las barras de escala blancas son de 20 m. También cabe destacar la fuerte expresión de TPC1 en vesículas intracelulares, conocida según los experimentos independientes para representar los endosomas tempranos y el reciclaje12. (B) Separación BN-PAGE preparativa de 2,5 mg de membranas enriquecidas con endosomaso solubilizado en un gel de gradiente de poliacrilamida del 1%-13% (p/v). Se cortó un carril estrecho (enmarcado en rojo) para el posterior análisis sDS-PAGE/western blot (panel superior), resolviendo diferentes complejos asociados a TPC1 y patrones de glicosilación (flechas rojas: anti-TPC1/anticonejo HRP/ECL prime; verde: posiciones y predichas masas [MDa] de complejos de proteínas marcadoras identificados por la tinción total de proteínas [mancha de manchas de rubí SYPRO]) de la mancha. De derecha a izquierda: Na+/K+-transporte ATPase, dimer complejo del citocromo b-c1, ATP sintasa, NADH:ubiquinona oxidoreductasa. Una sección de 3 cm de interés del carril de gel fue extirpada, incrustada en el medio de incrustación de tejido, montada y cortada en 101 secciones (0,25 mm) a lo largo del frente de migración de proteínas utilizando un criomicrotome (panel inferior; ver enlace de vídeo). Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 2: Parámetros clave que determinan la precisión de la asignación y cuantificación de la señal MS, así como la profundidad del análisis. (A) Distribución de errores de masa relativos (en ppm) después de la calibración m/z de señales de péptido ssecuenciadas MS/MS (barras rojas) e indirectamente asignadas (es decir, basadas en tiempos de masa y retención estrechamente coincidentes, ver protocolo; barras azules) señales de péptidos. Esto sugiere un error de masa final de <1 ppm (para el 95% de las señales/PV asignados) y una tasa muy baja de asignaciones de falsos positivos. (B) Distribución de las desviaciones del tiempo de retención nano-HPLC del promedio total después de la alineación del tiempo de elución de las señales de péptido, utilizando la regresión de Loess (ver protocolo) y la codificación de color como se utiliza en (A). El error de tiempo es inferior a 30 s para >95% de las señales de péptido/PV asignados. (C) Variación de ejecución a ejecución de las intensidades totales de LA MS trazadas en relación con el promedio de las dos muestras vecinas. Estos factores de escala se aplicaron a las tablas fotovoltaicas sin procesar para minimizar los errores técnicos sistemáticos. (D) Información de péptido utilizada para calcular perfiles de abundancia relativa de proteínas. Después de filtrar los péptidos específicos de proteínas para valores atípicos, mal puntuados o identificaciones individuales (ver protocolo), se determinaron 2.545 perfiles de abundancia de proteínas, >75% de los cuales se basaron en al menos tres péptidos con confianza razonable. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3: Impacto crítico del tamaño del paso en el muestreo de gel en la resolución de complexome de TPC1. Los conjuntos de datos se unieron sumando las intensidades de la señal en grupos de 1, 2, 3 y 4 sectores consecutivos (A-D, respectivamente) y se procesaron de forma idéntica para simular diferentes tamaños de paso en el corte de gel como se indica. El perfil TPC1 muestra algo de ruido (sobremuestreo) a 0,25 mm, pero una resolución de buen tamaño de tres poblaciones complejas (véase también la figura 1B),que se conserva en gran medida con un ancho de paso de 0,5 mm. La discriminación de estas poblaciones se pierde a medida que se aborda 1 mm. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 4: Determinación del peso molecular aparente. Se utilizaron 23 complejos de marcadores con composición molecular definida (como se indica, según UniProtKB/Swiss-Prot) como marcadores de tamaño. Los valores logarítmicos de sus pesos moleculares esperados (en kDa) se trazaron frentea . el índice máximo de rebanada del perfil de la subunidad de proteína representativa indicada (círculos rellenos en negro). La regresión lineal ajustada a estos datos (línea roja) proporcionaba una función que convertía los valores de índice de sectores en pesos moleculares aparentes. Los máximos máximos máximos se determinaron mediante un conjunto gaussiano automatizado que se ajusta a los picos de perfil de proteína, como se muestra en el recuadro (derecha) de la proteína de chaperona BCS1 (datos primarios en azul, límites de ajuste indicados por líneas naranjas, función de ajuste en rojo). Además, se ajustan a los anchos medios máximos de pico determinados (línea verde, 6,5 rodajas o 1,6 mm para el ejemplo mostrado) con los complejos de enfoque más nítidos que abarcan alrededor de un gel de 1,5 mm. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 5: Ejemplos de perfiles de subunidades complejas proteicas. Abundancia relativa de proteínas vs. peso molecular aparente trazado para la cadena pesada y ligera de ferritina (A) revelando heterogeneidad molecular de la estequiometría subunidad de ferritina, más claramente visible después de reescalar las abundancias (inset). Las flechas rellenas y las flechas abiertas denotan el complejo completo (440 kDa) y dos subcomplejos, respectivamente. Las subunidades gamma-secretase (B) se integran cuantitativamente en una población compleja de un solo núcleo. Los subcomplejos de vacuolar H+-ATPases(C) exhibieron múltiples conjuntos con composición de subunidades distintas, todos expresados en endosomas. Las proteínas nomo1 y nicalina(D)formaban un complejo exclusivo (la GPI-transamidasa), que es una maquinaria enzimática multisubunidad que forma varios complejos. (E) El complejo de núcleo de proteasoma 20S que muestra (F) un patrón sutil de subcomplejocon dos poblaciones indicadas por flechas en gris, todas originadas por otras localizaciones subcelulares. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Discusión
El estudio presentado se basó en la técnica csBN-MS previamente comparada con una preparación mitocondrial11 e incorporó mejoras en la preparación de muestras, procesamiento de gel y evaluación de datos de MS. El análisis centrado de una sección del gel BN-PAGE de separación a gran escala proporcionó un conjunto completo de datos que muestran medidas de calidad comparables al estudio con membranas mitocondriales. Los errores de masa y tiempo de retención, así como las variaciones de ejecución a ejecución, se mantuvieron muy bajos y proporcionaron la base para determinar perfiles confiables de abundancia de proteínas. La resolución del tamaño parecía ser buena, con anchos máximos de media máxima tan bajos como seis sectores (correspondientes a 1,5 mm, Figura 4) y diferencias de tamaño relativas de menos del 10% resueltas(Figura 3, Figura 5A). Estos valores no cumplían plenamente con la calidad de resolución de tamaño del análisis anterior de csBN-MS de las mitocondrias (a pesar del tamaño de paso de muestreo de gel más pequeño elegido), pero son significativamente mejores que el rendimiento del BN-MS convencional o la 20 que recientemente se han popularidos.
La importancia de una resolución de tamaño complejo efectiva alta se subraya en el experimento de simulación de la Figura 3 (utilizando los complejos asociados a TPC1) que difícilmente se puede resolver mediante el análisis de la mancha occidental 2D BN/SDS-PAGE(Figura 1B). Estos resultados sugieren que el corte de 0,25 mm en este caso dio lugar a un cierto sobremuestreo, pero esto todavía resultó ser útil para la eliminación del "ruido de cuantificación" sin comprometer la resolución efectiva del tamaño. Por lo tanto, en línea con los resultados anteriores11, un tamaño de paso de muestreo de 0,3 mm es generalmente recomendable.
En particular, la discriminación de los complejos asociados a TPC1 se pierde por completo con el muestreo de gel de 1 mm, que es el tamaño de paso más pequeño proporcionado por la división manual en BN-MS5convencional,6. Esto puede explicar el hecho de que a pesar de que las potentes tecnologías de LA MS están disponibles, muy pocos complejos proteicos y subunidades han sido identificados de novo por perfiles de complexome. Además de su buena potencia de resolución, csBN-MS ofrece una alta versatilidad. Los complejos unidos a membranas y los complejos de proteínas solubles que van desde 50 kDa a varios MDa se pueden resolver eficazmente en un solo experimento con un sesgo mínimo11. Esto contrasta con técnicas de separación alternativas utilizadas para perfiles de complexome como la exclusión de tamaño o la cromatografía de intercambio iónico, que funcionan con subconjuntos de proteínas solubles con ciertos rangos de tamaño o propiedades de carga. En el lado negativo, csBN-MS es menos escalable (carga máxima de 3 mg de proteína por gel), puede ser técnicamente difícil y no se puede automatizar.
En general, los resultados demuestran que la elaboración de perfiles de complexoma basados en csBN-MS se puede aplicar con éxito a objetivos no mitocondriales, pero también indican algunos desafíos asociados. Por lo tanto, la extracción eficiente y la estabilidad bioquímica de los complejos proteicos requieren más optimización, y pasos de limpieza y todavía pueden ser limitados. Dentro de la ventana de tamaño investigada, el número de complejos de proteínas monodispersas bien enfocados fue de hecho considerablemente menor (datos no mostrados) en comparación con una muestra mitocondrial. También se recomienda reducir las cargas de muestra BN-PAGE para obtener una separación de gel aceptable. Las cargas más altas pueden requerir carriles de gel más amplios que son más difíciles de procesar correctamente para el corte (ver vídeo adjunto). Además, la complejidad proteica de las muestras era mayor (alrededor de dos veces) que los digeridos derivados de las mitocondrias, lo que llevó a que falten más valores fotovoltaicos y un rango dinámico reducido. De hecho, algunas proteínas pequeñas que se espera que formen parte de los complejos mostrados en la Figura 5 faltaban en los análisis. Estos problemas se pueden resolver en el futuro mediante el uso de instrumentos de EM más rápidos y sensibles o modos de adquisición independientes de datos.
La preparación de muestras es muy crítica para la recuperación de complejos proteicos, la estabilidad y la calidad de la separación del gel. Los parámetros y procedimientos deben optimizarse para cada tejido fuente, lisato celular, membrana (fracción) y complejo proteico de interés. Se proporcionan las siguientes recomendaciones generales que pueden ayudar a ampliar las aplicaciones de csBN-MS:
(i) Preparación de muestras frescas y evitar calentamiento/congelación, diluciones fuertes, cambios en las condiciones de amortiguación y retrasos innecesarios;
(ii) Usar tampones que estén esencialmente desprovistos de sales (reemplazar con 500-750 mM de betaína o ácido aminocaproico), alrededor de un pH neutro, y que contenga hasta un 1% (p/v) de detergente no desnaturalizante (relación proteína:detergente entre 1:4-1:10 para la solubilización de la membrana complejos proteicos, sin detergente necesario para los complejos proteicos solubles);
(iii) Pruebas cuidadosas y ajuste de las condiciones del detergente mediante BN-PAGE analítico, ya que estos pueden afectar fuertemente a la eficiencia de la solubilización compleja, la representación de complejos proteicos de membrana en la muestra, la estabilidad y la homogeneidad de micelas de proteínas y detergentes. Estos últimos son requisitos previos para que las proteínas se enfoquen como bandas distintas/poblaciones complejas en geles BN-PAGE. La literatura anterior ofrece una amplia gama de detergentes neutros. Sin embargo, DDM (n-dodecílalo-d-maltoside)1,2,4,5,6 y digitalina3,5,7,8, 9,10,13,18 han sido las opciones más populares para los análisis BN-MS hasta el momento. Debe subrayarse que cualquier condición detergente representa necesariamente un compromiso entre la eficiencia de la solubilización y la preservación de las interacciones proteicas y puede no ser igualmente adecuada para todos los tipos de proteína objetivo y material de origen;
(iv) Extracción de polímeros cargados como fibrillas, filamentos, polilisina, ADN y abundantes componentes de menor peso molecular (es decir, metabolitos, lípidos o péptidos). Esto se puede lograr mediante ultracentrifugación, filtración de gel o diálisis. Esto es particularmente importante para los lisados celulares o tisulares totales;
(v) Adición de Coomassie G-250 (concentración final 0,05%-0,1%) y sacarosa (para aumentar la densidad de carga, concentración final 10%-20% [p/v]) a la muestra justo antes de la carga, para borrar por ultracentrifugación corta, cargar la muestra sin perturbación e iniciar la carrera inmediatamente después.
Como perspectiva futura, el perfilado de complexoma basado en csBN-MS ofrece opciones de multiplexación para estudiar dinámicas complejas de proteínas o cambios relacionados con condiciones biológicas específicas. La separación combinada de muestras etiquetadas metabólicamente como propuesta para la elaboración de perfiles21 basada en la exclusión de tamaño parece sencilla, pero puede verse obstaculizada por el intercambio espontáneo de subunidades en complejos que se producen independientemente de la separación utilizada Método. Alternativamente, las muestras etiquetadas se pueden resolver en carriles de gel vecinos, que luego se pueden co-cortar o combinar después de la digestión para el análisis diferencial con alta sensibilidad y robustez.
Divulgaciones
El autor Uwe Schulte es un empleado y accionista de Logopharm GmbH que produce ComplexioLyte 47 utilizado en este estudio. La compañía proporciona reactivos ComplexioLyte a instituciones académicas sin fines de lucro.
Agradecimientos
Este estudio fue apoyado por la Deutsche Forschungsgemeinschaft (DFG, Fundación Alemana de Investigación) – Project-ID 403222702 – SFB 1381 y bajo la Estrategia de Excelencia de Alemania CIBSS - EXC-2189 - Project ID 390939984. Agradecemos a Katja Zappe por la asistencia técnica.
Materiales
| Name | Company | Catalog Number | Comments |
| 30% Acrylamide/Bis Solution, 37.5:1 | Bio Rad | #1610158 | Recommended for acrylamide gradient gel solutions up to 13% |
| 30% Acrylamide/Bis Solution, 19:1 | Bio Rad | #1610154 | Recommended for acrylamide gradient gel solutions >13% |
| SYPRO Ruby Protein Blot Stain | Bio Rad | #1703127 | Total protein stain on blot membranes; sensitive and compatible with immunodetection |
| Coomassie Brilliant Blue G-250 | Serva | no. 35050 | Centrifugate stock solutions prior to use |
| ComplexioLyte 47 | Logopharm | CL-47-01 | Ready-to-use detergent buffer (1%) for mild solubilization of membrane proteins |
| Embedding Medium / Tissue Freezing Medium | Leica Biosystems | 14020108926 | Embedding medium for gel sections to be sliced by a cryo-microtome |
| Immobilon-P Membrane, PVDF, 0,45 µm | Merck | IPVH00010 | |
| ECL Prime Western Blotting Detection Reagent | GE Healthcare | RPN2232 | |
| Plastic syringe with rubber stopper, 20-30 ml | n.a. | n.a. | any supplier, important for making gel section embedding tool |
| broad razor blade | n.a. | n.a. | any supplier, for BN-PAGE gel trimming / excision of lanes |
| metal tube / cylinder, ca. 4 cm long | n.a. | n.a. | mold for embedding and freezing of gel samples |
| Protein LoBind Tubes, 1.5 ml | Eppendorf | Nr. 0030108116 | highly recommended to minimize protein/peptide loss due to absorption |
| sequencing-grade modified trypsin | Promega | V5111 | |
| C18 PepMap100 precolumn, particle size 5 µm | Dionex / Thermo Scientific | P/N 160454 | |
| PicoTip emitter (i.d. 75 µm; tip 8 µm) | New Objective | FS360-75-8 | |
| ReproSil-Pur 120 ODS-3 (C18, 3 µm) | Dr. Maisch GmbH | r13.93. | columns packed manually |
| rabbit anti-TPC1 antibody | Gramsch Laboratories | custom production | described in Castonguay, et al., 2017 (Reference 12) |
| Cy3-biotinylated goat anti-rabbit IgG | Vector Laboratories | CY-1300 | described in Castonguay, et al., 2017 (Reference 12) |
| biotinylated Lotus tetragonolobus lectin, FITC-conjugated | Vector Laboratories | #B1325 | described in Castonguay, et al., 2017 (Reference 12) |
| cryo-microtome Leica CM1950 | Leica Biosystems | 14047743905 | |
| Mini Protean II Cell with wetblot unit | Bio Rad | n.a. | for SDS-PAGE and Westernblot (not sold any more) |
| Penguin Midi Gel Electrophoresis System | PeqLab | n.a. | for BN-PAGE (not sold any more) |
| Zeiss Axiovert 200 M microscope + Photometrics Coolsnap 2 digital camera | Zeiss / Photometrics | n.a. | |
| peristaltic pump (IP high precision multichannel) | Ismatec | ISM940 | for casting of gradient polyacrylamide gels |
| gradient mixer with stirring (two chambers) | selfmade, alternatively Bio Rad | 1652000 or 1652001 | for casting of gradient polyacrylamide gels, manual provides instructions to cast linear or hyperbolic gradient gels (http://www.bio-rad.com/webroot/web/pdf/lsr/literature/M1652000.pdf) |
| ultracentrifuge Sorvall M120 with S80 AT3 rotor | Sorvall / Thermo Scientific | n.a. | for sample preparation (not sold any more) |
| UltiMate 3000 RSLCnano HPLC | Dionex / Thermo Scientific | ULTIM3000RSLCNANO | |
| Orbitrap Elite mass spectrometer | Thermo Scientific | IQLAAEGAAPFADBMAZQ |
Referencias
- Majeran, W., et al. Consequences of C4 Differentiation for Chloroplast Membrane Proteomes in Maize Mesophyll and Bundle Sheath Cells. Molecular & Cellular Proteomics. 7, 1609-1638 (2008).
- Wessels, H. J., et al. LC-MS/MS as an alternative for SDS-PAGE in blue native analysis of protein complexes. Proteomics. 9 (17), 4221-4228 (2009).
- Heide, H., et al. Complexome profiling identifies TMEM126B as a component of the mitochondrial complex I assembly complex. Cell Metabolism. 16 (4), 538-549 (2012).
- Wessels, H. J., et al. Analysis of 953 human proteins from a mitochondrial HEK293 fraction by complexome profiling. PLoS ONE. 8 (7), e68340 (2013).
- Wöhlbrand, L., et al. Analysis of membrane-protein complexes of the marine sulfate reducer Desulfobacula toluolica Tol2 by 1D blue native-PAGE complexome profiling and 2D blue native-/SDS-PAGE. Proteomics. 16 (6), 973-988 (2016).
- Takabayashi, A., et al. PCoM-DB Update: A Protein Co-Migration Database for Photosynthetic Organisms. Plant and Cell Physiology. 58 (1), e10 (2017).
- Senkler, J., et al. The mitochondrial complexome of Arabidopsis thaliana. The Plant Journal. 89 (6), 1079-1092 (2017).
- de Almeida, N. M., et al. Membrane-bound electron transport systems of an anammox bacterium: A complexome analysis. Biochimica et Biophysica Acta. 1857 (10), 1694-1704 (2016).
- Anand, R., Strecker, V., Urbach, J., Wittig, I., Reichert, A. S. Mic13 Is Essential for Formation of Crista Junctions in Mammalian Cells. PLoS ONE. 11 (8), e0160258 (2016).
- Eydt, K., Davies, K. M., Behrendt, C., Wittig, I., Reichert, A. S. Cristae architecture is determined by an interplay of the MICOS complex and the F1FO ATP synthase via Mic27 and Mic10. Microbial Cell. 4 (8), 259-272 (2017).
- Müller, C. S., et al. Cryoslicing Blue Native-Mass Spectrometry (csBN-MS), a Novel Technology for High Resolution Complexome Profiling. Molecular & Cellular Proteomics. 15 (2), 669-681 (2016).
- Castonguay, J., et al. The two-pore channel TPC1 is required for efficient protein processing through early and recycling endosomes. Scientific Reports. 7 (1), 10038 (2017).
- Schägger, H., Pfeiffer, K. Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. The EMBO Journal. 19 (8), 1777-1783 (2000).
- Banyard, S. H., Stammers, D. K., Harrison, P. M. Electron density map of apoferritin at 2.8-A resolution. Nature. 271 (5642), 282-284 (1978).
- Dettmer, U., et al. Transmembrane protein 147 (TMEM147) is a novel component of the Nicalin-NOMO protein complex. The Journal of Biological Chemistry. 285 (34), 26174-26181 (2010).
- Kimberly, W. T., et al. Gamma-secretase is a membrane protein complex comprised of presenilin, nicastrin Aph-1, and Pen-2. Proceedings of the National Academy of Sciences of the United States of America. 100 (11), 6382-6387 (2003).
- Hong, Y., et al. Human PIG-U and yeast Cdc91p are the fifth subunit of GPI transamidase that attaches GPI-anchors to proteins. Molecular Biology of the Cell. 14 (5), 1780-1789 (2003).
- Van Damme, T., et al. Mutations in ATP6V1E1 or ATP6V1A Cause Autosomal-Recessive Cutis Laxa. The American Journal of Human Genetics. 100 (2), 216-227 (2017).
- Budenholzer, L., Cheng, C. L., Li, Y., Hochstrasser, M. Proteasome Structure and Assembly. Journal of Molecular Biology. 429 (22), 3500-3524 (2017).
- Heusel, M., et al. Complex-centric proteome profiling by SEC-SWATH-MS. Molecular Systems Biology. 15 (1), e8438 (2019).
- Kristensen, A. R., Gsponer, J., Forster, L. J. A high-throughput approach for measuring temporal changes in the interactome. Nature Methods. 9 (9), 907-919 (2012).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados