
Method Article
Profilazione complesso ad alta risoluzione di Cryoslicing BN-MS Analysis
In questo articolo
Riepilogo
Per la profilazione complesso ad alta risoluzione viene presentato un versatile protocollo BN-MS crioscillante utilizzando un microtoma.
Abstract
Le proteine generalmente esercitano funzioni biologiche attraverso interazioni con altre proteine, sia in gruppi proteici dinamici che come parte di complessi formati stabilmente. Quest'ultimo può essere elegantemente risolto in base alle dimensioni molecolari utilizzando l'elettroforesi gel poliacrilamide nativo (BN-PAGE). L'accoppiamento di tali separazioni alla spettrometria di massa sensibile (BN-MS) è stato ben consolidato e teoricamente consente una valutazione esaustiva del complesso estraibile in campioni biologici. Tuttavia, questo approccio è piuttosto laborioso e fornisce una risoluzione e una sensibilità limitate e complesse. Inoltre, la sua applicazione è rimasta limitata ad abbondanti proteine mitocondriali e plastidi. Pertanto, per la maggior parte delle proteine, mancano ancora informazioni sull'integrazione in complessi proteici stabili. Presentato qui è un approccio ottimizzato per la profilazione complessoma che comprende la separazione BN-PAGE in scala puntuale, il campionamento sub-millimetrico di ampie corsie gel mediante affettare il criomicrotomi e l'analisi spettrometrica di massa con quantificazione delle proteine prive di etichette. Le procedure e gli strumenti per i passaggi critici sono descritti in dettaglio. Come applicazione, il rapporto descrive l'analisi complessoma di una frazione di membrana soluttivo arricchita da reni di topo, con 2.545 proteine profilate in totale. I risultati dimostrano l'identificazione di proteine della membrana uniformi e a bassa abbondanza, come i canali ionici intracellulari, nonché modelli di assemblaggio di proteine complessi e ad alta risoluzione, comprese le isoforme della glicosilazione. I risultati sono in accordo con analisi biochimiche indipendenti. In sintesi, questa metodologia consente un'identificazione completa e imparziale delle proteine (super)complessi e della loro composizione di sottounità, fornendo una base per studiare la stoichiometria, l'assemblaggio e la dinamica di interazione dei complessi proteici in qualsiasi sistema biologico.
Introduzione
La separazione BN-PAGE è stata prima accoppiata direttamente all'analisi LC-MS (BN-MS) dai gruppi di ricerca Majeran1 e Wessels2 utilizzando il taglio manuale delle corsie di gel BN-PAGE. Le loro analisi hanno identificato una serie di abbondanti complessi proteici di membrana con composizione nota di sottounità da plastidi vegetali e mitocondri cellulari HEK, rispettivamente. Tuttavia, queste analisi erano tutt'altro che complete e non consentivano l'identificazione imparziale di nuove assemblee. Da allora, le prestazioni degli spettrometri di massa e dei metodi di quantificazione senza etichette sono notevolmente migliorate, il che ha permesso analisi BN-MS complete. Questo ha coniato il termine "profilazione complesso". Ad esempio, Heide e colleghi hanno analizzato i mitocondri cardiaci del ratto identificando e raggruppando 464 proteine mitocondriali, confermando così molti gruppi noti. Inoltre, hanno trovato TMEM126B una nuova e cruciale sottounità di un complesso di assemblaggio specifico3. Risultati comparabili (con 437 profili di proteine mitocondriali) sono stati ottenuti in uno studio parallelo dei mitocondri a cellule HEK4.
Nonostante questi miglioramenti, sono rimasti diversi problemi che limitano il pieno potenziale di BN-MS per la profilazione complesso. Una delle principali limitazioni è l'effettiva risoluzione delle dimensioni dei complessi che è determinata da due fattori: la qualità (i) della separazione BN-PAGE, che dipende dall'uniformità del gradiente dei pori a matrice di gel, nonché dalla stabilità/solubilità dei complessi campione, e (ii) dimensione del passaggio del campionamento del gel, che è al massimo 1 mm quando si utilizza il taglio manuale convenzionale5,6. La scarsa risoluzione delle dimensioni non solo manca di sottili isoforme complesse ed eterogeneità, ma influisce anche negativamente sulla gamma dinamica e sulla fiducia dell'assegnazione e della quantificazione di sottounità imparziali e de novo.
Altre sfide includono la precisione della quantificazione delle proteine e la copertura dell'effettiva gamma dinamica di abbondanza di proteine nel campione mediante analisi spettrometrica di massa. Pertanto, l'applicazione della profilazione del complesso BN-MS è rimasta in gran parte limitata a campioni biologici con minore complessità, alta espressione dei complessi bersaglio e proprietà di solubilizzazione favorevoli (ad esempio, plastidi, mitocondri e microrganismi)6,7,8,9,10.
Recentemente abbiamo introdotto IL criomicrotome BN-MS (csBN-MS), che combina un preciso campionamento sub-millimetrico di corsie gel BN-PAGE con un'analisi completa della Ms e un'elaborata elaborazione dei dati MS per la determinazione dei profili proteici con fiducia11. L'applicazione di una preparazione della membrana mitocondriale da cervelli di ratto ha dimostrato una risoluzione effettiva delle dimensioni complesse in precedenza non soddisfatta e la massima copertura delle sottounità complesse della catena respiratoria ossidativa (OXPHOS) (cioè 90 di 90 MS accessibili). Questo esempio ha anche identificato una serie di nuovi gruppi proteici.
Di seguito sono descritte le procedure ottimizzate per la separazione BN-PAGE su scala punziose dei complessi proteici (non limitati a una particolare fonte biologica), la colata di grandi gel BN-PAGE preparativi, la suddivisione criomicrotomi di ampie corsie di gel e i dati MS processo. Le prestazioni della profilazione ad alta risoluzione sono dimostrate per una preparazione complessa proteica dalle membrane arricchite con endosomi rene di topo. Infine, vengono discussi i vantaggi derivanti dall'aumento della risoluzione e della precisione della quantificazione spettrometrica di massa.
Protocollo
1. Preparativo BN-PAGE
- Preparazione del gel
- Utilizzare un sistema di elettroforesi verticale a gel di formato medio-grande (>10 cm distanza di separazione gel; 14 cm x 11 cm, 1,5 mm distanziatore) con un raffreddamento efficace impostato su 10 .
- Lanciare gel a gradiente poro lineare o iperbolico (1,5-3,0 mm) utilizzando un miscelatore a due camere guidato da una pompa (vedere Tabella dei materiali e reagenti). Nell'esempio presentato (gel a gradiente lineare 1%-13%):
- Preparare una soluzione da 13 mL per la camera anteriore (miscelazione) costituita da: 13% acrilammide (dal 30% di soluzione stock, 37.5:1.0 acrilammide:bisacrilamide), 0.75 M acido aminocaproproico, 50 mM Bis-Tris (pH - 7,0), e 10% gislyol.
- Preparare una soluzione da 10 mL per la camera del serbatoio costituita da: 1% acrilammide (dalla soluzione di magazzino 30%, 37.5.1.0 acrilammide:bisacrilamide), 0,75 M di acido aminocaproproico, 50 mM Bis-Tris (pH - 7,0) e 0,2% CL-47 detergenti.
- Avviare lo staffe e aggiungere 30 microlitri di APS (ammonio peroxodisulfate, 10% soluzione stock) e 2,5 L di TEMED (N,N',N'-tetramethyl ethylediamine) e 2,5 microlitri di TEMED alla soluzione nella camera anteriore. Avviare la pompa e aprire la valvola anteriore (il flusso deve essere regolato per completare la colata in 10 min). Dopo 1 min aggiungere 90 l di APS e 5 L di TEMED alla soluzione nella camera del serbatoio e aprire la connessione della camera.
- Lasciare che il gel sia polimerizzato lentamente ma accuratamente per almeno 24 h a temperatura ambiente (RT) per generare un gradiente omogeneo delle dimensioni dei pori. Se mantenuto umido, il gel polimerizzato può essere conservato in posizione verticale a 4 gradi centigradi per un massimo di 1 settimana.
NOTA: Intenzionalmente, la parte superiore del gel avrà una consistenza morbida / sottile. Questo sarà successivamente rimosso, ma permette l'ingresso agevole di proteine nel gel, con un rischio minimo di precipitazioni proteiche che altrimenti potrebbero portare a manufatti di migrazione (cioè striature o precipitazioni proteiche).
- Preparazione e caricamento dei campioni
- Preparare gli slot di carico inserendo distanziali appropriati (ad esempio, tubi di silicio) tra le lastre di vetro per separare 0,5-2,0 mg di proteine. Le fessure devono essere fatte ad almeno 3 cm di larghezza (o meglio, 5-6 cm di larghezza).
- Solubilizzare 2,5 mg di membrana (preparazione arricchita endosomico rene di topo) in 2 mL di tampone di solubilità contenente 1% (w/v) detergente non denaturante (ComplexioLyte CL-47) per 30 min sul ghiaccio. Ultracentrifugate (sedimentazione cut-off 200 S o meno; 130.000 x g/11 min viene utilizzato qui).
- Concentrare il solubilisato su un breve 50%/20% (w/v, 0,3 ml ciascuno) sfumatura passo di sucrosi per ultracentrifugazione per 1 h a 400.000 x g. La resa proteica finale dovrebbe essere di almeno 1 mg.
- Aggiungere lo 0,05% (w/v) Coomassie G-250 al solulubile e caricare il campione sul gel. Limitare il carico proteico a una sezione trasversale della corsia di gel 10-15g/mm/mm per ottenere un'alta risoluzione ed evitare manufatti risultanti da precipitazioni proteiche.
- Condizioni di funzionamento BN-PAGE
- Per l'esecuzione di buffer, preparare un buffer catodo standard composto da 50 m tricine, 15 mM Bis-Tris e 0.01% Coomassie G-250. Preparare un buffer anodo standard costituito da 50 mM Bis-Tris (pH - 7,0).
- Eseguire un BN-PAGE costruttivo a 10 gradi durante la notte utilizzando un protocollo di tensione in tre fasi13 composto da: una fase di 30 min a 100 V, quindi una rampa lenta (3 h) a tensione massima (lunghezza gel 40-50 V/cm) che viene infine mantenuta per almeno 6 h per l'endpoint che si concentra sulle proteine.
NOTA: Si consiglia di mettere in pausa l'elettroforesi quando il fronte migratorio ha raggiunto il centro del gel e scambiare il buffer catodo con buffer fresco senza Coomassie G250. Questo aiuta a evitare artefatti di precipitazioni nel gel risultante dal collasso locale della struttura dei pori della matrice.
2. Campionamento e digestione di gel
- Escissione di corsie gel
- Dopo la corsa, scansionare i gel per scopi di documentazione mantenendoli tra le lastre di vetro.
- Smontare le piastre e accigliare le sezioni di corsia di interesse.
- Prendere una striscia campione della corsia per l'analisi da 2D BN/SDS-PAGE e colorazione proteica o gonfiore occidentale (come mostrato nella Figura 1B) per determinare le regioni di interesse, la risoluzione efficace delle dimensioni complesse e l'abbondanza proteica.
- Fissare due volte le corsie gel selezionate per almeno 30 min con 30% (v/v) di etanolo e 15% (v/v) acido acetico.
- Trasferire il campione al mezzo di incorporamento e lasciarlo in ammollo ed equilibrato per almeno 2 h a 4 gradi centigradi, mantenendo la lastra di gel al rallentatore su uno shaker orbitale.
NOTA: La separazione del gel deve essere attentamente ispezionata per la qualità complessiva della separazione e gli artefatti di migrazione. Le bande gel che rappresentano proteine dominanti devono essere prive di distorsione e omogenee in intensità. Gli artefatti locali sul gel devono essere asandati o esclusi dall'analisi.
- Incorporamento e taglio criomicrotoma
NOTA: Questa è una versione migliorata della procedura di incorporamento descritta e foto documentata in precedenza che consente l'incorporamento e il taglio di corsie gel più ampie fino a 8 cm11.- In primo luogo, tagliare le corsie gel fisse in sezioni (qui, 3 cm) esattamente parallelamente al modello anteriore/banda di migrazione delle proteine. Per una maneggevolezza più semplice, posizionare ogni sezione su un supporto pellicola di plastica con dimensioni uguali.
- Trasferire le corsie in un tubo aperto con tappi (chiuso sul fondo, centralmente perforato sulla parte superiore, entrambi perfettamente allineati con le estremità superiore e inferiore della sezione gel).
- Immergere brevemente il cilindro in azoto liquido per avviare rapidamente la solidificazione. Il mezzo di incorporamento trasparente si solidifica in pochi secondi e diventa di colore bianco.
- Riempire la cavità con il mezzo di incorporamento, immergerla brevemente in azoto liquido e congelare il cilindro a -20 gradi centigradi per diverse ore.
NOTA: Raffreddare rapidamente il cilindro immergendolo in azoto liquido aiuta a evitare lo spostamento della lastra di gel all'interno del tubo. La distorsione deve essere evitata per garantire un'elevata risoluzione nella seguente analisi degli Stati membri. - Dopo lo smontaggio, rimuovere la pellicola di plastica e trasferire il blocco con la sezione gel incorporato in un cilindro metallico raffreddato, di diametro più grande, posto su un supporto piatto (cioè piatto piatto) e sigillato con supporto di incorporamento all'esterno del cilindro. Riempire il cilindro con il supporto di incorporamento e congelare accuratamente.
- Ripetere questa procedura con l'altro lato del cilindro per ottenere un blocco solido con superfici a planari.
- Rimuovere il blocco dal cilindro, incollarlo con un supporto di incorporamento su un supporto metallico preraffreddato e inserire il supporto nella macchina crioslicing (criotome). La superficie del blocco deve essere accuratamente allineata rispetto al piano di sezionamento. Lasciare eclacazione alla temperatura ottimale per il processo di taglio (qui, -15 gradi centigradi).
NOTA: Utilizzare un ciclo di taglio manuale che progredisce lentamente di dimensioni di 0,1 mm fino a colpire la superficie della sezione gel incorporata per garantire il corretto posizionamento. - Raccogliere le fette di gel una dopo l'altra, con uno spessore finale desiderato di 0,25 mm di dimensione del passo, e trasferirle singolarmente in tubi di reazione con proprietà di legame a bassa proteina.
NOTA: In questo set-up, le fette di gel uniformi possono essere facilmente ottenute sottili come 0,1 mm e spesse come 0,5 mm.
- Digestione tryptica
- Eseguire la digestione triptica in gel dopo un ampio lavaggio delle fette di gel (si raccomandano almeno tre ulteriori cicli di lavaggio per rimuovere i componenti polimerici del mezzo di incorporamento) seguendo una procedura standard11.
- Peptidi eluiti a spirami e si dissolvono in acido trifluoroacetico dello 0,5% (v/v) scuotendo a 37 gradi centigradi (10 min) seguiti dalla sonicazione del bagno (5 min) e dalla breve centrifugazione.
3. Spettrometria di massa
- nanoHPLC e SM
- Caricare campioni digestione su una precolonna C18 (dimensione delle particelle - 5 m; diametro - 300 m) con 0,05% (v/v) acido trifluoroacetico utilizzando un nano-HPLC (privo di dividendi) accoppiato a uno spettrometro di massa con alta risoluzione.
- Peptidi catturati in eluturo con un gradiente acquoso-organico (eluente A): 5 min 3% B, 120 min dal 3% B al 30% B, 20 min dal 30% B al 99% B, 5 min 99% B, 5 min dal 99% B al 3% B, 15 min 3% B (velocità di flusso - 300 nL/min).
NOTA: le fette di gel csBN-MS in genere si traducono in campioni con abbondanza di peptidi da bassa a intermedia e un grado limitato di complessità. L'analisi nanoLC-MS/MS dovrebbe quindi essere eseguita con un set-up che fornisca una ragionevole sensibilità e velocità di sequenziamento, alta risoluzione di massa (>100,000) e un intervallo dinamico massimo (efficacemente 3-4 ordini di grandezza). Tuttavia, non richiede quote di colonna lunghe o gradienti di eluizione estesi oltre 3 h. - Peptidi eluiti separati in un emettitore (i.d. 75 m; punta 8 m) imballati manualmente circa 20 cm con materiale C18 (dimensione delle particelle - 3 m). Elettrospray i campioni a 2,3 kV (modalità iosone positivo) nel capillare di trasferimento riscaldato (250 gradi centigradi) dello spettrometro di massa.
- Eseguire analisi con le seguenti impostazioni dello strumento11: tempo massimo di iniezione MS/MS - 400 ms; durata dell'esclusione: 60 s; soglia minima del segnale: 5.000 conteggi, primi 10 precursori frammentati; larghezza di isolamento: 1,0 m/z).
NOTA: per facilitare la calibrazione della massa, il tempo di conservazione e l'assegnazione dei segnali dei peptidi in un gran numero di set di dati o misurazioni, si consiglia di eseguire le rispettive serie di misurazioni MS senza interruzioni o variazioni nei parametri e nell'hardware ( cioè sulla stessa colonna C18/emitter).
- Identificazione delle proteine (dati SM valutati come descritto in precedenza11)
- Estrarre le liste di picco dagli spettri di ioni di frammento utilizzando lo strumento "msconvert.exe" (parte di ProteoWizard).
- Spostare tutti i valori precursori di m/z per ogni set di dati in base all'offset m/z mediano di tutti i peptidi assegnati alle proteine in una ricerca preliminare nel database con tolleranza di massa peptide di 50 ppm.
- Cerca nelle liste di picco corrette con un motore di ricerca adatto (qui, Mascot 2.6.2) su tutte le voci del mouse del database UniProtKB/Swiss-Prot (release 2018_11).
- Selezionare "Acetil (Protein N-term)", "Carbamidomethyl (C)", "Gln piro-Glu (N-termine Q), Glu piro-Glu (N-termine E)", "Ossidazione (M)" e "Propionamide (C)" come modifiche variabili.
- Impostare la tolleranza di massa del peptide e della frammentazione rispettivamente su 5 ppm e 0,8 Da e consentire una cleavage a prova persa. Impostare il valore previsto cut-off per l'identificazione del peptide a 0,5 o meno. Utilizzare una ricerca nel database esca per determinare il tasso di individuazione dei falsi positivi (FDR). Impostare l'FDR su 1% o applicare criteri di qualità aggiuntivi per garantire un'identificazione affidabile.
NOTA: L'esperimento presentato ha identificato più di 3.500 proteine, con un Peptide FDR medio di 4,4 x 0,77% (n campioni di 101 fette), o 3.000 proteine quando il peptide FDR è stato impostato sull'1%. È importante sottolineare che sono stati utilizzati criteri più rigorosi per la selezione delle proteine profilate (2.568). Includeva tutte le proteine identificate con almeno due peptidi, almeno uno dei quali è specifico per le proteine, in almeno uno dei 101 campioni di fetta.
- Quantificazione delle proteine
- Utilizzare l'intensità del segnale peptide (volumi di picco [PV]) per la quantificazione delle proteine ottenute dalle scansioni complete FT e corrette per i tempi di ritenzione e gli spostamenti di massa utilizzando un software appropriato (qui, MaxQuant v1.6.3).
- Allineare i set di dati MS uno alla volta per fare riferimento (media totale) ai tempi di eluizione dei peptidi utilizzando la regressione LOESS. Assegnare i PV ai peptidi direttamente (identificazione basata su MS/MS) o indirettamente (cioè, in base al loro tempo di corrispondenza m/z e al tempo di eluizione entro tolleranze molto strette).
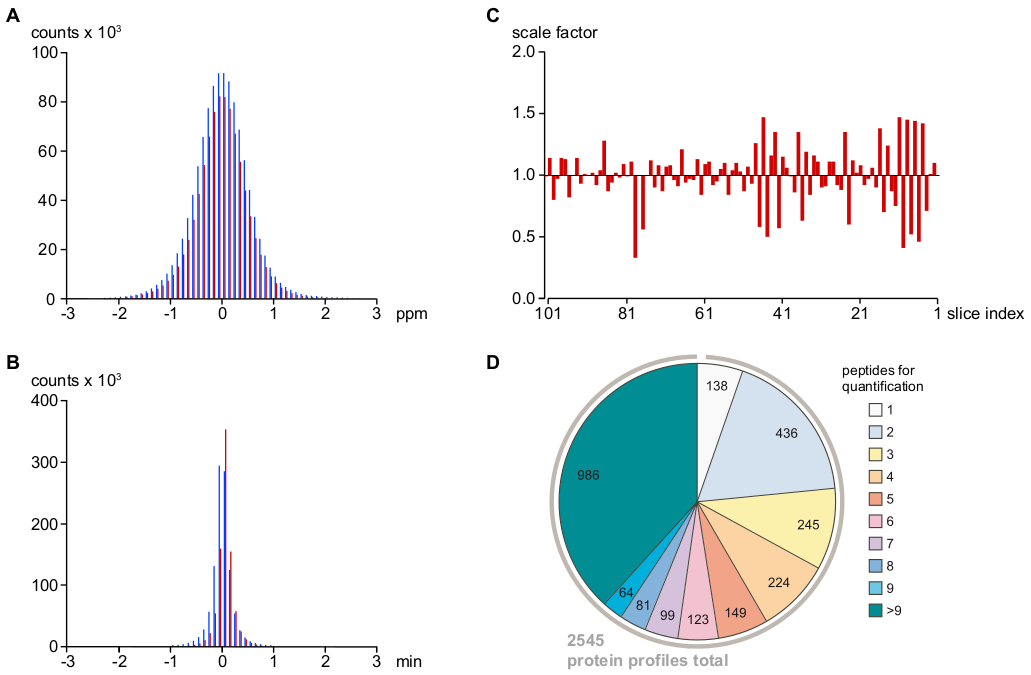
NOTA: Questo protocollo utilizza software interno per l'assegnazione dei peptidi "inseriti". I parametri set generano tolleranze di corrispondenza effettive m/z e tempo di elusione rispettivamente di 2 ppm e 1 min (vedere La Figura 2A,B). - Corretto per le variazioni sistematiche run-to-run nel carico di peptidi e nell'efficienza di ionizzazione da intensità del peptide riscalare calcolate dalle differenze mediane di intensità relative dei peptidi tra i campioni di fette vicine (Figura 2C).
- Filtrare i dati fotovoltaici per gli outlier e le rimanenti assegnazioni di falsi positivi identificate dall'analisi di coerenza PV interna.
- Normalizza i PV di ogni peptide ai loro valori massimi su tutti i set di dati delle sezioni, producendo profili relativi di abbondanza di peptidi.
- Infine, calcolare i profili relativi di abbondanza proteica come medie di almeno due (e fino a sei o 50%, qualunque valore sia maggiore) dei migliori profili peptidici correlati su una finestra di tre fette consecutive. Ciò consente di colmare i valori fotovoltaici mancanti e di ridurre il rumore.
NOTA: Questo ha infine portato a 2.545 (di 2.568 preselezionati) profiliproteici( Figura 2D).
- Caratterizzazione dei complessi proteici
- Analizzare i profili delle proteine eseguendo prima il rilevamento dei picchi utilizzando il metodo maxima locale e adattare consecutivamente le distribuzioni normali a questi picchi, producendo la posizione (cioè, indice di sezione o dimensione complessa apparente) del loro massimo e FWHM (larghezza completa a valori di intensità massima (inset della figura 4).
NOTA: nel set di dati, i profili vengono analizzati automaticamente utilizzando script personalizzati. I valori FWHM più piccoli sono indicativi della risoluzione effettiva delle dimensioni dell'approccio (qui, 6 x 0,25 x 1,5 mm). - Utilizzare picchi complessi di proteine di riferimento con massa molecolare definita (come riportato nel database UniProtKB/Swiss-Prot) per l'analisi della regressione lineare dei valori log10(massa molecolare prevista) per convertire gli indici dei numeri di sezione in dimensioni molecolari apparenti (ad esempio, apparente dimensione complessa in kDa).
NOTA: in questo studio sono stati selezionati 23 complessi di marcatori nel campione (Figura 4) sulla base di forme (i) monodisperse di picchi di profilo, (ii) supporto sperimentale dei pesi molecolari e (iii) distribuzioni lungo le sezioni del gel BN-PAGE studiate.
- Analizzare i profili delle proteine eseguendo prima il rilevamento dei picchi utilizzando il metodo maxima locale e adattare consecutivamente le distribuzioni normali a questi picchi, producendo la posizione (cioè, indice di sezione o dimensione complessa apparente) del loro massimo e FWHM (larghezza completa a valori di intensità massima (inset della figura 4).
Risultati
La stragrande maggioranza degli studi BN-MS convenzionali e l'approccio csBN-MS ad alta risoluzione recentemente stabilito sono stati applicati a i preparati mitocondriali e plastidi che sono (i) prontamente disponibili, (ii) hanno una complessità limitata e (iii) espresso complessi proteici bersaglio (membrana) ad alta densità. Questo protocollo estende l'applicazione della profilazione complessoma ad alta risoluzione alle membrane non mitocondriali che esprimono proteine a basso abbondanza, di cui sono disponibili poche informazioni sulla loro integrazione nei complessi. A scopo dimostrativo, abbiamo scelto una preparazione a membrana arricchita di endosomi dal rene di topo ottenuto dalla centrifugazione del gradiente di densità.
L'ottimizzazione di questa preparazione è stata guidata dalla proteina marcatore TPC1 che forma canali ionici intracellulari prevalentemente localizzati nei primi e negli endosomi di riciclaggio12. È anche altamente espresso nelle cellule tubolari procaliche renali, come dimostrato dall'analisi immunohistochimica delle sezioni del tessuto renale (Figura 1A). Queste membrane erano delicatamente solubili (ComplexioLyte 47 a basso rapporto proteine:detergent ratio di 1:8) e si concentravano su un cuscino di sucrosi per ultracentrifugazione. Quest'ultimo si è rivelato un passo importante per rimuovere i componenti di peso molecolare inferiore in eccesso (ad esempio, detergenti, lipidi, sali, polimeri organici e metaboliti) che tendono ad avere un impatto negativo sulla risoluzione delle separazioni CE-PAGE preparative.
La separazione complessa su un gel a gradiente poliacrilamide nativo dell'1%-13% (w/v)(Figura 1B, pannello centrale) ha mostrato bande proteiche fortemente colorate con pochissimi artefatti di migrazione. La separazione SDS-PAGE di una striscia stretta di gel BN-PAGE(Figura 1B, telaio in scatola in rosso) come seconda dimensione seguita dall'analisi delle macchie occidentali ha mostrato un modello ben risolto di distinte popolazioni complesse associate a TPC1(Figura 1B) , pannello superiore, contrassegnato da frecce rosse), molto probabilmente risultante dall'associazione con sottounità proteiche aggiuntive e/o modifiche post-traduzionali (come la glicosilazione12). Una sezione di 3 cm di interesse è stata ascisa, fissa e lavorata per la taglio criomicrotoma come descritto11. Le singole fasi di questa procedura (in particolare, l'allineamento preciso dell'ampia sezione del gel), che è di fondamentale importanza per preservare la risoluzione durante il campionamento, sono documentate nel video di accompagnamento. La sezione gel incorporata è stata infine tagliata in 101 fette di gel con uno spessore uniforme di 0,25 mm (Figura 1B, pannello inferiore), che sono state digerite separatamente e analizzate da spettrometria di massa accoppiata l'LC ad alte prestazioni.
Oltre alla risoluzione delle dimensioni, la qualità della quantificazione delle proteine è fondamentale per una profilazione complesso di successo. Con l'impostazione e le impostazioni ms utilizzate, l'analisi dei campioni è stata abbastanza completa, con conseguente identificazione media di più di 1.000 proteine e 10.000 peptidi (8.200 dei quali erano specifici delle proteine) per fetta, e circa 3.000 proteine e 43.000 peptidi (38.500 dei quali erano specifici delle proteine) in totale. Tuttavia, a causa della natura stocastica del sequenziamento MS/MS dipendente dai dati e dei suoi limiti nella gamma dinamica, le informazioni sull'intensità erano ancora frammentarie per le proteine meno abbondanti. Pertanto, è stata eseguita un'elaborata procedura di elaborazione dei dati MS11che si basa sull'assegnazione precisa di segnali peptidi (volumi di picco [PV] - intensità di segnale correlate ai peptidi integrate su m/z e tempo) su tutta la serie di set di dati.
Come illustrato nella Figura 2A,B, le deviazioni dei segnali peptidi nel tempo di massa e di conservazione rimasti dopo la calibrazione erano identiche per i VM in sequenza MS e per i PV assegnate indirettamente (con tolleranze molto strette di <1 ppm e <0,5 min per il 95% del PV), indicativo di un tasso molto basso per l'assegnazione di PV falsi positivi. Gli outlier rimanenti sono stati filtrati in base alla loro coerenza con altri PV correlati. Poiché tutte le misurazioni della SM sono state eseguite consecutivamente sullo stesso set-up LC-MS senza modifiche nei parametri o nei componenti hardware, le variazioni run-to-run (determinate come mediana di tutte le intensità fotovoltaiche in un campione relative a quelle nelle sezioni vicine) sono stati piccoli e facilmente eliminati con il ridimensionamento dei set di dati fotovoltaici (Figura 2C). Le informazioni sull'intensità dei peptidi risultanti sono state poi utilizzate per ricostruire 2.545 profili di abbondanza relativa delle proteine. Come mostrato nella Figura 2D,più del 75% di questi profili proteici si basava su almeno tre peptidi indipendenti specifici delle proteine.
Successivamente, il protocollo ha valutato la pertinenza della dimensione del passaggio del campionamento del gel BN-PAGE per la risoluzione dei complessi proteici. A tale scopo, i set di dati delle sezioni sono stati uniti sommando le informazioni fotovoltaiche da due, tre o quattro sezioni consecutive, simulando così il risultato per le dimensioni dei gradini di 0,5 mm, 0,75 mm e 1 mm (rispetto al campionamento originale di 0,25 mm). La figura 3 illustra i profili di abbondanza risultanti per la proteina TPC1 come esempio (A-D). A 0,25 mm, intensità relative e separazione delle dimensioni delle popolazioni complesse associate a TPC1 (Figura 3A) erano ben d'accordo con i risultati dell'analisi delle macchie occidentali (Figura 1B, pannello superiore); anche se il profilo mostrava un certo rumore, per lo più derivante da valori mancanti ("gaps") nella matrice fotovoltaica utilizzata per la quantificazione.
L'unione di due sezioni corrispondenti a 0,5 mm ha mantenuto le intensità corrette e la separazione dei complessi associati a TPC1 e ha rimosso il rumore di quantificazione (Figura 3B). Al contrario, dimensioni di gradino più grandi di 0,75 mm e 1 mm(Figura 3C,D) hanno portato a una perdita della risoluzione delle dimensioni e hanno abolito la discriminazione delle sottopopolazioni complesse TPC1. Va notato che la stragrande maggioranza delle analisi BN-MS convenzionali pubblicate utilizza manualmente tagliare fette da 2 mm (circa 60 per coprire l'intera corsia gel)7,8,9,10.
La conversione della distanza di migrazione o dell'indice delle fette in dimensioni molecolari è generalmente basata su marcatori, proteine native disponibili in commercio o complessi proteici endogeni ben caratterizzati con composizione nota di sottounità (per lo più [super] complessi della catena respiratoria ossidativa mitocondriale [OXPHOS])13. Tuttavia, poiché la separazione BN-PAGE si basa sull'effettiva sezione molecolare determinata non solo dalla massa molecolare, ma anche dalla struttura 3D e dal numero di molecole di lipidi, detergenti e coomassie associate, le singole proteine possono deviazioni maggiori. Pertanto, è stato scelto di utilizzare più grandi set di complessi proteici come marcatori11. Il grafico in Figura 4 mostra 23 marcatori selezionati con una sottounità rappresentativa mostrata come un cerchio nero, che indica i valori log10 della massa molecolare prevista (secondo il database UniProtKB/Swiss-Prot) rispettoa . l'indice della sezione del picco di profilo corrispondente. Questi ultimi sono stati ottenuti da automatizzato gaussiano si adatta ai dati relativi abbondanza, come mostrato nell'insetto della Figura 4 che mostra l'esempio con chaperone BCS1. La regressione lineare (linea rossa) ha fornito una funzione per convertire i valori dell'indice delle sezioni in dimensioni molecolari apparenti, che variavano da 160-630 kDa, lungo la sezione gel studiata.
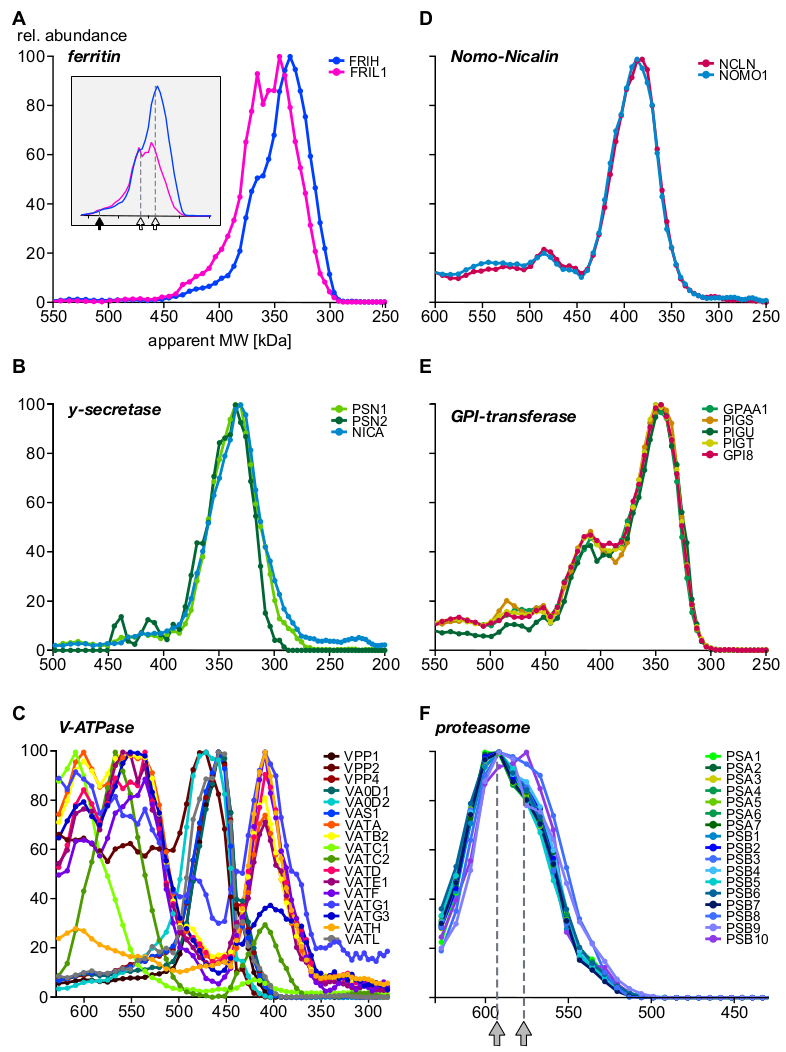
Infine, l'analisi ha fornito informazioni su complessi ben caratterizzati e ha dimostrato l'esistenza di nuove sottounità e assiemi complessi. Esempi che evidenziano diversi aspetti del complesso sono mostrati nella Figura 5 (A-C: proteine espresse o preferibilmente situate in compartimenti endosomici; D-F: complessi di altre localizzazioni subcellulari). La ferritina proteica di trasporto del ferro è nota per formare complessi da 24 sottounità leggere (FRIL1) e/o pesanti (FRIH), con un peso molecolare totale di 440 kDa14 (Figura 5A, freccia piena). I profili delle sottounità (Figura 5A) suggeriscono l'esistenza di almeno due forme più piccole del complesso (con una massa apparente di 360 kDa e 340 kDa; frecce aperte) con stoichiometrie a catena pesante/leggera distinti (meglio visibile dopo il ridimensionamento abbondanza, inset di Figura 5A) che sono abbondantemente presenti negli endosomi.
Al contrario, i carilini-nomo115 (Figura 5D), il complesso principale gamma-secretase16 (Figura 5B) e i macchinari GPI-transamidase17 (Figura 5E) mostrano fisso l'abbondanza delle loro sottounità fondamentali su tutta la gamma di dimensioni e indipendente dall'associazione con proteine aggiuntive. Ciò indica che le relative sottounità sono esclusive l'una dell'altra. VacuolarH -ATPases sono complessi multiproteici assemblati da una piscina di oltre 20 sottounità in modo modulare con un peso molecolare totale di circa 900 kDa. La figura 5C rivela sottocomplessi con composizioni distinte da almeno 17 sottounità, che rappresentano intermedi biologici (dis)assemblaggio o sottocomplessi generati dalle condizioni sperimentali, alcune delle quali sono state anch'essi osservato in un recente studio BN-MS18. Un altro esempio complesso multi-proteina è il proteasome19 (Figura 5F). Un'attenta ispezione dei profili di abbondanza delle sottounità alfa e beta che formano il nucleo proteasome 20S suggerisce due grandi popolazioni complesse con sottili differenze di dimensioni (590 kDa e 575 kDa, indicate da frecce grigie) e l'integrazione di tre sottounità beta.
In sintesi, la profilazione complessa csBN-MS delle membrane renali arricchite di endosomie fornisce risultati completi e dettagliati per quanto riguarda l'integrazione (i) di proteine bersaglio uniformi e a basso abbondanza in complessi, (ii) composizione generale di sottounità complesse e stoichiometria e (iii) eterogeneità complesse, sottostrutture e (intermedi) di assemblaggio.

Figura 1: Separazione Preparativa BN-PAGE delle membrane solubilizzate arricchite di endosomi dal rene di topo utilizzando il canale intracellulare TPC1 come marcatore. (A) Localizzazione immunoistochimica della proteina TPC1 in tubuli coniugali prossimali mediante microscopia confocale. Verde: anti-TPC1 anticorpo12 colorazione visualizzata con secondaria Cy3-biotinylated capra anti-coniglio IgG; rosso: lectina biomutata del loto tetragonolobus (LTL, 10 g/mL, coniugata FITC) che contrassegna la superficie luminosa delle cellule del tubulus prossimale. L'insetto mostra la colorazione di una sezione corrispondente da un rene TPC1-KO come controllo negativo. Le barre bianche della scala sono di 20 m. Da notare anche la forte espressione di TPC1 nelle vescicle intracellulari, note da esperimenti indipendenti per rappresentare le prime e riciclare gli endosomi12. (B) Setsetativo BN-PAGE separazione di 2,5 mg di membrane solubili arricchite di endosomiconi su un gel a gradiente poliacrilamide 1%-13% (w/v). Una corsia stretta (incorniciata in rosso) è stata tagliata per la successiva analisi delle macchie SDS-PAGE/western (pannello superiore), risolvendo diversi complessi associati a TPC1 e modelli di glicosilazione (frecce rosse: anti-TPC1/anti-coniglio HRP/ECL prime; verde: posizioni e previsioni HRP/ECL prime; verde: posizioni e previsioni HRP/ECL prime; verde: posizioni e previsioni HRP/ECL prime; verde: posizioni e previsioni masse [MDa] di complessi proteici marker identificati dalla colorazione proteica totale [SYPRO Ruby macchia macchia macchia macchia macchia di macchia della macchia]) della macchia. Da destra a sinistra: Na:/K-trasporto ATPase, Cytochrome b-c1 complesso dimero, synthase ATP, NADH:ubiquinone oxidoreductase. Una sezione di 3 cm di interesse dalla corsia gel è stata ascisa, incorporata nel mezzo di incorporamento dei tessuti, montata e tagliata in 101 sezioni (0,25 mm) lungo il fronte di migrazione delle proteine utilizzando un criomicrotoma (pannello inferiore; vedi collegamento video). Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 2: Parametri chiave che determinano l'accuratezza dell'assegnazione e della quantificazione del segnale MS, nonché la profondità dell'analisi. (A) Distribuzione degli errori di massa relativi (in ppm) dopo la calibrazione m/z di MS/MS sequenziati (barre rosse) e assegnati indirettamente (cioè, in base ai tempi di massa e ritenzione strettamente corrispondenti, vedi protocollo; barre blu) segnali peptidici. Ciò suggerisce un errore di massa finale di <1 ppm (per il 95% dei segnali/PV assegnati) e un tasso molto basso di assegnazioni false positive. (B) Distribuzione delle deviazioni del tempo di ritenzione nano-HPLC dalla media totale dopo l'allineamento del tempo di eluizione dei segnali peptidici, utilizzando la regressione di Loess (vedi protocollo) e la codifica dei colori come usato in (A). L'errore di tempo è inferiore a 30 s per >95% di segnali peptidi/ PV assegnati (C) Variazione run-to-run delle intensità sMF totali tracciate rispetto alla media dei due campioni vicini. Questi fattori di scala sono stati applicati alle tabelle fotovoltaiche grezze per ridurre al minimo gli errori tecnici sistematici. (D) Informazioni sui peptidi utilizzate per calcolare i profili di abbondanza relativa delle proteine. Dopo aver filtrato i peptidi specifici delle proteine per gli outlier, i punteggi scarsi o le singole identificazioni (vedi protocollo), sono stati determinati 2.545 profili di abbondanza di proteine, >75% dei quali erano basati su almeno tre peptidi con ragionevole fiducia. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 3: Impatto critico della dimensione del gradino nel campionamento del gel sulla risoluzione complexome di TPC1. I set di dati sono stati uniti sommando l'intensità del segnale in gruppi di 1, 2, 3 e 4 sezioni consecutive (rispettivamente A-D) ed elaborati in modo identico per simulare diverse dimensioni del passo nel gel slicing come indicato. Il profilo TPC1 mostra alcuni (sovracampionamento) di rumore a 0,25 mm ma una risoluzione di buone dimensioni di tre popolazioni complesse (vedere anche Figura 1B), che viene in gran parte conservato a una larghezza di 0,5 mm. La discriminazione di queste popolazioni si perde con l'avvicinarsi di 1 mm. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 4: Determinazione del peso molecolare apparente. Secondo UniProtKB/Swiss-Prot, sono stati utilizzati 23 complessi marcatori con composizione molecolare definita (come indicato, secondo UniProtKB/Swiss-Prot). I valori logaritmici dei loro pesi molecolari attesi (in kDa) sono stati tracciati contro. l'indice di sezione massima del picco del profilo della sottounità proteica rappresentativa indicata (cerchi riempiti in nero). La regressione lineare adatta a questi dati (linea rossa) ha fornito una funzione che converte i valori dell'indice della sezione in pesi molecolari apparenti. Il massimo massimo è stato determinato dall'automatizzato Gaussian si adatta ai picchi del profilo proteico, come mostrato nell'incasso (a destra) per la proteina chaperone BCS1 (dati primari in blu, confini di adattamento indicati da linee arancioni, funzione di adattamento in rosso). Inoltre, questi adattamenti determinano larghezze massime di picco (linea verde, 6,5 fette o 1,6 mm per l'esempio mostrato) con i complessi di messa a fuoco più nitidi che si estendono intorno a un gel di 1,5 mm. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 5: Esempi di profili di sottounità complessi di proteine. Abbondanza di proteine relativa vs. apparente peso molecolare tracciato per la catena pesante e leggera di ferritina (A) rivelando l'eterogeneità molecolare della stoichiometria della sottounità di ferritina, più chiaramente visibile dopo il ridimensionamento dell'abbondanza. Freccia piena e frecce aperte indicano il complesso completo (440 kDa) e due sottocomplessi, rispettivamente. Le sottounità gamma-secretasi (B) sono quantitativamente integrate in una popolazione complessa mononucleo. I sottocomplessi del vacuolar H -ATPases (C) esibivano più assiemi con composizione distinta di sottounità, tutti espressi in endosomi. Le proteine nomo1 e nicalina (D) formavano un complesso esclusivo (il GPI-transamidase), che è un macchinario enzimatico multi-subunità che forma diversi complessi. (E) Il complesso del nucleo proteasome 20S che mostra (F) un sottile pattern subcomplesso con due popolazioni indicate da frecce in grigio, tutte provenienti da altre localizzazioni subcellulari. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
Discussione
Lo studio presentato si è basato sulla tecnica csBN-MS precedentemente confrontata con una preparazione mitocondriale11 e ha incorporato miglioramenti nella preparazione dei campioni, nell'elaborazione dei gel e nella valutazione dei dati MS. L'analisi mirata di una sezione del gel BN-PAGE di separazione su larga scala ha fornito una serie completa di dati che mostrano misure di qualità paragonabili allo studio con membrane mitocondriali. Gli errori relativi ai tempi di massa e di ritenzione e le variazioni run-to-run sono stati mantenuti molto bassi e hanno fornito la base per determinare profili affidabili di abbondanza di proteine. La risoluzione delle dimensioni sembrava essere buona, con larghezze di picco semimassime a partire da sei fette (corrispondenti a 1,5 mm, Figura 4) e differenze di dimensioni relative inferiori al 10% risolte (Figura 3, Figura 5A). Questi valori non soddisfacevano pienamente la qualità di risoluzione delle dimensioni della precedente analisi csBN-MS dei mitocondri (nonostante la dimensione del passo di campionamento del gel più piccola scelta), ma sono significativamente migliori rispetto alle prestazioni del BN-MS convenzionale o della SM di esclusione delle dimensioni si avvicinaa 20 che sono recentemente diventati popolari.
L'importanza di un'elevata risoluzione di dimensioni complesse effettive è sottolineata dall'esperimento di simulazione nella figura 3 (utilizzando i complessi associati a TPC1) che difficilmente possono essere risolti dall'analisi della macchia occidentale 2D BN/SDS-PAGE (Figura 1B). Questi risultati suggeriscono che il taglio di 0,25 mm in questo caso ha provocato un sovracampionamento, ma questo si è comunque rivelato utile per l'eliminazione del "rumore di quantificazione" senza compromettere la risoluzione effettiva delle dimensioni. Pertanto, in linea con i risultati precedenti11, è generalmente consigliata una dimensione del passaggio di campionamento di 0,3 mm.
In particolare, la discriminazione dei complessi associati a TPC1 è completamente persa con il campionamento di gel di 1 mm, che è la più piccola dimensione del passo fornita dal taglio manuale nel convenzionale BN-MS5,6. Questo può spiegare il fatto che, nonostante le potenti tecnologie MS disponibili, pochissimi complessi proteici e sottounità sono stati identificati de novo mediante profilazione di complexome. Oltre alla buona potenza di risoluzione, csBN-MS offre un'elevata versatilità. Complessi legati alla membrana e complessi proteici solubili che vanno da 50 kDa a diversi MDa possono essere efficacemente risolti in un unico esperimento con la distorsione minima11. Ciò contrasta con le tecniche di separazione alternative utilizzate per la profilazione di complessioma come l'esclusione delle dimensioni o la cromatografia dello scambio di ioni, che operano con sottoinsiemi di proteine solubili con determinate gamme di dimensioni o proprietà di carica. Al rovescio della medaglia, csBN-MS è meno scalabile (carico massimo di 3 mg di proteine per gel), può essere tecnicamente impegnativo, e non può essere automatizzato.
Nel complesso, i risultati dimostrano che la profilatura complessoma basata su csBN-MS può essere applicata con successo a obiettivi non mitocondriali, ma indicano anche alcune sfide associate. Pertanto, l'estrazione efficiente e la stabilità biochimica dei complessi proteici richiedono una maggiore ottimizzazione e fasi di pulizia e possono ancora essere limitate. All'interno della finestra delle dimensioni studiata, il numero di complessi proteici monodispersi (dati non mostrati) ben focalizzati era notevolmente inferiore (dati non mostrati) rispetto a un campione mitocondriale. Si raccomanda inoltre di ridurre i carichi di campioni di BN-PAGE per ottenere una separazione accettabile del gel. Carichi più elevati possono richiedere corsie gel più ampie che sono più difficili da elaborare correttamente per affettare (vedi video di accompagnamento). Inoltre, la complessità proteica dei campioni era maggiore (circa due volte) rispetto ai digest a fette derivate dai mitocondri, portando a più valori fotovoltaici mancanti e a una gamma dinamica ridotta. Infatti, alcune piccole proteine che dovrebbero far parte dei complessi illustrati nella Figura 5 mancavano nelle analisi. Questi problemi possono essere risolti in futuro utilizzando strumenti MS più veloci e sensibili o modalità di acquisizione indipendenti dai dati.
La preparazione del campione è altamente fondamentale per il recupero complesso proteico, la stabilità e la qualità della separazione del gel. I parametri e le procedure devono essere ottimizzati per ogni tessuto di origine, lisato cellulare, membrana (frazione) e complesso proteico di interesse. Vengono fornite le seguenti raccomandazioni generali che possono aiutare a estendere le applicazioni di csBN-MS:
(i) Preparare campioni freschi ed evitare il riscaldamento/congelamento, forti diluizioni, cambiamenti nelle condizioni del buffer e ritardi inutili;
(ii) Utilizzando tamponi essenzialmente privi di sali (sostituire con 500-750 mm di betaina o acido aminocaproico), circa un pH neutro e contenente fino all'1% (w/v) di detergenti non denatura (proteina:detergent ratio tra 1:4-1:10 per la solubilizzazione della membrana complessi proteici, nessun detersivo necessario per i complessi proteici solubili);
(iii) Test accurati e regolazione delle condizioni detergenti mediante BN-PAGE analitico, poiché queste possono avere un forte impatto sull'efficienza della solubilità complessa, la rappresentazione dei complessi proteici della membrana nel campione, la stabilità e l'omogeneità le micelle proteiche detergenti. Questi ultimi sono prerequisiti per la messa a fuoco delle proteine come bande distinte/popolazioni complesse su gel BN-PAGE. La letteratura precedente offre un'ampia gamma di detergenti neutri. Tuttavia, il DDM (n-dodecyl z-d-maltoside)1,2,4,5,6 e digitonin3,5,7,8, 9,10,13,18 sono state le scelte più popolari per le analisi BN-MS finora. Va sottolineato che qualsiasi condizione detergente rappresenta necessariamente un compromesso tra l'efficienza di solubilizzazione e la conservazione delle interazioni proteiche e potrebbe non essere ugualmente adatta a tutti i tipi di proteine e materiali di origine target;
(iv) Rimozione di polimeri carichi come fibrille, filamenti, polisina, DNA e abbondanti componenti di peso molecolare inferiore (ad esempio, metaboliti, lipidi o peptidi). Questo può essere realizzato da ultracentrifugazione, filtrazione gel, o dialisi. Ciò è particolarmente importante per le lismi totali delle cellule o dei tessuti;
(v) Aggiunta di Coomassie G-250 (concentrazione finale 0,05%-0,1%) e saccarosio (per aumentare la densità per il carico, concentrazione finale 10%-20% [w/v]) al campione appena prima del caricamento, per cancellare con una breve ultracentrifugazione, caricare il campione senza perturbazione, e avviare la corsa immediatamente dopo.
Come prospettiva futura, la profilazione complessa basata su csBN-MS offre opzioni per il multiplexing per studiare le dinamiche complesse di proteine o i cambiamenti relativi a specifiche condizioni biologiche. La combinazione combinata di campioni con etichetta metabolicamente come proposto per la profilatura basata sull'esclusione delle dimensioni21 appare semplice, ma può essere ostacolata dallo scambio spontaneo di sottounità in complessi che si verificano indipendentemente dalla separazione utilizzata metodo. In alternativa, i campioni etichettati possono essere risolti nelle corsie di gel vicine, che possono quindi essere co-fette o combinate post-digestione per l'analisi differenziale con elevata sensibilità e robustezza.
Divulgazioni
L'autore Uwe Schulte è un dipendente e azionista di Logopharm GmbH che produce ComplexioLyte 47 utilizzato in questo studio. L'azienda fornisce reagenti ComplexioLyte alle istituzioni accademiche senza scopo di lucro.
Riconoscimenti
Questo studio è stato sostenuto dalla Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Project-ID 403222702 – SFB 1381 e nell'ambito della strategia di eccellenza tedesca CIBSS - EXC-2189 - Project ID 390939984. Ringraziamo Katja sappe per l'assistenza tecnica.
Materiali
| Name | Company | Catalog Number | Comments |
| 30% Acrylamide/Bis Solution, 37.5:1 | Bio Rad | #1610158 | Recommended for acrylamide gradient gel solutions up to 13% |
| 30% Acrylamide/Bis Solution, 19:1 | Bio Rad | #1610154 | Recommended for acrylamide gradient gel solutions >13% |
| SYPRO Ruby Protein Blot Stain | Bio Rad | #1703127 | Total protein stain on blot membranes; sensitive and compatible with immunodetection |
| Coomassie Brilliant Blue G-250 | Serva | no. 35050 | Centrifugate stock solutions prior to use |
| ComplexioLyte 47 | Logopharm | CL-47-01 | Ready-to-use detergent buffer (1%) for mild solubilization of membrane proteins |
| Embedding Medium / Tissue Freezing Medium | Leica Biosystems | 14020108926 | Embedding medium for gel sections to be sliced by a cryo-microtome |
| Immobilon-P Membrane, PVDF, 0,45 µm | Merck | IPVH00010 | |
| ECL Prime Western Blotting Detection Reagent | GE Healthcare | RPN2232 | |
| Plastic syringe with rubber stopper, 20-30 ml | n.a. | n.a. | any supplier, important for making gel section embedding tool |
| broad razor blade | n.a. | n.a. | any supplier, for BN-PAGE gel trimming / excision of lanes |
| metal tube / cylinder, ca. 4 cm long | n.a. | n.a. | mold for embedding and freezing of gel samples |
| Protein LoBind Tubes, 1.5 ml | Eppendorf | Nr. 0030108116 | highly recommended to minimize protein/peptide loss due to absorption |
| sequencing-grade modified trypsin | Promega | V5111 | |
| C18 PepMap100 precolumn, particle size 5 µm | Dionex / Thermo Scientific | P/N 160454 | |
| PicoTip emitter (i.d. 75 µm; tip 8 µm) | New Objective | FS360-75-8 | |
| ReproSil-Pur 120 ODS-3 (C18, 3 µm) | Dr. Maisch GmbH | r13.93. | columns packed manually |
| rabbit anti-TPC1 antibody | Gramsch Laboratories | custom production | described in Castonguay, et al., 2017 (Reference 12) |
| Cy3-biotinylated goat anti-rabbit IgG | Vector Laboratories | CY-1300 | described in Castonguay, et al., 2017 (Reference 12) |
| biotinylated Lotus tetragonolobus lectin, FITC-conjugated | Vector Laboratories | #B1325 | described in Castonguay, et al., 2017 (Reference 12) |
| cryo-microtome Leica CM1950 | Leica Biosystems | 14047743905 | |
| Mini Protean II Cell with wetblot unit | Bio Rad | n.a. | for SDS-PAGE and Westernblot (not sold any more) |
| Penguin Midi Gel Electrophoresis System | PeqLab | n.a. | for BN-PAGE (not sold any more) |
| Zeiss Axiovert 200 M microscope + Photometrics Coolsnap 2 digital camera | Zeiss / Photometrics | n.a. | |
| peristaltic pump (IP high precision multichannel) | Ismatec | ISM940 | for casting of gradient polyacrylamide gels |
| gradient mixer with stirring (two chambers) | selfmade, alternatively Bio Rad | 1652000 or 1652001 | for casting of gradient polyacrylamide gels, manual provides instructions to cast linear or hyperbolic gradient gels (http://www.bio-rad.com/webroot/web/pdf/lsr/literature/M1652000.pdf) |
| ultracentrifuge Sorvall M120 with S80 AT3 rotor | Sorvall / Thermo Scientific | n.a. | for sample preparation (not sold any more) |
| UltiMate 3000 RSLCnano HPLC | Dionex / Thermo Scientific | ULTIM3000RSLCNANO | |
| Orbitrap Elite mass spectrometer | Thermo Scientific | IQLAAEGAAPFADBMAZQ |
Riferimenti
- Majeran, W., et al. Consequences of C4 Differentiation for Chloroplast Membrane Proteomes in Maize Mesophyll and Bundle Sheath Cells. Molecular & Cellular Proteomics. 7, 1609-1638 (2008).
- Wessels, H. J., et al. LC-MS/MS as an alternative for SDS-PAGE in blue native analysis of protein complexes. Proteomics. 9 (17), 4221-4228 (2009).
- Heide, H., et al. Complexome profiling identifies TMEM126B as a component of the mitochondrial complex I assembly complex. Cell Metabolism. 16 (4), 538-549 (2012).
- Wessels, H. J., et al. Analysis of 953 human proteins from a mitochondrial HEK293 fraction by complexome profiling. PLoS ONE. 8 (7), e68340 (2013).
- Wöhlbrand, L., et al. Analysis of membrane-protein complexes of the marine sulfate reducer Desulfobacula toluolica Tol2 by 1D blue native-PAGE complexome profiling and 2D blue native-/SDS-PAGE. Proteomics. 16 (6), 973-988 (2016).
- Takabayashi, A., et al. PCoM-DB Update: A Protein Co-Migration Database for Photosynthetic Organisms. Plant and Cell Physiology. 58 (1), e10 (2017).
- Senkler, J., et al. The mitochondrial complexome of Arabidopsis thaliana. The Plant Journal. 89 (6), 1079-1092 (2017).
- de Almeida, N. M., et al. Membrane-bound electron transport systems of an anammox bacterium: A complexome analysis. Biochimica et Biophysica Acta. 1857 (10), 1694-1704 (2016).
- Anand, R., Strecker, V., Urbach, J., Wittig, I., Reichert, A. S. Mic13 Is Essential for Formation of Crista Junctions in Mammalian Cells. PLoS ONE. 11 (8), e0160258 (2016).
- Eydt, K., Davies, K. M., Behrendt, C., Wittig, I., Reichert, A. S. Cristae architecture is determined by an interplay of the MICOS complex and the F1FO ATP synthase via Mic27 and Mic10. Microbial Cell. 4 (8), 259-272 (2017).
- Müller, C. S., et al. Cryoslicing Blue Native-Mass Spectrometry (csBN-MS), a Novel Technology for High Resolution Complexome Profiling. Molecular & Cellular Proteomics. 15 (2), 669-681 (2016).
- Castonguay, J., et al. The two-pore channel TPC1 is required for efficient protein processing through early and recycling endosomes. Scientific Reports. 7 (1), 10038 (2017).
- Schägger, H., Pfeiffer, K. Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. The EMBO Journal. 19 (8), 1777-1783 (2000).
- Banyard, S. H., Stammers, D. K., Harrison, P. M. Electron density map of apoferritin at 2.8-A resolution. Nature. 271 (5642), 282-284 (1978).
- Dettmer, U., et al. Transmembrane protein 147 (TMEM147) is a novel component of the Nicalin-NOMO protein complex. The Journal of Biological Chemistry. 285 (34), 26174-26181 (2010).
- Kimberly, W. T., et al. Gamma-secretase is a membrane protein complex comprised of presenilin, nicastrin Aph-1, and Pen-2. Proceedings of the National Academy of Sciences of the United States of America. 100 (11), 6382-6387 (2003).
- Hong, Y., et al. Human PIG-U and yeast Cdc91p are the fifth subunit of GPI transamidase that attaches GPI-anchors to proteins. Molecular Biology of the Cell. 14 (5), 1780-1789 (2003).
- Van Damme, T., et al. Mutations in ATP6V1E1 or ATP6V1A Cause Autosomal-Recessive Cutis Laxa. The American Journal of Human Genetics. 100 (2), 216-227 (2017).
- Budenholzer, L., Cheng, C. L., Li, Y., Hochstrasser, M. Proteasome Structure and Assembly. Journal of Molecular Biology. 429 (22), 3500-3524 (2017).
- Heusel, M., et al. Complex-centric proteome profiling by SEC-SWATH-MS. Molecular Systems Biology. 15 (1), e8438 (2019).
- Kristensen, A. R., Gsponer, J., Forster, L. J. A high-throughput approach for measuring temporal changes in the interactome. Nature Methods. 9 (9), 907-919 (2012).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati