
Method Article
High-Resolution Complexome Profiling by Cryoslicing BN-MS Analysis
In diesem Artikel
Zusammenfassung
Für die hochauflösende Complexome-Profilierung wird ein vielseitiges kryolicing BN-MS-Protokoll mit einem Mikrotom vorgestellt.
Zusammenfassung
Proteine üben in der Regel biologische Funktionen durch Wechselwirkungen mit anderen Proteinen aus, entweder in dynamischen Proteinbaugruppen oder als Teil stabil geformter Komplexe. Letzteres lässt sich mit nativer Polyacrylamid-Gelelektrophorese (BN-PAGE) elegant nach molekularer Größe auflösen. Die Kopplung solcher Trennungen an die empfindliche Massenspektrometrie (BN-MS) ist gut etabliert und ermöglicht theoretisch eine erschöpfende Beurteilung des ausziehbaren Complexoms in biologischen Proben. Dieser Ansatz ist jedoch ziemlich mühsam und bietet eine begrenzte Auflösung und Empfindlichkeit in komplexer Größe. Auch, seine Anwendung ist auf reichlich mitochondriale und plastid Proteine beschränkt geblieben. So fehlen für die meisten Proteine noch Informationen über die Integration in stabile Proteinkomplexe. Präsentiert wird hier ein optimierter Ansatz für die komplexe Profilierung von Komplexen, die eine präparative BN-PAGE-Trennung, eine Submillimeter-Probenahme breiter Gelspuren durch Kryomikrotom-Schneiden und eine massenspektrometrische Analyse mit etikettenfreier Proteinquantifizierung umfassen. Die Verfahren und Tools für kritische Schritte werden ausführlich beschrieben. Als Anwendung beschreibt der Bericht komplexe Analyse einer solubilisierten endosome-angereicherten Membranfraktion aus Mausnieren, wobei insgesamt 2.545 Proteine profiliert werden. Die Ergebnisse zeigen die Identifizierung einheitlicher, überflussarmer Membranproteine wie intrazellulärer Ionenkanäle sowie hochauflösender, komplexer Protein-Montagemuster, einschließlich Glykosylierungs-Isoformen. Die Ergebnisse stehen im Einvernehmen mit unabhängigen biochemischen Analysen. Zusammenfassend lässt sich sagen, dass diese Methode eine umfassende und unvoreingenommene Identifizierung von Protein-(Super-)Komplexen und deren Zusammensetzung der Untereinheit ermöglicht und eine Grundlage für die Untersuchung der Stoichiometrie, -montage und -interaktionsdynamik von Proteinkomplexen in biologisches System.
Einleitung
Die BN-PAGE-Trennung wurde zunächst direkt an die LC-MS-Analyse (BN-MS) durch die Forschungsgruppen Majeran1 und Wessels2 gekoppelt, indem BN-PAGE-Gelspuren manuell getrennt wurden. Ihre Analysen identifizierten eine Reihe von reichlich vorhandenen Membranproteinkomplexen mit bekannter Untereinheitszusammensetzung aus Pflanzlichen Plasmiden bzw. HEK-Zellmitochondrien. Diese Analysen waren jedoch alles andere als umfassend und ließen keine unvoreingenommene Identifizierung neuartiger Versammlungen zu. Die Leistung von Massenspektrometern und etikettenfreien Quantifizierungsmethoden hat sich seitdem deutlich verbessert, was umfassende BN-MS-Analysen ermöglicht hat. Dies hat den Begriff "complexome profiling" geprägt. So analysierten Heide und Kollegen rattenherzige Mitochondrien, die 464 mitochondriale Proteine identifizierten und gruppierten, wodurch viele bekannte Baugruppen bestätigt wurden. Darüber hinaus fanden sie TMEM126B als eine neuartige und entscheidende Untereinheit eines spezifischen Montagekomplexes3. Vergleichbare Ergebnisse (mit 437 mitochondrialen Proteinprofilen) wurden in einer parallelen Studie mit HEK-Zellmitochondrien4erzielt.
Trotz dieser Verbesserungen sind mehrere Probleme geblieben, die das volle Potenzial von BN-MS für complexome Profiling einschränken. Eine wesentliche Einschränkung ist die effektive Größenauflösung von Komplexen, die durch zwei Faktoren bestimmt wird: die (i) Qualität der BN-PAGE-Trennung, die von der Homogenität des Gelmatrix-Porengradienten sowie der Stabilität/Löslichkeit der Probenkomplexe abhängt, und (ii) Schrittgröße der Gelprobenahme, die bestenfalls 1 mm beträgt, wenn herkömmlichemanuelles Schneiden5,6verwendet wird. Eine schlechte Größenauflösung verfehlt nicht nur subtile komplexe Isoformen und Heterogenitäten, sondern wirkt sich auch negativ auf den Dynamikumfang und das Vertrauen von unvoreingenommener, de novo Subunit-Zuweisung und Quantifizierung aus.
Weitere Herausforderungen sind die Präzision der Proteinquantifizierung und die Abdeckung des tatsächlichen Dynamikbereichs von Proteinüberfluss in der Probe durch massenspektrometrische Analyse. Daher ist die Anwendung von BN-MS-Komplexprofiling weitgehend auf biologische Proben mit geringerer Komplexität, hoher Expression von Zielkomplexen und günstigen Löslichkeitseigenschaften (d. h. Plastiden, Mitochondrien und Mikroorganismen)6,7,8,9,10.
Kürzlich haben wir kryomikrotome-slicing-assisted BN-MS (csBN-MS) eingeführt, die präzise Submillimeter-Probenahme von BN-PAGE-Gelspuren mit umfassender MS-Analyse und aufwendiger MS-Datenverarbeitung zur Bestimmung von Proteinprofilen mit hohem Vertrauen11. Die Anwendung auf eine mitochondriale Membranpräparation von Rattengehirnen zeigte bisher eine unerfüllte effektive komplexe Größenauflösung und maximale Abdeckung von oxidativen Atemkettenkomplexen (OXPHOS) (d. h. 90 von 90 MS-zugänglichen). In diesem Beispiel wurden auch eine Reihe neuartiger Proteinassemblys identifiziert.
Beschrieben werden hier optimierte Verfahren für die präparative BN-PAGE-Trennung von Proteinkomplexen (nicht auf eine bestimmte biologische Quelle beschränkt), das Gießen großer präparativer BN-PAGE-Gele, das Kryomikrotom-Schneiden breiter Gelspuren und MS-Daten. verarbeitung. Die Leistungsfähigkeit der hochauflösenden Profilierung wird für ein proteinkomplexes Präparat aus mit Mausnieren endosomeangereicherten Membranen demonstriert. Schließlich werden die Vorteile einer zunehmenden Auflösung und Präzision der massenspektrometrischen Quantifizierung diskutiert.
Protokoll
1. Präparative BN-PAGE
- Gelzubereitung
- Verwenden Sie ein mittelgroßes vertikales Gelelektrophoresesystem (>10 cm Geltrennabstand; 14 cm x 11 cm, 1,5 mm Abstandsabstand) mit effektiver Kühlung auf 10 °C.
- Gegossenes lineares oder hyperbolisches Porengradientengel (1,5-3,0 mm Abstandshalter) mit einem rührenden Zweikammer-Gradientenmischer, der von einer Pumpe angetrieben wird (siehe Tabelle der Materialien und Reagenzien). Im vorgestellten Beispiel (lineares Gradientengel 1%-13%):
- Bereiten Sie eine 13 ml Lösung für die vordere (Misch-)Kammer vor, die aus: 13% Acrylamid (aus 30% Stammlösung, 37,5:1,0 Acrylamid:Bisacrylamid), 0,75 M Aminocaproinsäure, 50 mM Bis-Tris (pH = 7,0) und 10% Glycerin besteht.
- Bereiten Sie eine 10 ml Lösung für die Reservoirkammer vor, die aus: 1% Acrylamid (aus 30% Stammlösung, 37,5:1,0 Acrylamid:Bisacrylamid), 0,75 M Aminocaproinsäure, 50 mM Bis-Tris (pH = 7,0) und 0,2% CL-47-Waschmittel besteht.
- Starten Sie den Rührer und fügen Sie 30 Mikroliter APS (Ammoniumperoxodisulfat, 10% Stammlösung) und 2,5 l TEMED (N,N,N',N'-Tetramethylethylendiamin) und 2,5 Mikroliter TEMED in die Lösung in der vorderen Kammer hinzu. Starten Sie die Pumpe und öffnen Sie das Vorderventil (der Durchfluss sollte in 10 min auf den kompletten Guss eingestellt werden). Nach 1 min fügen Sie der Lösung in der Reservoirkammer 90 l APS und 5 l TEMED hinzu und öffnen Sie den Kammeranschluss.
- Lassen Sie das Gel langsam, aber gründlich für mindestens 24 h bei Raumtemperatur (RT) polymerisieren, um einen homogenen Porengrößengradienten zu erzeugen. Bei Feuchtgehalten kann das polymerisierte Gel bis zu 1 Woche lang bei 4 °C aufrecht gelagert werden.
HINWEIS: Absichtlich wird die Oberseite des Gels eine weiche/schleimige Konsistenz haben. Dies wird später entfernt, ermöglicht aber einen reibungslosen Eintritt von Proteinen in das Gel, mit minimalem Risiko von Proteinfällungen, die andernfalls zu Migrationsartefakten (d. h. Streifen oder Proteinfällungen) führen könnten.
- Probenvorbereitung und -beladung
- Vorbereiten von Ladeschlitzen durch Einsetzen geeigneter Abstandshalter (z. B. Siliziumrohre) zwischen die Glasplatten zum Trennen von 0,5-2,0 mg Protein. Die Schlitze sollten mindestens 3 cm breit (oder besser, 5-6 cm breit) sein.
- Löslichkeits-2,5 mg Membran (Mausnieren-Endosome-angereichertes Präparat) in 2 ml Löslichkeitspuffer mit 1% (w/v) nicht-denaturing Waschmittel (ComplexioLyt CL-47) für 30 min auf Eis. Ultrazentrifugate (Sedimentationsabschaltung = 200 S oder weniger; 130.000 x g/11 min wird hier verwendet).
- Konzentrieren Sie das Solubilisate auf einen kurzen 50%/20% (w/v, je 0,3 ml) Saccharoseschrittgradient durch Ultrazentrifugation für 1 h bei 400.000 x g. Die endgültige Proteinausbeute sollte mindestens 1 mg betragen.
- Fügen Sie 0,05% (w/v) Coomassie G-250 zum Solubilisate hinzu und laden Sie die Probe auf das Gel. Beschränken Sie die Proteinbelastung auf einen 2-G-Gel-Spurquerschnitt von 10-15 g/mm, um eine hohe Auflösung zu erzielen und Artefakte zu vermeiden, die durch Proteinfällungen entstehen.
- BN-PAGE Laufbedingungen
- Für laufende Puffer einen Standardkathodenpuffer aus 50 mM Tricin, 15 mM Bis-Tris und 0,01% Coomassie G-250 vorbereiten. Bereiten Sie einen Standard-Anodenpuffer vor, der aus 50 mM Bis-Tris (pH = 7.0) besteht.
- Führen Sie eine präparative BN-PAGE bei 10 °C über Nacht mit einem dreistufigen Spannungsprotokoll13 aus: einer Ausgleichsphase für 30 min bei 100 V, dann einer langsamen (3 h) Rampe bis zur maximalen Spannung (40-50 V/cm Gellänge), die schließlich für mindestens 6 h für Endpunktfokussierung von Proteinen.
HINWEIS: Es wird empfohlen, die Elektrophorese zu unterbrechen, wenn die Migrationsfront die Mitte des Gels erreicht hat, und den Kathodenpuffer ohne Coomassie G250 gegen einen frischen Puffer auszutauschen. Dies hilft, Niederschlagsartefakte im Gel zu vermeiden, die durch den lokalen Zusammenbruch der Matrixporenstruktur entstehen.
2. Gelprobenahme und -verdauung
- Exzision von Gelspuren
- Scannen Sie die Gele nach dem Lauf zu Dokumentationszwecken, während Sie sie zwischen den Glasplatten aufbewahren.
- Zerlegen Sie die Platten und verbrauchen Sie die Fahrspurabschnitte von Interesse.
- Nehmen Sie einen Probenstreifen der Spur zur Analyse durch 2D BN/SDS-PAGE und Proteinfärbung oder Western Blotting (siehe Abbildung 1B), um Interessenbereiche, effektive komplexe Größenauflösung und Proteinreichtum zu bestimmen.
- Fix ausgewählte Gelspuren zweimal für mindestens 30 min mit 30% (v/v) Ethanol und 15% (v/v) Essigsäure.
- Übertragen Sie die Probe auf das Einbettmedium und lassen Sie sie mindestens 2 h bei 4 °C einweichen und aushalten, während die Gelplatte in Zeitlupe auf einem Orbital-Shaker gehalten wird.
HINWEIS: Die Geltrennung sollte sorgfältig auf die Gesamttrennqualität und Migrationsartefakte überprüft werden. Gelbänder, die dominante Proteine darstellen, sollten verzerrungsfrei und homogen in der Intensität sein. Lokale Artefakte auf dem Gel sollten ausgeschnitten oder aus der Analyse herausgelassen werden.
- Einbettung und Kryomikrotome Schneiden
HINWEIS: Dies ist eine verbesserte Version des zuvor beschriebenen und fotodokumentierten Einbettungsverfahrens, das das Einbetten und Schneiden breiterer Gelspuren von bis zu 8 cm11ermöglicht.- Schneiden Sie zunächst feste Gelspuren in Abschnitte (hier 3 cm) genau parallel zum Proteinmigrationsfront-/Bandmuster. Zur einfacheren Handhabung legen Sie jeden Abschnitt auf eine Kunststofffolienstütze mit gleichen Abmessungen.
- Übertragen Sie die Fahrspuren in ein offenes Rohr mit Stopfen (auf der Unterseite geschlossen, zentral perforiert auf der Oberseite, beide präzise mit dem oberen und unteren Ende des Gelabschnitts ausgerichtet).
- Tauchen Sie den Zylinder kurz in flüssigen Stickstoff, um die Erstarrung schnell zu initiieren. Das transparente Einbettmedium verfestigt sich innerhalb von Sekunden und wird weiß in der Farbe.
- Füllen Sie den Hohlraum mit einbettendem Medium, tauchen Sie ihn kurz in flüssigen Stickstoff und frieren Sie den Zylinder mehrere Stunden bei -20 °C ein.
HINWEIS: Eine schnelle Kühlung des Zylinders durch Eintauchen in flüssigen Stickstoff hilft, eine Verschiebung der Gelplatte innerhalb des Rohres zu vermeiden. Verzerrungen sollten vermieden werden, um eine hohe Auflösung in der folgenden MS-Analyse zu gewährleisten. - Nach der Demontage die Kunststofffolie entfernen und den Block mit dem eingebetteten Gelabschnitt auf einen gekühlten, größeren Durchmesser übertragen, Metallzylinder auf einem flachen Träger (d.h. Petrischale) platziert und mit Einbettmedium an der Außenseite des Zylinders versiegelt. Füllen Sie den Zylinder mit einbettendem Medium und einfrieren Sie gründlich.
- Wiederholen Sie diesen Vorgang mit der anderen Seite des Zylinders, um einen festen Block mit koplanaren Flächen zu erhalten.
- Entfernen Sie den Block aus dem Zylinder, kleben Sie ihn mit einbettendem Medium auf einen vorgekühlten Metallhalter und stecken Sie den Halter in die Kryoilicing-Maschine (Kryotome). Die Oberfläche des Blocks muss sorgfältig in Bezug auf die Schnittebene ausgerichtet werden. Lassen Sie es bei der optimalen Temperatur für den Schneidprozess ausdemivieren (hier -15 °C).
HINWEIS: Verwenden Sie einen langsam fortschreitenden manuellen Schneidzyklus von 0,1 mm Schrittgröße, bis Sie die Oberfläche des eingebetteten Gelabschnitts treffen, um eine korrekte Positionierung zu gewährleisten. - Die Gelscheiben nacheinander mit einer endgültigen gewünschten Dicke von 0,25 mm Schrittgröße ernten und einzeln in Reaktionsröhrchen mit geringen Proteinbindungseigenschaften übertragen.
HINWEIS: Bei diesem Aufbau können gleichmäßige Gelscheiben leicht bis zu 0,1 mm und bis zu 0,5 mm dicke Gelscheiben erhalten werden.
- Tryptische Verdauung
- Führen Sie die tryptische Verdauung in-Gel nach dem ausgiebigen Waschen der Gelscheiben (mindestens drei zusätzliche Waschrunden werden empfohlen, um polymere Komponenten des Einbettmediums zu entfernen) nach einem Standardverfahren11vor.
- Vakuumtrocken eluierte Peptide und lösen sich in 0,5% (v/v) Trifluoressigsäure durch Schütteln bei 37 °C (10 min) auf, gefolgt von Badbeschallung (5 min) und kurzer Zentrifugation.
3. Massenspektrometrie
- nanoHPLC und MS-Einrichtung
- Last verdaute Proben auf eine C18-Vorspalte (Partikelgröße = 5 m; Durchmesser = 300 m) mit 0,05% (v/v) Trifluoressigsäure mit einem (split-free) Nano-HPLC gekoppelt an ein Massenspektrometer mit hoher Auflösung.
- Elute erfasste Peptide mit wässrig-organischem Gradient (Eluent A): 5 min 3% B, 120 min von 3% B bis 30% B, 20 min von 30% B bis 99% B, 5 min 99% B, 5 min von 99% B bis 3% B, 15 min 3% B (Durchflussrate = 300 nL/min).
HINWEIS: csBN-MS-Gelscheiben führen in der Regel zu Proben mit geringer bis mittlerer Peptidfülle und einem begrenzten Grad an Komplexität. NanoLC-MS/MS-Analysen sollten daher mit einer Einrichtung durchgeführt werden, die eine angemessene Empfindlichkeit und Sequenzierungsgeschwindigkeit, hohe Massenauflösung (>100.000) und maximalen Dynamikbereich (effektiv 3-4 Größenordnungen) bietet. Es sind jedoch keine langen Säulenabmessungen oder Elutionsgradienten erforderlich, die über 3 h hinaus verlängert werden. - Getrennte eluierte Peptide in einem Emitter (d.h. 75 m; Spitze = 8 m) manuell verpackt ca. 20 cm mit C18-Material (Partikelgröße = 3 m). Elektrospray die Proben bei 2,3 kV (positiver Ionenmodus) in die beheizte Transferkapillare (250 °C) des Massenspektrometers.
- Durchführen von Analysen mit den folgenden Instrumenteneinstellungen11: maximale MS/MS-Injektionszeit = 400 ms; Ausschlussdauer = 60 s; Mindestsignalschwelle = 5.000 Zählungen, Top-10-Vorläufer fragmentiert; Isolationsbreite = 1,0 m/z).
ANMERKUNG: Um die Kalibrierung von Masse, Retentionszeit und Zuweisung von Peptidsignalen in einer großen Anzahl von Datensätzen oder Messungen zu erleichtern, wird empfohlen, die entsprechenden MS-Messreihen ohne Unterbrechungen oder Änderungen von Parametern und Hardware durchzuführen ( d. h. auf derselben C18-Säule/Emitter).
- Proteinidentifikation (MS-Daten, wie zuvor beschrieben11)
- Extrahieren Sie Spitzenlisten aus Fragmentionenspektren mit dem Werkzeug "msconvert.exe" (Teil von ProteoWizard).
- Verschieben Sie alle Vorläufer-m/z-Werte für jedes Dataset durch den medianen m/z-Offset aller Peptide, die Proteinen in einer vorläufigen Datenbanksuche mit 50 ppm Peptidmassentoleranz zugeordnet sind.
- Suchen Sie die korrigierten Spitzenlisten mit einer geeigneten Suchmaschine (hier Maskottchen 2.6.2) mit allen Mauseinträgen der UniProtKB/Swiss-Prot Datenbank (Release 2018_11).
- Wählen Sie "Acetyl (Protein N-term)", "Carbamidomethyl (C)", "Gln | pyro-Glu (N-Term Q), Glu | pyro-Glu (N-Term E)", "Oxidation (M)" und "Propionamid (C)" als variable Modifikationen.
- Stellen Sie die Peptid- und Fragmentmassentoleranz auf 5 ppm bzw. 0,8 Da fest, und lassen Sie eine verpasste tryptische Spaltung zu. Legen Sie den Erwartungswert-Cut-off für die Peptididentifikation auf 0,5 oder weniger fest. Verwenden Sie eine Decoy-Datenbanksuche, um die falsch positive Ermittlungsrate (FDR) zu ermitteln. Legen Sie die FDR auf 1 % fest oder wenden Sie zusätzliche Qualitätskriterien an, um eine zuverlässige Identifizierung zu gewährleisten.
HINWEIS: Das vorgestellte Experiment identifizierte mehr als 3.500 Proteine mit einer durchschnittlichen Peptid-FDR von 4,4 x 0,77 % (n = 101 Scheibenproben) oder 3.000 Proteinen, wenn Peptid FDR auf 1 % eingestellt wurde. Wichtig ist, dass strengere Kriterien für die Auswahl von profilierten Proteinen verwendet wurden (2.568). Es umfasste alle Proteine, die mit mindestens zwei Peptiden identifiziert wurden, von denen mindestens eines proteinspezifisch ist, in mindestens einer der 101-Slice-Proben.
- Proteinquantifizierung
- Verwenden Sie Peptidsignalintensitäten (Spitzenvolumen [PVs]) für die Proteinquantifizierung, die aus FT-Vollscans gewonnen und mit entsprechender Software auf Retentionszeit und Massenverschiebungen korrigiert werden (hier MaxQuant v1.6.3).
- Richten Sie MS-Datasets nacheinander aus, um die Peptid-Elutionszeiten (Gesamtdurchschnitt) mithilfe der LOESS-Regression zu referenzieren. Weisen Sie PVs Peptiden entweder direkt (MS/MS-basierte Identifikation) oder indirekt (d. h. basierend auf ihrer übereinstimmenden m/z- und Elutionszeit innerhalb sehr enger Toleranzen) zu.
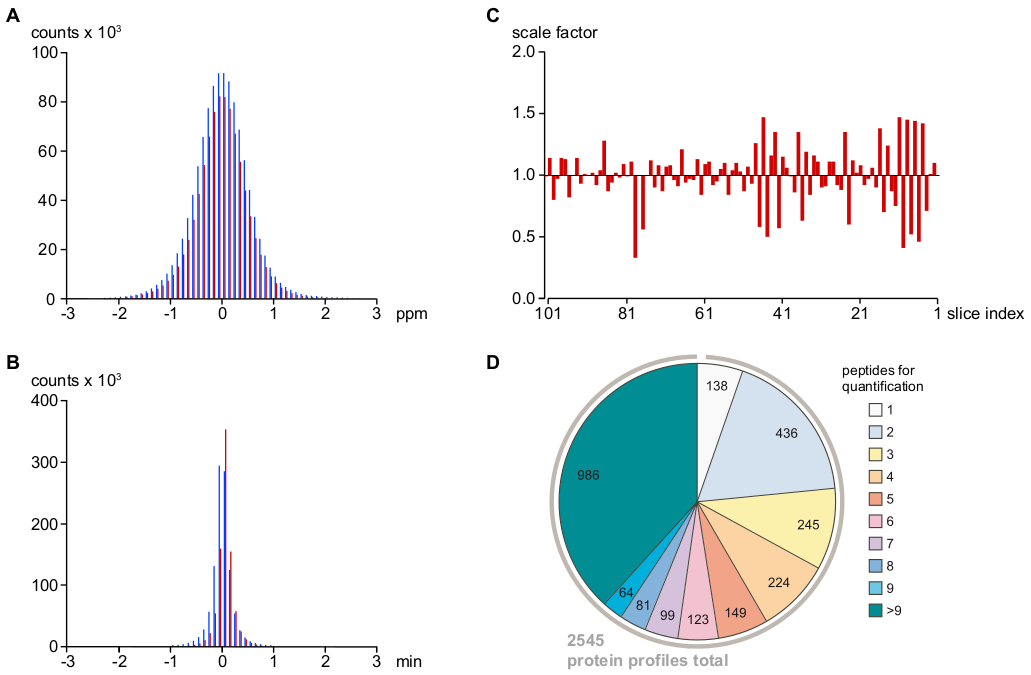
HINWEIS: Dieses Protokoll verwendet interne Software für die Zuweisung der "eingefügten" sogenannten Peptide. Eingestellte Parameter führen zu effektiven m/z- und Elutionszeit-Matching-Toleranzen von 2 ppm bzw. 1 min (siehe Abbildung 2A,B). - Korrekt für systematische Run-to-Run-Variationen der Peptidlast und Ionisationseffizienz durch Peptidintensitätsneuskalierung, berechnet aus den medianen Unterschieden der relativen Peptidintensitäten zwischen benachbarten Slice-Proben (Abbildung 2C).
- Filtern Sie PV-Daten nach Ausreißern und verbleibenden falsch positiven Zuordnungen, die durch interne PV-Konsistenzanalysen identifiziert wurden.
- Normalisieren Sie PVs jedes Peptids auf ihre maximalen Werte über alle Slice-Datasets, die relative Peptid-Flussprofile ergeben.
- Schließlich berechnen Sie relative Proteinhäufigkeitsprofile als Durchschnittswerte von mindestens zwei (und bis zu sechs oder 50 %, welcher Wert höher ist) der am besten korrelierenden Peptidprofile über ein Fenster von drei aufeinanderfolgenden Scheiben. Dies ermöglicht die Überbrückung fehlender PV-Werte und die Reduzierung von Geräuschen.
HINWEIS: Dies führte schließlich zu 2.545 (von 2.568 vorgewählten) Proteinprofilen (Abbildung 2D).
- Charakterisierung von Proteinkomplexen
- Analysieren Sie Proteinprofile, indem Sie zuerst die Spitzenerkennung mit der lokalen Maxima-Methode durchführen, und passen Sie nacheinander Normalverteilungen an diese Spitzen an, wodurch die Position (d. h. Slice-Index oder scheinbar komplexe Größe) ihrer Maxima und FWHM (volle Breite bei Halbmaximale Intensität) (Einset von Abbildung 4).
HINWEIS: Im Dataset werden Profile automatisch mithilfe benutzerdefinierter Skripts analysiert. Die kleinsten FWHM-Werte sind bezeichnend für die effektive Größenauflösung des Ansatzes (hier 6 x 0,25 = 1,5 mm). - Verwenden Sie Referenzproteinkomplexspitzen mit definierter Molekularmasse (wie in der UniProtKB/Swiss-Prot-Datenbank berichtet) für die lineare Regressionsanalyse der log10(vorhergesagte Molekularmasse)-Werte, um Slice-Zahlenindizes in scheinbare molekulare Größen (d. h. scheinbare komplexe Größe in kDa).
ANMERKUNG: In dieser Studie wurden 23 Markerkomplexe in der Stichprobe ausgewählt (Abbildung 4), basierend auf (i) monodispersen Formen von Profilspitzen, (ii) experimenteller Unterstützung von Molekulargewichten und (iii) Verteilungen entlang der untersuchten BN-PAGE-Gelabschnitte.
- Analysieren Sie Proteinprofile, indem Sie zuerst die Spitzenerkennung mit der lokalen Maxima-Methode durchführen, und passen Sie nacheinander Normalverteilungen an diese Spitzen an, wodurch die Position (d. h. Slice-Index oder scheinbar komplexe Größe) ihrer Maxima und FWHM (volle Breite bei Halbmaximale Intensität) (Einset von Abbildung 4).
Ergebnisse
Die überwiegende Mehrheit der herkömmlichen BN-MS-Studien sowie der kürzlich eingeführte hochauflösende csBN-MS-Ansatz wurden auf mitochondriale und plastidische Präparate angewendet, die (i) leicht verfügbar sind, (ii) eine begrenzte Komplexität aufweisen und (iii) Express- (Membran-)Proteinkomplexe bei hoher Dichte. Dieses Protokoll erweitert die Anwendung von hochauflösendem Complexome-Profiling auf nicht-mitochondriale Membranen, die wenig reichlich eiternde Proteine exdrücken, über die nur wenige Informationen über ihre Integration in Komplexe verfügbar sind. Zu Demonstrationszwecken haben wir uns für ein endosome-angereichertes Membranpräparat aus der Mausniere entschieden, das durch Dichtegradientenzentrifugation gewonnen wird.
Die Optimierung dieser Zubereitung wurde durch das Markerprotein TPC1 geleitet, das intrazelluläre Ionenkanäle bildet, die überwiegend auf frühe und recycelte Endosomen lokalisiert sind12. Es ist auch stark in renalen proximalen röhrenförmigen Zellen exprimiert, wie die immunhistochemische Analyse von Nierengewebeabschnitten zeigt (Abbildung 1A). Diese Membranen wurden sanft löslich (ComplexioLyt 47 bei einem niedrigen Protein-Waschmittel-Verhältnis von 1:8) und konzentrierten sich durch Ultrazentrifugation auf ein Saccharosekissen. Letzteres erwies sich als wichtiger Schritt zur Entfernung von überschüssigen Bestandteilen mit niedrigerem Molekulargewicht (z. B. Waschmittel, Lipide, Salze, organische Polymere und Metaboliten), die sich tendenziell negativ auf die Auflösung präparativer BN-PAGE-Trennungen auswirken.
Komplexe Trennung auf einem nativen 1%-13% (w/v) Polyacrylamid Gradientgel (Abbildung 1B, mittleres Panel) zeigte stark gefärbte Proteinbänder mit sehr wenig Migrationsartefakten. SDS-PAGE Trennung eines schmalen BN-PAGE-Gelstreifens(Abbildung 1B, in rot geschachtelter Rahmen) als zweite Dimension gefolgt von western blot analysis zeigte ein gut aufgelöstes Muster unterschiedlicher TPC1-assoziierter komplexer Populationen (Abbildung 1B , oberes Panel, gekennzeichnet durch rote Pfeile), höchstwahrscheinlich aufgrund der Verbindung mit zusätzlichen Proteinuntereinheiten und/oder posttranslationalen Modifikationen (z. B. Glykosylierung12). Ein 3 cm großer Abschnitt wurde wie beschrieben11für kryomikrotome slicing ausgeschnitten, fixiert und verarbeitet. Die einzelnen Schritte dieses Verfahrens (insbesondere die präzise Ausrichtung des breiten Gelabschnitts), die für die Aufrechterhaltung der Auflösung während der Probenahme von entscheidender Bedeutung ist, sind im begleitenden Video dokumentiert. Der eingebettete Gelteil wurde schließlich in 101 Gelscheiben mit einer gleichmäßigen Dicke von 0,25 mm geschnitten(Abbildung 1B, untere Platte), die separat verdaut und durch Hochleistungs-LC-gekoppelte Massenspektrometrie analysiert wurden.
Neben der Größenauflösung ist die Qualität der Proteinquantifizierung der Schlüssel für eine erfolgreiche Complexome-Profilierung. Mit der MS-Einrichtung und den verwendeten Einstellungen war die Analyse der Proben recht umfassend, was zu einer durchschnittlichen Identifizierung von mehr als 1.000 Proteinen und 10.000 Peptiden (davon 8.200 proteinspezifisch) pro Scheibe und rund 3.000 Proteinen und 43.000 Proteinen und 43.000 Proteinen führte. Peptide (38.500 davon waren proteinspezifisch) insgesamt. Aufgrund der stochastischen Natur der datenabhängigen MS/MS-Sequenzierung und ihrer Einschränkungen im Dynamikbereich waren die Intensitätsinformationen für weniger reichlich etolische Proteine jedoch noch fragmentarisch. Daher wurde ein ausgeklügeltes MS-Datenverarbeitungsverfahren11durchgeführt, das auf der präzisen Zuordnung von Peptidsignalen (Spitzenvolumen [PVs] = peptidbezogene Signalintensitäten, die über m/z und Zeit integriert sind) über die gesamte Datensatzreihe basiert.
Wie in Abbildung 2A,Bdargestellt, waren Abweichungen von Peptidsignalen in Masse und Retentionszeit, die nach der Kalibrierung verblieben waren, identisch für MS-sequenzierte und indirekt zugewiesene PVs (mit sehr engen Toleranzen von <1 ppm und <0,5 min für 95% der pVs), was auf eine sehr niedrige Rate für falsch-positive PV-Zuweisung hindeutet. Verbleibende Ausreißer wurden basierend auf ihrer Konsistenz mit anderen verwandten PVs gefiltert. Da alle MS-Messungen nacheinander an demselben LC-MS-Setup ohne Änderungen an Parametern oder Hardwarekomponenten durchgeführt wurden, wurden Durchlaufabweichungen (bestimmt als Median aller PV-Intensitäten in einer Stichprobe relativ zu denen in den benachbarten Slices) durch neu skalierende PV-Datensätze (Abbildung 2C) leicht eliminiert wurden. Die resultierenden Peptidintensitätsinformationen wurden dann verwendet, um 2.545 Protein-relative Häufigkeitsprofile zu rekonstruieren. Wie in Abbildung 2Ddargestellt, basierten mehr als 75 % dieser Proteinprofile auf mindestens drei unabhängigen proteinspezifischen Peptiden.
Als nächstes bewertete das Protokoll die Relevanz der Schrittgröße der BN-PAGE Gelprobenahme für die Auflösung von Proteinkomplexen. Zu diesem Zweck wurden Slice-Datensätze durch Summieren der PV-Informationen aus zwei, drei oder vier aufeinanderfolgenden Slices verbunden, wodurch das Ergebnis für Schrittgrößen von 0,5 mm, 0,75 mm und 1 mm simuliert wurde (im Vergleich zur ursprünglichen Probenahme von 0,25 mm). Abbildung 3 zeigt die resultierenden Häufigkeitsprofile für Protein TPC1 als Beispiel (A-D). Mit 0,25 mm waren die relativen Intensitäten und die Größentrennung der TPC1-assoziierten komplexen Populationen (Abbildung 3A) gut mit den Ergebnissen der Western Blot-Analyse übereinstimmend (Abbildung 1B, Oberteil); obwohl das Profil etwas Rauschen zeigte, das hauptsächlich auf fehlende Werte ("Lücken") in der PV-Matrix zur Quantifizierung zurückzuführen ist.
Das Verbinden von zwei Scheiben, die 0,5 mm entsprechen, hielt die korrekten Intensitäten und die Trennung der TPC1-assoziierten Komplexe bei und entfernte Quantifizierungsgeräusche (Abbildung 3B). Größere Schrittgrößen von 0,75 mm und 1 mm(Abbildung 3C,D) führten dagegen zu einem Verlust der Größenauflösung und der Abschaffung der Diskriminierung komplexer TPC1-Subpopulationen. Es sei darauf hingewiesen, dass die überwiegende Mehrheit der veröffentlichten konventionellen BN-MS-Analysen manuell geschnitten 2 mm Scheiben (ca. 60, um die gesamte Gelspur abdecken)7,8,9,10.
Die Umwandlung des Migrationsentfernungs- oder Slice-Index in molekulare Größe basiert im Allgemeinen auf Markern, entweder kommerziell erhältlichen nativen Standardproteinen oder gut charakterisierten endogenen Proteinkomplexen mit bekannter Untereinheitszusammensetzung (meist [super] komplexe der mitochondrialen oxidativen Atmungskette [OXPHOS])13. Da die BN-PAGE-Trennung jedoch auf dem effektiven molekularen Querschnitt basiert, der nicht nur durch die Molekularmasse, sondern auch durch die 3D-Struktur und die Anzahl der zugehörigen Lipide, Waschmittel und Coomassie-Moleküle bestimmt wird, können einzelne Proteine größeren Abweichungen. Daher wurde ausgewählt, größere Sätze von Proteinkomplexen als Marker11zu verwenden. Das Diagramm in Abbildung 4 zeigt 23 ausgewählte Marker mit einer repräsentativen Untereinheit, die als schwarzer Kreis dargestellt ist und die Log10-Werte ihrer vorhergesagten Molekularmasse (gemäß der UniProtKB/Swiss-Prot-Datenbank) im Vergleichangibt. den Slice-Index des entsprechenden Profilspitzenmaximums. Letztere wurden aus automatisierten Gaußschen Anpassungen an die relativen Überflussdaten gewonnen, wie im Einset von Abbildung 4 gezeigt, das das Beispiel mit Chaperon BCS1 zeigt. Die lineare Regression (rote Linie) bot eine Funktion zum Konvertieren von Slice-Indexwerten in scheinbare molekulare Größen, die von 160-630 kDa entlang des untersuchten Gelabschnitts reichten.
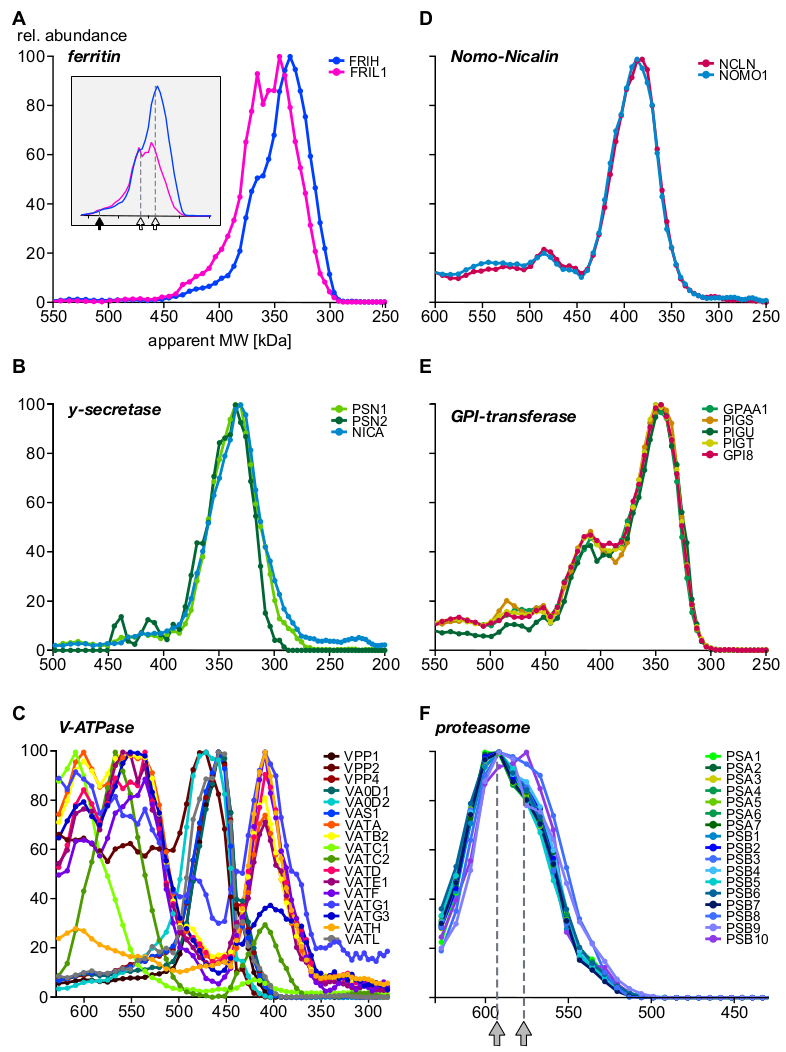
Schließlich lieferte die Analyse Informationen über gut charakterisierte Komplexe und zeigte die Existenz neuartiger Untereinheiten und komplexer Baugruppen auf. Beispiele, die verschiedene Aspekte des Complexoms hervorheben, sind in Abbildung 5 dargestellt (A-C: Proteine, die exprimiert oder vorzugsweise in endosomalen Kompartimenten lokalisiert werden; D-F: Komplexe aus anderen subzellulären Lokalisationen). Das eisentransportierende Protein Ferritin bildet bekanntermaßen Komplexe aus 24 leichten (FRIL1) und/oder schweren (FRIH) Untereinheiten mit einem Gesamtmolekulargewicht von 440 kDa14 (Abbildung 5A,gefüllter Pfeil). Die Untereinheitsprofile (Abbildung 5A) deuten auf die Existenz von mindestens zwei kleineren Formen des Komplexes (mit einer scheinbaren Masse von 360 kDa und 340 kDa; offene Pfeile) mit deutlichen schweren/leichten Kettenstoichiometrien (besser sichtbar nach Häufigkeiten, Einsetz von Abbildung 5A), die reichlich in Endosomen vorhanden sind.
Nicalin-Nomo1-Komplexe15 (Abbildung 5D), der Gamma-Sekretase-Kernkomplex16 (Abbildung 5B) und die GPI-Transamidase-Maschine17 (Abbildung 5E) zeigen Häufigkeitsverhältnisse ihrer Kernuntereinheiten über den gesamten Größenbereich und unabhängig von der Zuordnung mit zusätzlichen Proteinen. Dies weist darauf hin, dass ihre Untereinheiten exklusiv füreinander sind. Vacuolar H+-ATPases sind Multiproteinkomplexe, die aus einem Pool von mehr als 20 Untereinheiten modular mit einem Gesamtmolekulargewicht von rund 900 kDa zusammengesetzt werden. Abbildung 5C zeigt Unterkomplexe mit unterschiedlichen Zusammensetzungen aus mindestens 17 Untereinheiten, die entweder biologische (Dis-)Baugruppenzwischenprodukte oder Unterkomplexe darstellen, die durch die experimentellen Bedingungen erzeugt wurden, von denen einige auch in einer aktuellen BN-MS-Studie18beobachtet. Ein weiteres Multiprotein-Komplex-Beispiel ist das Proteasom19 (Abbildung 5F). Eine genaue Untersuchung der Häufigkeitsprofile der Alpha- und Beta-Untereinheiten, die den 20S-Proteasomkern bilden, legt nahe, dass zwei große komplexe Populationen mit subtilen Größenunterschieden (590 kDa und 575 kDa, angezeigt durch graue Pfeile) und der Integration von drei Beta-Untereinheiten vorhanden sind.
Zusammenfassend lässt sich sagen, dass csBN-MS-Komplexomeprofilierung endosome-angereicherter Nierenmembranen umfassende und detaillierte Ergebnisse zur (i) Integration einheitlicher, wenig vorhandener Zielproteine in Komplexe, (ii) der allgemeinen komplexen Zusammensetzung der Untereinheit und Stoichiometrie und (iii) komplexe Heterogenitäten, Unterstrukturen und (Dis-)Montagezwischenprodukte.

Abbildung 1: Präparative BN-PAGE-Trennung von solubilisierten endosome-angereicherten Membranen von der Mausniere unter Verwendung des intrazellulären Kanals TPC1 als Marker. (A) Immunhistochemische Lokalisation des TPC1-Proteins in renalen proximalen Tubuli durch konfokale Mikroskopie. Grün: Anti-TPC1-Antikörper12 Färbung visualisiert mit sekundären Cy3-biotinylierten Ziege Anti-Kaninchen IgG; rot: biotinyliertes Lotus tetragonolobus lectin (LTL, 10 g/ml, FITC-konjugiert), das die luminale Oberfläche proximaler Tubuluszellen markiert. Der Einset zeigt die Färbung eines entsprechenden Abschnitts aus einer TPC1-KO-Niere als Negativkontrolle. Weiße Schuppenbalken sind 20 m. Bemerkenswert ist auch die starke TPC1-Expression in intrazellulären Vesikeln, die aus unabhängigen Experimenten bekannt sind, um frühe und recycelnde Endosomen darzustellen12. (B) Präparative BN-PAGE-Trennung von 2,5 mg solubilisierten endosom angereicherten Membranen auf einem 1%-13% (w/v) Polyacrylamid-Gradientengel. Für die nachfolgende SDS-PAGE/Western Blot Analyse (Oberpanel) wurde eine schmale Spur (rot eingerahmt) geschnitten, die verschiedene TPC1-assoziierte Komplexe und Glykosylierungsmuster auflöste (rote Pfeile: Anti-TPC1/Anti-Kaninchen HRP/ECL prime; grün: Positionen und vorhergesagt Massen [MDa] von Markerproteinkomplexen, die durch die gesamte Proteinfärbung [SYPRO Ruby Blot Stain]) des Fleckens identifiziert werden. Von rechts nach links: Na+/K+-transporting ATPase, Cytochrome b-c1 komplexer Dimer, ATP-Synthase, NADH:Ubiquinon oxidoreductase. Ein 3 cm großer Abschnitt von Interesse aus der Gelspur wurde entfernt, eingebettet in Gewebeeinbettungsmedium, montiert und in 101 Abschnitte (0,25 mm) entlang der Proteinmigrationsfront mit einem Kryomikrotom (untere Platte; siehe Videolink) geschnitten. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 2: Wichtige Parameter zur Bestimmung der Genauigkeit der MS-Signalzuweisung und -quantifizierung sowie der Analysetiefe. (A) Verteilung der relativen Massenfehler (in ppm) nach m/z Kalibrierung von MS/MS sequenziert (rote Balken) und indirekt zugewiesen (d. h. basierend auf eng übereinstimmenden Massen- und Retentionszeiten, siehe Protokoll; blaue Balken) Peptidsignale. Dies deutet auf einen endgültigen Massenfehler von <1 ppm (für 95% der Signale/zugewiesenen PVs) und eine sehr niedrige Rate falsch positiver Zuordnungen hin. (B) Verteilung der Nano-HPLC-Retentionszeitabweichungen vom Gesamtdurchschnitt nach der Elutionszeitausrichtung von Peptidsignalen unter Verwendung der Loess-Regression (siehe Protokoll) und der Farbcodierung gemäß (A). Der Zeitfehler beträgt weniger als 30 s für >95% der Peptidsignale/zugewiesenen PVs. (C) Ausführungsvariation der gesamten MS-Intensitäten, die relativ zum Durchschnitt der beiden benachbarten Samples dargestellt werden. Diese Skalenfaktoren wurden auf die rohen PV-Tabellen angewendet, um systematische technische Fehler zu minimieren. (D) Peptidinformationen, die zur Berechnung der relativen Häufigkeitsprofile von Proteinen verwendet werden. Nach der Filterung proteinspezifischer Peptide für Ausreißer, schlecht bewertete oder Einzelidentifikationen (siehe Protokoll) wurden 2.545 Protein-Überflussprofile ermittelt, von denen >75% mit angemessener Zuversicht auf mindestens drei Peptiden basierten. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 3: Kritische Auswirkungen der Schrittgröße bei der Gelprobenahme auf die komplexe Auflösung von TPC1. Die Datensätze wurden durch Summierung der Signalintensitäten in Gruppen von 1, 2, 3 und 4 aufeinander folgenden Slices (A-D) ergänzt und identisch verarbeitet, um verschiedene Schrittgrößen beim Gelschneiden wie angegeben zu simulieren. Das TPC1-Profil zeigt einige (Oversampling-)Rauschen bei 0,25 mm, aber eine gute Größenauflösung von drei komplexen Populationen (siehe auch Abbildung 1B), die bei einer Schrittbreite von 0,5 mm weitgehend erhalten bleibt. Die Diskriminierung dieser Bevölkerungsgruppen geht verloren, wenn 1 mm näher rückt. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 4: Bestimmung des sichtbaren Molekulargewichts. Als Größenmarker wurden 23 Markerkomplexe mit definierter molekularer Zusammensetzung (wie angegeben, nach UniProtKB/Swiss-Prot) verwendet. Logarithmische Werte ihrer erwarteten Molekulargewichte (in kDa) wurden im Vergleichdargestellt. der Profilspitzenindex für maximale Seimen der angegebenen repräsentativen Proteinuntereinheit (gefüllte Kreise in Schwarz). Die lineare Regression, die zu diesen Daten passt (rote Linie), stellte eine Funktion bereit, die Slice-Indexwerte in scheinbare Molekulargewichte konvertierte. Spitzenmaxima wurden durch automatisierte Gaußsche Passungen an Proteinprofilspitzen bestimmt, wie im Einset (rechts) für das Chaperonprotein BCS1 gezeigt (Primärdaten in blau, Passgrenzen, die durch orange Linien angezeigt werden, Passfunktion in Rot). Darüber hinaus passen diese passt bestimmte Spitzenhalbmaximalbreiten (grüne Linie, 6,5 Scheiben oder 1,6 mm für das gezeigte Beispiel) mit den schärfsten Fokussierkomplexen, die sich um ein 1,5 mm Gel erstrecken. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 5: Beispiele für Proteinkomplexe Untereinheitenprofile. Relative Proteinfülle vs. sichtbares Molekulargewicht, das für schwere und leichte Kette von Ferritin (A) gezeichnet wird und die molekulare Heterogenität der Ferritin-Untereinheitssstosprache offenbart, deutlicher sichtbar nach der Neuskalierung der Häufigkeiten (Einschub). Ausgefüllte Pfeile und offene Pfeile bezeichnen den gesamten Komplex (440 kDa) bzw. zwei Unterkomplexe. Die Gamma-Sekretase (B) untereinheiten quantitativ in eine einkernige komplexe Population integriert. Die Unterkomplexe von Vacuolar H+-ATPases (C) wiesen mehrere Baugruppen mit unterschiedlicher Untereinheitszusammensetzung auf, die alle in Endosomen ausgedrückt waren. Die Nomo1- und Nicalin-Proteine (D) bildeten einen exklusiven Komplex (die GPI-Transamidase), eine enzymatische Maschinerie mit mehreren Untereinheiten, die mehrere Komplexe bildet. (E) Der 20S-Proteasom-Kernkomplex zeigt (F) ein subtiles subkomplexes Muster mit zwei Populationen, die durch Pfeile in Grau gekennzeichnet sind, die alle aus anderen subzellulären Lokalisationen stammen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Diskussion
Die vorgestellte Studie basiert auf der csBN-MS-Technik, die zuvor mit einer mitochondrialen Zubereitung11 bewertet wurde, und beinhaltete Verbesserungen bei der Probenvorbereitung, Gelverarbeitung und MS-Datenauswertung. Die fokussierte Analyse eines Abschnitts des großflächigen Separations-BN-PAGE-Gels lieferte einen umfassenden Satz von Daten, die Qualitätskennzahlen aufzeigten, die mit der Studie mit mitochondrialen Membranen vergleichbar sind. Massen- und Retentionszeitfehler sowie Lauf-zu-Lauf-Variationen wurden sehr gering gehalten und lieferten die Grundlage für die Bestimmung zuverlässiger Proteinhäufigkeitsprofile. Die Größenauflösung schien gut zu sein, mit halbmaximalen Spitzenbreiten von nur sechs Scheiben (entsprechend 1,5 mm, Abbildung 4) und relativen Größenunterschieden von weniger als 10 % gelöst (Abbildung 3, Abbildung 5A). Diese Werte entsprachen nicht vollständig der Größenauflösungsqualität der vorherigen csBN-MS-Analyse von Mitochondrien (trotz der ausgewählten kleineren Gel-Sampling-Schrittgröße), aber sie sind deutlich besser als die Leistung herkömmlicher BN-MS- oder Größenausschluss-MS nähert sich 20, die in letzter Zeit populär geworden sind.
Die Bedeutung einer hohen effektiven komplexen Größenauflösung wird durch das Simulationsexperiment in Abbildung 3 (mit DenPc1-assoziierten Komplexen) unterstrichen, das durch die 2D-BN/SDS-PAGE Western Blot-Analyse kaum aufgelöst werden kann (Abbildung 1B). Diese Ergebnisse deuten darauf hin, dass das 0,25 mm-Schneiden in diesem Fall zu einigen Überstichproben geführt hat, aber es erwies sich dennoch als nützlich für die Beseitigung von "Quantifizierungsgeräuschen", ohne die effektive Größenauflösung zu beeinträchtigen. Daher ist im Einklang mit den bisherigen Ergebnissen11generell eine Stichprobenstufengröße von 0,3 mm zu empfehlen.
Insbesondere die Diskriminierung von TPC1-assoziierten Komplexen geht mit 1 mm Gelprobenahme vollständig verloren, was die kleinste Schrittgröße ist, die durch manuelles Schneiden in herkömmlichen BN-MS5,6bereitgestellt wird. Dies könnte die Tatsache erklären, dass trotz der Bereitstellung leistungsfähiger MS-Technologien nur sehr wenige Proteinkomplexe und Untereinheiten de novo durch Complexome-Profiling identifiziert wurden. Neben seiner guten Auflösungsleistung bietet csBN-MS eine hohe Vielseitigkeit. Membrangebundene Komplexe und lösliche Proteinkomplexe von 50 kDa bis zu mehreren MDa können in einem einzigen Experiment mit minimaler Vorspannung effektiv gelöst werden11. Dies steht im Gegensatz zu alternativen Trenntechniken, die für komplexe Profilierungen wie Größenausschluss oder Ionenaustauschchromatographie verwendet werden, die mit Teilmengen löslicher Proteine mit bestimmten Größenbereichen oder Ladungseigenschaften arbeiten. Auf der anderen Seite ist csBN-MS weniger skalierbar (maximale Belastung von 3 mg Protein pro Gel), kann technisch anspruchsvoll sein und kann nicht automatisiert werden.
Insgesamt zeigen die Ergebnisse, dass csBN-MS-basiertes Complexome-Profiling erfolgreich auf nicht-mitochondriale Ziele angewendet werden kann, aber auch einige damit verbundene Herausforderungen aufzeigen. Daher erfordern eine effiziente Extraktion und biochemische Stabilität von Proteinkomplexen mehr Optimierung und Reinigungsschritte und können dennoch begrenzt sein. Innerhalb des untersuchten Größenfensters war die Anzahl der gut fokussierten, monodispersen Proteinkomplexe im Vergleich zu einer mitochondrialen Probe in der Tat deutlich geringer (Daten nicht dargestellt). Es wird auch empfohlen, die BN-PAGE-Probenlasten zu senken, um eine akzeptable Geltrennung zu erhalten. Höhere Belastungen erfordern möglicherweise breitere Gelspuren, die für das Schneiden schwieriger zu verarbeiten sind (siehe begleitendes Video). Darüber hinaus war die Proteinkomplexität der Proben höher (etwa zweifach) als die mitochondrienabgeleiteten Scheibenverdauten, was zu mehr fehlenden PV-Werten und einem reduzierten Dynamikbereich führte. Tatsächlich fehlten einige kleine Proteine, die zu den in Abbildung 5 dargestellten Komplexen gehören sollten, in den Analysen. Diese Probleme können in Zukunft durch den Einsatz schnellerer und sensiblerer MS-Instrumente oder datenunabhängiger Erfassungsmodi gelöst werden.
Die Probenvorbereitung ist sehr wichtig für die Protein-Komplex-Retrieval-, Stabilitäts- und Geltrennungsqualität. Parameter und Verfahren sollten für jedes Quellgewebe, Zelllysat, Membran (Fraktion) und Proteinkomplex von Interesse optimiert werden. Es werden die folgenden allgemeinen Empfehlungen bereitgestellt, die zur Erweiterung von Anwendungen von csBN-MS beitragen können:
i) Die Vorbereitung von Proben frisch und die Vermeidung von Erwärmung/Einfrieren, starke Verdünnungen, Änderungen der Pufferbedingungen und unnötige Verzögerungen;
ii) Verwendung von Puffern, die im Wesentlichen salzfrei sind (ersetzen durch 500-750 mM Betain oder Aminocaproinsäure), etwa einen neutralen pH-Wert und einen Gehalt an bis zu 1 % (w/v) nicht abbauendem Reinigungsmittel (Protein:Waschmittel-Verhältnis zwischen 1:4-1:10 für die Löslichkeit der Membran Proteinkomplexe, kein Reinigungsmittel für lösliche Proteinkomplexe erforderlich);
iii) Sorgfältige Prüfung und Anpassung der Waschmittelbedingungen durch analytische BN-PAGE, da diese die Effizienz komplexer Löslichkeit, Darstellung von Membranproteinkomplexen in der Probe, Stabilität und Homogenität der Protein-Waschmittel-Mizellen. Letztere sind Voraussetzungen dafür, dass Proteine sich als unterschiedliche Bänder/komplexe Populationen auf BN-PAGE-Gele konzentrieren. Frühere Literatur bietet eine breite Palette von neutralen Waschmitteln. DDM (n-dodecyl -d-maltoside)1,2,4,5,6 und digitonin3,5,7,8, 9,10,13,18 waren bisher die beliebtesten Wahlmöglichkeiten für BN-MS-Analysen. Es muss betont werden, dass jeder Waschmittelzustand notwendigerweise einen Kompromiss zwischen der Löslichkeitseffizienz und der Erhaltung von Proteinwechselwirkungen darstellt und möglicherweise nicht für alle Arten von Zielprotein und Ausgangsmaterial gleichermaßen geeignet ist;
iv) Entfernen geladener Polymere wie Fibrillen, Filamente, Polylysin, DNA und reichlich niedermolekulare Bestandteile (d. h. Metaboliten, Lipide oder Peptide). Dies kann durch Ultrazentrifugation, Gelfiltration oder Dialyse erreicht werden. Dies ist besonders wichtig für Gesamtzell- oder Gewebelysate;
v) Zugabe von Coomassie G-250 (Endkonzentration 0,05%-0,1%) und Saccharose (zur Erhöhung der Beladungsdichte, Endkonzentration 10%-20% [w/v]) zur Probe kurz vor dem Laden, um durch kurze Ultrazentrifugation zu löschen, die Probe ohne Störung zu beladen und den Lauf unmittelbar danach zu starten.
Als Zukunftsperspektive bietet csBN-MS-basiertes Complexome-Profiling Optionen für Multiplexing, um Protein-Komplexe Dynamiken oder Veränderungen im Zusammenhang mit spezifischen biologischen Bedingungen zu untersuchen. Die kombinierte Trennung von metabolisch gekennzeichneten Proben, wie sie für die größenausschlussbasierte Profilierung21 vorgeschlagen wird, erscheint einfach, kann jedoch durch einen spontanen Austausch von Untereinheiten in Komplexen behindert werden, die unabhängig von der verwendeten Trennung auftreten. system. Alternativ können markierte Proben in benachbarten Gelspuren gelöst werden, die dann für die Differentialanalyse mit hoher Empfindlichkeit und Robustheit mitgeschnitten oder kombiniert werden können.
Offenlegungen
Der Autor Uwe Schulte ist Mitarbeiter und Gesellschafter der Logopharm GmbH, die in dieser Studie verwendete ComplexioLyte 47 herstellt. Das Unternehmen stellt komplexe Reagenzien für akademische Einrichtungen auf gemeinnütziger Basis zur Verfügung.
Danksagungen
Diese Studie wurde von der Deutschen Forschungsgemeinschaft (DFG) – Projekt-ID 403222702 – SFB 1381 und im Rahmen der Exzellenzstrategie CIBSS - EXC-2189 - Projekt-ID 390939984 unterstützt. Wir danken Katja Zappe für die technische Unterstützung.
Materialien
| Name | Company | Catalog Number | Comments |
| 30% Acrylamide/Bis Solution, 37.5:1 | Bio Rad | #1610158 | Recommended for acrylamide gradient gel solutions up to 13% |
| 30% Acrylamide/Bis Solution, 19:1 | Bio Rad | #1610154 | Recommended for acrylamide gradient gel solutions >13% |
| SYPRO Ruby Protein Blot Stain | Bio Rad | #1703127 | Total protein stain on blot membranes; sensitive and compatible with immunodetection |
| Coomassie Brilliant Blue G-250 | Serva | no. 35050 | Centrifugate stock solutions prior to use |
| ComplexioLyte 47 | Logopharm | CL-47-01 | Ready-to-use detergent buffer (1%) for mild solubilization of membrane proteins |
| Embedding Medium / Tissue Freezing Medium | Leica Biosystems | 14020108926 | Embedding medium for gel sections to be sliced by a cryo-microtome |
| Immobilon-P Membrane, PVDF, 0,45 µm | Merck | IPVH00010 | |
| ECL Prime Western Blotting Detection Reagent | GE Healthcare | RPN2232 | |
| Plastic syringe with rubber stopper, 20-30 ml | n.a. | n.a. | any supplier, important for making gel section embedding tool |
| broad razor blade | n.a. | n.a. | any supplier, for BN-PAGE gel trimming / excision of lanes |
| metal tube / cylinder, ca. 4 cm long | n.a. | n.a. | mold for embedding and freezing of gel samples |
| Protein LoBind Tubes, 1.5 ml | Eppendorf | Nr. 0030108116 | highly recommended to minimize protein/peptide loss due to absorption |
| sequencing-grade modified trypsin | Promega | V5111 | |
| C18 PepMap100 precolumn, particle size 5 µm | Dionex / Thermo Scientific | P/N 160454 | |
| PicoTip emitter (i.d. 75 µm; tip 8 µm) | New Objective | FS360-75-8 | |
| ReproSil-Pur 120 ODS-3 (C18, 3 µm) | Dr. Maisch GmbH | r13.93. | columns packed manually |
| rabbit anti-TPC1 antibody | Gramsch Laboratories | custom production | described in Castonguay, et al., 2017 (Reference 12) |
| Cy3-biotinylated goat anti-rabbit IgG | Vector Laboratories | CY-1300 | described in Castonguay, et al., 2017 (Reference 12) |
| biotinylated Lotus tetragonolobus lectin, FITC-conjugated | Vector Laboratories | #B1325 | described in Castonguay, et al., 2017 (Reference 12) |
| cryo-microtome Leica CM1950 | Leica Biosystems | 14047743905 | |
| Mini Protean II Cell with wetblot unit | Bio Rad | n.a. | for SDS-PAGE and Westernblot (not sold any more) |
| Penguin Midi Gel Electrophoresis System | PeqLab | n.a. | for BN-PAGE (not sold any more) |
| Zeiss Axiovert 200 M microscope + Photometrics Coolsnap 2 digital camera | Zeiss / Photometrics | n.a. | |
| peristaltic pump (IP high precision multichannel) | Ismatec | ISM940 | for casting of gradient polyacrylamide gels |
| gradient mixer with stirring (two chambers) | selfmade, alternatively Bio Rad | 1652000 or 1652001 | for casting of gradient polyacrylamide gels, manual provides instructions to cast linear or hyperbolic gradient gels (http://www.bio-rad.com/webroot/web/pdf/lsr/literature/M1652000.pdf) |
| ultracentrifuge Sorvall M120 with S80 AT3 rotor | Sorvall / Thermo Scientific | n.a. | for sample preparation (not sold any more) |
| UltiMate 3000 RSLCnano HPLC | Dionex / Thermo Scientific | ULTIM3000RSLCNANO | |
| Orbitrap Elite mass spectrometer | Thermo Scientific | IQLAAEGAAPFADBMAZQ |
Referenzen
- Majeran, W., et al. Consequences of C4 Differentiation for Chloroplast Membrane Proteomes in Maize Mesophyll and Bundle Sheath Cells. Molecular & Cellular Proteomics. 7, 1609-1638 (2008).
- Wessels, H. J., et al. LC-MS/MS as an alternative for SDS-PAGE in blue native analysis of protein complexes. Proteomics. 9 (17), 4221-4228 (2009).
- Heide, H., et al. Complexome profiling identifies TMEM126B as a component of the mitochondrial complex I assembly complex. Cell Metabolism. 16 (4), 538-549 (2012).
- Wessels, H. J., et al. Analysis of 953 human proteins from a mitochondrial HEK293 fraction by complexome profiling. PLoS ONE. 8 (7), e68340 (2013).
- Wöhlbrand, L., et al. Analysis of membrane-protein complexes of the marine sulfate reducer Desulfobacula toluolica Tol2 by 1D blue native-PAGE complexome profiling and 2D blue native-/SDS-PAGE. Proteomics. 16 (6), 973-988 (2016).
- Takabayashi, A., et al. PCoM-DB Update: A Protein Co-Migration Database for Photosynthetic Organisms. Plant and Cell Physiology. 58 (1), e10 (2017).
- Senkler, J., et al. The mitochondrial complexome of Arabidopsis thaliana. The Plant Journal. 89 (6), 1079-1092 (2017).
- de Almeida, N. M., et al. Membrane-bound electron transport systems of an anammox bacterium: A complexome analysis. Biochimica et Biophysica Acta. 1857 (10), 1694-1704 (2016).
- Anand, R., Strecker, V., Urbach, J., Wittig, I., Reichert, A. S. Mic13 Is Essential for Formation of Crista Junctions in Mammalian Cells. PLoS ONE. 11 (8), e0160258 (2016).
- Eydt, K., Davies, K. M., Behrendt, C., Wittig, I., Reichert, A. S. Cristae architecture is determined by an interplay of the MICOS complex and the F1FO ATP synthase via Mic27 and Mic10. Microbial Cell. 4 (8), 259-272 (2017).
- Müller, C. S., et al. Cryoslicing Blue Native-Mass Spectrometry (csBN-MS), a Novel Technology for High Resolution Complexome Profiling. Molecular & Cellular Proteomics. 15 (2), 669-681 (2016).
- Castonguay, J., et al. The two-pore channel TPC1 is required for efficient protein processing through early and recycling endosomes. Scientific Reports. 7 (1), 10038 (2017).
- Schägger, H., Pfeiffer, K. Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. The EMBO Journal. 19 (8), 1777-1783 (2000).
- Banyard, S. H., Stammers, D. K., Harrison, P. M. Electron density map of apoferritin at 2.8-A resolution. Nature. 271 (5642), 282-284 (1978).
- Dettmer, U., et al. Transmembrane protein 147 (TMEM147) is a novel component of the Nicalin-NOMO protein complex. The Journal of Biological Chemistry. 285 (34), 26174-26181 (2010).
- Kimberly, W. T., et al. Gamma-secretase is a membrane protein complex comprised of presenilin, nicastrin Aph-1, and Pen-2. Proceedings of the National Academy of Sciences of the United States of America. 100 (11), 6382-6387 (2003).
- Hong, Y., et al. Human PIG-U and yeast Cdc91p are the fifth subunit of GPI transamidase that attaches GPI-anchors to proteins. Molecular Biology of the Cell. 14 (5), 1780-1789 (2003).
- Van Damme, T., et al. Mutations in ATP6V1E1 or ATP6V1A Cause Autosomal-Recessive Cutis Laxa. The American Journal of Human Genetics. 100 (2), 216-227 (2017).
- Budenholzer, L., Cheng, C. L., Li, Y., Hochstrasser, M. Proteasome Structure and Assembly. Journal of Molecular Biology. 429 (22), 3500-3524 (2017).
- Heusel, M., et al. Complex-centric proteome profiling by SEC-SWATH-MS. Molecular Systems Biology. 15 (1), e8438 (2019).
- Kristensen, A. R., Gsponer, J., Forster, L. J. A high-throughput approach for measuring temporal changes in the interactome. Nature Methods. 9 (9), 907-919 (2012).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten