
Method Article
Descifrar los efectos estructurales de la activación de mutaciones somáticas de EGFR con simulación de dinámica molecular
En este artículo
Resumen
El objetivo de este protocolo es utilizar simulaciones de dinámica molecular para examinar los cambios estructurales dinámicos que se producen debido a la activación de mutaciones de la proteína quinasa EGFR.
Resumen
Numerosas mutaciones somáticas que ocurren en la familia del receptor del factor de crecimiento epidérmico (EGFR) (ErbB) de las tirosina quinasas del receptor (RTK) se han notificado de pacientes con cáncer, aunque relativamente pocos han sido probados y demostrados para causar cambios funcionales en ErbBs. Los receptores ErbB son dimerizados y activados sobre la unión de ligandos, y los cambios de conformación dinámica de los receptores son inherentes a la inducción de la señalización aguas abajo. Para dos mutaciones mostradas experimentalmente para alterar la función EGFR, A702V y la mutación de eliminación DE746ELREA750, ilustramos en el siguiente protocolo cómo las simulaciones de dinámica molecular (MD) pueden sondear la (1) estabilidad conformacional de la estructura de tirosina quinasa mutante en comparación con el EGFR de tipo salvaje; 2) consecuencias estructurales y transiciones conformacionales y su relación con los cambios funcionales observados; 3) los efectos de las mutaciones en la fuerza del ATP de unión, así como para la unión entre los dominios quinasa en el dimer asimétrico activado; y (4) los efectos de las mutaciones en interacciones clave dentro del sitio de unión al EGFR asociado con la enzima activada. El protocolo proporciona un procedimiento detallado paso a paso, así como una guía que puede ser más generalmente útil para la investigación de estructuras proteicas utilizando simulaciones MD como un medio para sondear la dinámica estructural y la relación con la función biológica.
Introducción
La familia del receptor del factor de crecimiento epidérmico humano (EGFR) (ErbB) de las quinasas de tirosina del receptor (RTK) incluye cuatro miembros - EGFR/ErbB1/HER1, ErbB2/HER2, ErbB3/HER3 y ErbB4/HER4. Los receptores ErbB regulan procesos celulares fundamentales como el crecimiento celular y la proliferación, diferenciación, migración y supervivencia1,2, y por lo tanto son potentes proto-oncogenes. La actividad aberrante de los receptores ErbB, especialmente EGFR y ErbB2, se ha asociado con frecuencia con cánceres humanos haciendo receptores ErbB objetivos clave para las terapias contra el cáncer2,,3.
Se han notificado varias alteraciones somáticas de los genes ERBB a partir de neoplasias malignas humanas3,4,5. Los ejemplos mejor caracterizados incluyen las mutaciones puntuales recurrentes y activantes y las deleciones cortas en el marco en el dominio de la quinasa egFR en el cáncer de pulmón de células no pequeñas (NSCLC). Estas mutaciones del EGFR representan los factores clave del crecimiento del cáncer y predicen la sensibilidad al EGFR dirigido a los medicamentos contra el cáncer6,,7,,8. Sin embargo, en la mayoría de los cánceres, las mutaciones somáticas en el EGFR ocurren fuera de estos "puntos calientes" recurrentes y se distribuyen a lo largo de todo el período de 1210 residuos del receptor. De hecho, la mayoría de los residuos a lo largo de la secuencia primaria del EGFR se han encontrado mutados en el cáncer humano9. Sin embargo, aparte de los pocos puntos críticos, la importancia funcional de la gran mayoría de las mutaciones de EGFR asociadas al cáncer sigue siendo desconocida.
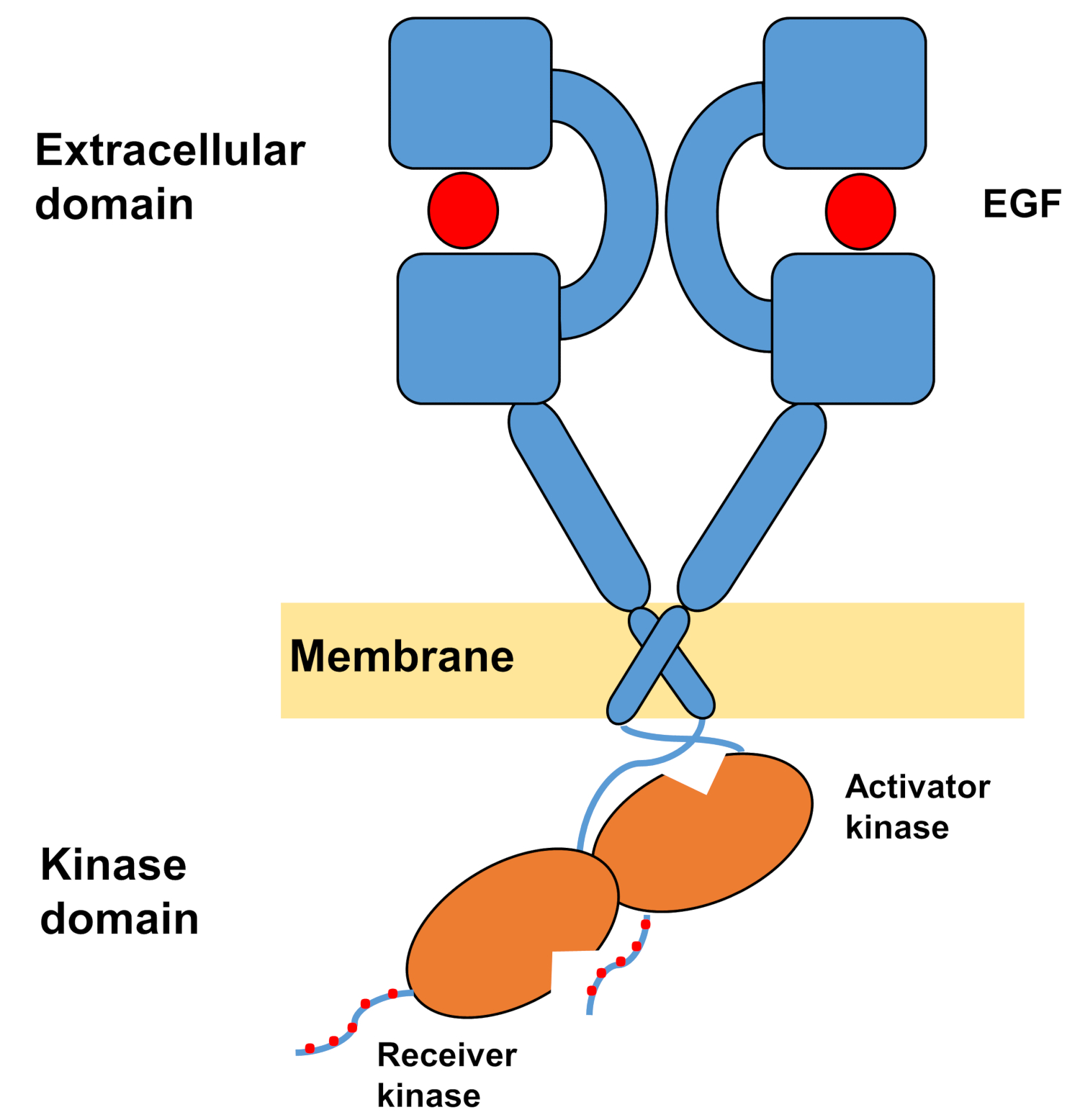
La estructura monomérica de ErbBs consiste en un gran dominio extracelular terminal amino, seguido de una sola hélice transmembrana que conduce al dominio intracelular de tirosina quinasa y región de cola C-terminal que contiene sitios de acoplamiento para proteínas de señalización intracelular. La unión ligando desencadena un cambio conformación dramático en el dominio extracelular, que facilita la formación de dimers receptores al exponer los brazos de dimerización que se cruzan simétricamente entre sí e interactúan con sus superficies aromáticas/hidrofóbicas. Tras la formación del dimer receptor, los dominios de tirosina quinasa entran en contacto de forma asimétrica(Figura 1),lo que da lugar a la activación de las quinasas que fosforilan las colas C-terminales de los monómeros receptores, y posteriormente en la activación de la señalización aguas abajo10,,11.

Figura 1: Estructura del dimer EGFR. DI EGFR dimerizes cuando los dominios extracelulares unen el factor de crecimiento (EGF, factor de crecimiento epidérmico). El dominio de la quinasa receptora se activa a través de la interacción asimétrica con el dominio de la quinasa activador, y las colas del terminal C se autofosforil se autofosforil se autofosforilan en los residuos de tirosina (modificado de Tamirat et al.12). Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Debido a los reordenamientos estructurales dinámicos que se producen durante las  transiciones de dimer monómero, junto con la activación de la quinasa que se asocia con la formación de un dimer asimétrico, las mutaciones a lo largo de toda la longitud de la estructura del receptor pueden tener potencialmente un efecto sobre la función del receptor. Aquí describimos varios ejemplos de nuestros estudios anteriores en los que el modelado de la mutación y la visualización fueron suficientes para explicar las consecuencias de la función.
transiciones de dimer monómero, junto con la activación de la quinasa que se asocia con la formación de un dimer asimétrico, las mutaciones a lo largo de toda la longitud de la estructura del receptor pueden tener potencialmente un efecto sobre la función del receptor. Aquí describimos varios ejemplos de nuestros estudios anteriores en los que el modelado de la mutación y la visualización fueron suficientes para explicar las consecuencias de la función.
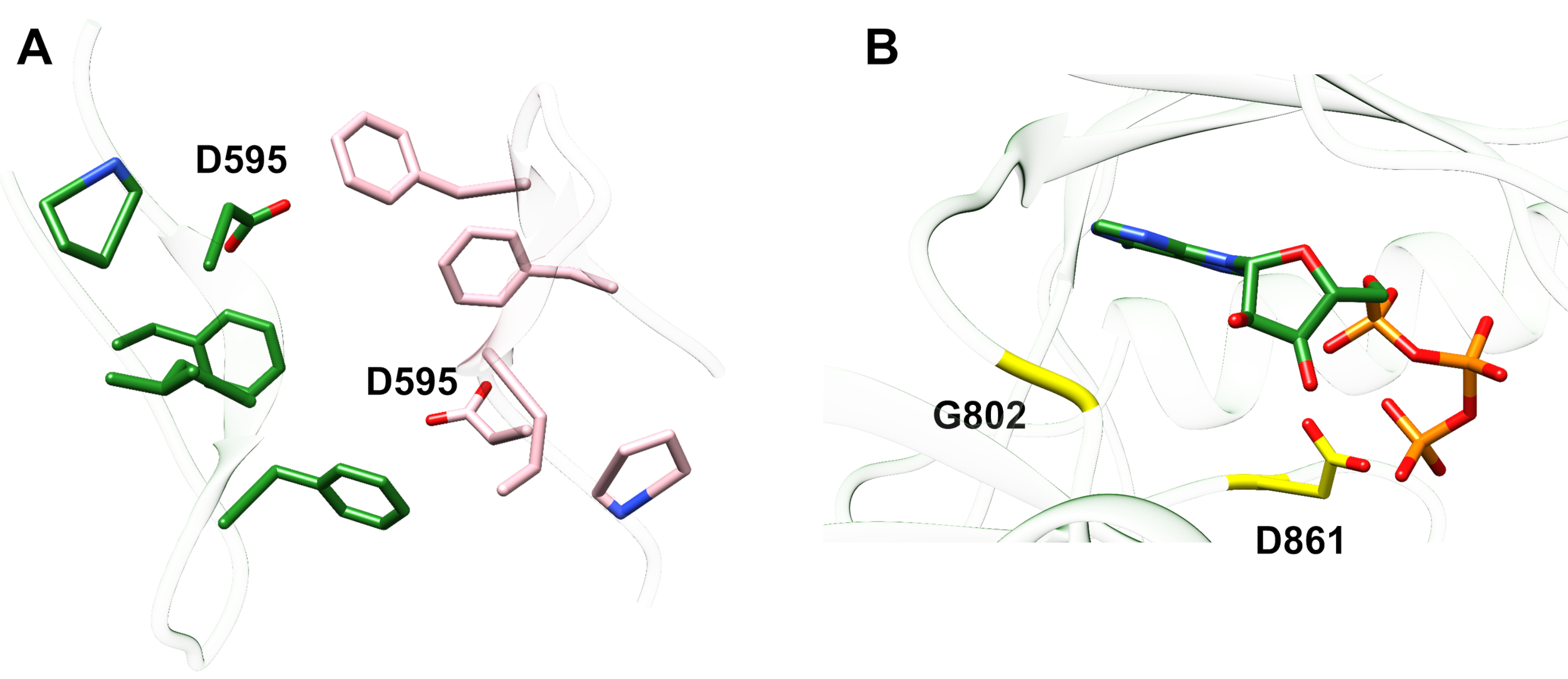
Ejemplo 1: Una mutación reportada, D595V en ErbB413,condujo a un aumento de la dimerización y fosforilación ErbB414. La visualización de la ubicación de la mutación fue un factor crítico para comprender los efectos funcionales observados: D595V ocurrió en el cruce simétrico de los brazos dimericos del ectodominio (Figura 2A). Los brazos son en gran parte aromáticos e hidrófobos, y se espera que la sustitución del ácido aspártico polar por valina aumente las interacciones hidrofóbicas "pegajosas", estabilizando el dimer y por lo tanto aumentar el tiempo cuando la fosforilación tiene lugar14. Fue una sorpresa al principio encontrar aspartato en cada brazo, pero en retrospectiva se podría pensar en él como un mecanismo de sincronización para la actividad, donde las cadenas laterales de ácido polar reducen la afinidad y la vida útil del dimer intacto y por lo tanto limitan la fosforilación y la señalización mediadas por la quinasa. La sustitución por valine eliminaría esta salvaguardia estabilizando aún más el dimer ErbB4.

Figura 2: Ubicación de una mutación activadoras De ErbB4 y mutaciones que producen ErbB4 muerto de quinasa. (A) D595 (activación de la mutación D595V) se encuentra en los brazos dimericos aromáticos/hidrofóbicos del modelo de ectodominio ErbB4; las armas se asocian a la unión al factor de crecimiento; (los residuos cercanos se muestran como palos). (B) En ErbB4, G802 (mutación inactivante G802dup) ayuda a formar el bolsillo de unión alrededor del anillo de adenina de ATP y el catalítico D861 (mutación inactivante D861Y) une tanto Mg2+ (no mostrado) como el grupo γ-fosfato de ATP. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Ejemplo 2: Uno podría anticipar que las mutaciones somáticas que apuntan al sitio de unión ATP del dominio de la quinasa alterarían o eliminarían la actividad enzimática que llevaría a un receptor deteriorado o muerto por quinasa incapaz de señalización. De nueve mutaciones notificadas de pacientes con mama, gástrica, colorrectal o NSCLC15,dos de las nueve mutaciones cuando se analizaron tuvieron una actividad de fosforilación muy disminuida16:G802dup (G → GG) y D861Y. Ambas mutaciones somáticas inactivadoras se encontraron dentro del sitio de unión ATP de la estructura de dominio de tirosina quinasa (Figura 2B): glicina flexible, duplicada, alteraría el sitio del anillo de adenina y el ácido aspártico pequeño reemplazado por la tirosina voluminosa cerca de los fosfatos terminales evitaría físicamente Mg2+-ATP de la unión. Sin embargo, ya que ErbB4 puede formar un heterodímero con ErbB2 - ErbB2 no une un factor de crecimiento y depende de la asociación con un ErbB que lo hace con el fin de heterodimerizar - el ErbB2 (activo)-ErbB4(kinase-dead) heterodimer estimularía la proliferación celular a través de la vía de señalización Erk/Akt, pero las células no se diferenciarían debido a ErbB4 muerto por quinasa y a la falta de activación de la vía STAT516.
En estudios más recientes, se hizo evidente que los movimientos dinámicos de ErbBs eran relevantes para entender los efectos de algunos mutantes en la función ErbB, especialmente las mutaciones que se producen dentro del dominio de la tirosina quinasa. El dominio de la tirosina quinasa consiste en un lóbulo N (principalmente hojas de β) y C-lóbulo (en gran parte alfa helicoidal), que están separados por el sitio catalítico donde ATP se une. El lóbulo N incluye la hélice Y el P-loop, mientras que la activación (bucle A) y los bucles catalíticos están presentes en el lóbulo C17,,18,,19. Las estructuras cristalinas del dominio de la tirosina quinasa revelaron dos conformaciones inactivas, la mayoría de las estructuras tienen el estado inactivo similar a Src. En la conformación activa, el aspartato catalítico del bucle A apunta hacia el sitio de unión ATP y la hélice de C está orientada hacia la conformación de la unión ATP (C-in), formando una fuerte interacción entre iones de iones de glutamato y lisina.
Debido a que los ErbB y el dominio de la quinasa de componentes son entidades altamente dinámicas, y especialmente en los casos en que es probable que los efectos de las mutaciones sobre la función y la actividad biológica estén estrechamente vinculados a los estados conformacionales de los ErBB, es importante evaluar las mutaciones con respecto al rango de cambios dinámicos que experimentarían. Las estructuras de cristal de rayos X de ErbBs proporcionan instantáneas estáticas de la estructura 3D, que pueden o no ser relevantes para comprender las consecuencias dinámicas de una mutación. Con el fin de sondear el rango de cambios dinámicos correspondientes al "paisaje energético" disponible para una estructura tridimensional (3D), las simulaciones de dinámica molecular (MD) son ampliamente utilizadas20. En el caso de mutaciones que conducirían a cambios de conformación local dentro del dominio de la tirosina quinasa o la estabilización de un complejo, las simulaciones del orden de 100 ns pueden ser suficientes. Sin embargo, los cambios de conformación de mayor escala (por ejemplo, transiciones entre las conformaciones activas e inactivas del dominio de la quinasa) requieren un tiempo de simulación más largo - en el orden de los microsegundos21.
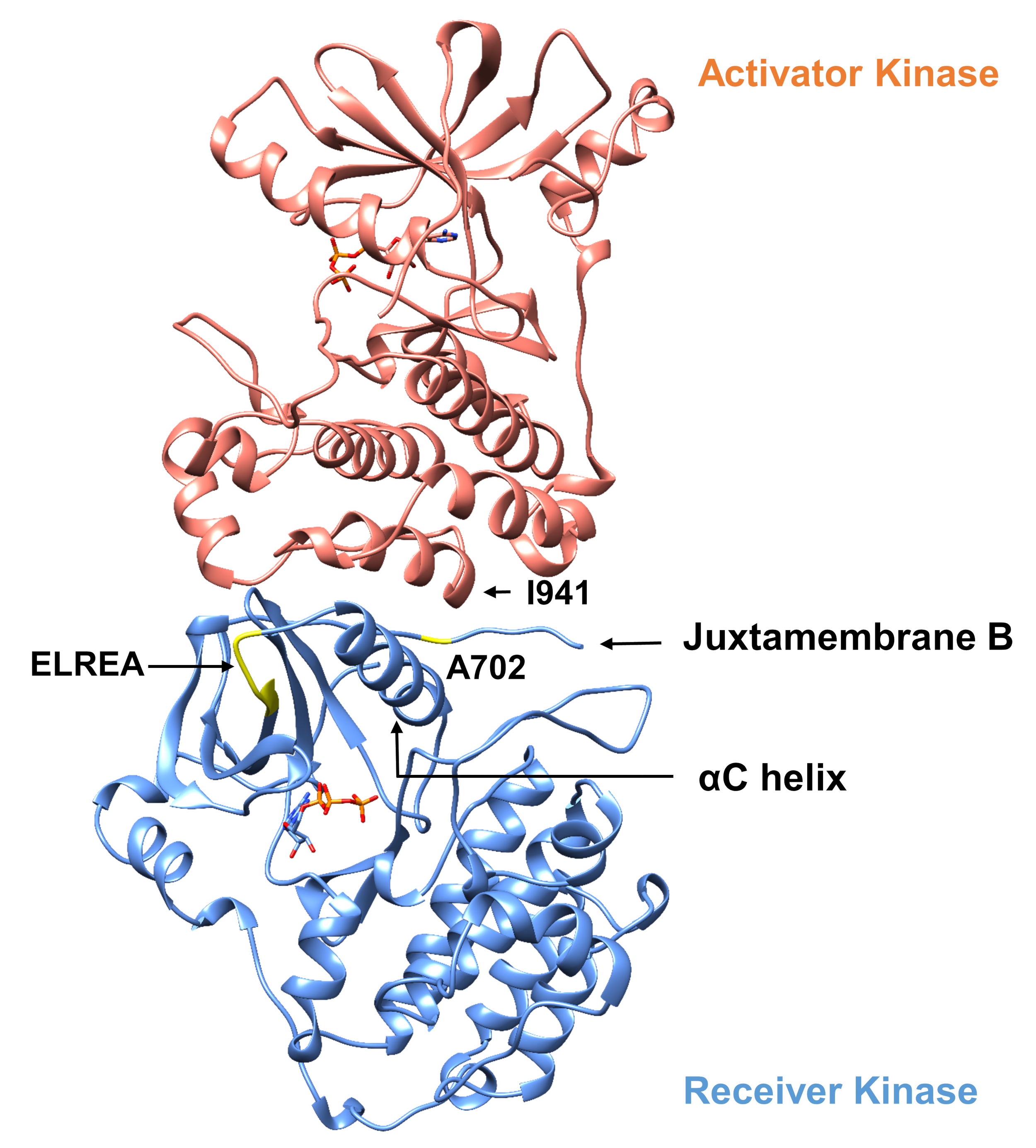
Con respecto al protocolo descrito a continuación, consideramos dos mutaciones activadoras dentro del dominio de la tirosina quinasa (Figura 3). Ambas mutaciones se encuentran dentro del dominio de la quinasa en ubicaciones que experimentan cambios conformacionales locales que dictan si la quinasa está activa o no, y por lo tanto las simulaciones MD se aplicaron en ambos casos. En el primer caso, consideramos los cambios que afectan directamente al sitio de enlace ATP y a la maquinaria catalítica del dominio de la quinasa receptora EGFR, examinando específicamente las consecuencias de una mutación de eliminación exón 19 que está ampliamente implicada en NSCLC4,,7. La mutaciónde 746ELREA750, que reduce la longitud del bucle de 3C que precede a la hélice -C" - la hélice que se mueve hacia la unión/sitio activo en la activación de la quinasa y participa en la formación de la interacción electrostática crítica entre E762 del hélice y K745 mediante la colocación de la lisina para la interacción con ATP - predispone el dominio para la activación12. En el segundo caso, consideramos que la mutación A702V del EGFR, que ha demostrado ser una nueva mutación activadora de ganancia de función revelada por la plataforma iScream9 e identificada en un paciente NSCLC22. La alanina-702 en el dominio de la quinasa receptora se encuentra en el segmento B de juxtamembrana en la interfaz del receptor y los dominios de la quinasa activador, en los que este complejo de dimer kinasa asimétrico y los cambios en la conformación de la quinasa son necesarios para la activación9.

Figura 3: El dimer de dominio de quinasa asimétrica de EGFR. La mutación A702V se ubicaría en la interfaz crítica de los dominios de la quinasa activador y receptora, adyacente a la hélice de la C y cerca de la isoleucina 941 del activador quinasa. Los cambios de conformación inducidos por la formación del dimer asimétrico conducen a la activación de la quinasa. El bucle de 3-C que contiene la secuencia ELREA precede directamente a la hélice de C; durante la activación, la hélice de C se mueve hacia adentro hacia el sitio de enlace DE ATP. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Protocolo
NOTA: Los pasos detallados tomados para examinar los efectos de la mutación DEELREA y A702V en la estructura del EGFR mediante simulaciones MD se discuten de la siguiente manera:
1. Preparación de la estructura
NOTA: Con el fin de estudiar los impactos estructurales de la mutación DEREA, las formas de tipo salvaje y mutantes de las estructuras de monómero EGFR activos, activos y apo inactivos de ATP se preparan de la siguiente manera.
- Abra el programa de visualización Quimera23 (https://www.cgl.ucsf.edu/chimera/) para preparar la estructura de quinasa EGFR activa de tipo salvaje. En el menú Archivo, haga clic en la opción Obtener por ID y seleccione la base de datos Banco de datos de proteínas (PDB24)y especifique el código PDB 2GS225 (resolución de 2,8o). El PDB es un repositorio de estructuras 3D resueltos por diferentes técnicas experimentales, incluyendo cristalografía de rayos X, espectroscopia de resonancia magnética nuclear, microscopía crioelectrófica y difracción de neutrones.
- Construir los elementos estructurales que faltan de 2GS2 tomando estos segmentos de las estructuras EGFR PDB 1M1426 (2,6o) y 3W2S27 (1,9o). Para ello, abra 1M14 y 3W2S y superpongalos en 2GS2 mediante la opción MatchMaker del menú de comparación Herramientas → Estructura.
- Recorte los segmentos que se agregarán desde 1M14 y 3W2S. Seleccione los átomos terminales del residuo antes de los huecos en 2GS2 y los átomos que se añadirán de 1M14 y 3W2S (para obtener información detallada sobre los segmentos añadidos, véase la Tabla 1). En la línea de comando, escriba bond sel y pulse enter. Esta estructura es la estructura de plantilla.
- Utilice la estructura de plantilla del paso 1.2 para construir la forma mutante de eliminación de LA ZONA DEErea del dominio egFR quinasa. Produzca la secuencia de formato FASTA de EGFR de LA EREA guardando la secuencia de la estructura de la plantilla (Archivo de → de secuencia de → favoritos → guardar como) y, a continuación, eliminando la secuencia ELREA en los números de residuo 746-750.
- Abra la secuencia EGFR de LA ZONA EN Quimera y alinee con la secuencia de la estructura de plantilla 2GS2 mediante el menú Secuencia. En la ventana de alineación seleccione la opción Estructura → Modelador (homología)28.
- En la ventana emergente, especifique la estructura compuesta 2GS2 como la plantilla y la secuencia mutante como la consulta que se va a modelar. A continuación, pulse OK. Seleccione un modelo mutante entre los modelos resultantes, en función de la puntuación zDOPE (normalmente la puntuación más baja) y la inspección visual.
- Para preparar la estructura quinasa EGFR de tipo salvaje ligada a ATP, utilice la estructura PDB 2ITX29 (2,98o) como estructura principal. Cree los segmentos que faltan (véase la Tabla 1) utilizando las estructuras 2GS625 (2,6o) y 3W2S siguiendo el procedimiento descrito en el paso 1.2. Convierta el ligando ANP en la estructura resultante en ATP abriendo el archivo PDB en un editor de texto y cambiando el átomo de nitrógeno N3B de ANP a un átomo de oxígeno.
- Abra la estructura en el paso 1.4 en Quimera como se indica en el paso 1.1. Añadir un ion de magnesio a esta estructura de la estructura PDB 2ITN29 (2,47o) con el fin de lograr un posicionamiento similar para el ion Mg2+.
- Con la estructura resultante del paso 1.5, modele la forma mutante de LA ZONA después del paso 1.3.
| EGFR activo Apo | EGFR inactivo Apo | EGFR activo con destino a ATP | |
| Estructura principal | 2GS2 | 2GS7 | 2ITX |
| Estructuras utilizadas para construir bucles que faltan | 1M14 (723-725) | 3W2S (958-984) | 2GS6 (862-865) |
| 3W2S (967-981) | 4HJO (848-850) | 3W2S (990-1001) |
Tabla 1: Estructuras utilizadas para construir modelos compuestos de estructuras activas apo activas, apo inactivas y estructuras activas enlazadas a ATP. Las regiones que faltaban (rango de aminoácidos entre paréntesis) en la estructura principal se construyeron a partir de las estructuras enumeradas.
- Para preparar la estructura de quinasa EGFR inactiva de tipo salvaje, abra la estructura PDB 2GS725 (2,6o) como en el paso 1.1 y elimine los ligandos enlazados y las aguas cristalográficas. Agregue los segmentos que faltan en 2GS7 (véase la Tabla 1) de las estructuras 3W2S y 4HJO30 (2,75o) utilizando el procedimiento del paso 1.2. Basándose en la estructura final inactiva de EGFR, prepare el modelo mutante utilizando el procedimiento del paso 1.3.
NOTA: Para el estudio de mutación A702V, se estudia la estructura asimétrica del dimer EGFR, ya que la mutación se encuentra en el segmento de juxtmembrana B del dominio quinasa que forma una gran parte de la interfaz dimer. Las estructuras de EGFR mutante de tipo salvaje y A702V se preparan de la siguiente manera: - La estructura de dimer asimétrico de tipo salvaje se construye a partir de la estructura PDB 2GS2, que inicialmente se muestra en la forma monomérica. Para convertir al conjunto biológico que contiene las quinasas activador y receptora en la disposición asimétrica, abra 2GS2 en Quimera como en (1.1) y realice cálculos de simetría haciendo clic en el menú Herramientas → Estructura de orden superior → Celda de unidad. Seleccione la estructura 2GS2 e introduzca Crear copias. Por último, seleccione y guarde un único dimer asimétrico de las múltiples copias del dimer resultantes de las operaciones de simetría.
- Usando la estructura EGFR asimétrica de tipo salvaje del paso 1.8, construye el mutante A702V reemplazando la alanina 702 por la valina usando la opción de edición Herramientas → Estructura → Rotamers en Quimera.
NOTA: Colectivamente, se preparan seis estructuras monóméricas y dos estructuras dimericas de EGFR para los estudios de mutación de LAREA y A702V, respectivamente. Cada estructura se procesa posteriormente para la simulación utilizando el asistente de preparación de proteínas en el programa Maestro31 y las simulaciones MD se realizan con el programa Amber32. - Abra la estructura en Maestro mediante la opción Estructura de importación de → archivo. A continuación, haga clic en el botón del asistente de preparación de proteínas y seleccione lo siguiente: añadir átomos de hidrógeno, construir átomos de cadena lateral que faltan, determinar los estados de protonación de residuos ionizables a pH 7.0 utilizando PROPKA, optimizar la orientación de la asparagina, la glutamina y los residuos de histidina para la unión de hidrógeno, y finalmente minimizar la estructura.
2. Configuración del sistema
- Abra el programa leap incluido en el paquete de software Amber. Importe el campo de fuerza ff14SB33 (fuente leaprc.protein.ff14SB) y las moléculas de agua TIP3P34 (fuente leaprc.water.tip3p). Para los sistemas enlazados a ATP también importar parámetros para ATP35 (loadamberparams frcmod.phosphate, loadamberprep ATP.prep). A continuación, cargue la estructura(mol - loadpdb structure.pdb).
- Solvate la estructura en una caja octahedral con moléculas de agua TIP3P explícitas que se extiende 10o en todas las direcciones desde los átomos superficiales de la proteína (solvateoct mol TIP3PBOX 10.0).
- Compruebe el sistema construido (Comprobar mol) y neutralízalo añadiendo los iones necesarios (addions mol Na+ 0). Para modelar suficientemente los sistemas biomoleculares, añadir átomos adicionales Na+/Cl- a la caja de simulación para llevar la concentración de sal del sistema a 0,15 M (adiciones mol Na + X, adiciones mol Cl- X), donde X se sustituye por el resultado de: concentración de sal deseada * número de moléculas de agua * volumen por molécula de agua * Número de Avogadro.
- Genere y guarde la topología y los archivos de coordenadas del sistema, que sirven como entradas para la simulación de producción posterior (saveamberparm mol X.prmtop X.inpcrd).
3. Simulación de dinámica molecular
- Usando Amber, inicialmente someta el sistema de simulación a 5000 ciclos de descenso más pronunciado y minimización de energía de gradiente conjugado para eludir configuraciones desfavorables. Llevar a cabo la minimización en múltiples pasos, reduciendo gradualmente la restricción aplicada en los átomos de soluto de 25 kcal mol-1 a-2 a 0 kcal mol-1 á-2.
- En el archivo de entrada de minimización, min.in, ajuste la variable maxcyc para el ciclo de minimización total (maxcyc á 5000) y ncyc para indicar el número de ciclos para el algoritmo de descenso más pronunciado. Utilice la variable restraint_wt para aplicar la fuerza de restricción a los átomos de soluto especificados por el parámetro de máscara de retención. A continuación, ejecute la minimización de la siguiente manera:
$AMBERHOME/bin/sander -O -i min.in -o min.out -p X.prmtop -c X.inpcrd -r min.rst -ref X.inpcrd
NOTA: La estrategia y los parámetros reales utilizados pueden variar según las propias preferencias. Los detalles y la orientación se pueden encontrar en el manual y sitio web de Amber (https://ambermd.org/index.php)
- En el archivo de entrada de minimización, min.in, ajuste la variable maxcyc para el ciclo de minimización total (maxcyc á 5000) y ncyc para indicar el número de ciclos para el algoritmo de descenso más pronunciado. Utilice la variable restraint_wt para aplicar la fuerza de restricción a los átomos de soluto especificados por el parámetro de máscara de retención. A continuación, ejecute la minimización de la siguiente manera:
- Calentar el sistema durante 100 ps de 0 K a 300 K ajustando una restricción de 10 kcal mol-1 a-2 en átomos de soluto. Para ello, establezca tempi en 0,0, temp0 a 300,0, dt a 0,002 ps, nstlim a 50000 y restraint_wt a 10 en el archivo de entrada de heat.in. Lleve a cabo la calefacción con el siguiente comando:
$AMBERHOME/bin/sander -O -i heat.in -o heat.out -p X.prmtop -c min.rst -r heat.rst -x heat.mdcrd -ref min.rst - Equilibrar el sistema durante 900 ps bajo un conjunto NPT; número constante de átomos, temperatura (temp0 a 300,0) y presión (ntp - 1), controlándolo con el método Berendsen (ntt - 1). Establezca un límite de distancia de 9o(corte a 9,0) para interacciones electrostáticas de largo alcance. Baje gradualmente la restricción del átomo de soluto a 0,1 kcal mol-1 a-2 (restraint_wt a 0,1). Ejecute el archivo de entrada de equilibrio equil.in que describe los parámetros anteriores de la siguiente manera:
$AMBERHOME/bin/sander -O -i equil.in -o equil.out -p X.prmtop -c heat.rst -r equil.rst -x equil.mdcrd -ref heat.rst - Finalizar el equilibrio con una simulación de 5 ns sin restricciones (set dt a 0,002 ps, ntslim a 2500000).
$AMBERHOME/bin/sander -O -i equil_final.in -o equil_final.out -p X.prmtop -c equil.rst -r equil_final.rst -x equil_final.mdcrd -ref equil.rst - Compruebe que el sistema se ha equilibrado examinando los valores de temperatura, presión, densidad y energía.
$AMBERHOME/bin/process_mdout.perl heat.out equil.out equil_final.out
xmgrace resumen. TEMP/DENSIDAD/ETOT/EPTOT/EKTOT - Llevar a cabo la simulación de producción durante 100 ns (set dt a 0,002 ps, ntslim a 50000000 en prod.in) y guardar las conformaciones cada 10 ps (ntwx a 5000).
$AMBERHOME/bin/sander -O -i prod.in -o prod.out -p X.prmtop -c equil_final.rst -r prod.rst -x prod.mdrcd -ref equil_final.rst
4. Análisis
- Inspección visual
- Visualice las conformaciones muestreadas durante las simulaciones de quinasa EGFR mutante y de tipo salvaje abriendo los archivos de topología ámbar X.prmtop y los archivos de trayectoria prod.mdcrd correspondientes en VMD36. Utilizando convenientes representaciones de estructura secundaria, analice la dinámica estructural general de las proteínas a partir de la trayectoria registrada. Vea interacciones específicas entre átomos/residuos de interés, como el puente de sal catalíticamente esencial K745 - E762.
- Alternativamente, guarde varias conformaciones muestreadas durante la simulación en formato PDB y ábralas utilizando el programa Quimera. Superponga las estructuras de la estructura inicial o mediana mediante la opción MatchMaker. Visualice la estructura inicial/mediana en sólido y el resto de las estructuras alineadas en blanco descolorido. Este enfoque permite visualizar los movimientos estructurales registrados con más claridad.
NOTA: Las sugerencias para representaciones efectivas y procesamiento de conjuntos conformacionales de MDS se pueden encontrar en Melvin et al.37.
- Análisis RMSD y RMSF
- Calcular los cálculos de desviación cuadrada media de la raíz (RMSD) y fluctuación cuadrada de la media de la raíz (RMSF) con el programa Cpptraj 38 para analizar la estabilidad global de las proteínas y examinar la flexibilidad de las diferentes unidades estructurales. En los archivos de entrada rmsd.in y rmsf.in, indique los átomos de estructura básica (para RMSD) y los átomos de C (para RMSF) de la estructura inicial como referencia para el ajuste RMS. En los archivos rmsd/rmsf.in, importe los archivos de topología ámbar (parm X.promtop) y los archivos de trayectoria correspondientes (trajin prod.mdcrd). A continuación, ejecute el comando Cpptraj -i rmsd/rmsf.in. Trazar los datos de salida para su análisis.
- Alternativamente, alinee los conjuntos conformacionales y coloree cada residuo en función del átomo de C. RMSD. Para ello, abra las conformaciones en Quimera y alinee con la opción Matchmaker.
- Vaya a Herramientas → Representación → Renderizar por atributo. Seleccione Residuos del conjunto conformacional y C-RMSD como atributos y haga clic en Aceptar. El trazado de cadena de las conformaciones se coloreará de azul → blanco → rojo, respectivamente reflejando regiones de alta, media y baja estabilidad estructural.
- Análisis de bonos de hidrógeno
- Analizar la interacción de los enlaces de hidrógeno entre ATP y los EGFRs de tipo salvaje/AELREA. Preparar un script Cpptraj, hbond.in, para llevar a cabo esta tarea. Definir un enlace de hidrógeno con una distancia donante-aceptante inferior o igual a 3,5o y un ángulo de unión mayor o igual a 135o. Especifique el análisis solo para los enlaces intermoleculares de hidrógeno con la variable nointramol, es decir, los enlaces de hidrógeno entre ATP y EGFR (hbond All nointramol dist 3.5 out nhb.agrout avghb.dat). Ejecute el script como Cpptraj -i hbond.in.
- Utilice este script para evaluar las interacciones intramoleculares, por ejemplo entre los residuos K745 y E762, que son residuos clave para la actividad quinasa de EGFR. Para ello, especifique K745 como donante de enlace de hidrógeno y E762 como el aceptador de enlace de hidrógeno en hbond.in y ejecute el script en consecuencia.
- Distancia de monitoreo entre átomos
- Mida la distancia entre K745 y E762 abriendo las trayectorias de tipo salvaje y de los EGFR de Apo en VMD. Seleccione la etiqueta de Glu762 y Nz de Lys745 haciendo clic en Ratón → etiqueta → enlace. Supervise la distancia durante la simulación trazando un gráfico con gráficos → etiquetas → enlace → gráfico.
- Cálculos de energía gratis
- Para calcular las energías libres de unión estimadas entre ATP y los EGFR de tipo salvaje/ELREA, y entre las quinasas activadoras y receptoras de los EGFR de tipo salvaje/A702V, utilice la mecánica molecular generalizada del módulo39 de superficie del Born (MM-GBSA) disponible en el paquete AMBER. Establecer ATP como el ligando y EGFR como el receptor en el estudio de LAREA. En el estudio A702V, especifique la quinasa receptora como ligando y la quinasa activador como receptor.
- Primero prepare los archivos PDB complejos ligando, receptor y ligando-receptor por separado en el programa de salto estableciendo el valor PBRadii en mbondi2. Para los archivos PDB, guarde los archivos de topología ámbar de fase gas (.prmtop) y coordenadas (.inpcrd).
- A continuación, en el archivo de entrada mmgbsa, mmgbsa.in, set igb á 2, saltcon a 0,1. Ejecute cálculos de energía vinculante utilizando las trayectorias de las simulaciones, los archivos ámbar receptor/ligand preparados y los parámetros en el mmgbsa.in con el script MMPBSA.py disponible en ámbar de la siguiente manera:
$AMBERHOME/bin/MMPBSA.py -O -i mmgbsa.in -o mmgbsa.dat -sp X.prmtop -cp complex.prmtop -rp receptor.prmtop -lp ligand.prmtop -y prod.mdcrd -eo output.csv - Analice los datos de salida, output.csv, trazando gráficos.
Resultados
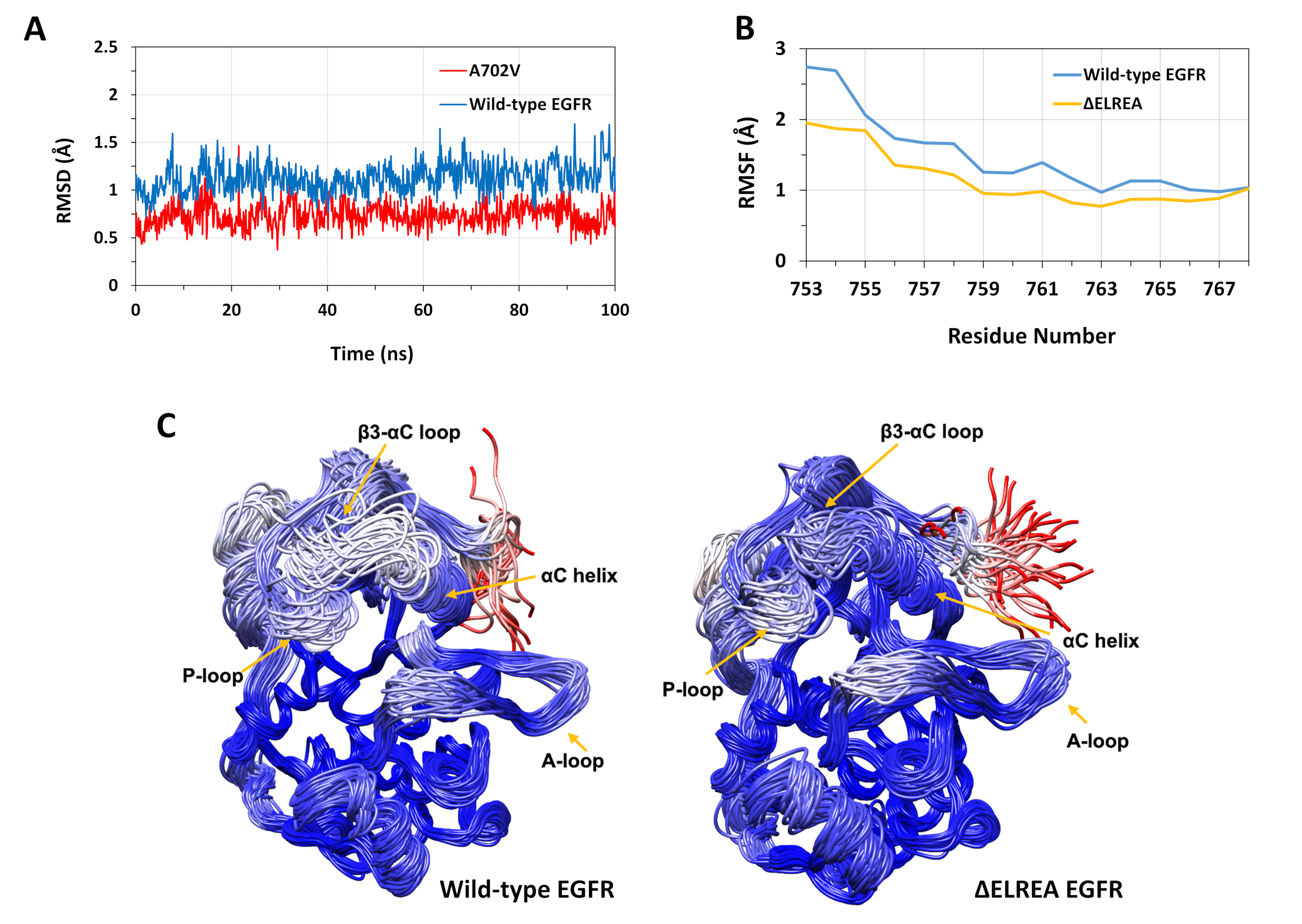
El protocolo descrito se utilizó para estudiar los efectos estructurales de las mutaciones de LAREA y A702V en la estructura de la quinasa egFR. Una aplicación del protocolo fue investigar el efecto de las mutaciones en la estabilidad estructural/conformación local mediante la computación de los valores RMSD y RMSF a partir de las simulaciones MD. Como la mutación A702V se encuentra en el segmento juxtmemembrane B, el RMSD de este segmento del receptor quinasa en relación con la estructura inicial se calculó tanto para el tipo silvestre como para los EGFR A702V. El resultado (Figura 4A) reveló que el segmento de juxtramembrana B del mutante ha aumentado la estabilidad conformacional durante la simulación de 100 ns (promedio de RMSD 0,7o - intervalo de confianza del 95% (CI) 0,009) en comparación con el dominio de quinasa EGFR de tipo salvaje (promedio de RMSD 1,1o - 95% CI 0,01). Esto es muy probable que sea el resultado de las interacciones hidrofóbicas más estrictas en la interfaz dimer debido a la sustitución de la alanina 702 (cadena lateral del grupo metilo) a un residuo hidrofóbico más voluminoso, valina (cadena lateral del grupo isopropílico), lo que conduce a un aumento de las interacciones hidrofóbicas de V702 en el dominio de la quinasa receptora con isoleucina 941 del dominio de la quinasa activadora.
La mutación de LAELREA se encuentra en el bucle de 3-C, adyacente a la hélice funcionalmente crítica de la C; la conformación de la hélice de la C es clave para los cambios entre los estados activos e inactivos de la quinasa EGFR. La estabilidad conformacional de la hélice de la C en el estado activo se evaluó examinando el RMSF sobre los átomos de C de los residuos dentro de la hélice durante las simulaciones MD(Figura 4B):en general, hay fluctuaciones más bajas en el mutante (promedio de RMSF 1,1 - 95% IC 0,4) en comparación con el tipo silvestre (promedio de RMSF 1,5 - 95% CI 0,57); con la mayor diferencia en las fluctuaciones registradas para los residuos del N-terminal. Las conformaciones muestreadas, respectivamente, superpuestas en la estructura mediana del dominio de la quinasa de tipo silvestre y del dominio de la quinasa de la EREA también soportan estos resultados(Figura 4C):tanto los dominios de tipo salvaje como los de la quinasa de LAREA tienen una estabilidad similar en general para las conformaciones superpuestas, excepto para el bucle C-C y el Estos hallazgos indican que la eliminación de la secuencia ELREA restringe el movimiento de la hélice del estado activo, por lo tanto, restringiendo y estabilizando así la conformación activa. Además, dado que la hélice de la C forma parte de la interfaz de dimer asimétrico, es muy probable que las restricciones de la hélice de la C en el mutante estabilicen el dimer asimétrico, prolongando la duración del estado activado.
Otra aplicación del protocolo es investigar el comportamiento de las interacciones clave intra e intermoleculares que tienen lugar durante la simulación. Por lo tanto, la interacción entre K745 y E762, que es fundamental para la actividad enzimática egFR, se analizó tanto para la forma activa de tipo salvaje como para la quinasa de EGFR de LAELREA midiendo el porcentaje de ocupación de los enlaces de hidrógeno formados entre los átomos polares de la cadena lateral de los dos residuos durante las simulaciones de MD(Figura 5A):esta interacción electrostática clave se formó con mayor frecuencia en el dominio de la quinasa de la cadena lateral en comparación con el dominio de la quinasa de tipo salvaje, debido a la hélice más estable que acomoda a E762. También se evaluaron las interacciones entre Mg2+-ATP y los dominios de tipo salvaje y éELREA EGFR quinasa (Figura 5B) durante la simulación(Figura 5C):el número de bonos de hidrógeno fue mayor para el EREA (valor medio 4,0 - 95% IC 0,03) que para el EGFR de tipo salvaje (valor medio 3,2 - 95% CI 0,04). Un análisis posterior de los enlaces de hidrógeno reveló que K745 interactúa con más frecuencia con los grupos de fosfatos de ATP en el EGFR de LA RASREA, que está vinculado a la interacción K745-E762 más estable que se observa en la simulación del dominio mutante EGFR quinasa de LA RAS-ELA.
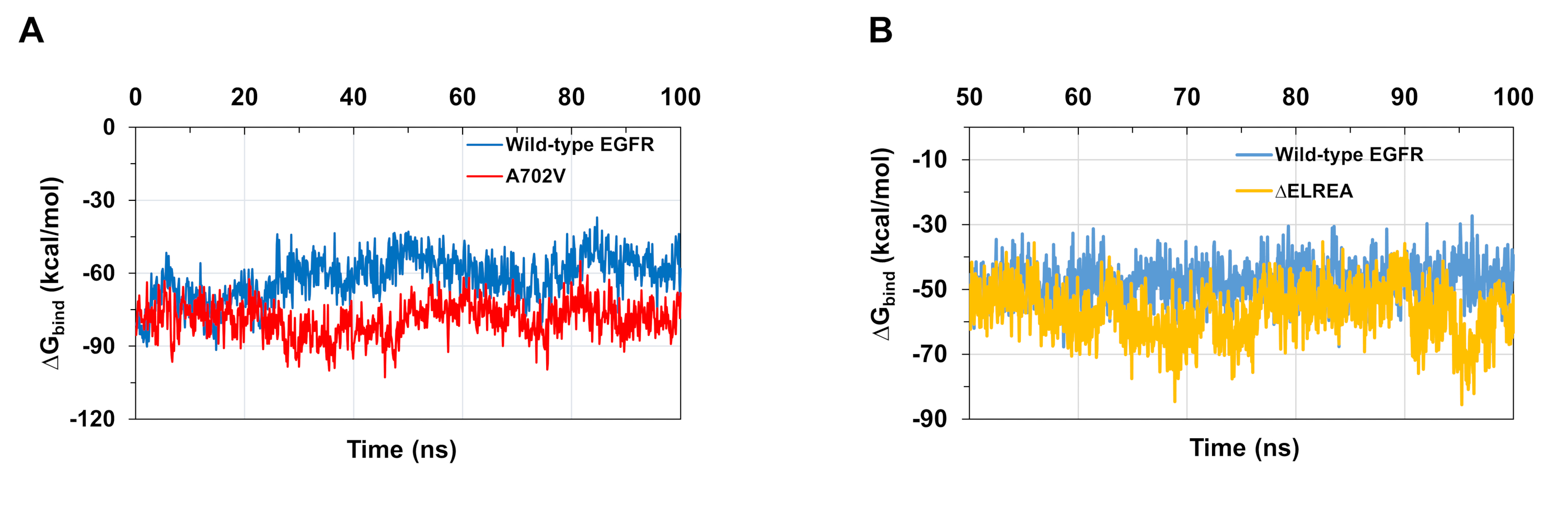
Las simulaciones MD como se describe en el protocolo también son útiles para evaluar la energía libre relativa de la unión para las interacciones proteína-proteína y proteína-ligand. Las energías de unión entre los dominios de la quinasa activadora y receptora de los EGFR de tipo salvaje y A702V, y entre los dominios DE LAFR quinasa de EGFR, y entre los dominios de tipo salvaje y mutante de LA EFR quinasa, se calcularon a partir de los cálculos de la superficie del Born generalizado (MMGBSA)(Figura 6A):el mutante A702V produjo un valor medio deunión más bajo (promedio deAglu a -76 kcal/mol - CI 0,47% de CI, que representa interacciones dimer más favorables, en contraste con el dominio EGFR de tipo salvaje(unión de G promedio de -61 kcal/mol - 95% CI 0,61). Esta observación es consistente con el segmento de juxtamembrana B más estable y una interfaz de dimer más estrecha debido al aumento de las interacciones hidrofóbicas observadas para el dominio A702V EGFR quinasa. En el caso de la unión de ATP a los dominios de tipo salvaje y éELREA EGFR quinasa (Figura 6B), los cálculos de MMGBSA predicen un enlace ATP más fuerte con el mutante de LA AELREA(unión media de G -57 kcal/mol - 95% CI 0.43) en comparación con el EGFR de tipo salvaje (promedio deenlace de G -48 kcal/mol - 95% CI 0.33). Este resultado está en línea con el mayor número de enlaces de hidrógeno registrados entre ATP y ELREA EGFR (Figura 5C) en comparación con el dominio de tipo salvaje.
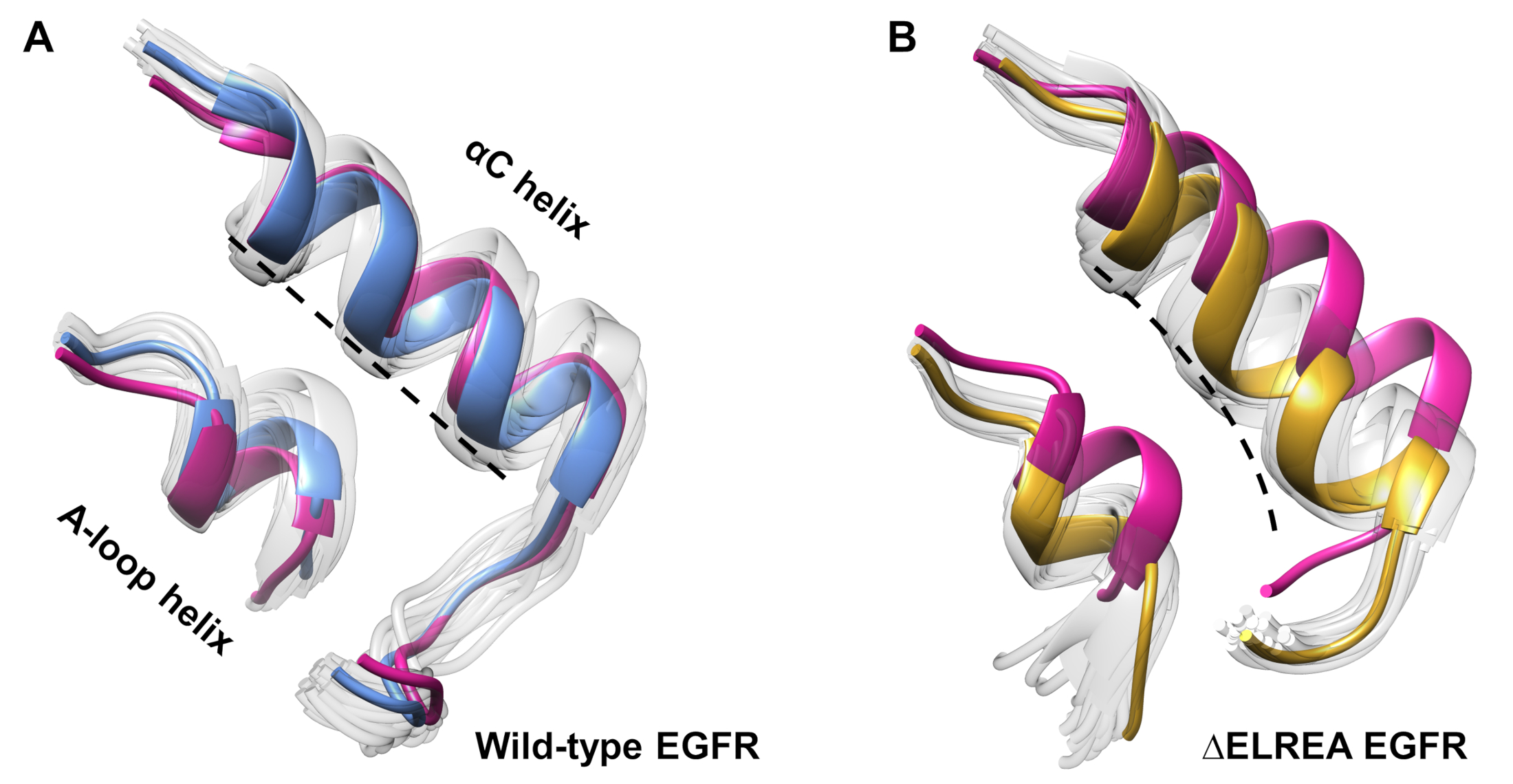
El protocolo también se puede emplear para investigar los cambios de conformación observados durante una simulación. En el estudio actual, los efectos de la mutación ÉELREA en la conformación inactiva del EGFR se estudiaron mediante la inspección visual y la superposición de las conformaciones muestreadas de la simulación. El análisis descubrió un movimiento hacia adentro de la hélice de la C en el dominio de la quinasa EGFR de LAC(Figura 7A),un cambio estructural esperado durante la transición al estado activo. Por el contrario, la hélice de tipo salvaje de EGFR, inactiva, mantuvo su conformación inicial(Figura 7B). Así, las simulaciones MD apoyan la propuesta de que la mutación de la deleción, mostrada experimentalmente para aumentar la actividad quinasa40,41, promueve un cambio de conformación de la quinasa inactiva hacia el estado activo.

Figura 4: Estabilidad de tipo salvaje y conformación mutante del dominio de la quinasa EGFR activo durante las simulaciones de MD. (A) RMSD (átomos de columna vertebral) sobre el segmento juxtmembrana B del dominio quinasa receptora de tipo salvaje (azul) y A702V (rojo). (B) RMSF (átomos de C) sobre los residuos de la hélice de C: tipo salvaje (azul) y ÉELREA (oro). (C) Conformaciones superpuestas de tipo salvaje (izquierda) y dominio de EGFR quinasa egFR (derecha); trazas de cadena coloreadas a base de RMSD (átomos de C) de cada residuo con respecto a la estructura mediana. El coloración varía de azul a blanco a rojo, representando regiones de alta a baja estabilidad conformal. Tenga en cuenta que las regiones N-terminal "libres" del dominio quinasa aislado, de color rojo, no exhibirían este nivel de movilidad en la estructura intacta del EGFR. Cifras adaptadas de Chakroborty et al.9 (Figura 4A reproducida con permiso de la Revista de Química Biológica) y Tamirat et al.12. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 5: Características clave observadas en el receptor activo quinasa durante las simulaciones de MD: el puente de sal K745-E762, la hélice de C y las interacciones con ATP. (A) Porcentaje de ocupación de la interacción K745-E762 durante la simulación de los dominios de tipo salvaje (azul) y ÉELREA (oro) EGFR quinasa. (B) Residuos del tipo salvaje y mutante de LAREA que interactúan con ATP (palos). Mg2+ (verde) coordenadas con ATP y D855. (C) El número de enlaces de hidrógeno formados por ATP con los dominios de tipo salvaje y éELREA EGFR quinasa durante las simulaciones MD. Figura de Tamirat et al.12. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 6: Se observan energías libres relativas más bajas de unión para los dominios de la quinasa mutante durante las simulaciones. (A) Energías vinculantes calculadas para la interacción entre los dominios de activador y quinasa receptora de los EGFR de tipo salvaje (azul) y A702V (rojo). (B)Enlace G de ATP a dominios de tipo salvaje (azul) y éELREA (oro) EGFR quinasa. Cifras adaptadas de Chakroborty et al.9 (Figura 6A reproducida con permiso de la Revista de Química Biológica) y Tamirat et al.12. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 7: Conformaciones superpuestas del dominio de la quinasa EGFR inte inactivo de tipo salvaje y de la EELREA. Conformación de la hélice y la hélice del bucle A de (A) de tipo salvaje (estructura mediana en azul) y (B) -LOSELREA (oro). Otras conformaciones muestreadas, de color blanco descolorido; estructuras iniciales antes de las simulaciones de MD, rosa. Figura de Tamirat et al.12. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Discusión
El protocolo descrito en este estudio se centra en el uso de simulaciones de dinámica molecular para investigar las alteraciones estructurales locales y globales que surgen de la activación de mutaciones somáticas del dominio de la quinasa EGFR. Aunque las estructuras de cristal de rayos X de los EGFR mutantes y de tipo salvaje proporcionan una visión estructural invaluable, representan una o algunas representaciones estáticas. Sin embargo, inherente a la función biológica de ErbBs es la transición necesaria entre la tirosina quinasa enzimáticamente inactiva y activa, invocando cambios dinámicos tanto en la estructura como en las interacciones intramoleculares entre los monómeros de la quinasa. Por lo tanto, se llevaron a cabo simulaciones md para sondear la naturaleza dinámica del dominio de la tirosina quinasa egFR, incluyendo la estructura de tipo salvaje, la mutación de eliminación de LAREA introducida y la mutación A702V. Estas simulaciones tuvieron éxito en el esclarecimiento del papel probable de estas mutaciones en las estructuras y cómo sus efectos en la conformación del dominio de la tirosina quinasa conducirían a los aumentos observados experimentalmente en la actividad quinasa del EGFR.
Un paso crucial en este protocolo es el uso de una estructura relevante para evaluar el impacto de la mutación. Una forma de seleccionar una estructura de entrada de simulación relevante es visualizar la ubicación de la mutación en la estructura 3D estática y examinar su posible impacto con respecto a los aminoácidos y unidades estructurales vecinas. En este estudio, por ejemplo, dado que la mutación EGFR A702V se encuentra en el segmento juxtamembrane B que forma la interfaz de dimer asimétrica, el uso de la estructura dimer para la simulación en lugar del monómero es crítico. El uso de una estructura monomérica habría expuesto el segmento juxtmembrana B del receptor quinasa al disolvente, privándolo de las interacciones estabilizadoras, potenciado por la mutación a un residuo hidrófobo más grande e interacciones con isoleucina 941 a partir de los residuos del lóbulo C de la quinasa activadora. Además, cabe destacar que la estructura 3D representada por las coordenadas en un archivo PDB no corresponde necesariamente a la estructura biológicamente relevante que debe utilizarse para el estudio. Por ejemplo, con la estructura de ErbB4, código PDB 3BCE, las coordenadas PDB corresponden a un trimer, pero esto se debe a contactos de cristal (pocos contactos entre los monómeros se ven al visualizar esta estructura). Las matrices dentro del archivo PDB se pueden utilizar (por ejemplo, dentro de Quimera) para reconstruir las estructuras cristalinamente relacionadas, que se pueden visualizar para identificar cadenas que corresponden a la estructura 3D biológicamente relevante como se indica en la publicación original42. Otro paso esencial del protocolo es preparar adecuadamente la estructura de entrada de simulación, como la construcción de aminoácidos que faltan en diferentes regiones de bucle, y especialmente donde se encuentra en las proximidades de la mutación. Aunque existen numerosas estructuras de EGFR de tipo salvaje en la PDB, sólo un número limitado de estructuras mutantes EGFR están disponibles. En consecuencia, las estructuras mutantes también necesitan ser modeladas; para una sola mutación de residuos como A702V, se utilizó Quimera para mutar el residuo; mientras que, para la mutación de la deleción de LAREA, se utilizó Modeller.
Los diversos parámetros utilizados en los archivos de entrada de simulación - por ejemplo, el número de ciclos de minimización, el calentamiento del sistema a la temperatura deseada de una sola vez o en su lugar calentando lentamente a través de varias temperaturas intermedias, el período de tiempo para el equilibrio y para las simulaciones de producción - se pueden modificar en función de la molécula de estudio, el objetivo de la obra y las propias preferencias. Al realizar simulaciones MD, también es común encontrarse con errores que pueden surgir de los archivos de entrada, problemas relacionados con el software de simulación en uso o incluso un error del usuario. Por lo tanto, es muy importante entender el origen de los errores examinando cuidadosamente cualquier mensaje de error. La mayoría de los programas de simulación tienen una lista de correo donde los usuarios pueden plantear preguntas a los desarrolladores de software y a otros usuarios por los que la mayoría de los problemas se pueden resolver. Además, los manuales de usuario proporcionan una asistencia significativa para comprender los detalles del protocolo de simulación, incluidas las suposiciones y limitaciones. Aunque la simulación MD es una herramienta importante para explorar las propiedades dinámicas de las moléculas, recuerde que los resultados computacionales deben evaluarse cuidadosamente junto con otras fuentes de información para evaluar su validez. Siempre que sea posible, trabajar junto con investigadores que son expertos en las proteínas en estudio, especialmente cuando se realizan estudios experimentales de laboratorio húmedo relevantes, que sirven para proporcionar resultados para la interpretación estructural, así como para sugerir experimentos que se pueden hacer sobre la base de observaciones estructurales para probar hipótesis.
En este estudio, el protocolo fue eficaz en el examen de los impactos estructurales dinámicos de las mutaciones de ÉELREA y A702V en las estructuras quinasas del EGFR. Las simulaciones revelaron que el AREA restringe la hélice funcionalmente esencial y promueve un cambio conformacional de la quinasa inactiva a una quinasa activa estabilizada. Los resultados de la simulación están respaldados de forma independiente por datos de respuesta a fármacos que demostraron los efectos de los inhibidores de la tirosina quinasa en las líneas celulares de cáncer de pulmón que tienen la mutación de la deleción de LAELREA y el EGFR de tipo salvaje, donde se notificó una mayor inhibición por fármacos que reconocen la conformación de la quinasa activa para el EELREA que para el EGFR12de tipo salvaje. Con la mutación A702V, las simulaciones MD indican, en comparación con el tipo salvaje, una mayor estabilización de la interfaz de la quinasa activador-receptor, así como una mayor afinidad del activador y la quinasa receptora entre sí, apoyando juntos el mantenimiento de la conformación activada de la quinasa EGFR. La mutación A702V, ubicada en el segmento juxtmembrana B del receptor quinasa, aumentaría las interacciones hidrofóbicas con el activador quinasa, funcionando para prolongar la duración del estado activado. La mutación A702V apoya la supervivencia celular en ausencia de factor de crecimiento y se identificó en un cribado in vitro de las mutaciones del EGFR9.
Divulgaciones
Los autores no tienen nada que revelar.
Agradecimientos
Esta investigación está financiada por subvenciones a M.S.J de la Academia de Finlandia (308317, 320005), Sigrid Juselius Foundation y Tor, Joe y Pentti Borg fondo conmemorativo, y a K.E. de la Academia de Finlandia (274728, 316796), la Fundación contra el Cáncer de Finlandia, y el Hospital Central de la Universidad de Turku. M.Z.T. está financiado por la Red de Doctorado de Biología Informativa y Estructural de la Akademi. Agradecemos al CSC IT Center for Science por los recursos informáticos y al Dr. Jukka Lehtonen por el apoyo de TI en el marco de la red bioinformática Biocenter Finland; y la red de infraestructura de biología estructural de Biocenter Finland.
Materiales
| Name | Company | Catalog Number | Comments |
| Amber software | University of California, San Francisco | Version 2018 | Executable |
| Chimera program | Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco | Version 1.13.1 | Executable |
| EGFR struture files | The Protein Data Bank | 3D coordinates of EGFR structures | |
| Maestro | Schrödinger LLC | Version 2018-3 | Executable |
| Modeller program | The Andrej Šali Lab, Departments of Biopharmaceutical Sciences and Pharmaceutical Chemistry, University of California San Francisco | Included in the Chimera program | |
| VMD software | Theoretical and Computational Biophysics Group, University of Illinois at Urbana-Champaign | Version 1.9.3 | Executable |
Referencias
- Yarden, Y., Sliwkowski, M. X. Untangling the ErbB signalling network. Nature Reviews Molecular Cell Biology. 2, 127-137 (2001).
- Lemmon, M. A., Schlessinger, J., Ferguson, K. M. The EGFR family: not so prototypical receptor tyrosine kinases. Cold Spring Harbor Perspectives in Biology. 6, a020768 (2014).
- Arteaga, C. L., Engelman, J. A. ERBB receptors: From oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell. 2, 282-303 (2014).
- Mishra, R., Hanker, A. B., Garrett, J. T. Genomic alterations of ERBB receptors in cancer: Clinical implications. Oncotarget. 8, 114371-114392 (2017).
- . cBioPortal for Cancer Genomics Available from: https://www.cbioportal.org (2020)
- Lynch, T. J., et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. New England Journal of Medicine. 350 (21), 2129-2139 (2004).
- Paez, J. G., et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 304 (5676), 1497-1500 (2004).
- Pao, W., et al. EGF receptor gene mutations are common in lung cancers from "never smokers" and are associated with sensitivity of tumors to gefitinib and erlotinib. Proceedings of the National Academy of Sciences U.S.A. 101 (36), 13306-13311 (2004).
- Chakroborty, D., et al. Unbiased in vitro screen for activating EGFR mutations. Journal of Biological Chemistry. 294 (24), 9377-9389 (2019).
- Leahy, D. J. Structure and Function of the Epidermal Growth Factor (EGF/ErbB) Family of Receptors. Advances in Protein Chemistry. 68, 1-27 (2004).
- Roskoski, R. ErbB/HER protein-tyrosine kinases: Structures and small molecule inhibitors. Pharmacological Research. 87, 42-59 (2014).
- Tamirat, M. Z., Koivu, M., Elenius, K., Johnson, M. S. Structural characterization of EGFR exon 19 deletion mutation using molecular dynamics simulation. PLoS ONE. 14 (9), e0222814 (2019).
- Ding, L., et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 455, 1069-1075 (2008).
- Kurppa, K. J., Denessiouk, K., Johnson, M. S., Elenius, K. Activating somatic ERBB4 mutations in non small-cell lung cancer. Oncogene. 35 (10), 1283-1291 (2016).
- Soung, Y. H., et al. Somatic mutations of the ERBB4 kinase domain in human cancers. International Journal of Cancer. 118, 1426-1429 (2006).
- Tvorogov, D., et al. Somatic mutations of ERBB4: selective loss-of-function phenotype affecting signal transduction pathways in cancer. Journal of Biological Chemistry. 284, 5582-5591 (2009).
- Hubbard, S. R., Till, J. H. Protein tyrosine kinase structure and function. Annual Review of Biochemistry. 69 (1), 373-398 (2000).
- Huse, M., Kuriyan, J. The conformational plasticity of protein kinases. Cell. 109 (3), 275-282 (2002).
- Jura, N., et al. Catalytic control in the EGF receptor and its connection to general kinase regulatory mechanisms. Molecular Cell. 42, 9-22 (2011).
- Karplus, M., Kuriyan, M., J, Molecular dynamics and protein function. Proceedings of the National Academy of Sciences U.S.A. 102 (19), 6679-6685 (2005).
- Shan, Y., Arkhipov, A., Kim, E. T., Pan, A. C., Shaw, D. E. Transitions to catalytically inactive conformations in EGFR kinase. Proceedings of the National Academy of Sciences U.S.A. 110 (18), 7270-7275 (2013).
- Reckamp, K. L., et al. A phase I trial to determine the optimal biological dose of celecoxib when combined with erlotinib in advanced non-small cell lung cancer. Clinical Cancer Research. 12 (11 Pt 1), 3381-3388 (2006).
- Pettersen, E. F., et al. UCSF Chimera-a visualization system for exploratory research and analysis. Journal of Computational Chemistry. 25 (13), 1605-1612 (2004).
- Berman, H. M., et al. The Protein Data Bank. Nucleic Acids Research. 28 (1), 235-242 (2000).
- Zhang, X., Gureasko, J., Shen, K., Cole, P. A., Kuriyan, J. An Allosteric Mechanism for Activation of the Kinase Domain of Epidermal Growth Factor Receptor. Cell. 125 (6), 1137-1149 (2006).
- Stamos, J., Sliwkowski, M. X., Eigenbrot, C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. Journal of Biological Chemistry. 277 (48), 46265-46272 (2002).
- Sogabe, S., et al. Structure-Based Approach for the Discovery of Pyrrolo[3,2-d]pyrimidine-Based EGFR T790M/L858R Mutant Inhibitors. ACS Medicinal Chemistry Letters. 4 (2), 201-205 (2013).
- Sali, A., Blundell, T. L. Comparative protein modelling by satisfaction of spatial restraints. Journal of Molecular Biology. 234 (3), 779-815 (1993).
- Yun, C. H., et al. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell. 11 (3), 217-227 (2007).
- Park, J. H., Liu, Y., Lemmon, M. A., Radhakrishnan, R. Erlotinib binds both inactive and active conformations of the EGFR tyrosine kinase domain. Biochemical Journal. 448 (3), 417-423 (2012).
- . Release 2018-3: Maestro Available from: https://www.schrodinger.com/maestro (2018)
- Case, D. A., et al. . AMBER 2018. , (2018).
- Maier, J. A., Martinez, C., Kasavajhala, K., Wickstrom, L., Hauser, K. E., Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. Journal of Chemical Theory and Computation. 11 (8), 3696-3713 (2015).
- Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W., Klein, M. L. Comparison of simple potential functions for simulating liquid water. Journal of Chemical Physics. 79 (2), 926-935 (1983).
- Meagher, K. L., Redman, L. T., Carlson, H. A. Development of polyphosphate parameters for use with the AMBER force field. Journal of Computational Chemistry. 24 (9), 1016-1025 (2003).
- Humphrey, W., Dalke, A., Schulten, K. VMD: Visual molecular dynamics. Journal of Molecular Graphics. 14 (1), 33-38 (1996).
- Melvin, R. L., Salsbury, F. R. Visualizing ensembles in structural biology. Journal of Molecular Graphics and Modelling. 67, 44-53 (2016).
- Roe, D. R., Cheatham, T. E. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. Journal of Chemical Theory and Computation. 9 (7), 3084-3095 (2013).
- Miller, B. R., et al. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. Journal of Chemical Theory and Computation. 8 (9), 3314-3321 (2012).
- Guha, U., et al. Comparisons of tyrosine phosphorylated proteins in cells expressing lung cancer-specific alleles of EGFR and KRAS. Proceedings of the National Academy of Sciences U.S.A. 105 (37), 14112-14117 (2008).
- Furuyama, K., et al. Sensitivity and kinase activity of epidermal growth factor receptor (EGFR) exon 19 and others to EGFR-tyrosine kinase inhibitors. Cancer Science. 104 (5), 584-589 (2013).
- Qiu, C., et al. Mechanism of Activation and Inhibition of the HER4/ErbB4 Kinase. Structure. 6 (3), 460-467 (2008).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados