
Method Article
Décryptage des effets structurels de l’activation des mutations somatiques EGFR avec simulation de dynamique moléculaire
Dans cet article
Résumé
L’objectif de ce protocole est d’utiliser des simulations de dynamique moléculaire pour examiner les changements structurels dynamiques qui se produisent en raison de l’activation des mutations de la protéine DEGFR kinase.
Résumé
De nombreuses mutations somatiques se produisant dans la famille du récepteur épidermique de facteur de croissance (EGFR) de la kinases de tyrosine de récepteur (RTK) ont été rapportées des patients de cancer, bien que relativement peu aient été éprouvés et montrés pour causer des changements fonctionnels dans ErbBs. Les récepteurs ErbB sont dimés et activés sur la liaison ligand, et les changements conformationnels dynamiques des récepteurs sont inhérents à l’induction de la signalisation en aval. Pour deux mutations montrées expérimentalement pour modifier la fonction d’EGFR, A702V et la mutation de suppression d’ELREA746 ELREA 750, nous illustrons dans le protocole suivant comment les simulations de dynamique moléculaire (MD) peuvent sonder la (1) stabilité conformationnelle de la structure mutante de la tyrosine kinase par rapport à l’EGFR de type sauvage ;746 (2) les conséquences structurelles et les transitions conformationnelles et leur relation avec les changements fonctionnels observés; (3) les effets des mutations sur la force de liaison de l’ATP ainsi que sur la liaison entre les domaines kinase dans le dimer asymétrique activé; et (4) les effets des mutations sur les interactions clés au sein du site de liaison EGFR associés à l’enzyme activée. Le protocole fournit une procédure détaillée étape par étape ainsi que des conseils qui peuvent être plus généralement utiles pour l’étude des structures protéiques en utilisant des simulations MD comme un moyen de sonder la dynamique structurelle et la relation à la fonction biologique.
Introduction
La famille des récepteurs du facteur de croissance épidermique humain (EGFR) des kinases de tyrosine de récepteur (RTKs) comprend quatre membres - EGFR/ErbB1/HER1, ErbB2/HER2, ErbB3/HER3 et ErbB4/HER4. Les récepteurs ErbB régulent les processus cellulaires fondamentaux tels que la croissance et la prolifération cellulaires, la différenciation, la migration et la survie1,2, et sont donc de puissants proto-oncogènes. L’activité aberrante des récepteurs ErbB, en particulier EGFR et ErbB2, a été fréquemment associée à des cancers humains faisant des récepteurs ErbB cibles clés pour les thérapeutiques du cancer2,3.
Plusieurs altérations somatiques des gènes ERBB ont été rapportées à partir de malignités humaines3,4,5. Les exemples les mieux caractérisés incluent les mutations récurrentes et activantes de point et les suppressions courtes dans le cadre dans le domaine de kinase d’EGFR dans le cancer du poumon non à petites cellules (NSCLC). Ces mutations EGFR représentent les principaux moteurs de la croissance du cancer, et prédisent la sensibilité à EGFR ciblant les médicaments contre le cancer6,7,8. Cependant, dans la plupart des cancers, les mutations somatiques de l’EGFR se produisent en dehors de ces « points chauds » récurrents et sont réparties sur l’ensemble de la portée de 1210 résidus du récepteur. En effet, la plupart des résidus le long de la séquence primaire EGFR se sont avérés mutés dans le cancer humain9. Néanmoins, en dehors des quelques points chauds, l’importance fonctionnelle de la grande majorité des mutations EGFR associées au cancer reste inconnue.
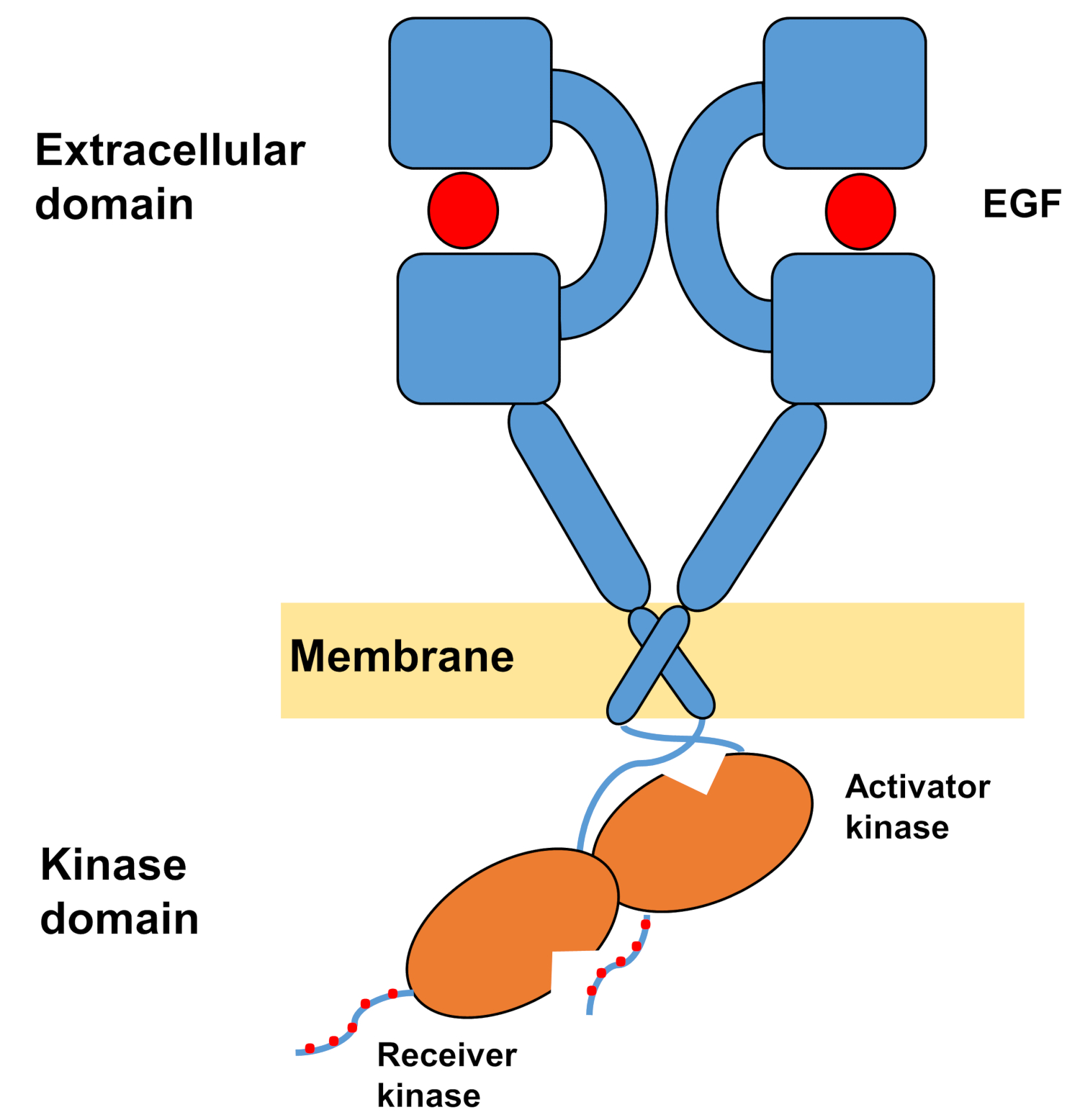
La structure monomérique des ErbBs se compose d’un grand domaine extracellulaire aminé, suivi d’une seule hélice transmembrane menant au domaine intracellulaire de la tyrosine kinase et à la région de queue c-terminal qui contient des sites d’amarrage pour les protéines de signalisation intracellulaire. La liaison ligand déclenche un changement conformationnel spectaculaire dans le domaine extracellulaire, ce qui facilite la formation des dimères récepteurs en exposant les bras de dimération qui se croisent symétriquement et interagissent avec leurs surfaces aromatiques/hydrophobes. Lors de la formation de dimères récepteurs, les domaines de la tyrosine kinase entrent en contact de façon asymétrique (figure 1), ce qui entraîne l’activation des kinases qui phosphoratent les queues c-terminales des monomères récepteurs, puis l’activation de la signalisation en aval10,11.

Figure 1 : Structure du dimrage EGFR. EGFR dimerizes lorsque les domaines extracellulaires lient le facteur de croissance (EGF, facteur de croissance épidermique). Le domaine kinase récepteur est ensuite activé par l’interaction asymétrique avec le domaine kinase activateur, et les queues c-terminal sont autophosphorylés à des résidus de tyrosine (Modifié à partir de Tamirat et al.12). Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}
En raison des réarrangements structurels dynamiques qui se produisent pendant les  transitions de dimer monomère, avec l’activation de kinase qui est associée à la formation d’un dimre asymétrique, les mutations le long de la pleine longueur de la structure de récepteur peuvent potentiellement avoir un effet sur la fonction de récepteur. Ici, nous décrivons plusieurs exemples de nos études précédentes dans lesquelles la modélisation de la mutation et la visualisation étaient suffisantes pour expliquer les conséquences pour la fonction.
transitions de dimer monomère, avec l’activation de kinase qui est associée à la formation d’un dimre asymétrique, les mutations le long de la pleine longueur de la structure de récepteur peuvent potentiellement avoir un effet sur la fonction de récepteur. Ici, nous décrivons plusieurs exemples de nos études précédentes dans lesquelles la modélisation de la mutation et la visualisation étaient suffisantes pour expliquer les conséquences pour la fonction.
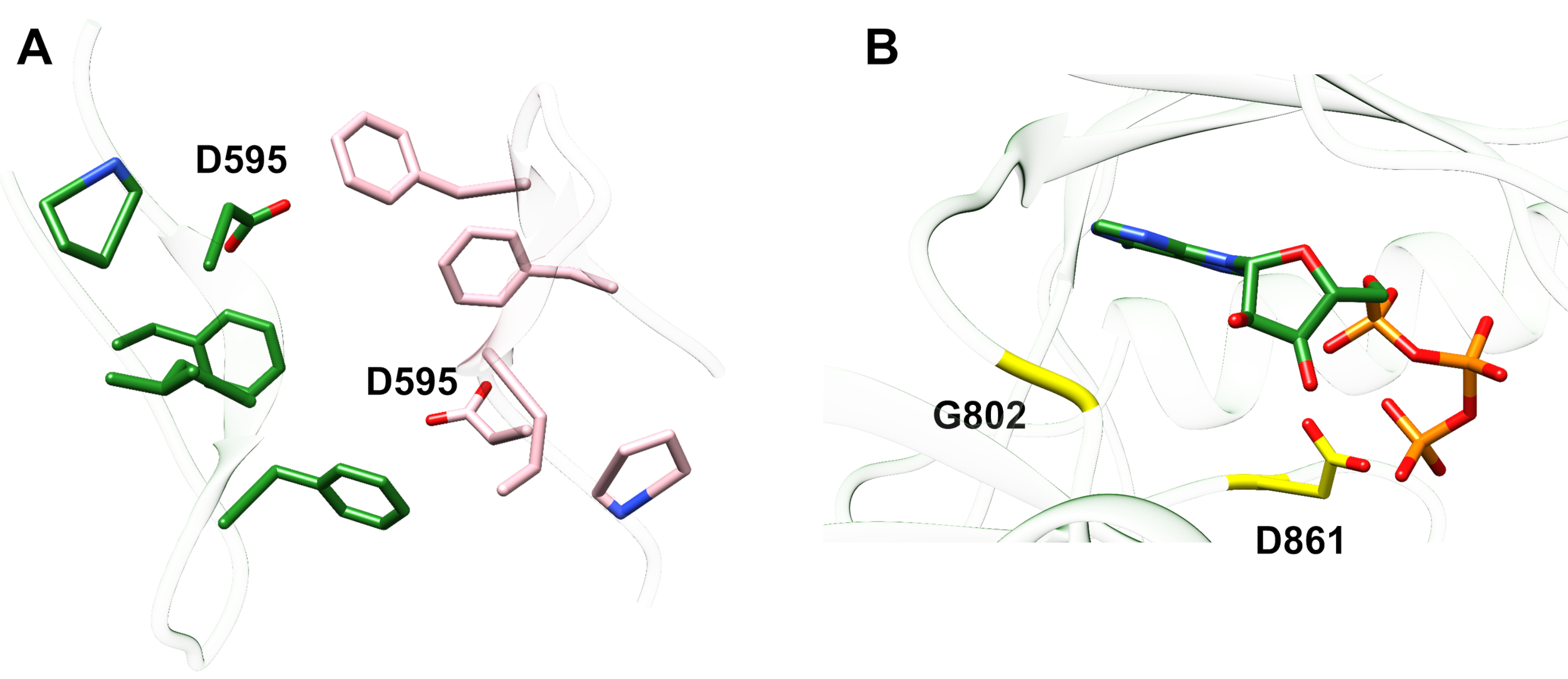
Exemple 1 : Une mutation rapportée, D595V dans ErbB413, a mené à la dimréisation et à la phosphorylation d’ErbB4accrues 14. La visualisation de l’emplacement de la mutation a été un facteur critique dans la compréhension des effets fonctionnels observés : D595V s’est produit au croisement symétrique des bras dimériques de l’ectodomain (Figure 2A). Les bras sont en grande partie aromatiques et hydrophobes, et le remplacement de l’acide aspartique polaire par la valine devrait augmenter les interactions hydrophobes « collantes », stabilisant le dimère et augmentant ainsi la durée pendant laquelle la phosphorylation a lieu14. C’était une surprise au début de trouver l’aspartate dans chaque bras, mais rétrospectivement on pourrait penser à lui comme un mécanisme de synchronisation pour l’activité, où les chaînes latérales d’acide polaire réduisent l’affinité et la durée de vie du dimère intact et limitent ainsi la phosphorylation et la signalisation kinase-mediated. Le remplacement par la valine supprimerait alors cette protection en stabilisant davantage le dimrébre ErbB4.

Figure 2 : Emplacement d’une mutation d’activation erbB4 et mutations produisant l’ErbB4 mort de kinase. (A) D595 (activation de la mutation D595V) est situé sur les bras dimeriques aromatiques/hydrophobes du modèle ectodomain ErbB4; l’association des armes sur la liaison des facteurs de croissance; (les résidus à proximité sont représentés sous forme de bâtons). (B) Dans ErbB4, G802 (mutation inactivante du G802dup) aide à former la poche de liaison autour de l’anneau adénine de l’ATP et catalytique D861 (mutation D861Y inactivante) lie à la fois Mg2+ (non montré) et le groupe γ-phosphate de l’ATP. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}
Exemple 2 : On peut prévoir que les mutations somatiques qui ciblent le site de liaison ATP du domaine kinase modifieraient ou élimineraient l’activité enzymatique conduisant à un récepteur altéré ou mort de kinase incapable de signaler. Des neuf mutations rapportées des patients présentant le sein, gastrique, colorectal, ou NSCLC15, deux des neuf mutations une fois testées ont eu l’activité fortement diminuée de phosphorylation16: G802dup (G → GG) et D861Y. Les deux mutations somatiques inactivantes ont été trouvées dans le site de liaison ATP de la structure de domaine de la tyrosine kinase (Figure 2B): la glycine flexible, dupliquée, modifierait le site de l’anneau d’adénine et le petit acide aspartique remplacé par la tyrosine encombrante près des phosphates terminaux empêcherait physiquement Mg2+-ATP de se lier. Toutefois, depuis ErbB4 peut former un hétérodimère avec ErbB2 - ErbB2 ne lie pas un facteur de croissance et dépend de l’association avec un ErbB qui fait pour hétérodimeriser - l’ErbB2(actif)-ErbB4(kinase-dead) hétérodimer stimulerait la prolifération cellulaire par la voie de signalisation Erk/Akt mais les cellules ne se différencieraient pas en raison de la kinase-mort ErbB4 et de l’absence d’activation de la voie STAT516.
Dans des études plus récentes, il est devenu évident que les mouvements dynamiques des ErbB étaient pertinents pour comprendre les effets de certains mutants sur la fonction ErbB, en particulier les mutations qui se produisent dans le domaine de la tyrosine kinase. Le domaine de la tyrosine kinase se compose d’un Lobe N (principalement β-feuilles) et de Lobe C (en grande partie alpha hélicoïdal), qui sont séparés par le site catalytique où l’ATP se lie. Le Lobe N comprend l’hélice αC et la boucle P, tandis que l’activation (boucle A) et les boucles catalytiques sont présentes dans le Lobe C17,18,19. Les structures cristallines du domaine de la tyrosine kinase ont révélé deux conformations inactives, la majorité des structures ont l’état inactif de genre src. Dans la conformation active, l’aspartate catalytique de la boucle A pointe vers le site de liaison ATP et l’hélice αC est orientée vers la poche de liaison ATP (conformation « C-in »), formant une forte interaction glutamate-lysine ion-paire.
Étant donné que les ErbB et le domaine de la kinase des composants sont des entités très dynamiques, et en particulier dans les cas où les effets des mutations sur la fonction et l’activité biologique sont susceptibles d’être étroitement liés aux états conformationnels des ErbB, il est important d’évaluer les mutations par rapport à la gamme de changements dynamiques qu’elles subiraient. Les structures cristallines à rayons X des ErbB fournissent des instantanés statiques de la structure 3D, qui peuvent ou non être pertinents pour comprendre les conséquences dynamiques d’une mutation. Afin de sonder la gamme de changements dynamiques correspondant au « paysage énergétique » disponible pour une structure tridimensionnelle (3D), les simulations de dynamique moléculaire (MD) sont largement utilisées20. Dans le cas de mutations qui conduiraient à des changements conformationnels locaux dans le domaine de la tyrosine kinase ou à la stabilisation d’un complexe, les simulations de l’ordre de 100 ns peuvent être suffisantes. Toutefois, les changements conformationnels à plus grande échelle (p. ex., les transitions entre les conformations actives et inactives du domaine kinase) nécessitent un temps de simulation plus long - de l’ordre des microsecondes21.
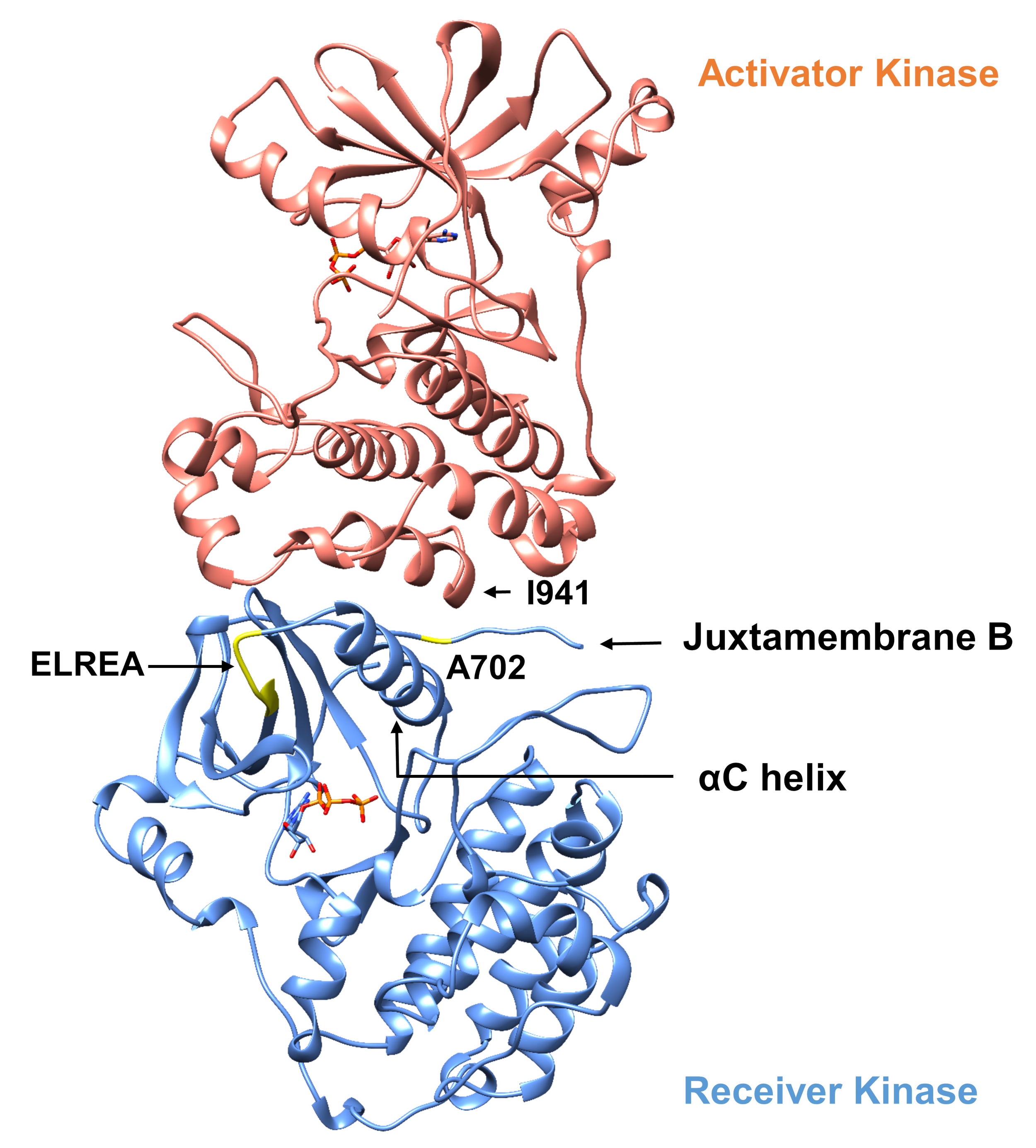
En ce qui concerne le protocole décrit ci-dessous, nous considérons deux mutations activantes dans le domaine de la tyrosine kinase (Figure 3). Les deux mutations sont situées dans le domaine kinase à des endroits qui connaissent des changements conformationnels locaux qui dictent si la kinase est active ou non, et donc des simulations MD ont été appliquées dans les deux cas. Dans le premier cas, nous considérons les changements qui affectent directement le site de liaison ATP et les machines catalytiques du domaine kinase du récepteur EGFR, en examinant spécifiquement les conséquences d’une mutation de suppression exon 19 qui est largement impliquée dans NSCLC4,7. La mutation Δ746ELREA750, qui réduit la longueur de la boucle β3-αC précédant l’hélice αC - l’hélice qui se déplace vers le site de liaison/actif sur l’activation de la kinase et participe à la formation de l’interaction électrostatique critique entre E762 de l’hélice et le K745 en positionnant la lysine pour l’interaction avec l’ATP - prédispose le domaine pour l’activation12. Dans le deuxième cas, nous considérons la mutation A702V d’EGFR, montrée pour être une nouvelle mutation d’activation de gain de fonction révélée par la plate-forme d’iScream9 et identifiée dans un patient de NSCLC22. Alanine-702 sur le domaine kinase récepteur est situé sur le segment B juxtamembrane à l’interface du récepteur et l’activateur kinase domaines, dans lequel ce complexe asymétrique kinase dimer et kinase changements conformationnels sont nécessaires pour l’activation9.

Figure 3 : Dimer de domaine de kinase asymétrique d’EGFR. La mutation A702V serait située à l’interface critique des domaines activateur et récepteur kinase, à côté de l’hélice αC et à proximité de l’isoleucine 941 de l’activateur kinase. Les changements conformationnels induits par la formation du dimber asymétrique conduisent à l’activation de kinase. La boucle β3-αC contenant la séquence ELREA précède directement l’hélice αC; pendant l’activation, l’hélice αC se déplace vers l’intérieur vers le site de liaison ATP. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}
Protocole
REMARQUE : Les mesures détaillées prises pour examiner les effets de la mutation ΔELREA et A702V sur la structure EGFR à l’aide de simulations MD sont discutées comme suit :
1. Préparation de la structure
REMARQUE : Afin d’étudier les impacts structurels de la mutation ΔELREA, les formes sauvages et mutantes des structures monomères EGFR actives, actives et apo liées à l’ATP sont préparées comme suit.
- Ouvrez le programme de visualisation Chimera23 (https://www.cgl.ucsf.edu/chimera/) pour préparer la structure active de kinase EGFR de type sauvage. Dans le menu Fichier, cliquez sur l’option Chercher par ID et sélectionnez la base de données De la Banque de données protéiques (PDB24)et spécifiez le code PDB 2GS225 (résolution 2.8 Å). Le PDB est un référentiel pour les structures 3D résolues par différentes techniques expérimentales, y compris la cristallographie aux rayons X, la spectroscopie par résonance magnétique nucléaire, la microscopie cryo-électronique et la diffraction des neutrons.
- Construire les éléments structurels manquants de 2GS2 en prenant ces segments des structures EGFR PBB 1M1426 (2,6 Å) et 3W2S27 (1,9 Å). Pour ce faire, ouvrez 1M14 et 3W2S et superposez-les sur 2GS2 à l’aide de l’option MatchMaker dans le menu comparaison Outils → Structure.
- Rogner les segments à ajouter à partir de 1M14 et 3W2S. Sélectionnez les atomes terminaux des résidus avant les lacunes de 2GS2 et les atomes à ajouter à partir de 1M14 et 3W2S (pour plus d’informations sur les segments ajoutés, voir le tableau 1). Sur la ligne de commande, tapez le lien sel et frappez entrez. Cette structure est la structure de modèle.
- Utilisez la structure de modèle à partir de l’étape 1.2 pour construire la forme mutante de suppression ΔELREA du domaine kinase EGFR. Produire la séquence de format FASTA de ΔELREA EGFR en sauvant la séquence de la structure de modèle (→ séquence → fichier → enregistrer sous)et en supprimant la séquence ELREA aux numéros de résidus 746-750.
- Ouvrez la séquence ΔELREA EGFR dans Chimera et alignez-la avec la séquence de la structure du modèle 2GS2 à l’aide du menu Séquence. Dans la fenêtre d’alignement, sélectionnez l’option Structure → Modeller (homologie)28.
- Sur la fenêtre contextuelle, spécifiez la structure composite 2GS2 comme modèle et la séquence mutante comme requête à modéliser. Puis appuyez sur OK. Sélectionnez un modèle mutant parmi les modèles résultants, en fonction du score zdope (généralement le score le plus bas) et de l’inspection visuelle.
- Pour préparer la structure de kinase DE KINase de type sauvage liée à l’ATP, utilisez la structure PDB2ITX 29 (2,98 Å) comme structure principale. Construire des segments manquants (voir le tableau 1) à l’aide des structures 2GS625 (2.6 Å) et 3W2S suivant la procédure à l’étape 1.2. Convertir l’ANP ligand dans la structure résultante en ATP en ouvrant le fichier PDB dans un éditeur de texte et en changeant l’atome d’azote N3B de l’ANP en atome d’oxygène.
- Ouvrez la structure à l’étape 1.4 de Chimera comme indiqué à l’étape 1.1. Ajoutez un ion de magnésium à cette structure de la structure PDB 2ITN29 (2.47 Å) afin d’atteindre un positionnement similaire pour l’ion Mg2+.
- Avec la structure résultant de l’étape 1.5, modèle le formulaire mutant ΔELREA suivant l’étape 1.3.
| Apo active EGFR | Apo inactif EGFR | EGFR actif lié à l’ATP | |
| Structure principale | 2GS2 | 2GS7 | 2ITX |
| Structures utilisées pour construire des boucles manquantes | 1M14 (723-725) | 3W2S (958-984) | 2GS6 (862-865) |
| 3W2S (967-981) | 4HJO (848-850) | 3W2S (990-1001) |
Tableau 1 : Structures utilisées pour construire des modèles composites de structures actives apo actives, inactives et actives liées à l’ATP. Les régions manquantes (gamme d’acides aminés entre parenthèses) dans la structure principale ont été construites à partir des structures énumérées.
- Pour préparer la structure inactive de kinase EGFR de type sauvage, ouvrez la structure PBB 2GS725 (2.6 Å) comme à l’étape 1.1 et supprimez les ligands liés et les eaux cristallines. Ajouter les segments manquants dans 2GS7 (voir le tableau 1) des structures 3W2S et 4HJO30 (2,75 Å) à l’aide de la procédure à l’étape 1.2. Sur la base de la structure EGFR inactive finale, préparer le modèle mutant en utilisant la procédure à l’étape 1.3.
NOTE : Pour l’étude de mutation D’A702V, la structure asymétrique de dimre d’EGFR est étudiée car la mutation est située au segment Juxtamembrane B du domaine de kinase qui forme une grande partie de l’interface de dimer. Les structures mutantes EGFR de type sauvage et A702V sont préparées comme suit : - La structure asymétrique de dimer de type sauvage est construite à partir de la structure 2GS2 de PBB, qui est initialement affichée sous forme monomérique. Pour convertir à l’assemblage biologique qui contient l’activateur et les kinases de récepteur dans l’arrangement asymétrique, ouvrez 2GS2 dans Chimera comme dans (1.1) et effectuez des calculs de symétrie en cliquant sur les outils → structure d’ordre supérieur → menu cellule unitaire. Sélectionnez la structure 2GS2 et entrez Faire des copies. Enfin, sélectionnez et enregistrez un seul dimer asymétrique à partir des multiples copies du dimer résultant des opérations de symétrie.
- En utilisant la structure asymétrique EGFR de type sauvage de l’étape 1.8, construire le mutant A702V en remplaçant alanine 702 par de la valine à l’aide de l’option Tools → Structure → l’option Rotamers dans Chimera.
NOTE : Collectivement, six structures monomériques et deux EGFR dimériques sont préparées pour les études de mutation de ΔELREA et d’A702V, respectivement. Chaque structure est ensuite traitée pour simulation à l’aide de l’assistant de préparation des protéines dans le programme Maestro31 et les simulations MD sont faites avec le programme Amber32. - Ouvrez la structure dans Maestro à l’aide de l’option Fichier → structure d’importation. Cliquez ensuite sur le bouton de l’Assistant préparation des protéines et sélectionnez ce qui suit : ajouter des atomes d’hydrogène, construire des atomes de chaîne latérale manquants, déterminer les états de protonation des résidus ionizables au pH 7.0 à l’aide de PROPKA, optimiser l’orientation des résidus d’asparagine, de glutamine et d’histidine pour la liaison de l’hydrogène, et enfin minimiser la structure.
2. Configuration du système
- Ouvrez le programme saut inclus dans le logiciel Amber. Importer les molécules d’eau ff14SB33 (source leaprc.protein.ff14SB) et les molécules d’eau TIP3P34 (source leaprc.water.tip3p). Pour les systèmes liés à l’ATP également les paramètres d’importation pour ATP35 (loadamberparams frcmod.phosphate, loadamberprep ATP.prep). Ensuite, chargez la structure (mol = loadpdb structure.pdb).
- Solvate la structure dans une boîte octaèdre avec des molécules d’eau TIP3P explicites qui s’étend 10 Å dans toutes les directions des atomes de surface de la protéine (solvateoct mol TIP3PBOX 10.0).
- Vérifiez le système construit (Vérifier mol) et neutralisez-le en ajoutant les ions nécessaires (addions mol Na+ 0). Pour modéliser suffisamment les systèmes biomoléculaires,- ajoutez des atomes de Na+/Cl supplémentaires à la boîte de simulation pour porter la concentration de sel du système à 0,15 M(ajouts mol Na+ X, addions mol Cl- X), où X est remplacé par le résultat de: concentration de sel souhaitée * nombre de molécules d’eau * volume par molécule d’eau * Nombre d’Avogadro.
- Générer et enregistrer les fichiers de topologie et de coordonnées du système, qui servent d’entrées pour la simulation de production ultérieure(saveamberparm mol X.prmtop X.inpcrd).
3. Simulation de dynamique moléculaire
- À l’aide de l’ambre, soumettez d’abord le système de simulation à 5000 cycles de descente la plus raide et de minimisation de l’énergie de gradient conjuguée pour contourner les configurations défavorables. Effectuer la minimisation en plusieurs étapes, en abaissant progressivement la retenue appliquée sur les atomes de soluté de 25 kcal mol-1 Å-2 à 0 kcal mol-1 Å-2.
- Dans le fichier d’entrée de minimisation, min.in, ajuster la variable maxcyc pour le cycle de minimisation totale (maxcyc = 5000) et ncyc pour indiquer le nombre de cycles pour l’algorithme de descente le plus raide. Utilisez la variable restraint_wt pour appliquer la force de retenue sur les atomes de soluté spécifiés par le paramètre de masque de retenue. Exécutez ensuite la minimisation comme suit :
$AMBERHOME/bin/sander -O -i min.in -o min.out -p X.prmtop -c X.inpcrd -r min.rst -ref X.inpcrd
REMARQUE : La stratégie et les paramètres réels utilisés peuvent varier en fonction de ses propres préférences. Les détails et les conseils peuvent être trouvés à partir du manuel Amber et le site Web (https://ambermd.org/index.php)
- Dans le fichier d’entrée de minimisation, min.in, ajuster la variable maxcyc pour le cycle de minimisation totale (maxcyc = 5000) et ncyc pour indiquer le nombre de cycles pour l’algorithme de descente le plus raide. Utilisez la variable restraint_wt pour appliquer la force de retenue sur les atomes de soluté spécifiés par le paramètre de masque de retenue. Exécutez ensuite la minimisation comme suit :
- Chauffer le système pendant 100 ps de 0 K à 300 K réglage d’une mol de 10 kcal-1 Å-2 retenue sur les atomes de soluté. Pour ce faire, définissez tempi = 0,0, temp0 = 300,0, dt = 0,002 ps, nstlim = 50000 et restraint_wt = 10 dans le fichier d’entrée heat.in. Effectuer le chauffage avec la commande suivante:
$AMBERHOME/bin/sander -O -i heat.in -o heat.out -p X.prmtop -c min.rst -r heat.rst -x heat.mdcrd -ref min.rst - Équilibrer le système pour 900 ps dans le cadre d’un ensemble TNP; nombre constant d’atomes, température (temp0 = 300,0) et pression (ntp = 1), le contrôlant avec la méthode Berendsen (ntt = 1). Réglez une coupure de distance de 9 Å(coupe = 9,0) pour les interactions électrostatiques à longue portée. Abaisser progressivement la retenue de l’atome soluté à 0,1 kcal mol-1 Å-2 (restraint_wt = 0,1). Exécutez le fichier d’entrée d’équilibre equil.in qui décrit les paramètres ci-dessus comme suit :
$AMBERHOME/bin/sander -O -i equil.in -o equiuil.out -p X.prmtop -c heat.rst -r equil.rst -x equil.mdcrd -ref heat.rst - Finaliser l’équilibre avec une simulation sans retenue de 5 ns (set dt = 0,002 ps, ntslim = 2500000).
$AMBERHOME/bin/sander -O -i equil_final.in -o equil_final.out -p X.prmtop -c equil.rst -r equil_final.rst -x equil_final.mdcrd -ref equil.rst - Vérifiez que le système a équilibré en examinant la température, la pression, la densité et les valeurs énergétiques.
$AMBERHOME/bin/process_mdout.perl heat.out equil.out equil_final.out
résumé xmgrace. TEMP/DENSITY/ETOT/EPTOT/EKTOT - Effectuer la simulation de production pour 100 ns (set dt = 0,002 ps, ntslim = 50000000 dans prod.in) et enregistrer les conformations tous les 10 ps (ntwx = 5000).
$AMBERHOME/bin/sander -O -i prod.in -o prod.out -p X.prmtop -c equil_final.rst -r prod.rst -x prod.mdrcd -ref equil_final.rst
4. Analyse
- Inspection visuelle
- Visualisez les conformations échantillonnées lors des simulations de kinase EGFR de type sauvage et mutant en ouvrant les fichiers de topologie ambre X.prmtop et les fichiers de trajectoire prod.mdcrd correspondants dans VMD36. À l’aide de représentations pratiques de la structure secondaire, analyser la dynamique structurelle globale des protéines à partir de la trajectoire enregistrée. Voir les interactions spécifiques entre les atomes/résidus d’intérêt, tels que le pont de sel K745 - E762, catalytiquement essentiel.
- Vous pouvez également enregistrer plusieurs conformations échantillonnées au cours de la simulation au format PDB et ouvrez-les à l’aide du programme Chimera. Superposez les structures sur la structure initiale ou médiane à l’aide de l’option MatchMaker. Affichez la structure initiale/médiane en solide et le reste des structures alignées en blanc fané. Cette approche permet de visualiser les mouvements structurels enregistrés avec plus de clarté.
REMARQUE : Les suggestions de représentations efficaces et de traitement des ensembles conformationnels de MDS se trouvent dans Melvin et al.37.
- Analyse RMSD et RMSF
- Calculer les calculs de la déviation carrée moyenne racine-moyenne (RMSD) et des fluctuations carrées moyennes des racines (RMSF) avec le programme38 de Cpptraj pour analyser la stabilité globale des protéines et examiner la flexibilité des différentes unités structurelles. Dans les fichiers d’entrée rmsd.in et rmsf.in, indiquez les atomes de colonne vertébrale (pour RMSD) et les atomes Cα (pour RMSF) de la structure initiale comme référence pour le montage RMS. Dans les fichiers rmsd/rmsf.in importer les fichiers de topologie ambre (parm X.promtop) et les fichiers de trajectoire correspondants (trajin prod.mdcrd). Puis exécutez la commande Cpptraj -i rmsd/rmsf.in. Tracez les données de sortie pour analyse.
- Alternativement, aligner les ensembles conformationnels et colorer chaque résidu basé sur l’atome Cα RMSD. Pour ce faire, ouvrez les conformations dans Chimera et alignez-les avec l’option Matchmaker.
- Accédez à Outils → Représentation → Render par attribut. Sélectionnez Résidus de l’ensemble conformationnel et Cα RMSD comme attributs et cliquez sur OK. La trace de chaîne des conformations sera alors colorée du bleu → blanc → rouge, reflétant respectivement des régions de haute, moyenne et faible stabilité structurelle.
- Analyse des liaisons hydrogène
- Analyser l’interaction de liaison hydrogène entre l’ATP et les EFFR de type sauvage/ΔELREA. Préparez un script Cpptraj, hbond.in, pour mener à bien cette tâche. Définir une liaison hydrogène avec une distance donneur-accepteur inférieure ou égale à 3,5 Å et un angle de liaison supérieur ou égal à 135°. Spécifier l’analyse uniquement pour les liaisons intermoléculaires d’hydrogène avec la variable nointramol c’est-à-dire les liaisons hydrogène entre ATP et EGFR (hbond All nointramol dist 3.5 out nhb.agr avgout avghb.dat). Exécuter le script comme Cpptraj -i hbond.in.
- Utilisez ce script pour évaluer les interactions intramoléculaires, par exemple entre les résidus K745 et E762, qui sont des résidus clés pour l’activité de kinase EGFR. Pour ce faire, spécifiez K745 comme donneur de liaison d’hydrogène et E762 comme l’accepteur de liaison d’hydrogène dans hbond.in et exécutez le script en conséquence.
- Distance de surveillance entre les atomes
- Mesurer la distance entre K745 et E762 en ouvrant les trajectoires de type sauvage et ΔELREA apo EGFRs dans VMD. Sélectionnez le Cδ de Glu762 et Nz de Lys745 en cliquant sur L’étiquette → de souris → obligation. Surveillez la distance pendant la simulation en traçant un graphique avec des étiquettes graphiques → → graphique → de liaison.
- Calculs d’énergie libres
- Pour calculer les énergies libres de liaison estimées entre les EFFR de type SAUVAGE/ΔELREA, et entre l’activateur et les kinases de récepteur des EGFR de type sauvage/A702V, utilisez le module39 de la surface de naissance généralisée de la mécanique moléculaire (MM-GBSA) disponible dans le package AMBER. Définir l’ATP comme le ligand et l’EGFR comme récepteur dans l’étude ΔELREA. Dans l’étude A702V, spécifiez le récepteur kinase comme le ligand et l’activateur kinase comme récepteur.
- Préparez d’abord les fichiers PDB du complexe ligand, récepteur et ligand-récepteur séparément dans le programme de saut définissant la valeur PBRadii à mbendi2. Pour les fichiers PDB, enregistrez les fichiers de topologie ambre de phase gazeuse (.prmtop) et coordonnez (.inpcrd).
- Ensuite, dans le fichier d’entrée mmgbsa, mmgbsa.in, définir igb = 2, saltcon = 0,1. Exécuter des calculs d’énergie de liaison à l’aide des trajectoires des simulations, des fichiers ambres récepteur/ligand préparés et des paramètres de la mmgbsa.in avec le script MMPBSA.py disponible dans Amber comme suit :
$AMBERHOME/bin/MMPBSA.py -O -i mmgbsa.in -o mmgbsa.dat -sp X.prmtop -cp complex.prmtop -rp receptor.prmtop -lp ligand.prmtop -y prod.mdcrd -eo output.csv - Analysez les données de sortie, output.csv, en traçant des graphiques.
Résultats
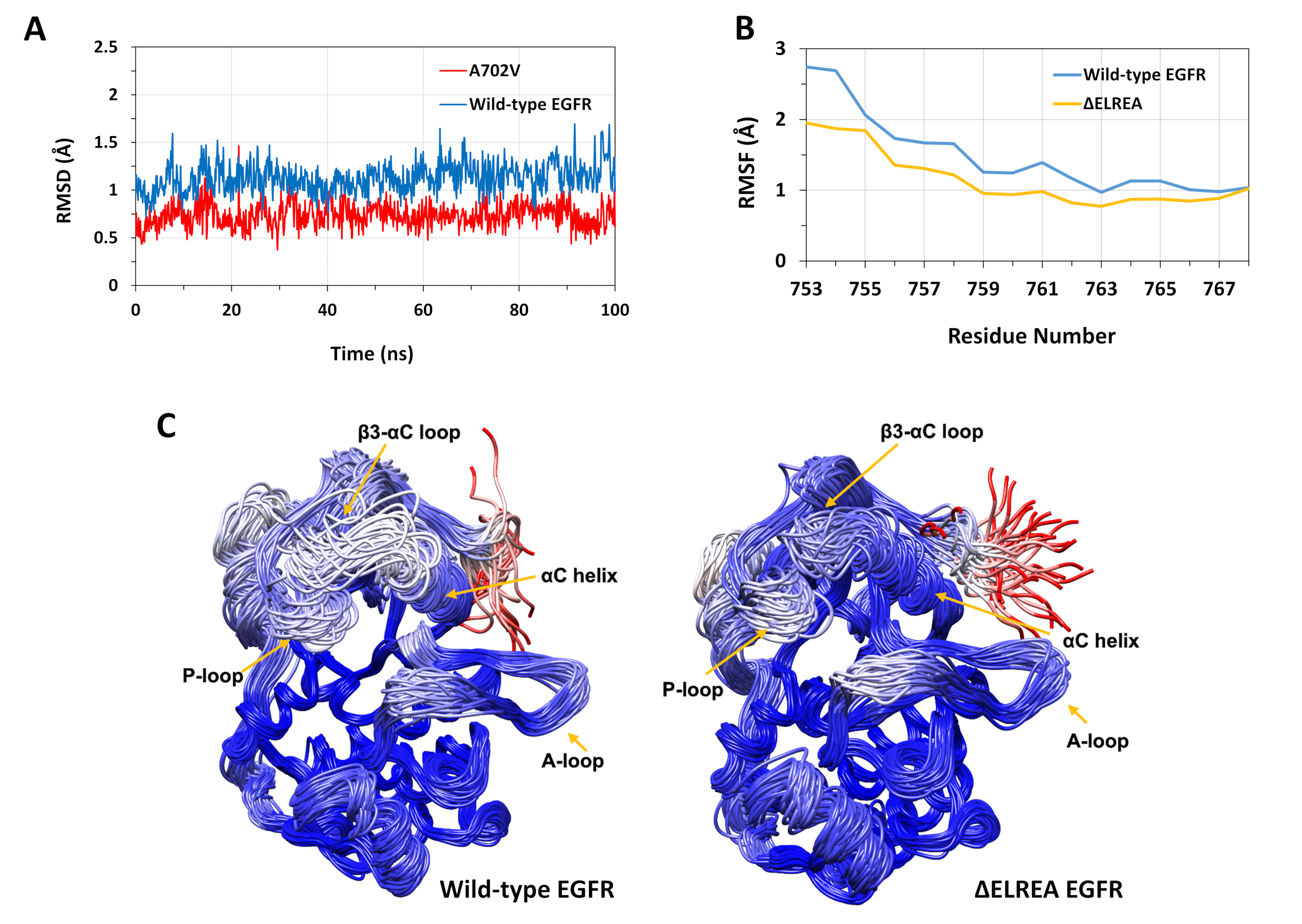
Le protocole décrit a été utilisé pour étudier les effets structurels des mutations ΔELREA et A702V sur la structure de kinase d’EGFR. Une application du protocole était d’étudier l’effet des mutations sur la stabilité structurelle/conformationnelle locale en calculant les valeurs RMSD et RMSF des simulations MD. Comme la mutation A702V est située sur le segment Juxtamemembrane B, le RMSD de ce segment du récepteur kinase par rapport à la structure de départ a été calculé pour les EGFR de type sauvage et A702V. Le résultat (Figure 4A) a révélé que le segment Juxtramembrane B du mutant a augmenté la stabilité conformationnelle au cours de la simulation de 100 ns (moyenne RMSD 0,7 Å - intervalle de confiance de 95% (CI) 0,009) par rapport au domaine de type sauvage EGFRase (moyenne RMSD 1,1 Å - 95% CI 0,01). C’est très probablement le résultat des interactions hydrophobes plus serrées à l’interface de dimère due au remplacement de l’alanine 702 (chaîne latérale de groupe de méthyle) à un résidu hydrophobe plus volumineux, la valine (chaîne latérale de groupe isopropyle), conduisant à des interactions hydrophobes accrues de V702 sur le domaine kinase de récepteur avec l’isoleucine 941 du domaine kinase d’activateur.
La mutation ΔELREA est située à la boucle β3-αC, adjacente à l’hélice αC fonctionnellement critique; la conformation de l’hélice αC est essentielle pour passer entre les états actifs et inactifs de l’EGFR kinase. La stabilité conformationnelle de l’hélice αC dans l’état actif a été évaluée en examinant le RMSF sur les atomes de Cα de résidus dans l’hélice au cours des simulations md (Figure 4B) : globalement, il y a des fluctuations plus faibles dans le mutant (moyenne RMSF 1.1 Å - 95% CI 0.4) par rapport au type sauvage (moyenne RMSF 1.5 Å - 95% CI 0.57); la plus grande différence de fluctuations enregistrées pour les résidus du terminal N. Les conformations échantillonnées respectivement superposées sur la structure médiane du domaine kinase de type sauvage et du domaine kinase de type ΔELREA appuient également ces résultats (figure 4C): les domaines de type sauvage et de kinase ΔELREA ont une stabilité globale similaire pour les conformations superposées, à l’exception de la boucle β3-αC et de l’hélice αC, qui sont nettement plus stables dans ΔELREA EGFR. Ces résultats indiquent que la suppression de la séquence ELREA restreint le mouvement de l’état actif αC helix, donc la restriction et donc la stabilisation de la conformation active. En outre, puisque l’hélice αC fait partie de l’interface de dimer asymétrique, les retenues sur l’hélice αC dans le mutant stabiliserait très probablement le dimer asymétrique, prolongeant la durée de l’état activé.
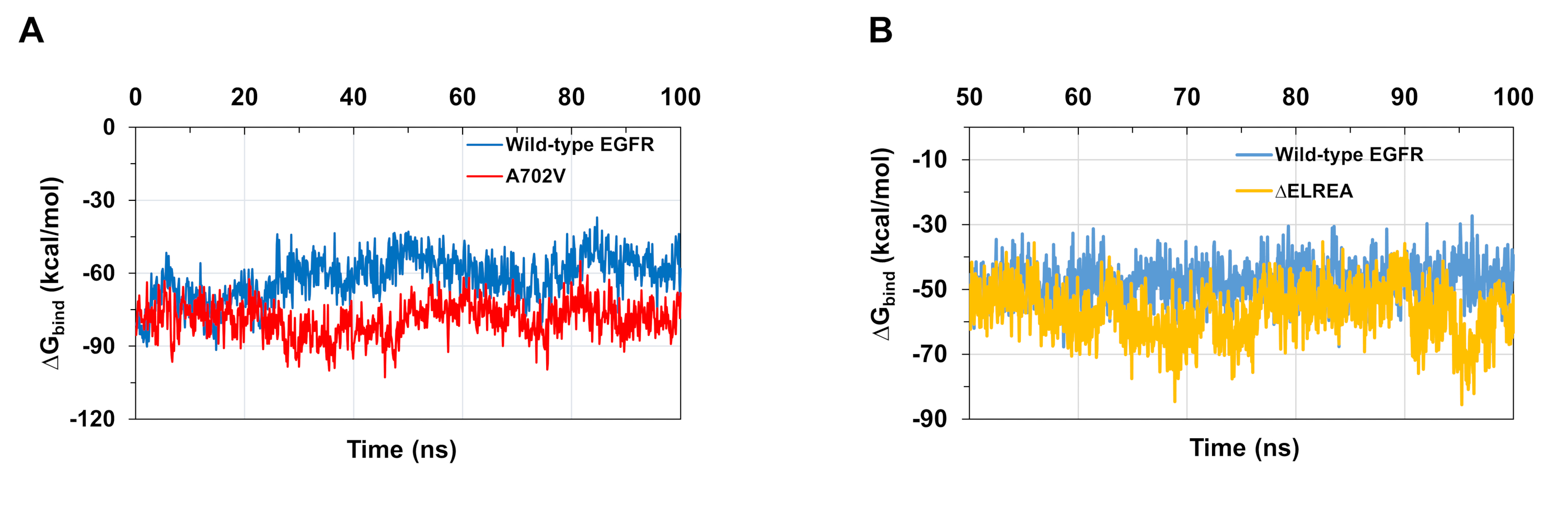
Une autre application du protocole consiste à étudier le comportement des interactions intra et intermoléculaires clés qui ont lieu au cours de la simulation. Ainsi, l’interaction entre K745 et E762, qui est fondamentale pour l’activité enzymatique EGFR, a été analysé à la fois pour la forme active de type sauvage et ΔELREA EGFR kinase en mesurant le pourcentage d’occupation des liaisons d’hydrogène formées entre les atomes polaires de la chaîne latérale des deux résidus au cours des simulations MD (Figure 5A):cette interaction électrostatique clé a été formée plus souvent dans le domaine kinase ΔELREA par rapport au domaine kinase de type sauvage, en raison de l’hélice αC plus stable qui accueille E762. Les interactions entre mg2+-ATP et les domaines de type sauvage et ΔELREA EGFR kinase (figure 5B) au cours de la simulation ont également été évaluées ( Figure5C): le nombre de liaisons hydrogène était plus élevé pour ΔELREA (valeur moyenne 4,0 - 95 % CI 0,03) que pour le type sauvage EGFR (valeur moyenne 3,2 à 95 % CI 0,04). Une analyse plus approfondie des liaisons hydrogène a révélé que K745 interagit plus fréquemment avec les groupes de phosphate de l’ATP dans ΔELREA EGFR, qui est liée à l’interaction K745-E762 plus stable notée dans la simulation du domaine mutant ΔELREA EGFR kinase.
Les simulations md telles que décrites dans le protocole sont également utiles pour évaluer l’énergie libre relative de liaison pour les interactions protéine-protéine et protéine-ligand. Les énergies de liaison entre les domaines kinase de type sauvage et de type A702V, ainsi qu’entre les domaines EGFR kinase mutants de type sauvage et ΔELREA, ont été calculés à partir de la mécanique moléculaire généralisée De la surface de naissance (MMGBSA) calculs (Figure 6A):le mutant A702V a produit une valeur deliaison moyenne inférieure ΔG(liaison moyenne ΔG = -76 kcal/mol - 9 5% CI 0,47), représentant des interactions plus favorables dimer, contrairement au domaine EGFR de type sauvage(liaison ΔG moyenne = -61 kcal/mol - 95% CI 0,61). Cette observation est compatible avec le segment B plus stable de juxtamembrane B et l’interface plus étroite de dimère en raison des interactions hydrophobes accrues observées pour le domaine de kinase d’EGFR D’A702V. Dans le cas de la liaison ATP aux domaines de la kinase de type sauvage et ΔELREA EGFR (figure 6B),les calculs mmgbsa prédisent une liaison ATP plus forte avec le mutant ΔELREA (liaison moyenne ΔGbind -57 kcal/mol - 95% CI 0,43) par rapport à l’EGFR de type sauvage (moyenne ΔGbind -48 kcal/mol - 95% CI 0,33). Ce résultat est en ligne avec le plus grand nombre de liaisons hydrogène enregistrées entre ATP et ΔELREA EGFR (Figure 5C) par rapport au domaine de type sauvage.
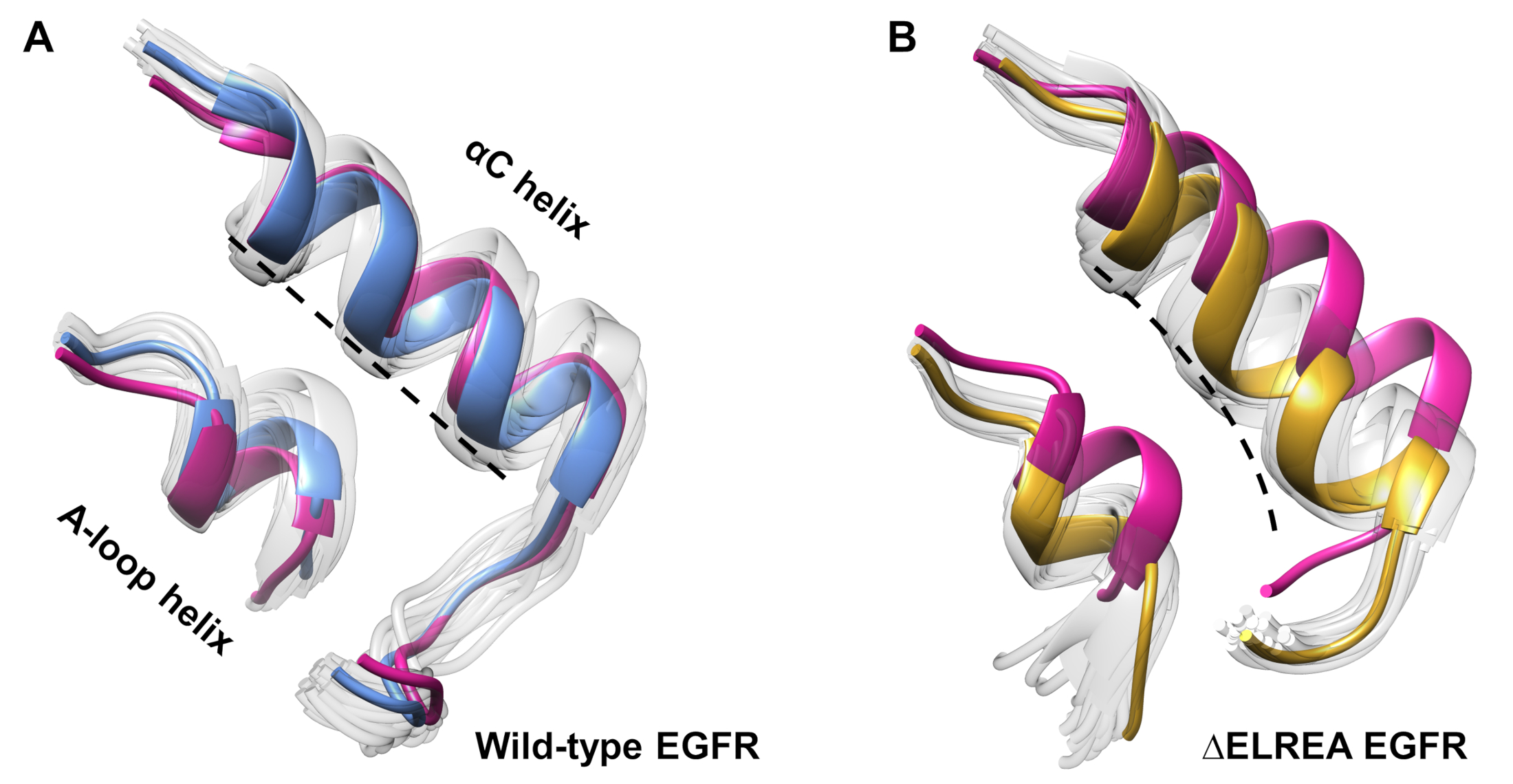
Le protocole peut également être utilisé pour étudier les changements conformationnels observés au cours d’une simulation. Dans la présente étude, les effets de la mutation ΔELREA sur la conformation inactive d’EGFR ont été étudiés par l’inspection visuelle et la superposition des conformations échantillonnées de la simulation. L’analyse a mis au jour un mouvement intérieur de l’hélice αC dans le domaine ΔELREA EGFR kinase (Figure 7A), un changement structurel attendu pendant la transition vers l’état actif. En revanche, l’hélice αC de type sauvage inactive EGFR a maintenu sa conformation initiale (figure 7B). Ainsi, les simulations md soutiennent la proposition que la mutation de suppression, montrée expérimentalement pour augmenter l’activité de kinase40,41, favorise un décalage conformationnel de la kinase inactive vers l’état actif.

Figure 4 : Stabilité conformationnelle de type sauvage et mutant du domaine actif de kinase EGFR pendant les simulations md. (A) RMSD (atomes de colonne vertébrale) sur le segment Juxtamembrane B du domaine kinase récepteur de type sauvage (bleu) et A702V (rouge). (B) RMSF (atomes de Cα) sur les résidus de l’hélice αC : type sauvage (bleu) et ΔELREA (or). (C) Conformations échantillonnées superposées de type sauvage (à gauche) et de domaine EGFR kinase de type sauvage (à droite); traces de chaîne colorées à partir de RMSD (atomes de Cα) de chaque résidu par rapport à la structure médiane. La coloration va du bleu au blanc en passant par le rouge, représentant des régions de haute à faible stabilité conformationnelle. Notez que les régions « ibr » en N-terminal du domaine de kinase isolé, rouge coloré, ne présenteraient pas ce niveau de mobilité dans la structure intacte egfr. Figures adaptées de Chakroborty et al.9 (Figure 4A reproduite avec la permission du Journal of Biological Chemistry) et de Tamirat et al.12. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 5 : Caractéristiques clés observées dans le récepteur actif kinase lors des simulations MD : le pont de sel K745-E762, l’hélice αC et les interactions avec l’ATP. (A)Pourcentage d’occupation de l’interaction K745-E762 lors de la simulation des domaines de kinase EGFR de type sauvage (bleu) et ΔELREA (or). (B) Résidus du mutant de type sauvage et ΔELREA interagissant avec l’ATP (bâtons). Mg2+ (vert) coordonne avec ATP et D855. (C) Nombre de liaisons hydrogène formées par l’ATP avec les domaines de type sauvage et ΔELREA EGFR kinase pendant les simulations MD. Figure de Tamirat et coll.12. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 6 : Des énergies libres relatives inférieures de liaison sont observées pour les domaines de kinase mutante pendant les simulations. (A) Énergies de liaison calculées pour l’interaction entre les domaines kinase de type sauvage (bleu) et A702V (rouge). (B) ΔGlier de l’ATP aux domaines de kinase EGFR de type sauvage (bleu) et ΔELREA (or). Figures adaptées de Chakroborty et al.9 (Figure 6A reproduite avec la permission du Journal of Biological Chemistry) et Tamirat et al.12. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 7 : Conformations superposées du domaine ΔELREA inactive EGFR kinase. Conformation de l’hélice αC et de l’hélice en boucle A de (A) de type sauvage (structure médiane en bleu) et (B) ΔELREA EGFRs (or). Autres conformations échantillonnées, blanc fané; structures initiales avant les simulations MD, rose. Figure de Tamirat et coll.12. Veuillez cliquer ici pour voir une version plus grande de ce chiffre.
{kind=link}
Discussion
Le protocole décrit dans cette étude se concentre sur l’utilisation de simulations de dynamique moléculaire pour étudier les altérations structurelles locales et mondiales qui découlent de l’activation des mutations somatiques du domaine de la kinase EGFR. Bien que les structures cristallines à rayons X des FEGF de type sauvage et mutants fournissent un aperçu structurel inestimable, elles représentent une ou quelques représentations statiques. Cependant, la fonction biologique des ErbB est inhérente aux transitions nécessaires entre la kinase enzymatiquement inactive et active de tyrosine, invoquant des changements dynamiques dans la structure et les interactions intramoléculaires entre les monomères kinase. Des simulations md ont ainsi été réalisées pour sonder la nature dynamique du domaine de la tyrosine kinase EGFR, y compris la structure de type sauvage, la mutation de suppression introduite par ΔELREA et la mutation A702V. Ces simulations ont réussi à élucider le rôle probable de ces mutations dans les structures et comment leurs effets sur la conformation du domaine de la tyrosine kinase conduiraient à l’augmentation expérimentalement observée de l’activité de kinase EGFR.
Une étape cruciale de ce protocole est l’utilisation d’une structure pertinente pour évaluer l’impact de la mutation. Une façon de sélectionner une structure d’entrée de simulation pertinente est de visualiser l’emplacement de la mutation dans la structure 3D statique et d’examiner son impact possible par rapport aux acides aminés voisins et les unités structurelles. Dans cette étude, par exemple, puisque la mutation EGFR de l’A702V est située sur le segment B juxtamembrane B qui forme l’interface asymétrique de dimer, l’utilisation de la structure de dimre pour la simulation par opposition au monomère est critique. L’utilisation d’une structure monomérique aurait exposé le segment juxtamembrane B du récepteur kinase au solvant, le privant des interactions stabilisatrices, renforcée par la mutation à un résidu hydrophobe plus grand et les interactions avec l’isoleucine 941 des résidus de C-lobe de la kinase activateur. En outre, il convient de noter que la structure 3D représentée par les coordonnées d’un fichier PDB ne correspond pas nécessairement à la structure biologiquement pertinente qui devrait être utilisée pour l’étude. Par exemple, avec la structure d’ErbB4, code PDB 3BCE, les coordonnées PDB correspondent à un trimer, mais cela est dû à des contacts en cristal (peu de contacts entre les monomères sont vus lors de la visualisation de cette structure). Des matrices dans le fichier PDB peuvent être utilisées (p. ex., dans la Chimère) pour reconstruire les structures cristallines, qui peuvent être visualisées pour identifier les chaînes qui correspondent à la structure 3D biologiquement pertinente telle qu’elle est rapportée dans la publication originale42. Une autre étape essentielle du protocole est de bien préparer la structure d’entrée de simulation, comme la construction d’acides aminés manquants dans différentes régions de boucle, et surtout lorsqu’il est situé à proximité de la mutation. Bien qu’il existe de nombreuses structures EGFR de type sauvage dans le PDB, seul un nombre limité de structures mutantes EGFR sont disponibles. Par conséquent, les structures mutantes doivent également être modélisées; pour une seule mutation de résidus comme A702V, la chimère a été utilisée pour muter les résidus; alors que, pour la mutation de suppression de ΔELREA, Modeller a été employé.
Les différents paramètres utilisés dans les fichiers d’entrée de simulation - par exemple, le nombre de cycles de minimisation, le chauffage du système à la température désirée en une seule fois ou au lieu de chauffage lentement à travers plusieurs températures intermédiaires, la période de temps pour l’équilibre et pour les simulations de production - peuvent être modifiés en fonction de la molécule d’étude, le but du travail et ses propres préférences. Lors de la réalisation de simulations MD, il est également courant de rencontrer des erreurs qui peuvent survenir à partir des fichiers d’entrée, des problèmes liés au logiciel de simulation en cours d’utilisation ou même une erreur de l’utilisateur. Par conséquent, il est très important de comprendre la source des erreurs en examinant attentivement les messages d’erreur. La plupart des programmes de simulation ont une liste de diffusion où les utilisateurs peuvent poser des questions aux développeurs de logiciels et aux autres utilisateurs par lesquels la plupart des problèmes peuvent être résolus. De plus, les manuels d’utilisation fournissent une aide importante pour comprendre les détails du protocole de simulation, y compris les hypothèses et les limitations. Bien que la simulation MD soit un outil important pour explorer les propriétés dynamiques des molécules, n’oubliez pas que les résultats computationnels doivent être soigneusement évalués en conjonction avec d’autres sources d’information pour évaluer leur validité. Dans la mesure du possible, travailler avec des chercheurs qui sont des experts sur les protéines à l’étude, en particulier lorsque des études expérimentales pertinentes en laboratoire humide sont faites, qui servent à fournir des résultats pour l’interprétation structurelle ainsi que pour suggérer des expériences qui peuvent être faites sur la base d’observations structurelles pour tester des hypothèses.
Dans cette étude, le protocole a été efficace dans l’examen des impacts structurels dynamiques des mutations ΔELREA et A702V sur les structures de kinase d’EGFR. Les simulations ont révélé que ΔELREA retient l’hélice αC fonctionnellement essentielle et favorise un passage conformationnel de la kinase inactive à une kinase active stabilisée. Les résultats de la simulation sont soutenus indépendamment par des données de réponse médicamenteuse qui ont démontré les effets des inhibiteurs de la tyrosine kinase sur les lignées cellulaires cancéreuses du poumon ayant la mutation de suppression de ΔELREA et le type sauvage EGFR, où une plus grande inhibition par des médicaments reconnaissant la conformation active de kinase a été rapportée pour ΔELREA que pour le type sauvage EGFR12. Avec la mutation A702V, les simulations md indiquent, par rapport au type sauvage, une stabilisation accrue de l’interface kinase activateur-récepteur ainsi qu’une plus grande affinité de l’activateur et du récepteur kinase les uns pour les autres, soutenant ensemble le maintien de la conformation activée de l’EGFRAse kinase. La mutation A702V, située sur le segment Juxtamembrane B du récepteur kinase, augmenterait les interactions hydrophobes avec l’activateur kinase, fonctionnant pour prolonger la durée de l’état activé. La mutation A702V soutient la survie cellulaire en l’absence de facteur de croissance et a été identifiée dans un dépistage in vitro des mutations EGFR9.
Déclarations de divulgation
Les auteurs n’ont rien à révéler.
Remerciements
Cette recherche est financée par des subventions à M.S.J de l’Académie de Finlande (308317, 320005), à la Fondation Sigrid Juselius et au fonds commémoratif Tor, Joe et Pentti Borg, et à K.E. de l’Académie de Finlande (274728, 316796), à la Fondation du cancer de Finlande et à l’Hôpital central de l’Université de Turku. M.Z.T. est financé par le Réseau doctoral de biologie informationnelle et structurelle d’Åbo Akademi. Nous remercions le Csc IT Center for Science pour les ressources informatiques et le Dr Jukka Lehtonen pour le soutien informatique dans le cadre du réseau bioinformatique Biocenter Finland; et Biocenter Finlande réseau d’infrastructures de biologie structurelle.
matériels
| Name | Company | Catalog Number | Comments |
| Amber software | University of California, San Francisco | Version 2018 | Executable |
| Chimera program | Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco | Version 1.13.1 | Executable |
| EGFR struture files | The Protein Data Bank | 3D coordinates of EGFR structures | |
| Maestro | Schrödinger LLC | Version 2018-3 | Executable |
| Modeller program | The Andrej Šali Lab, Departments of Biopharmaceutical Sciences and Pharmaceutical Chemistry, University of California San Francisco | Included in the Chimera program | |
| VMD software | Theoretical and Computational Biophysics Group, University of Illinois at Urbana-Champaign | Version 1.9.3 | Executable |
Références
- Yarden, Y., Sliwkowski, M. X. Untangling the ErbB signalling network. Nature Reviews Molecular Cell Biology. 2, 127-137 (2001).
- Lemmon, M. A., Schlessinger, J., Ferguson, K. M. The EGFR family: not so prototypical receptor tyrosine kinases. Cold Spring Harbor Perspectives in Biology. 6, a020768 (2014).
- Arteaga, C. L., Engelman, J. A. ERBB receptors: From oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell. 2, 282-303 (2014).
- Mishra, R., Hanker, A. B., Garrett, J. T. Genomic alterations of ERBB receptors in cancer: Clinical implications. Oncotarget. 8, 114371-114392 (2017).
- . cBioPortal for Cancer Genomics Available from: https://www.cbioportal.org (2020)
- Lynch, T. J., et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. New England Journal of Medicine. 350 (21), 2129-2139 (2004).
- Paez, J. G., et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 304 (5676), 1497-1500 (2004).
- Pao, W., et al. EGF receptor gene mutations are common in lung cancers from "never smokers" and are associated with sensitivity of tumors to gefitinib and erlotinib. Proceedings of the National Academy of Sciences U.S.A. 101 (36), 13306-13311 (2004).
- Chakroborty, D., et al. Unbiased in vitro screen for activating EGFR mutations. Journal of Biological Chemistry. 294 (24), 9377-9389 (2019).
- Leahy, D. J. Structure and Function of the Epidermal Growth Factor (EGF/ErbB) Family of Receptors. Advances in Protein Chemistry. 68, 1-27 (2004).
- Roskoski, R. ErbB/HER protein-tyrosine kinases: Structures and small molecule inhibitors. Pharmacological Research. 87, 42-59 (2014).
- Tamirat, M. Z., Koivu, M., Elenius, K., Johnson, M. S. Structural characterization of EGFR exon 19 deletion mutation using molecular dynamics simulation. PLoS ONE. 14 (9), e0222814 (2019).
- Ding, L., et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 455, 1069-1075 (2008).
- Kurppa, K. J., Denessiouk, K., Johnson, M. S., Elenius, K. Activating somatic ERBB4 mutations in non small-cell lung cancer. Oncogene. 35 (10), 1283-1291 (2016).
- Soung, Y. H., et al. Somatic mutations of the ERBB4 kinase domain in human cancers. International Journal of Cancer. 118, 1426-1429 (2006).
- Tvorogov, D., et al. Somatic mutations of ERBB4: selective loss-of-function phenotype affecting signal transduction pathways in cancer. Journal of Biological Chemistry. 284, 5582-5591 (2009).
- Hubbard, S. R., Till, J. H. Protein tyrosine kinase structure and function. Annual Review of Biochemistry. 69 (1), 373-398 (2000).
- Huse, M., Kuriyan, J. The conformational plasticity of protein kinases. Cell. 109 (3), 275-282 (2002).
- Jura, N., et al. Catalytic control in the EGF receptor and its connection to general kinase regulatory mechanisms. Molecular Cell. 42, 9-22 (2011).
- Karplus, M., Kuriyan, M., J, Molecular dynamics and protein function. Proceedings of the National Academy of Sciences U.S.A. 102 (19), 6679-6685 (2005).
- Shan, Y., Arkhipov, A., Kim, E. T., Pan, A. C., Shaw, D. E. Transitions to catalytically inactive conformations in EGFR kinase. Proceedings of the National Academy of Sciences U.S.A. 110 (18), 7270-7275 (2013).
- Reckamp, K. L., et al. A phase I trial to determine the optimal biological dose of celecoxib when combined with erlotinib in advanced non-small cell lung cancer. Clinical Cancer Research. 12 (11 Pt 1), 3381-3388 (2006).
- Pettersen, E. F., et al. UCSF Chimera-a visualization system for exploratory research and analysis. Journal of Computational Chemistry. 25 (13), 1605-1612 (2004).
- Berman, H. M., et al. The Protein Data Bank. Nucleic Acids Research. 28 (1), 235-242 (2000).
- Zhang, X., Gureasko, J., Shen, K., Cole, P. A., Kuriyan, J. An Allosteric Mechanism for Activation of the Kinase Domain of Epidermal Growth Factor Receptor. Cell. 125 (6), 1137-1149 (2006).
- Stamos, J., Sliwkowski, M. X., Eigenbrot, C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. Journal of Biological Chemistry. 277 (48), 46265-46272 (2002).
- Sogabe, S., et al. Structure-Based Approach for the Discovery of Pyrrolo[3,2-d]pyrimidine-Based EGFR T790M/L858R Mutant Inhibitors. ACS Medicinal Chemistry Letters. 4 (2), 201-205 (2013).
- Sali, A., Blundell, T. L. Comparative protein modelling by satisfaction of spatial restraints. Journal of Molecular Biology. 234 (3), 779-815 (1993).
- Yun, C. H., et al. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell. 11 (3), 217-227 (2007).
- Park, J. H., Liu, Y., Lemmon, M. A., Radhakrishnan, R. Erlotinib binds both inactive and active conformations of the EGFR tyrosine kinase domain. Biochemical Journal. 448 (3), 417-423 (2012).
- . Release 2018-3: Maestro Available from: https://www.schrodinger.com/maestro (2018)
- Case, D. A., et al. . AMBER 2018. , (2018).
- Maier, J. A., Martinez, C., Kasavajhala, K., Wickstrom, L., Hauser, K. E., Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. Journal of Chemical Theory and Computation. 11 (8), 3696-3713 (2015).
- Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W., Klein, M. L. Comparison of simple potential functions for simulating liquid water. Journal of Chemical Physics. 79 (2), 926-935 (1983).
- Meagher, K. L., Redman, L. T., Carlson, H. A. Development of polyphosphate parameters for use with the AMBER force field. Journal of Computational Chemistry. 24 (9), 1016-1025 (2003).
- Humphrey, W., Dalke, A., Schulten, K. VMD: Visual molecular dynamics. Journal of Molecular Graphics. 14 (1), 33-38 (1996).
- Melvin, R. L., Salsbury, F. R. Visualizing ensembles in structural biology. Journal of Molecular Graphics and Modelling. 67, 44-53 (2016).
- Roe, D. R., Cheatham, T. E. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. Journal of Chemical Theory and Computation. 9 (7), 3084-3095 (2013).
- Miller, B. R., et al. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. Journal of Chemical Theory and Computation. 8 (9), 3314-3321 (2012).
- Guha, U., et al. Comparisons of tyrosine phosphorylated proteins in cells expressing lung cancer-specific alleles of EGFR and KRAS. Proceedings of the National Academy of Sciences U.S.A. 105 (37), 14112-14117 (2008).
- Furuyama, K., et al. Sensitivity and kinase activity of epidermal growth factor receptor (EGFR) exon 19 and others to EGFR-tyrosine kinase inhibitors. Cancer Science. 104 (5), 584-589 (2013).
- Qiu, C., et al. Mechanism of Activation and Inhibition of the HER4/ErbB4 Kinase. Structure. 6 (3), 460-467 (2008).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.