
Method Article
Decifrando os efeitos estruturais da ativação de mutações somáticas do EGFR com simulação de dinâmica molecular
Neste Artigo
Resumo
O objetivo deste protocolo é usar simulações de dinâmica molecular para examinar as mudanças estruturais dinâmicas que ocorrem devido à ativação de mutações da proteína quinase EGFR.
Resumo
Numerosas mutações somáticas que ocorrem na família do receptor de fator de crescimento epidérmico (EGFR) da quinase receptora tyrosina (RTK) foram relatadas de pacientes com câncer, embora relativamente poucas tenham sido testadas e mostradas para causar alterações funcionais nos ErbBs. Os receptores ErbB são dimerizados e ativados na ligação de ligantes, e mudanças conformacionais dinâmicas dos receptores são inerentes à indução da sinalização a jusante. Para duas mutações mostradas experimentalmente para alterar a função EGFR, A702V e a mutação de exclusão Δ746ELREA750, ilustramos no protocolo a seguir como simulações de dinâmica molecular (MD) podem sondar a (1) estabilidade conformacional da estrutura mutante de quinase tyrosina em comparação com o EGFR tipo selvagem; (2) consequências estruturais e transições conformais e sua relação com as alterações funcionais observadas; (3) efeitos das mutações sobre a força da VINCULação ATP, bem como para a vinculação entre os domínios da quinase no dimer assimétrico ativado; e (4) efeitos das mutações nas interações-chave dentro do site de ligação EGFR associados à enzima ativada. O protocolo fornece um procedimento passo a passo detalhado, bem como orientação que pode ser mais geralmente útil para a investigação de estruturas proteicas usando simulações de MD como um meio de sondar a dinâmica estrutural e a relação com a função biológica.
Introdução
A família do receptor de fator de crescimento epidérmico humano (EGFR) (ErbB) de quinases receptoras de tyrosina (RTKs) inclui quatro membros - EGFR/ErbB1/HER1, ErbB2/HER2, ErbB3/HER3 e ErbB4/HER4. Os receptores ErbB regulam processos celulares fundamentais, como crescimento e proliferação celular, diferenciação, migração e sobrevivência1,2, e são, portanto, proto-oncogenes potentes. A atividade aberrante dos receptores ErbB, especialmente EGFR e ErbB2, tem sido frequentemente associada a cânceres humanos fazendo dos receptores ErbB alvos-chave para a terapêutica do câncer2,,3.
Várias alterações somáticas dos genes ERBB foram relatadas a partir de malignidades humanas3,,4,5. Os melhores exemplos caracterizados incluem as mutações de ponto recorrentes, ativando pontos e exclusões de curto quadro no domínio EGFR quinaase em câncer de pulmão de células não pequenas (NSCLC). Essas mutações EGFR representam os principais fatores de crescimento do câncer e prevêem sensibilidade ao EGFR direcionado aos medicamentos contra o câncer6,,7,,8. No entanto, na maioria dos cânceres, mutações somáticas no EGFR ocorrem fora desses "hotspots" recorrentes e são distribuídas ao longo de todo o período de 1210 resíduos do receptor. De fato, a maioria dos resíduos ao longo da sequência primária do EGFR foram encontrados como mutados no câncer humano9. No entanto, além dos poucos pontos quentes, a significância funcional da grande maioria das mutações EGFR associadas ao câncer permanece desconhecida.
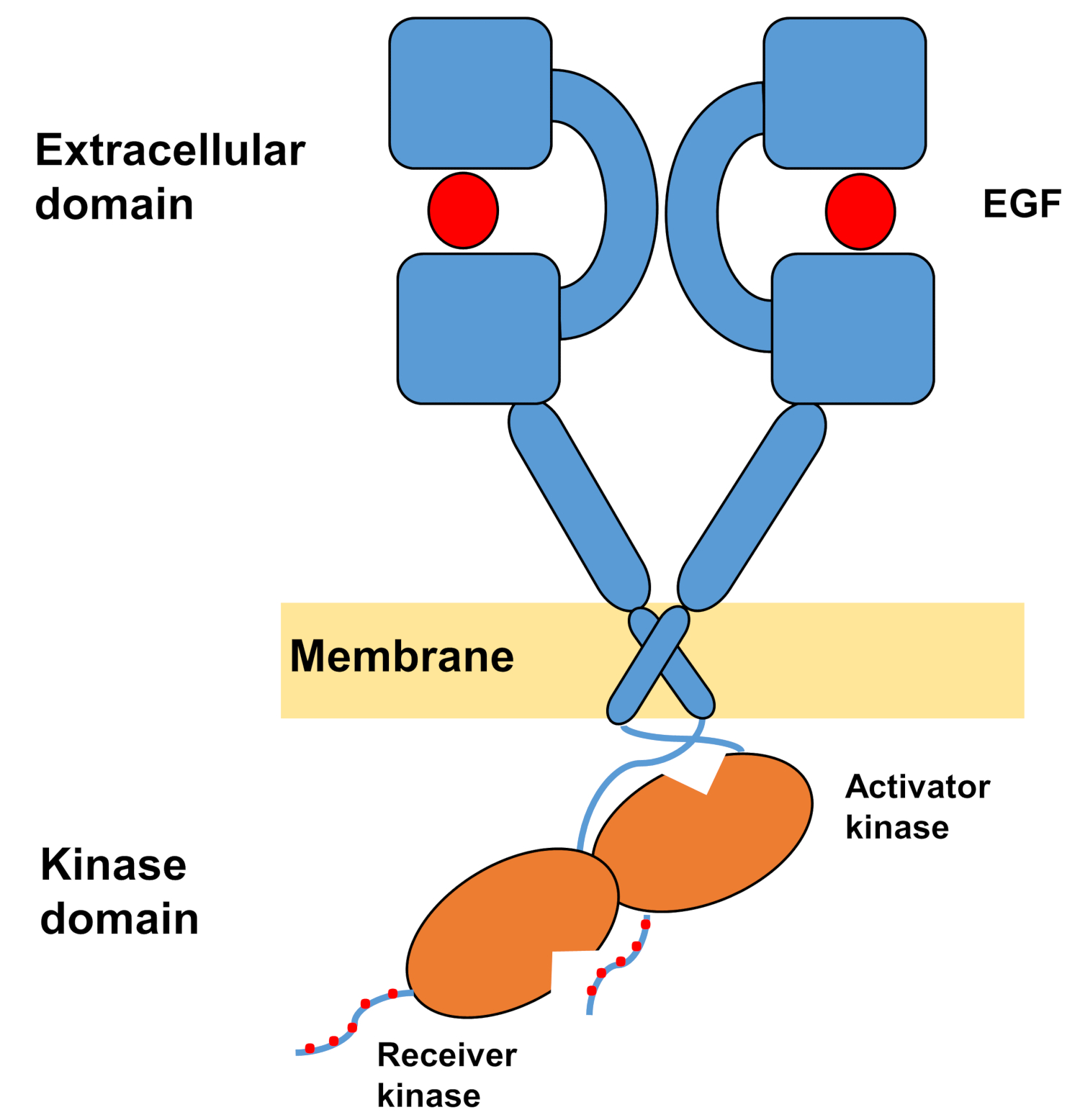
A estrutura monomérica dos ErbBs consiste em um grande domínio extracelular terminal amino, seguido por uma única hélice transmembrana que leva ao domínio de tyrosina quinase intracelular e região de cauda terminal C que contém locais de acoplamento para proteínas de sinalização intracelular. A ligação ligante desencadeia uma mudança conformacional dramática no domínio extracelular, o que facilita a formação de dimers receptores expondo os braços de dimerização que se cruzam simetricamente uns sobre os outros e interagem com suas superfícies aromáticas/hidrofóbicas. Após a formação do receptor dimer, os domínios de tyrosina quinase entram em contato assimetricamente(Figura 1),resultando na ativação das quinases que fosforilam as caudas terminais C dos monômeros receptores e, posteriormente, na ativação da sinalização a jusante10,,11.

Figura 1: Estrutura do dimer EGFR. O EGFR escurece quando os domínios extracelulares vinculam o fator de crescimento (EGF, fator de crescimento epidérmico). O domínio da quinase receptora é então ativado através da interação assimétrica com o domínio da quinase ativadora, e as caudas do terminal C são autofosforiladas em resíduos de tyrosina (Modificadas de Tamirat et al.12). Clique aqui para ver uma versão maior desta figura.
{kind=link}
Devido aos rearranjos estruturais dinâmicos que ocorrem durante as transições de  monómero, juntamente com a ativação da quinase que está associada à formação de um dimer assimétrico, mutações ao longo do comprimento total da estrutura receptora podem potencialmente ter um efeito sobre a função receptora. Aqui descrevemos vários exemplos de nossos estudos anteriores em que a modelagem da mutação e visualização foram suficientes para explicar as consequências para a função.
monómero, juntamente com a ativação da quinase que está associada à formação de um dimer assimétrico, mutações ao longo do comprimento total da estrutura receptora podem potencialmente ter um efeito sobre a função receptora. Aqui descrevemos vários exemplos de nossos estudos anteriores em que a modelagem da mutação e visualização foram suficientes para explicar as consequências para a função.
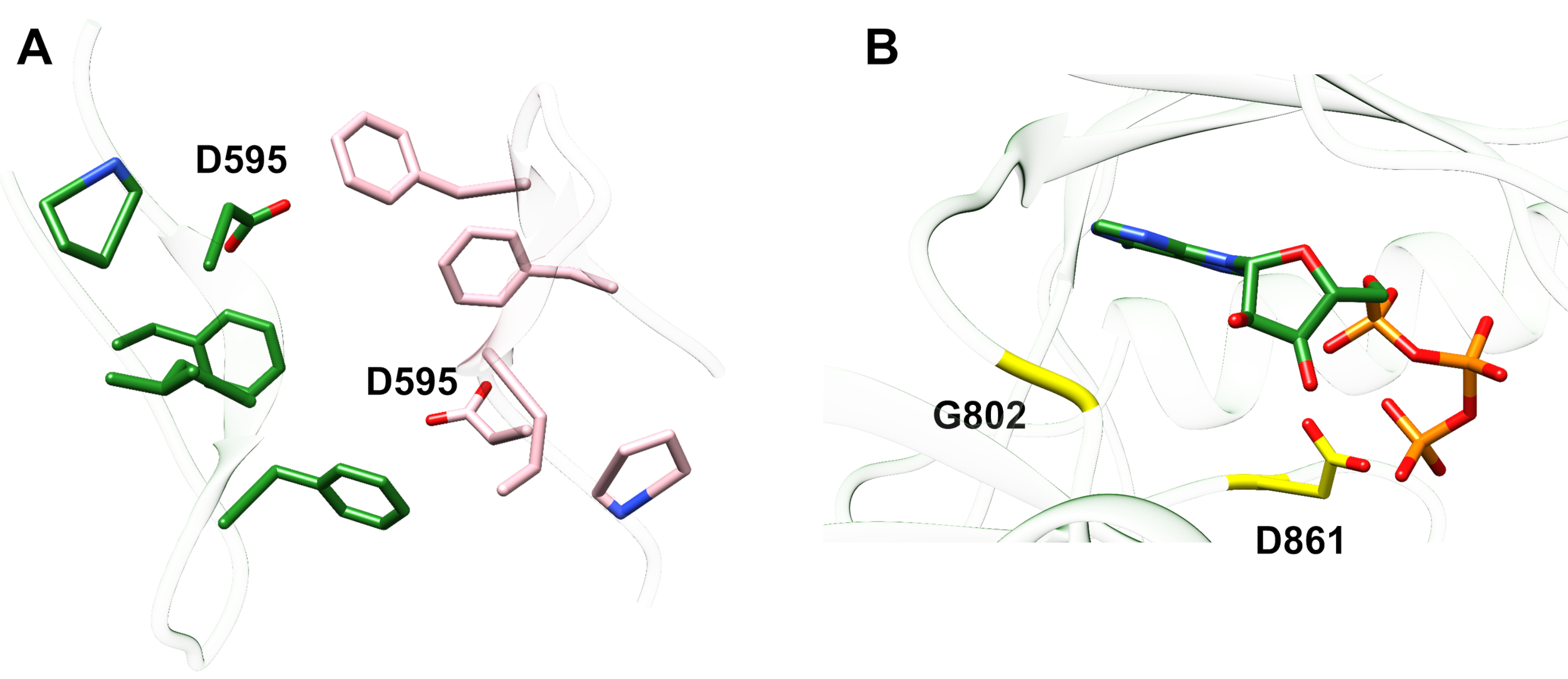
Exemplo 1: Uma mutação relatada, D595V em ErbB413, levou ao aumento da dimerização e fosforilação ErbB4 e fosforilação14. A visualização da localização da mutação foi um fator crítico na compreensão dos efeitos funcionais observados: O D595V ocorreu no cruzamento simétrico dos braços diméricos da ectodomína (Figura 2A). Os braços são em grande parte aromáticos e hidrofóbicos, e a substituição do ácido aspartico polar por valina seria esperada para aumentar as interações hidrofóbicas "pegajosas", estabilizando o mais fraco e, portanto, aumentar o tempo quando a fosforilação ocorre14. Foi uma surpresa no início encontrar aspartato em cada braço, mas em retrospectiva pode-se pensar nisso como um mecanismo de tempo para a atividade, onde as cadeias laterais de ácido polar reduzem a afinidade e a vida útil do dimer intacto e, portanto, limitam a fosforilação e sinalização mediadas pela quinase. A substituição por valina removeria essa salvaguarda estabilizando ainda mais o estonteante ErbB4.

Figura 2: Localização de uma mutação ativada ErbB4 e mutações produzindo ErbB4 morto pela quinase. (A) D595 (ativando a mutação D595V) está localizado nos braços diméricos aromáticos/hidrofóbicos do modelo ectodomínio ErbB4; os braços associam-se à vinculação do fator de crescimento; (resíduos próximos são mostrados como varas). (B) Em ErbB4, g802 (inativação da mutação G802dup) ajuda a formar o bolso de ligação em torno do anel de adenina de ATP e d861 catalítico (mutação D861Y inativante) liga tanto Mg2+ (não mostrado) quanto o grupo γ-fosfato de ATP. Clique aqui para ver uma versão maior desta figura.
{kind=link}
Exemplo 2: Pode-se prever que mutações somáticas que visam o local de vinculação ATP do domínio quinase alterariam ou eliminariam a atividade enzimática levando a um receptor prejudicado ou morto pela quinase incapaz de sinalizar. Das nove mutações relatadas de pacientes com mama, gástrico, colorretal ou NSCLC15, duas das nove mutações quando testadas apresentaram atividade de fosforilação altamente diminuída16: G802dup (G → GG) e D861Y. Ambas as mutações somáticas inativantes foram encontradas dentro do local de ligação ATP da estrutura de domínio de tyrosine quinase (Figura 2B): glicina flexível, duplicada, alteraria o local do anel de adenina e pequeno ácido aspânico substituído pela tyrosina volumosa perto dos fosfatos terminais impediria fisicamente mg2+-ATP de se amarrar. No entanto, uma vez que o ErbB4 pode formar um heterodimer com ErbB2 - ErbB2 não vincula um fator de crescimento e depende da associação com um ErbB que faz a fim de heterodimerizar - o ErbB2 (ativo)-E Heterodimer (quinase-morto) estimularia a proliferação celular através da via de sinalização Erk/Akt, mas as células não se diferenciariam devido à kinase-morta ErbB4 e à falta de ativação da viaSTAT5 16.
Em estudos mais recentes, tornou-se evidente que os movimentos dinâmicos dos ErbBs eram relevantes para entender os efeitos de alguns mutantes na função ErbB, especialmente mutações que ocorrem dentro do domínio da tyrosina quinase. O domínio de tyrosine quinase consiste em um N-lobe (principalmente β-folhas) e C-lobe (em grande parte alfa helicoso), que são separados pelo local catalítico onde o ATP se liga. O N-lobe inclui a hélice αC e o loop P, enquanto a ativação (A-loop) e os loops catalíticos estão presentes no C-lobe17,,18,,19. Estruturas cristalinas do domínio da quinase de tirasina revelaram duas conformações inativas, a maioria das estruturas tem o estado inativo semelhante ao SRC. Na conformação ativa, o aspartato catalítico do loop A aponta para o local de ligação ATP e a hélice αC é orientada para o bolso de ligação ATP ("αC-in" conformação), formando uma forte interação glutamato-ion-par de íons-ion-ion.
Como os ErbBs e o domínio da quinase componente são entidades altamente dinâmicas, e especialmente para os casos em que os efeitos das mutações na função e na atividade biológica provavelmente estão fortemente ligados aos estados conformacionais dos ErbBs, é importante avaliar mutações em relação ao alcance das mudanças dinâmicas que experimentariam. Estruturas de cristal de raios-X dos ErbBs fornecem instantâneos estáticos da estrutura 3D, que podem ou não ser relevantes para entender as consequências dinâmicas de uma mutação. Para sondar o alcance de mudanças dinâmicas correspondentes à "paisagem energética" disponível para uma estrutura tridimensional (3D), simulações de dinâmica molecular (MD) são amplamente utilizadas20. No caso de mutações que levariam a alterações conformais locais dentro do domínio de quinase tyrosina ou estabilização de um complexo, simulações na ordem de 100 ns podem ser suficientes. No entanto, mudanças conformais em escala maior (por exemplo, transições entre as conformações ativas e inativas do domínio da quinase) requerem um tempo de simulação mais longo - na ordem dos microsegundos21.
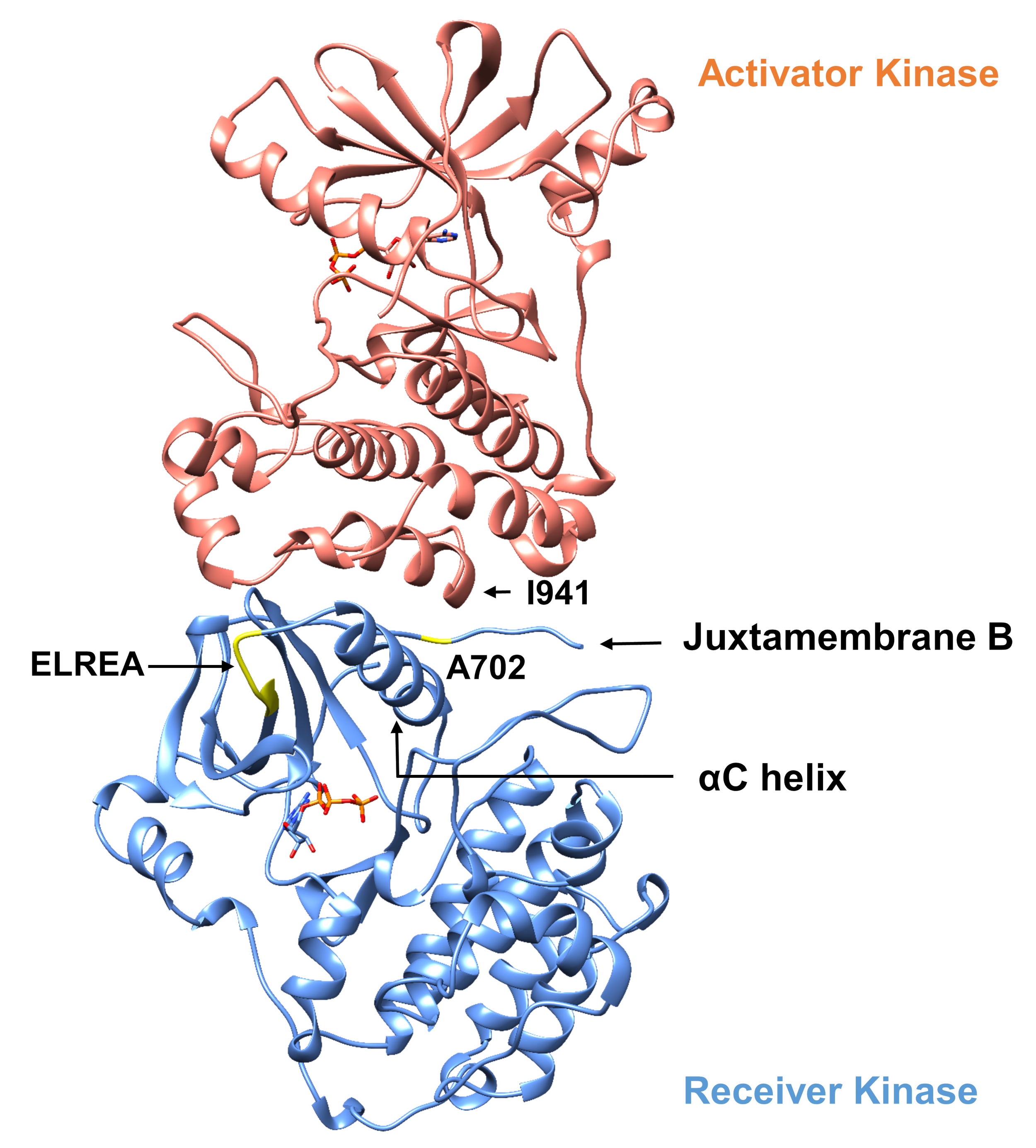
Com relação ao protocolo descrito abaixo consideramos duas mutações ativantes dentro do domínio tyrosine quinase(Figura 3). Ambas as mutações estão localizadas dentro do domínio da quinase em locais que experimentam alterações conformais locais que ditam se a quinase está ativa ou não, e, portanto, simulações de MD foram aplicadas em ambas as instâncias. No primeiro caso, consideramos alterações que afetam diretamente o site de vinculação ATP e o maquinário catalítico do domínio quinase receptor EGFR, examinando especificamente as consequências de uma mutação de exclusão exon 19 que está amplamente implicada no NSCLC4,7. A mutação Δ746ELREA750, que reduz o comprimento do loop β3-αC anterior à hélice αC - a hélice que se move em direção ao local de ligação/ativo na ativação da quinase e participa na formação da interação eletrostática crítica entre E762 da hélice e K745, posicionando a lise para interação com ATP - predispõe o domínio para ativação12. No segundo caso, consideramos a mutação A702V do EGFR, mostrada como uma nova mutação ativadora de ganho de função revelada pela plataforma iScream9 e identificada em um paciente NSCLC22. Alanine-702 no domínio quinase receptor está localizada no segmento B da justaxtamembrana na interface do receptor e dos domínios de quinase ativadora, nos quais este complexo er dimer assimétrico de quinase e alterações conformais de quinase são necessárias para a ativação9.

Figura 3: O dimer de domínio assimétrico da quinase do EGFR. A mutação A702V estaria localizada na interface crítica dos domínios ativador e quinase receptora, adjacente à hélice αC e perto da isoleucina 941 do ativador quinase. Alterações conformais induzidas pela formação do dimer assimétrico levam à ativação da quinase. O loop β3-αC contendo a sequência ELREA precede diretamente a hélice αC; durante a ativação, a hélice αC move-se para dentro em direção ao local de ligação ATP. Clique aqui para ver uma versão maior desta figura.
{kind=link}
Protocolo
NOTA: As medidas detalhadas tomadas para examinar os efeitos da mutação ΔELREA e A702V na estrutura EGFR usando simulações de MD são discutidas da seguinte forma:
1. Preparação da estrutura
NOTA: Para estudar os impactos estruturais da mutação ΔELREA, as formas de apo ativos, atp-bound ativo e apo inativo e apo inativas estruturas monômeros EGFR são preparadas da seguinte forma.
- Abra o programa de visualização Quimera23 (https://www.cgl.ucsf.edu/chimera/)para preparar a estrutura de quinase Apo ativa do tipo selvagem EGFR. No menu Arquivo clique na opção Buscar por ID e selecione o banco de dados Do Banco de Dados de Proteínas (PDB24) e especifique o código PDB 2GS225 (resolução 2.8 Å). O PDB é um repositório para estruturas 3D resolvido por diferentes técnicas experimentais, incluindo cristalografia de raios-X, espectroscopia de ressonância magnética nuclear, microscopia crio-elétron e difração de nêutrons.
- Construa os elementos estruturais ausentes do 2GS2, tirando esses segmentos das estruturas EGFR PDB 1M1426 (2.6 Å) e 3W2S27 (1,9 Å). Para isso, abra 1M14 e 3W2S e sobreponha-os no 2GS2 usando a opção MatchMaker no menu de comparação Ferramentas → Estrutura.
- Crop out os segmentos a serem adicionados a partir de 1M14 e 3W2S. Selecione os átomos terminais do resíduo antes das lacunas em 2GS2 e os átomos a serem adicionados a partir de 1M14 e 3W2S (para obter informações detalhadas sobre os segmentos adicionados, consulte Tabela 1). Na linha de comando tipo bond sel e hit enter. Esta estrutura é a estrutura do modelo.
- Use a estrutura do modelo a partir da etapa 1.2 para construir a forma mutante de exclusão ΔELREA do domínio de quinase EGFR. Produza a sequência de formato FASTA de ΔELREA EGFR salvando a sequência da estrutura de modelo (Favorite → Sequence → File → salvar como) e, em seguida, excluindo a sequência ELREA em números de resíduo 746-750.
- Abra a sequência ΔELREA EGFR na Quimera e alinhe-a com a sequência da estrutura do modelo 2GS2 usando o menu Sequência. Na janela de alinhamento selecione a opção Estrutura → Modelador (28).28
- Na janela pop-up, especifique a estrutura composta 2GS2 como o modelo e a sequência mutante como a consulta a ser modelada. Em seguida, pressione OK. Selecione um modelo mutante entre os modelos resultantes, com base na pontuação zDOPE (tipicamente a pontuação mais baixa) e inspeção visual.
- Para preparar a estrutura de quinase EGFR de tipo selvagem ligada ao ATP, use a estrutura PDB 2ITX29 (2.98 Å) como estrutura principal. Construa segmentos ausentes (ver Tabela 1) utilizando as estruturas 2GS625 (2.6 Å) e 3W2S seguindo o procedimento na etapa 1.2. Converta o ligante ANP na estrutura resultante para ATP abrindo o arquivo PDB em um editor de texto e alterando o átomo de nitrogênio N3B de ANP para um átomo de oxigênio.
- Abra a estrutura na etapa 1.4 em Quimera, conforme indicado na etapa 1.1. Adicione um íon de magnésio a esta estrutura da estrutura PDB 2ITN29 (2.47 Å) a fim de alcançar um posicionamento semelhante para o íon Mg2+.
- Com a estrutura resultante da etapa 1.5, modele a forma mutante ΔELREA seguindo o passo 1.3.
| Apo ativo EGFR | Apo inativo EGFR | EGFR ativo vinculado à ATP | |
| Estrutura principal | 2GS2 | 2GS7 | 2ITX |
| Estruturas usadas para construir laços perdidos | 1M14 (723-725) | 3W2S (958-984) | 2GS6 (862-865) |
| 3W2S (967-981) | 4HJO (848-850) | 3W2S (990-1001) |
Tabela 1: Estruturas utilizadas para construir modelos compostos de estruturas ativas apo ativas, apo inativas e ligadas a ATP. Foram construídas regiões ausentes (aminoácidos entre parênteses) na estrutura principal.
- Para preparar a estrutura de quinase Apo inativa EGFR, abra a estrutura PDB 2GS725 (2.6 Å) como na etapa 1.1 e exclua os ligantes ligados e as águas cristalográficas. Adicione os segmentos ausentes em 2GS7 (ver Tabela 1) das estruturas 3W2S e 4HJO30 (2,75 Å) utilizando o procedimento na etapa 1.2. Com base na estrutura final inativa do EGFR, prepare o modelo mutante utilizando o procedimento na etapa 1.3.
NOTA: Para o estudo da mutação A702V, a estrutura assimétrica do dimer EGFR é estudada à medida que a mutação está localizada no segmento de justxtamembrano B do domínio da quinase que forma grande parte da interface mais fraca. As estruturas EGFR mutantes do tipo selvagem e A702V são preparadas da seguinte forma: - A estrutura assimétrica do tipo selvagem é construída a partir da estrutura PDB 2GS2, que inicialmente é exibida na forma monomérica. Para converter para o conjunto biológico que contém o ativador e o receptor kinases no arranjo assimétrico, abra 2GS2 na Quimera como em (1.1) e realize cálculos de simetria clicando nas ferramentas → estrutura de ordem superior → menu Unit Cell. Selecione a estrutura 2GS2 e digite Fazer cópias. Finalmente, selecione e salve um único dimer assimétrico das múltiplas cópias do dimer resultante das operações de simetria.
- Usando a estrutura EGFR assimétrica do tipo selvagem a partir da etapa 1.8, construa o mutante A702V substituindo alanina 702 por valina usando a opção de edição de ferramentas → Estrutura → Rotamers na Quimera.
NOTA: Coletivamente, seis estruturas monoméricas e duas EGFR dimeric são preparadas para os estudos de mutação ΔELREA e A702V, respectivamente. Cada estrutura é posteriormente processada para simulação usando o assistente de preparação de proteínas no programa Maestro31 e simulações de MD são feitas com o programa Âmbar32. - Abra a estrutura no Maestro usando a opção arquivo → estrutura de importação. Em seguida, clique no botão do assistente de preparação de proteínas e selecione o seguinte: adicione átomos de hidrogênio, construa átomos de cadeia lateral ausentes, determine estados de protonação de resíduos ionizáveis no pH 7.0 usando PROPKA, otimize a orientação de resíduos de asparagina, glutamina e histidina para ligação de hidrogênio, e finalmente minimize a estrutura.
2. Configuração do sistema
- Abra o programa de salto incluído no pacote de software Amber. Importe as moléculas de água ff14SB33 (fonte leaprc.protein.ff14SB) e tip3P moléculas de água34 (fonte leaprc.water.tip3p). Para os sistemas vinculados à ATP também importam parâmetros para ATP35 (loadamberparams frcmod.fosphate, loadamberprep ATP.prep). Em seguida, carregue a estrutura(mol = estrutura loadpdb.pdb).
- Solvate a estrutura em uma caixa octaédral com moléculas explícitas de água TIP3P que se estende 10 Å em todas as direções a partir dos átomos superficiais da proteína(solvateoct mol TIP3PBOX 10.0).
- Verifique o sistema construído (Verifique mol) e neutralize-o adicionando íons necessários (adendos mol Na+ 0). Para modelar suficientemente sistemas biomoleculares, adicione átomos adicionais na+/Cl- à caixa de simulação para levar a concentração de sal do sistema para 0,15 M(adendos mol Na+ X, adendos mol Cl-X),onde X é substituído pelo resultado de: concentração de sal desejada * número de moléculas de água * volume por molécula de água * número de Avogadro.
- Gerar e salvar os arquivos de topologia e coordenação do sistema, que servem como entradas para a simulação de produção subsequente (saveamberparm mol X.prmtop X.inpcrd).
3. Simulação de dinâmica molecular
- Usando âmbar, inicialmente submete o sistema de simulação a 5000 ciclos de descida mais íngreme e minimização de energia de gradiente conjugado para contornar configurações desfavoráveis. Realizar a minimização em múltiplas etapas, reduzindo gradualmente a contenção aplicada em átomos solutos de 25 kcal mol-1 Å-2 a 0 kcal mol-1 Å-2.
- No arquivo de entrada de minimização, min.in, ajuste a variável maxcyc para o ciclo de minimização total (maxcyc = 5000) e ncyc para afirmar o número de ciclos para o algoritmo de descida mais íngreme. Use a variável restraint_wt para aplicar a força de contenção em átomos solutos especificados pelo parâmetro de máscara de contenção. Em seguida, execute a minimização da seguinte forma:
$AMBERHOME/bin/sander -O -i min.in -o min.out -p X.prmtop -c X.inpcrd -r min.rst -ref X.inpcrd
NOTA: A estratégia e os parâmetros reais utilizados podem variar de acordo com as próprias preferências. Detalhes e orientações podem ser encontrados no manual e site amber(https://ambermd.org/index.php)
- No arquivo de entrada de minimização, min.in, ajuste a variável maxcyc para o ciclo de minimização total (maxcyc = 5000) e ncyc para afirmar o número de ciclos para o algoritmo de descida mais íngreme. Use a variável restraint_wt para aplicar a força de contenção em átomos solutos especificados pelo parâmetro de máscara de contenção. Em seguida, execute a minimização da seguinte forma:
- Aqueça o sistema por 100 ps de 0 K a 300 K estabelecendo uma contenção de 10 kcal mol-1 Å-2 em átomos solutos. Para isso, defina tempi = 0,0, temp0 = 300,0, dt = 0,002 ps, nstlim = 50000 e restraint_wt = 10 no arquivo de entrada heat.in. Realize o aquecimento com o seguinte comando:
$AMBERHOME/bin/sander -O -i heat.in -o heat.out -p X.prmtop -c min.rst -r heat.rst -x heat.mdcrd -ref min.rst - Equilibre o sistema para 900 ps sob um conjunto NPT; número constante de átomos, temperatura(temperatura 300,0) e pressão(ntp = 1),controlando-o com o método Berendsen(ntt = 1). Defina um corte de distância de 9 Å (corte = 9,0) para interações eletrostáticas de longo alcance. Reduza gradualmente a contenção do átomo soluto para 0,1 kcal mol-1 Å-2 (restraint_wt = 0,1). Execute o arquivo de entrada de equilíbrio equil.in que descreve os parâmetros acima da seguinte forma:
$AMBERHOME/bin/sander -O -i equil.in -o equil.out -p X.prmtop -c heat.rst -r equil.rst -x equil.mdcrd -ref heat.rst - Finalize o equilíbrio com uma simulação de 5 ns sem restrições (conjunto dt = 0,002 ps, ntslim = 25000000).
$AMBERHOME/bin/sander -O -i equil_final.in -o equil_final.out -p X.prmtop -c equil.rst -r equil_final.rst -x equil_final.mdcrd -equil.rst - Verifique se o sistema se equilibrou examinando os valores de temperatura, pressão, densidade e energia.
$AMBERHOME/bin/process_mdout.perl heat.out equil.out equil_final.out
xmgrace resumo. TEMP/DENSIDADE/ETOT/EPTOT/EKTOT - Realizar a simulação de produção para 100 ns (conjunto dt = 0,002 ps, ntslim = 500000000 em prod.in) e salvar conformações a cada 10 ps(ntwx = 5000).
$AMBERHOME/bin/sander -O -i prod.in -o prod.out -p X.prmtop -c equil_final.rst -r prod.rst -x prod.mdrcd -ref equil_final.rst
4. Análise
- Inspeção visual
- Visualize as conformações amostradas durante as simulações de quinase EGFR do tipo selvagem e mutante, abrindo os arquivos de topologia âmbar X.prmtop e os arquivos de trajetória prod.mdcrd correspondentes em VMD36. Utilizando representações convenientes de estrutura secundária, analise a dinâmica estrutural geral das proteínas a partir da trajetória registrada. Veja interações específicas entre átomos/resíduos de interesse, como a catalítica essencial ponte de sal K745 - E762.
- Alternativamente, salve várias conformações amostradas durante a simulação no formato PDB e abra-as usando o programa Quimera. Sobreponha as estruturas na estrutura inicial ou mediana usando a opção MatchMaker. Exibir a estrutura inicial/mediana em sólido e o resto das estruturas alinhadas em branco desbotado. Essa abordagem permite visualizar os movimentos estruturais registrados com mais clareza.
NOTA: Sugestões para representações efetivas e processamento de conjuntos conformais de MDS podem ser encontradas em Melvin et al.37.
- Análise de RMSD e RMSF
- Calcular o desvio quadrado médio (RMSD) e os cálculos de flutuação quadrada média-raiz (RMSF) com o programa Cpptraj 38 para analisar a estabilidade global das proteínas e examinar a flexibilidade das diferentes unidades estruturais. Nos rmsd.in e rmsf.in arquivos de entrada, indique os átomos de espinha dorsal (para RMSD) e os átomos de Cα (para RMSF) da estrutura inicial como referência para o encaixe RMS. Nos arquivos rmsd/rmsf.in importa os arquivos de topologia âmbar(parm X.promtop) e os arquivos de trajetória correspondentes(trajin prod.mdcrd). Em seguida, execute o comando Cpptraj -i rmsd/rmsf.in. Plote os dados de saída para análise.
- Alternativamente, alinhe conjuntos conformais e colora cada resíduo com base no átomo de Cα RMSD. Para isso, abra as conformações na Quimera e alinhe-as com a opção Matchmaker.
- Vá para Ferramentas → Representação → Render render por atributo. Selecione Resíduos do conjunto conformacional e Cα RMSD como atributos e clique em OK. O traço de corrente das conformações será então colorido de azul → branco → vermelho, respectivamente refletindo regiões de alta, média e baixa estabilidade estrutural.
- Análise de ligação de hidrogênio
- Analise a interação de ligação de hidrogênio entre ATP e EGFRs tipo selvagem/ΔELREA. Prepare um script cpptraj, hbond.in,para realizar essa tarefa. Defina uma ligação de hidrogênio com uma distância aceitadora de doadores menor ou igual a 3,5 Å e um ângulo de ligação maior ou igual a 135°. Especifique a análise apenas para ligações de hidrogênio intermolecular com a variável nointramol, ou seja, ligações de hidrogênio entre ATP e EGFR (hbond All nointramol dist 3.5 out nhb.agr avgout avghb.dat). Execute o script como Cpptraj -i hbond.in.
- Use este script para avaliar interações intramoleculares, por exemplo, entre os resíduos K745 e E762, que são resíduos-chave para a atividade de quinase EGFR. Para isso, especifique o K745 como o doador de títulos de hidrogênio e e762 como o aceitador de ligação de hidrogênio em hbond.in e execute o script em conformidade.
- Distância de monitoramento entre átomos
- Meça a distância entre K745 e E762 abrindo as trajetórias de EGFRs de apo de tipo selvagem e ΔELREA em VMD. Selecione o Cδ de Glu762 e Nz de Lys745 clicando em mouse → rótulo → bond. Monitore a distância durante a simulação plotando um gráfico com etiquetas de → gráficos → gráfico de → de ligação.
- Cálculos de energia grátis
- Para calcular as energias livres de ligação estimadas entre ATP e EGFRs tipo selvagem/ΔELREA, e entre o ativador e o receptor de EGFRs tipo selvagem/A702V, use o módulo39 de superfície de born (MM-GBSA) de mecânica molecular. Definir ATP como o ligante e EGFR como o receptor no estudo ΔELREA. No estudo A702V, especifique a quinase receptora como o ligante e o ativador quinase como receptor.
- Primeiro prepare os arquivos PDB complexos de ligante, receptor e receptor de ligante separadamente no programa de salto que define o valor PBRadii para mbondi2. Para os arquivos PDB, salve os arquivos de topologia âmbar da fase de gás (.prmtop) e coordene (.inpcrd).
- Em seguida, no arquivo de entrada mmgbsa, mmgbsa.in, definir igb = 2, saltcon = 0,1. Execute cálculos de energia vinculativos usando as trajetórias das simulações, os arquivos âmbar receptor/ligante preparados e os parâmetros no mmgbsa.in com o script MMPBSA.py disponível em Âmbar da seguinte forma:
$AMBERHOME/bin/MMPBSA.py -O -i mmgbsa.in -o mmgbsa.dat -sp X.prmtop -cp complex.prmtop -rp receptor.prmtop -lp ligand.prmtop -y prod.mdcrd -eo output.csv - Analise os dados de saída, output.csv,plotando gráficos.
Resultados
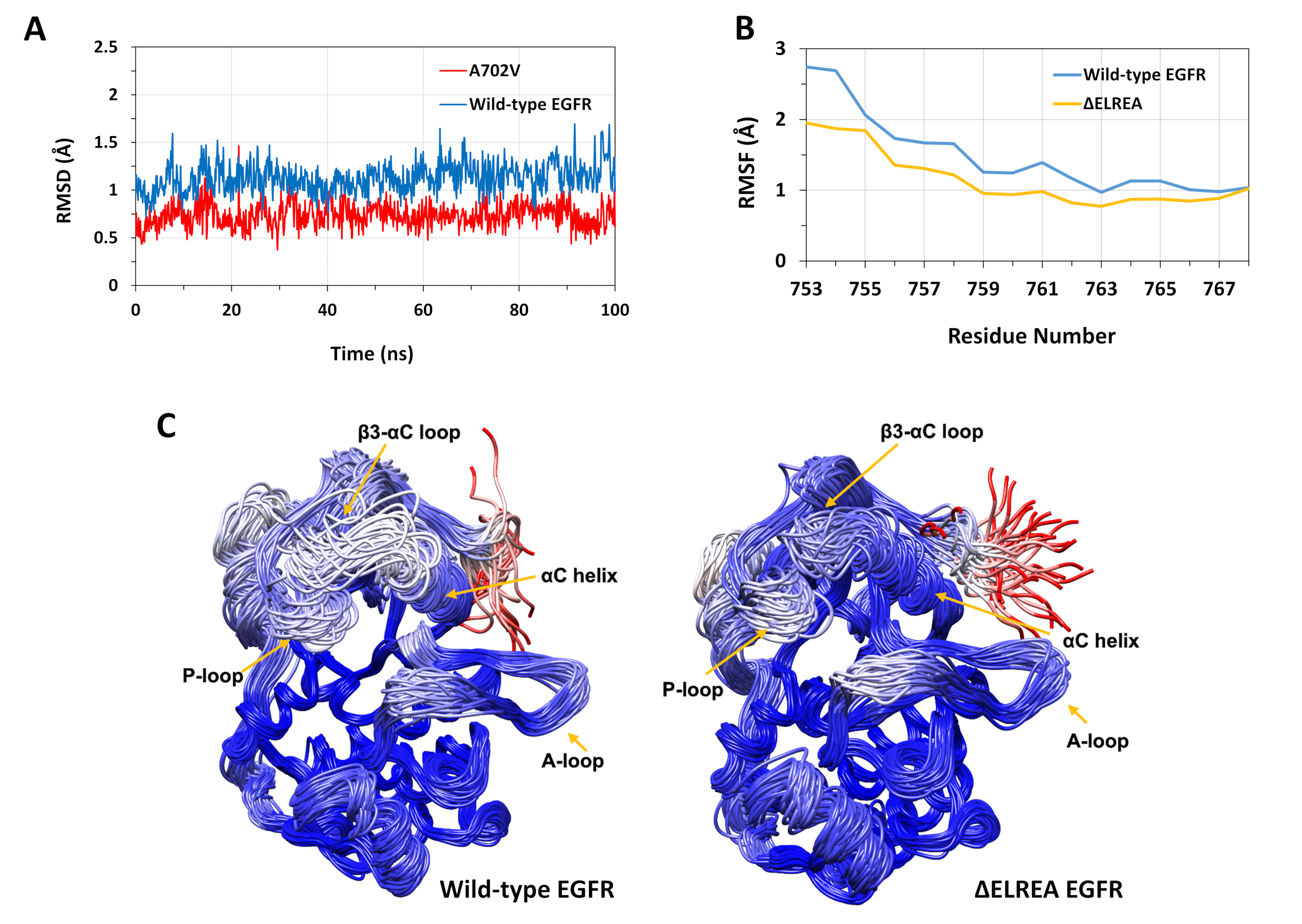
O protocolo descrito foi utilizado para estudar os efeitos estruturais das mutações ΔELREA e A702V na estrutura de quinase EGFR. Uma aplicação do protocolo foi investigar o efeito das mutações na estabilidade estrutural/conformacional local, computando os valores de RMSD e RMSF das simulações de MD. Como a mutação A702V está localizada no segmento B justotamembrane, o RMSD deste segmento da quinase receptora em relação à estrutura inicial foi calculado tanto para os EGFRs do tipo selvagem quanto para a A702V. O resultado (Figura 4A) revelou que o segmento de juxtramembrano B do mutante aumentou a estabilidade conformacional durante a simulação de 100 ns (RMSD médio 0,7 Å - intervalo de confiança de 95% (IC) 0,009) em comparação com o domínio de quinase EGFR tipo selvagem (RMSD médio 1,1 Å - 95% CI 0,01). Isso é muito provavelmente o resultado das interações hidrofóbicas mais apertadas na interface dimer devido à substituição da alanina 702 (cadeia lateral do grupo metil) por um resíduo hidrofóbico mais volumoso, valina (cadeia lateral do grupo isopropílico), levando ao aumento das interações hidrofóbicas do V702 no domínio da quinase receptora com isoleucina 941 do domínio ativador kinase.
A mutação ΔELREA está localizada no loop β3-αC, adjacente à hélice αC funcionalmente crítica; a conformação da hélice αC é fundamental para mudanças entre os estados ativos e inativos da quinase EGFR. A estabilidade conformacional da hélice αC no estado ativo foi avaliada examinando o RMSF sobre os átomos de Cα de resíduos dentro da hélice durante simulações de MD(Figura 4B): no geral, há flutuações mais baixas no mutante (RMSF médio 1,1 Å - 95% IC 0,4) em comparação com o tipo selvagem (RMSF médio 1,5 Å - 95% CI 0,57); com a maior diferença de flutuações registradas para os resíduos do terminal N. As conformações amostradas, respectivamente, sobrepostas à estrutura mediana do domínio quinase do tipo selvagem e do domínio ΔELREA quinaase também suportam esses resultados (Figura 4C): tanto os domínios de quinase selvagem quanto ΔELREA têm estabilidade global semelhante para as conformações sobrepostas, exceto pelo loop β3-αC e a hélice αC, que são claramente mais estáveis em ΔELREA EGFR. Esses achados indicam que a exclusão da sequência elrea restringe o movimento da hélice αC do estado ativo, restringindo e, assim, estabilizando a conformação ativa. Além disso, uma vez que a hélice αC faz parte da interface assimétrica dimer, as restrições na hélice αC no mutante provavelmente estabilizariam o dimer assimétrico, prolongando a duração do estado ativado.
Outra aplicação do protocolo é investigar o comportamento das principais interações intra e intermolecular que ocorrem durante a simulação. Assim, a interação entre K745 e E762, que é fundamental para a atividade enzimática EGFR, foi analisada tanto para a forma ativa do tipo selvagem quanto para a quinase ΔELREA EGFR medindo a ocupação percentual de ligações de hidrogênio formadas entre os átomos polares de cadeia lateral dos dois resíduos durante as simulações de MD(Figura 5A): esta interação eletrostática chave foi formada com mais frequência no domínio ΔELREA quinasase em comparação com o domínio de quinase do tipo selvagem, devido ao helix αC mais estável que acomoda E762. As interações entre mg2+-ATP e os domínios de quinase wild-type e ΔELREA EGFR(Figura 5B) durante a simulação também foram avaliadas (Figura 5C): o número de ligações de hidrogênio foram maiores para 2 ΔELREA (valor médio 4,0 - IC 95% 0,03) do que para o EGFR tipo selvagem (valor médio 3,2 - IC 95% 0,04). Uma análise mais aprofundada das ligações de hidrogênio revelou que o K745 interage com mais frequência com os grupos fosfatos de ATP em ΔELREA EGFR, que está ligado à interação K745-E762 mais estável observada na simulação do domínio mutante ΔELREA EGFR quinasase.
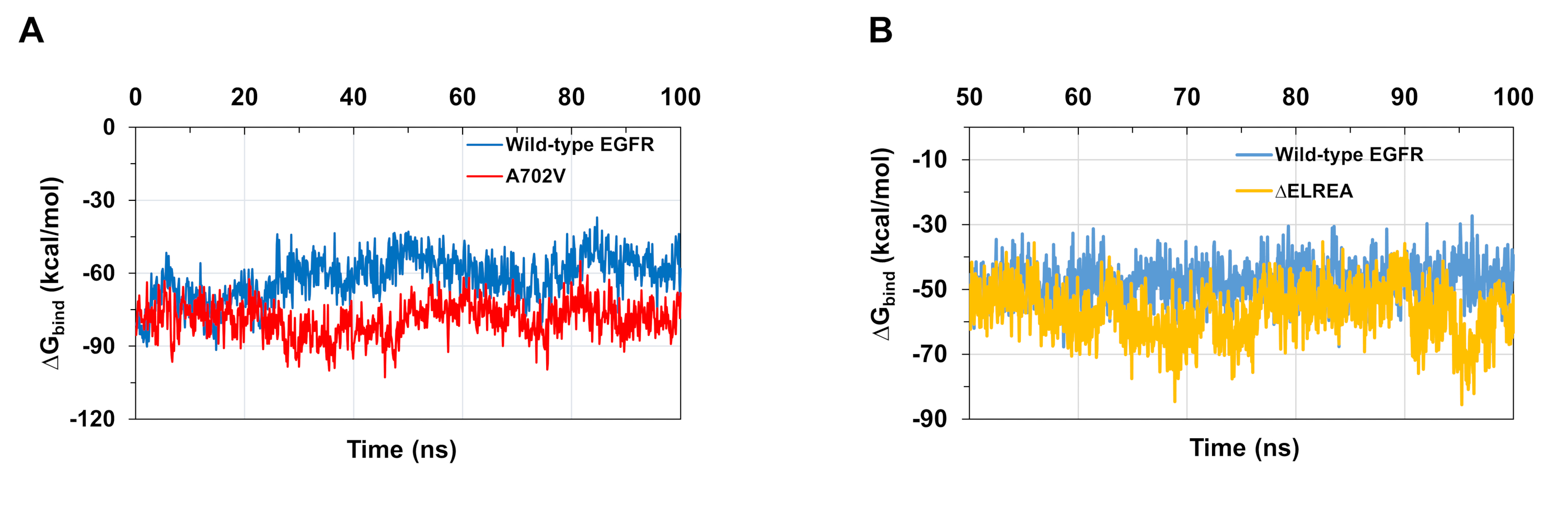
Simulações de MD como descrito no protocolo também são úteis na avaliação da energia relativa livre de ligação para interações proteína-proteína-ligand. As energias de ligação entre os domínios ativador e quinase receptor de EGFRs de tipo selvagem e A702V, e entre os domínios ATP e o tipo selvagem e os domínios mutantes ΔELREA EGFR quinase, foram computados a partir de cálculos generalizados da área de superfície nascida (MMGBSA)(Figura 6A):o mutante A702V produziu um valor médio deligação ΔG mais baixo(ligação média ΔG = -76 kcal/mol - 2 95% IC 0,47), representando interações mais favoráveis de dimer, em contraste com o domínio EGFR tipo selvagem(ligação ΔG média = -61 kcal/mol - 95% CI 0,61). Esta observação é consistente com o segmento mais estável da justaxtamembrana B e uma interface mais apertada devido ao aumento das interações hidrofóbicas observadas para o domínio de quinase A702V EGFR. No caso da vinculação ATP aos domínios de quinase ΔELREA EGFR(Figura 6B),os cálculos mmGBSA prevêem uma ligação ATP mais forte com o mutante ΔELREA(ligação média ΔG -5 7 kcal/mol - 95% CI 0,43) em comparação com o EGFR tipo selvagem(ligação média ΔG -48 kcal/mol - 95% IC 0,33). Este resultado está em linha com o maior número de ligações de hidrogênio registradas entre ATP e ΔELREA EGFR (Figura 5C) em comparação com o domínio do tipo selvagem.
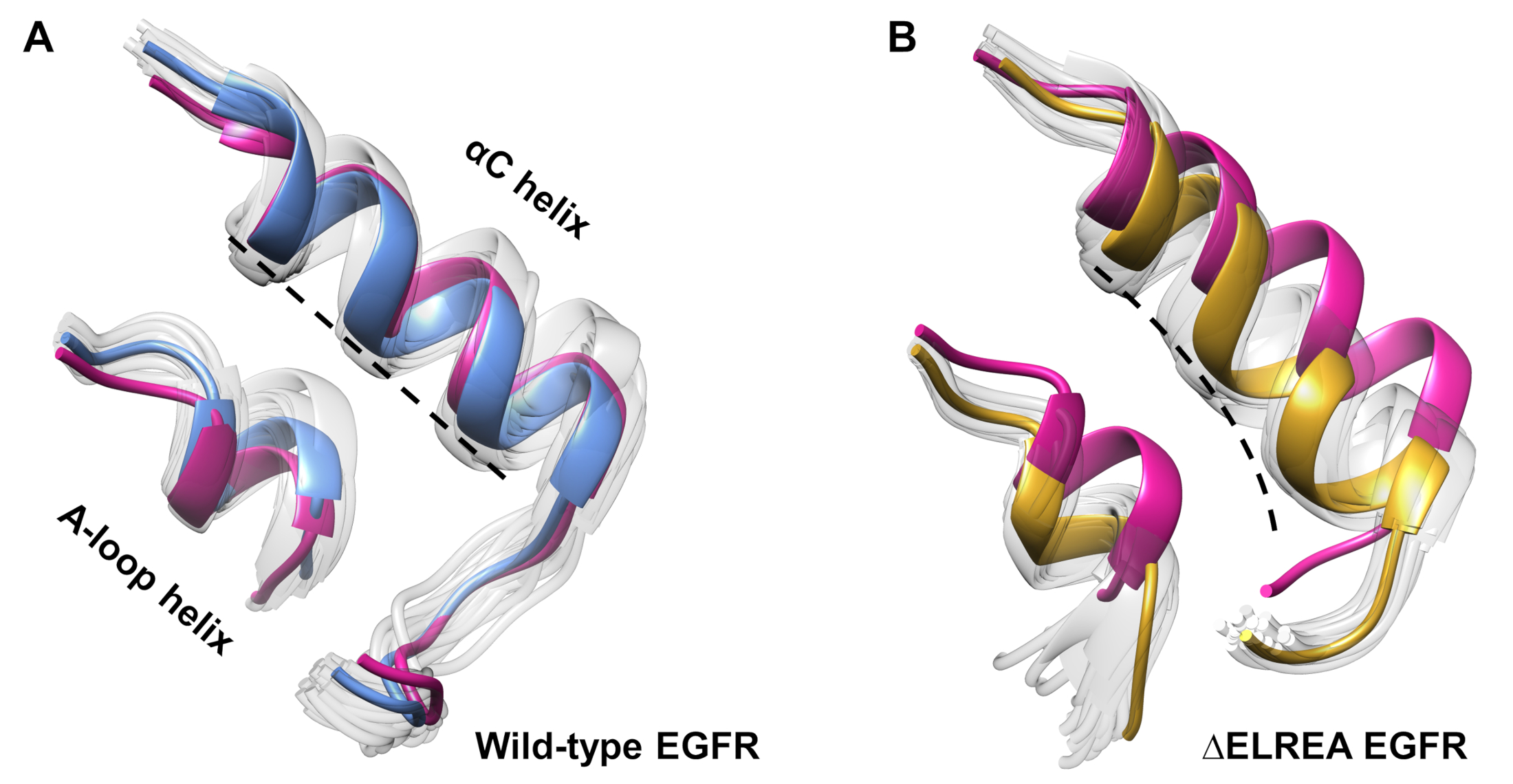
O protocolo também pode ser empregado para investigar alterações conformais observadas durante uma simulação. No presente estudo, os efeitos da mutação ΔELREA na conformação inativa do EGFR foram estudados por inspeção visual e superposicionamento das conformações amostradas a partir da simulação. A análise descobriu um movimento interno da hélice αC no domínio ΔELREA EGFR quinasase(Figura 7A),uma mudança estrutural esperada durante a transição para o estado ativo. Em contrapartida, a hélice αC do EGFR inativo do tipo selvagem manteve sua conformação inicial (Figura 7B). Assim, as simulações de DM apoiam a proposta de que a mutação de exclusão, demonstrada experimentalmente para aumentar a atividade de quinase40,41,promove uma mudança conformacional da quinase inativa em relação ao estado ativo.

Figura 4: Estabilidade conformacional do tipo selvagem e mutante do domínio ativo EGFR quinaase durante simulações de MD. (A) RMSD (átomos de espinha dorsal) sobre o segmento justtamembrano B do domínio quinase do receptor wild-type (azul) e A702V (vermelho). (B) RMSF (átomos de Cα) sobre resíduos da hélice αC: tipo selvagem (azul) e ΔELREA (ouro). (C) Conformações amostradas sobrepostas do tipo selvagem (esquerda) e domínio de quinase ΔELREA (direita); traços de cadeia coloridos com base em RMSD (átomos de Cα) de cada resíduo em relação à estrutura mediana. A coloração varia de azul a branco a vermelho, representando regiões de alta a baixa estabilidade conformacional. Note que as regiões "livres" do terminal N do domínio isolado da quinase, de cor vermelha, não exibiriam esse nível de mobilidade na estrutura EGFR intacta. Figuras adaptadas de Chakroborty et al.9 (Figura 4A reproduzida com permissão do Journal of Biological Chemistry) e Tamirat et al.12. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 5: Principais características vistas no receptor ativo quinase durante simulações de MD: a ponte de sal K745-E762, a hélice αC e interações com ATP. (A) Ocupação percentual da interação K745-E762 durante a simulação dos domínios de quinase EGFR (azul) e ΔELREA (ouro). (B) Resíduos do mutante ΔELREA e ΔELREA interagindo com ATP (varas). Mg2+ (verde) coordenadas com ATP e D855. (C) O número de ligações de hidrogênio formadas pela ATP com os domínios de quinase wild-type e ΔELREA EGFR durante simulações de MD. Figura de Tamirat et al.12. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 6: Energias relativas livres mais baixas de ligação são observadas para os domínios mutantes da quinase durante as simulações. (A) Energias de ligação calculadas para a interação entre os domínios ativador e quinase receptor dos EGFRs tipo selvagem (azul) e A702V (vermelho). (B) ΔGliga de ATP a domínios de tipo selvagem (azul) e ΔELREA (ouro) EGFR quinase. Figuras adaptadas de Chakroborty et al.9 (Figura 6A reproduzida com permissão do Journal of Biological Chemistry) e Tamirat et al.12. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 7: Conformações sobrepostas do domínio de quinase inativa ΔELREA e ΔELREA. Conformação da hélice αC e hélice de loop A de (A) tipo selvagem (estrutura mediana em azul) e (B) ΔELREA EGFRs (ouro). Outras conformações amostradas, brancas desbotadas; estruturas iniciais antes das simulações de MD, rosa. Figura de Tamirat et al.12. Clique aqui para ver uma versão maior desta figura.
{kind=link}
Discussão
O protocolo descrito neste estudo se concentra no uso de simulações de dinâmica molecular para investigar alterações estruturais locais e globais que surgem da ativação de mutações somáticas do domínio da quinase EGFR. Embora as estruturas de cristal de raios-X de EGFRs de tipo selvagem e mutantes forneçam uma visão estrutural inestimável, elas retratam uma ou algumas representações estáticas. No entanto, inerentes à função biológica dos ErbBs são as transições necessárias entre a quinase de tyrosina enzimática e ativa, invocando mudanças dinâmicas tanto na estrutura quanto nas interações intramoleculares entre monômeros de quinase. Assim, foram realizadas simulações de MD para sondar a natureza dinâmica do domínio de tyrosina quinase EGFR, incluindo a estrutura do tipo selvagem, a mutação introduzida de exclusão ΔELREA e a mutação A702V. Essas simulações foram bem sucedidas em elucidar o provável papel dessas mutações nas estruturas e como seus efeitos na conformação do domínio da quinase de tirasina levariam ao aumento experimentalmente observado na atividade de quinase EGFR.
Um passo crucial neste protocolo é o uso de uma estrutura relevante para avaliar o impacto da mutação. Uma maneira de selecionar uma estrutura de entrada de simulação relevante é visualizar a localização da mutação na estrutura estática 3D e examinar seu possível impacto em relação aos aminoácidos e unidades estruturais vizinhas. Neste estudo, por exemplo, uma vez que a mutação A702V EGFR está localizada no segmento justxtamembrano B que forma a interface assimétrica dimer, o uso da estrutura mais fraca para a simulação em oposição ao monômero é crítico. O uso de uma estrutura monomérica teria exposto o segmento de justoxtamembrano B da quinase receptora ao solvente, privando-o das interações estabilizadoras, potencializadas pela mutação a um resíduo hidrofóbico maior e interações com isoleucina 941 dos resíduos C-lóbulos do ativador quinase. Além disso, destaca-se que a estrutura 3D representada pelas coordenadas em um arquivo PDB não corresponde necessariamente à estrutura biologicamente relevante que deve ser utilizada para estudo. Por exemplo, com a estrutura do ErbB4, código PDB 3BCE, as coordenadas PDB correspondem a um aparador, mas isso se deve a contatos de cristal (poucos contatos entre os monômeros são vistos ao visualizar essa estrutura). Podem ser utilizadas matrizes dentro do arquivo PDB (por exemplo, dentro da Quimera) para reconstruir as estruturas cristalograficamente relacionadas, que podem ser visualizadas para identificar cadeias que correspondem à estrutura 3D biologicamente relevante, conforme relatado na publicação original42. Outra etapa essencial do protocolo é preparar adequadamente a estrutura de entrada de simulação, como a construção de aminoácidos ausentes em diferentes regiões de loop, e especialmente onde está localizada nas proximidades da mutação. Embora existam numerosas estruturas EGFR do tipo selvagem no PDB, apenas um número limitado de estruturas mutantes de EGFR estão disponíveis. Consequentemente, as estruturas mutantes também precisam ser modeladas; para uma única mutação de resíduos como A702V, Quimera foi usada para mutar o resíduo; que, para a mutação de exclusão ΔELREA, Modeller foi usado.
Os vários parâmetros utilizados nos arquivos de entrada de simulação - por exemplo, o número de ciclos de minimização, aquecendo o sistema à temperatura desejada de uma só vez ou, em vez disso, aquecendo lentamente através de várias temperaturas intermediárias, o período de tempo para o equilíbrio e para as simulações de produção - podem ser modificados com base na molécula de estudo, no objetivo do trabalho e nas próprias preferências. Ao realizar simulações de MD, também é comum encontrar erros que podem surgir dos arquivos de entrada, problemas relacionados ao software de simulação em uso ou até mesmo um erro do usuário. Por isso, é muito importante entender a origem dos erros examinando cuidadosamente quaisquer mensagens de erro. A maioria dos programas de simulação tem uma lista de discussão onde os usuários podem fazer perguntas aos desenvolvedores de software e a outros usuários pelos quais a maioria dos problemas pode ser resolvida. Além disso, os manuais do usuário fornecem assistência significativa para entender os detalhes do protocolo de simulação, incluindo suposições e limitações. Embora a simulação de MD seja uma ferramenta importante para explorar as propriedades dinâmicas das moléculas, lembre-se que os resultados computacionais precisam ser cuidadosamente avaliados em conjunto com outras fontes de informação para avaliar sua validade. Sempre que possível, trabalhe em conjunto com pesquisadores especialistas em proteínas em estudo, especialmente quando são feitos estudos experimentais relevantes em laboratório molhado, que servem para fornecer resultados para interpretação estrutural, bem como para sugerir experimentos que podem ser feitos com base em observações estruturais para testar hipóteses.
Neste estudo, o protocolo foi eficaz na análise dos impactos estruturais dinâmicos das mutações ΔELREA e A702V nas estruturas de quinase EGFR. As simulações revelaram que ΔELREA restringe a hélice αC funcionalmente essencial e promove uma mudança conformacional da quinase inativa para uma quinase ativa estabilizada. Os resultados da simulação são apoiados independentemente por dados de resposta a medicamentos que demonstraram os efeitos dos inibidores da quinase tyrosina nas linhas de células cancerígenas de pulmão com a mutação de exclusão ΔELREA e o EGFR do tipo selvagem, onde maior inibição por drogas que reconhecem a conformação da quinase ativa foi relatada para ΔELREA do que para o EGFR12do tipo selvagem . Com a mutação A702V, as simulações de MD indicam, em comparação com o tipo selvagem, aumento da estabilização da interface quinase ativador-receptor, bem como maior afinidade entre o ativador e a quinase receptora entre si, apoiando juntos a manutenção da conformação ativada da quinase EGFR. A mutação A702V, localizada no segmento justxtamembrano B da quinase receptora, aumentaria as interações hidrofóbicas com o ativador quinase, funcionando para prolongar a duração do estado ativado. A mutação A702V suporta a sobrevivência celular na ausência de fator de crescimento e foi identificada em uma triagem in vitro para mutações EGFR9.
Divulgações
Os autores não têm nada a revelar.
Agradecimentos
Esta pesquisa é financiada por subsídios para M.S.J da Academia da Finlândia (308317, 320005), Fundação Sigrid Juselius e Tor, Joe e Pentti Borg, e para K.E. da Academia da Finlândia (274728, 316796), da Fundação do Câncer da Finlândia e do Hospital Central da Universidade de Turku. M.Z.T. é financiado pela Rede de Doutorado Åbo Akademi de Biologia Informacional e Estrutural. Agradecemos ao CSC IT Center for Science pelos recursos de computação e ao Dr. Jukka Lehtonen pelo suporte de TI sob a rede bioinformática Biocenter Finland; e rede de infraestrutura estrutural da Biocenter Finlândia.
Materiais
| Name | Company | Catalog Number | Comments |
| Amber software | University of California, San Francisco | Version 2018 | Executable |
| Chimera program | Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco | Version 1.13.1 | Executable |
| EGFR struture files | The Protein Data Bank | 3D coordinates of EGFR structures | |
| Maestro | Schrödinger LLC | Version 2018-3 | Executable |
| Modeller program | The Andrej Šali Lab, Departments of Biopharmaceutical Sciences and Pharmaceutical Chemistry, University of California San Francisco | Included in the Chimera program | |
| VMD software | Theoretical and Computational Biophysics Group, University of Illinois at Urbana-Champaign | Version 1.9.3 | Executable |
Referências
- Yarden, Y., Sliwkowski, M. X. Untangling the ErbB signalling network. Nature Reviews Molecular Cell Biology. 2, 127-137 (2001).
- Lemmon, M. A., Schlessinger, J., Ferguson, K. M. The EGFR family: not so prototypical receptor tyrosine kinases. Cold Spring Harbor Perspectives in Biology. 6, a020768 (2014).
- Arteaga, C. L., Engelman, J. A. ERBB receptors: From oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell. 2, 282-303 (2014).
- Mishra, R., Hanker, A. B., Garrett, J. T. Genomic alterations of ERBB receptors in cancer: Clinical implications. Oncotarget. 8, 114371-114392 (2017).
- . cBioPortal for Cancer Genomics Available from: https://www.cbioportal.org (2020)
- Lynch, T. J., et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. New England Journal of Medicine. 350 (21), 2129-2139 (2004).
- Paez, J. G., et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 304 (5676), 1497-1500 (2004).
- Pao, W., et al. EGF receptor gene mutations are common in lung cancers from "never smokers" and are associated with sensitivity of tumors to gefitinib and erlotinib. Proceedings of the National Academy of Sciences U.S.A. 101 (36), 13306-13311 (2004).
- Chakroborty, D., et al. Unbiased in vitro screen for activating EGFR mutations. Journal of Biological Chemistry. 294 (24), 9377-9389 (2019).
- Leahy, D. J. Structure and Function of the Epidermal Growth Factor (EGF/ErbB) Family of Receptors. Advances in Protein Chemistry. 68, 1-27 (2004).
- Roskoski, R. ErbB/HER protein-tyrosine kinases: Structures and small molecule inhibitors. Pharmacological Research. 87, 42-59 (2014).
- Tamirat, M. Z., Koivu, M., Elenius, K., Johnson, M. S. Structural characterization of EGFR exon 19 deletion mutation using molecular dynamics simulation. PLoS ONE. 14 (9), e0222814 (2019).
- Ding, L., et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 455, 1069-1075 (2008).
- Kurppa, K. J., Denessiouk, K., Johnson, M. S., Elenius, K. Activating somatic ERBB4 mutations in non small-cell lung cancer. Oncogene. 35 (10), 1283-1291 (2016).
- Soung, Y. H., et al. Somatic mutations of the ERBB4 kinase domain in human cancers. International Journal of Cancer. 118, 1426-1429 (2006).
- Tvorogov, D., et al. Somatic mutations of ERBB4: selective loss-of-function phenotype affecting signal transduction pathways in cancer. Journal of Biological Chemistry. 284, 5582-5591 (2009).
- Hubbard, S. R., Till, J. H. Protein tyrosine kinase structure and function. Annual Review of Biochemistry. 69 (1), 373-398 (2000).
- Huse, M., Kuriyan, J. The conformational plasticity of protein kinases. Cell. 109 (3), 275-282 (2002).
- Jura, N., et al. Catalytic control in the EGF receptor and its connection to general kinase regulatory mechanisms. Molecular Cell. 42, 9-22 (2011).
- Karplus, M., Kuriyan, M., J, Molecular dynamics and protein function. Proceedings of the National Academy of Sciences U.S.A. 102 (19), 6679-6685 (2005).
- Shan, Y., Arkhipov, A., Kim, E. T., Pan, A. C., Shaw, D. E. Transitions to catalytically inactive conformations in EGFR kinase. Proceedings of the National Academy of Sciences U.S.A. 110 (18), 7270-7275 (2013).
- Reckamp, K. L., et al. A phase I trial to determine the optimal biological dose of celecoxib when combined with erlotinib in advanced non-small cell lung cancer. Clinical Cancer Research. 12 (11 Pt 1), 3381-3388 (2006).
- Pettersen, E. F., et al. UCSF Chimera-a visualization system for exploratory research and analysis. Journal of Computational Chemistry. 25 (13), 1605-1612 (2004).
- Berman, H. M., et al. The Protein Data Bank. Nucleic Acids Research. 28 (1), 235-242 (2000).
- Zhang, X., Gureasko, J., Shen, K., Cole, P. A., Kuriyan, J. An Allosteric Mechanism for Activation of the Kinase Domain of Epidermal Growth Factor Receptor. Cell. 125 (6), 1137-1149 (2006).
- Stamos, J., Sliwkowski, M. X., Eigenbrot, C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. Journal of Biological Chemistry. 277 (48), 46265-46272 (2002).
- Sogabe, S., et al. Structure-Based Approach for the Discovery of Pyrrolo[3,2-d]pyrimidine-Based EGFR T790M/L858R Mutant Inhibitors. ACS Medicinal Chemistry Letters. 4 (2), 201-205 (2013).
- Sali, A., Blundell, T. L. Comparative protein modelling by satisfaction of spatial restraints. Journal of Molecular Biology. 234 (3), 779-815 (1993).
- Yun, C. H., et al. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell. 11 (3), 217-227 (2007).
- Park, J. H., Liu, Y., Lemmon, M. A., Radhakrishnan, R. Erlotinib binds both inactive and active conformations of the EGFR tyrosine kinase domain. Biochemical Journal. 448 (3), 417-423 (2012).
- . Release 2018-3: Maestro Available from: https://www.schrodinger.com/maestro (2018)
- Case, D. A., et al. . AMBER 2018. , (2018).
- Maier, J. A., Martinez, C., Kasavajhala, K., Wickstrom, L., Hauser, K. E., Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. Journal of Chemical Theory and Computation. 11 (8), 3696-3713 (2015).
- Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W., Klein, M. L. Comparison of simple potential functions for simulating liquid water. Journal of Chemical Physics. 79 (2), 926-935 (1983).
- Meagher, K. L., Redman, L. T., Carlson, H. A. Development of polyphosphate parameters for use with the AMBER force field. Journal of Computational Chemistry. 24 (9), 1016-1025 (2003).
- Humphrey, W., Dalke, A., Schulten, K. VMD: Visual molecular dynamics. Journal of Molecular Graphics. 14 (1), 33-38 (1996).
- Melvin, R. L., Salsbury, F. R. Visualizing ensembles in structural biology. Journal of Molecular Graphics and Modelling. 67, 44-53 (2016).
- Roe, D. R., Cheatham, T. E. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. Journal of Chemical Theory and Computation. 9 (7), 3084-3095 (2013).
- Miller, B. R., et al. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. Journal of Chemical Theory and Computation. 8 (9), 3314-3321 (2012).
- Guha, U., et al. Comparisons of tyrosine phosphorylated proteins in cells expressing lung cancer-specific alleles of EGFR and KRAS. Proceedings of the National Academy of Sciences U.S.A. 105 (37), 14112-14117 (2008).
- Furuyama, K., et al. Sensitivity and kinase activity of epidermal growth factor receptor (EGFR) exon 19 and others to EGFR-tyrosine kinase inhibitors. Cancer Science. 104 (5), 584-589 (2013).
- Qiu, C., et al. Mechanism of Activation and Inhibition of the HER4/ErbB4 Kinase. Structure. 6 (3), 460-467 (2008).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados