
Method Article
Decifrare gli effetti strutturali dell'attivazione delle mutazioni somatiche EGFR con la simulazione della dinamica molecolare
In questo articolo
Riepilogo
L'obiettivo di questo protocollo è quello di utilizzare simulazioni di dinamica molecolare per esaminare i cambiamenti strutturali dinamici che si verificano a causa dell'attivazione delle mutazioni della proteina chinasi EGFR.
Abstract
Numerose mutazioni somatiche che si verificano nella famiglia del recettore del fattore di crescita epidermico (EGFR) della chinasi della tirosina recettore (RTK) sono state segnalate da pazienti oncologici, anche se relativamente pochi sono stati testati e hanno dimostrato di causare cambiamenti funzionali negli ErbB. I recettori ErbB sono dimerizzati e attivati al momento del legame del ligando, e i cambiamenti conformazionali dinamici dei recettori sono inerenti all'induzione della segnalazione a valle. Per due mutazioni mostrate sperimentalmente per alterare la funzione EGFR, A702V e la mutazione di delezione ELREA750 di746,illustremo nel seguente protocollo come le simulazioni di dinamica molecolare (MD) possono sondare la (1) stabilità conformazionale della struttura della chinasi della tirosina mutante rispetto all'EGFR di tipo selvatico; (2) conseguenze strutturali e transizioni conformazionali e il loro rapporto con i cambiamenti funzionali osservati; (3) effetti delle mutazioni sulla forza dell'ATP vincolante e per il legame tra i domini della chinasi nel dimer asimmetrico attivato; e (4) effetti delle mutazioni sulle interazioni chiave all'interno del sito di legame EGFR associato all'enzima attivato. Il protocollo fornisce una procedura dettagliata passo-passo e una guida che può essere più generalmente utile per lo studio delle strutture proteiche utilizzando simulazioni MD come mezzo per sondare la dinamica strutturale e la relazione con la funzione biologica.
Introduzione
Il recettore del fattore di crescita epidermico umano (EGFR) famiglia (ErbB) di recettori tirorosina chinasi (RTKs) comprende quattro membri - EGFR/ErbB1/HER1, ErbB2/HER2, ErbB3/HER3 e ErbB4/HER4. I recettori ErbB regolano i processi cellulari fondamentali come la crescita e la proliferazione delle cellule, la differenziazione, la migrazione e lasopravvivenza 1,2, e sono quindi potenti proto-oncogeni. L'attività aberrante dei recettori ErbB, in particolare EGFR ed ErbB2, è stata spesso associata a tumori umani che hanno reso i recettori ErbB obiettivi chiave per le terapie oncologiche2,3.
Diverse alterazioni somatiche dei geni ERBB sono state segnalate da neoplasie umane3,4,5. Gli esempi più caratterizzati includono le mutazioni ricorrenti, che attivano i punti e le brevi delezioni in-frame nel dominio della chinasi EGFR nel cancro del polmone non a piccole cellule (NSCLC). Queste mutazioni EGFR rappresentano i fattori chiave della crescita del cancro e predicono la sensibilità ai farmaciantitumoreEGFR mirati 6,7,8. Tuttavia, nella maggior parte dei tumori, le mutazioni somatiche nell'EGFR si verificano al di fuori di questi "hotspot" ricorrenti e sono distribuite nell'intera durata del recettore con 1210 residui. Infatti, la maggior parte dei residui lungo la sequenza primaria EGFR sono stati trovati per essere mutato nel cancro umano9. Tuttavia, a parte i pochi punti caldi, il significato funzionale della stragrande maggioranza delle mutazioni EGFR associate al cancro rimane sconosciuto.
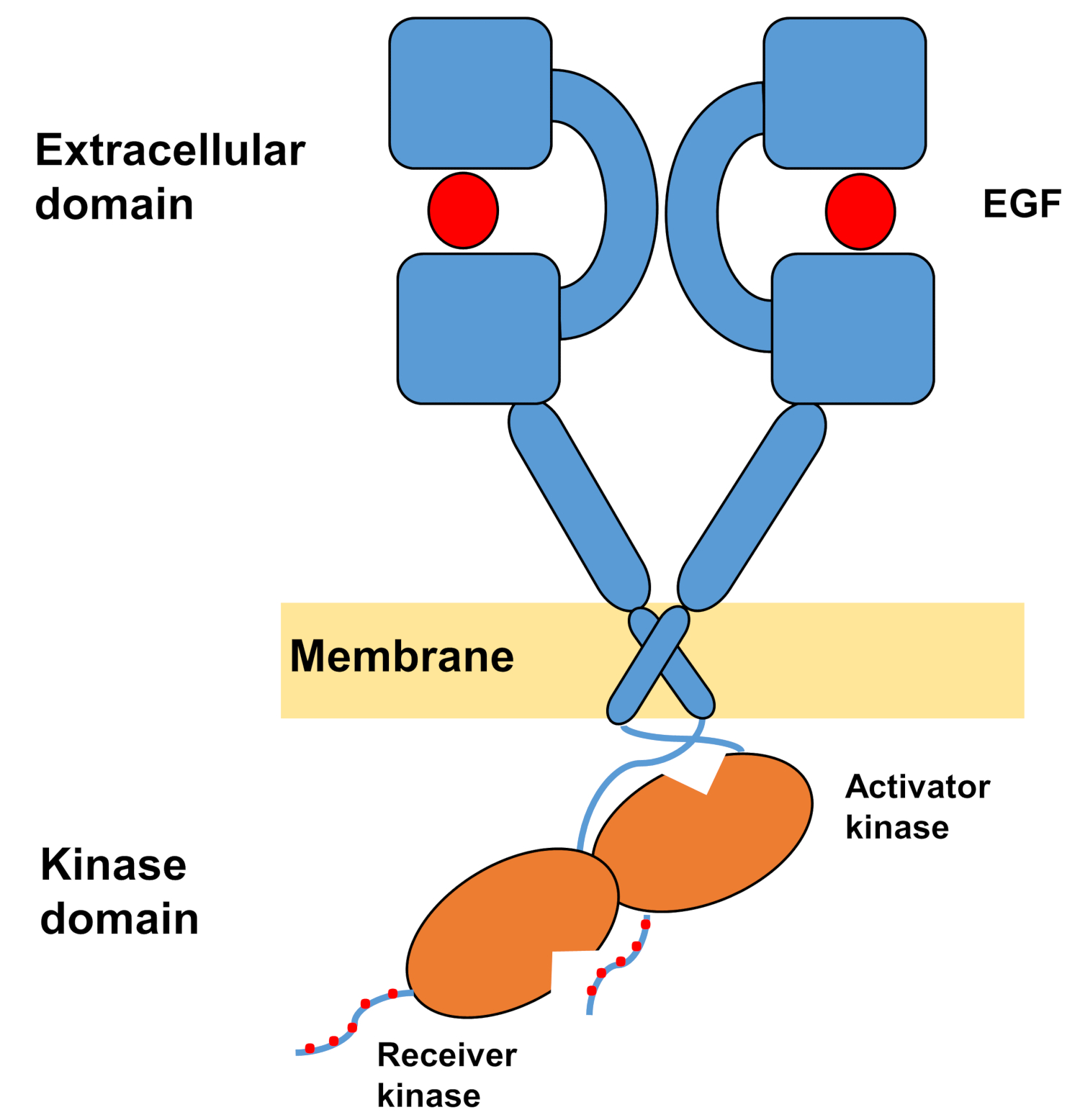
La struttura monomeccatica degli ErbB è costituita da un grande dominio extracellulare amino terminale, seguito da una singola elica transmembrana che conduce al dominio della chinasi della tirosina intracellulare e alla regione della coda C-terminal che contiene siti di attracco per le proteine di segnalazione intracellulari. L'associazione Ligand innesca un drammatico cambiamento conformazionale nel dominio extracellulare, che facilita la formazione dei dimer del recettore esponendo i bracci di dimerizzazione che si incrociano simmetricamente e interagiscono con le loro superfici aromatiche/idrofobiche. Sulla formazione del recettore dei dimer i domini della tirosina chinasi entrano in contatto asimmetrico (Figura 1), con conseguente attivazione delle chinasi che fosforilate le code C-terminali dei monomeri recettori, e successivamente in attivazione della segnalazione a valle10,11.

Figura 1: Struttura del dimer EGFR. EGFR si dimezza quando i domini extracellulari legano il fattore di crescita (EGF, fattore di crescita epidermico). Il dominio della chinasi del ricevitore viene quindi attivato attraverso l'interazione asimmetrica con il dominio chinasi dell'attivatore, e le code del terminale C sono autofosforilate a residui di tirosina (modificato da Tamirat et al.12). Si prega di fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
A causa dei riarrangiamenti strutturali dinamici che si verificano durante  le transizioni del dimero monomero, insieme all'attivazione della chinasi associata alla formazione di un dimer asimmetrico, le mutazioni lungo l'intera lunghezza della struttura del recettore possono potenzialmente avere un effetto sulla funzione del recettore. Qui descriviamo diversi esempi dei nostri studi precedenti in cui la modellazione della mutazione e della visualizzazione erano sufficienti a spiegare le conseguenze per la funzione.
le transizioni del dimero monomero, insieme all'attivazione della chinasi associata alla formazione di un dimer asimmetrico, le mutazioni lungo l'intera lunghezza della struttura del recettore possono potenzialmente avere un effetto sulla funzione del recettore. Qui descriviamo diversi esempi dei nostri studi precedenti in cui la modellazione della mutazione e della visualizzazione erano sufficienti a spiegare le conseguenze per la funzione.
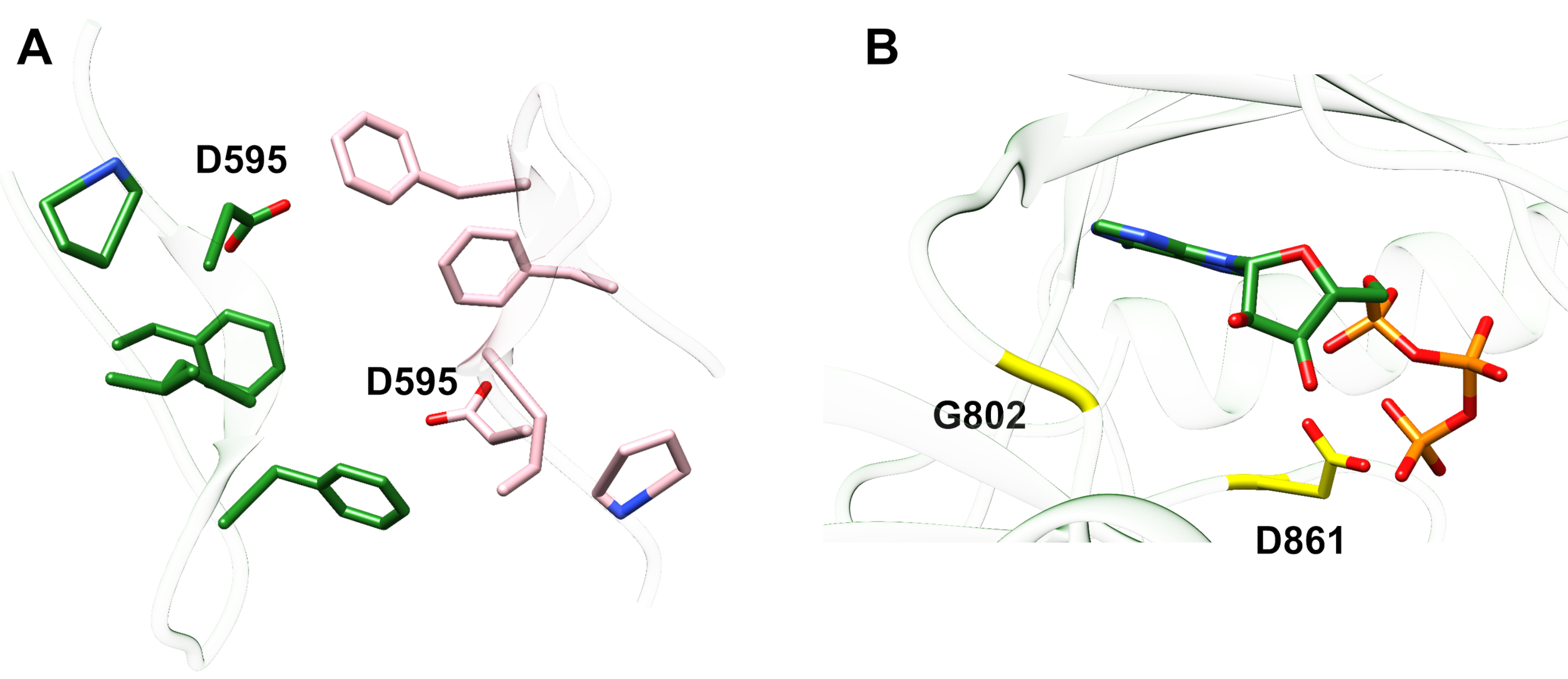
Esempio 1: Una mutazione riportata, D595V in ErbB413, ha portato ad una maggiore dimerizzazione ErbB4 e fosforilazione14. La visualizzazione della posizione della mutazione è stato un fattore critico per comprendere gli effetti funzionali osservati: D595V si è verificato al crossover simmetrico dei bracci dimerici dell'ectodomain (Figura 2A). Le braccia sono in gran parte aromatiche e idrofobiche, e la sostituzione dell'acido aspartico polare con valine dovrebbe aumentare le interazioni idrofobiche "appiccicose", stabilizzando il dimer e quindi aumentare la lunghezza del tempo in cui avviene la fosforilazione14. All'inizio è stata una sorpresa trovare l'aspartate in ogni braccio, ma in retrospettiva si potrebbe pensare ad esso come un meccanismo di temporizzazione per l'attività, dove le catene laterali dell'acido polare riducono l'affinità e la durata del dimero intatto e quindi limitano la fosforilazione mediata dalla chinasi e la segnalazione. La sostituzione con valine rimuoverebbe quindi questa salvaguardia stabilizzando ulteriormente il dimerE ErbB4.

Figura 2: Posizione di una mutazione e mutazioni che attivano e di attivare la chinasi. (A) D595 (attivazione della mutazione D595V) si trova sui bracci dimerici aromatici/idrofobici del modello ectodomain ErbB4; le armi si associano al legame del fattore di crescita; (i residui vicini sono mostrati come bastoni). (B) In ErbB4, G802 (mutazione inattiva G802dup) aiuta a formare la tasca di legame intorno all'anello di adenina di ATP e D861 catalitico (mutazione D861Y inattiva) lega sia Mg2 (non mostrato) e il gruppo γ-fosfato di ATP. Si prega di fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
Esempio 2: Si potrebbe prevedere che le mutazioni somatiche che prendono di mira il sito di legame ATP del dominio della chinasi altererebbero o eliminerebbero l'attività ezimatica che porta a un recettore alterato o morto in chinasi incapace di segnalare. Delle nove mutazioni segnalate da pazienti con seno, gastrico, colorettale o NSCLC15, due delle nove mutazioni al momento del test avevano un'attività di fosforilazionealtamente diminuita 16: G802dup (G → GG) e D861Y. Entrambe le mutazioni somatiche inattivanti sono state trovate all'interno del sito di legame ATP della struttura di dominio della chinasi della tirosina(Figura 2B): la glicina flessibile, duplicata, altererebbe il sito dell'anello di adenina e il piccolo acido aspartico sostituito dalla tirosina ingombrante vicino ai fosfati terminali impedirebbe fisicamente la legatura di Mg2-ATP. Tuttavia, dal momento che ErbB4 può formare un eterodimero con ErbB2 - ErbB2 non lega un fattore di crescita e dipende dall'associazione con un ErbB che fa al fine di eterodimerizzare - l'ErbB2(attivo)-ErbB4(chinasi-morto) eterodimero stimolerebbe la proliferazione cellulare attraverso il percorso di segnalazione Erk/Akt, ma le cellule non si differenziano a causa della chinasi-morto ErbB4 e la mancanza di attivazione del percorso STAT516.
In studi più recenti, è diventato evidente che i movimenti dinamici degli ErbB erano rilevanti per comprendere gli effetti di alcuni mutanti sulla funzione ErbB, in particolare le mutazioni che si verificano all'interno del dominio della chinasi tirosina. Il dominio della chinasi della tirosina è costituito da un N-lobo (principalmente fogli β) e C-lobo (in gran parte alfa elicoidale), che sono separati dal sito catalitico dove si lega l'ATP. Il lobo N include l'elica e il ciclo P, mentre i loop di attivazione (A-loop) e catalitici sono presenti nel C-lobo17,18,19. Le strutture cristalline del dominio della chinasi della tirosina hanno rivelato due conformazioni inattive, la maggior parte delle strutture hanno lo stato inattivo simile a Src. Nella conformazione attiva l'aspartato catalitico dell'A-loop punta verso il sito di rilegatura ATP e l'elica di C è orientata verso la tasca di legame ATP (conformazione "C-in"), formando una forte interazione glutammato-lysina ione-coppia.
Poiché gli ErbB e il dominio della chinasi dei componenti sono entità altamente dinamiche, e soprattutto per i casi in cui gli effetti delle mutazioni sulla funzione e sull'attività biologica sono probabilmente strettamente collegati agli stati conformazionali degli ErbB, è importante valutare le mutazioni rispetto alla gamma di cambiamenti dinamici che sperimenteranno. Le strutture cristalline a raggi X degli ErbB forniscono istantanee statiche della struttura 3D, che può o non può essere rilevante per comprendere le conseguenze dinamiche di una mutazione. Al fine di sondare la gamma di cambiamenti dinamici corrispondenti al "paesaggio energetico" disponibile per una struttura tridimensionale (3D), le simulazioni di dinamiche molecolari (MD) sonoampiamente utilizzate 20. Nel caso di mutazioni che porterebbero a cambiamenti conformazionali locali all'interno del dominio della chinasi della tirosina o alla stabilizzazione di un complesso, possono essere sufficienti simulazioni dell'ordine di 100 ns. Tuttavia, le modifiche conformazionali su scala più ampia (ad esempio, le transizioni tra le conformazioni attive e inattive del dominio chinasi) richiedono un tempo di simulazione più lungo - nell'ordine dei microsecondi21.
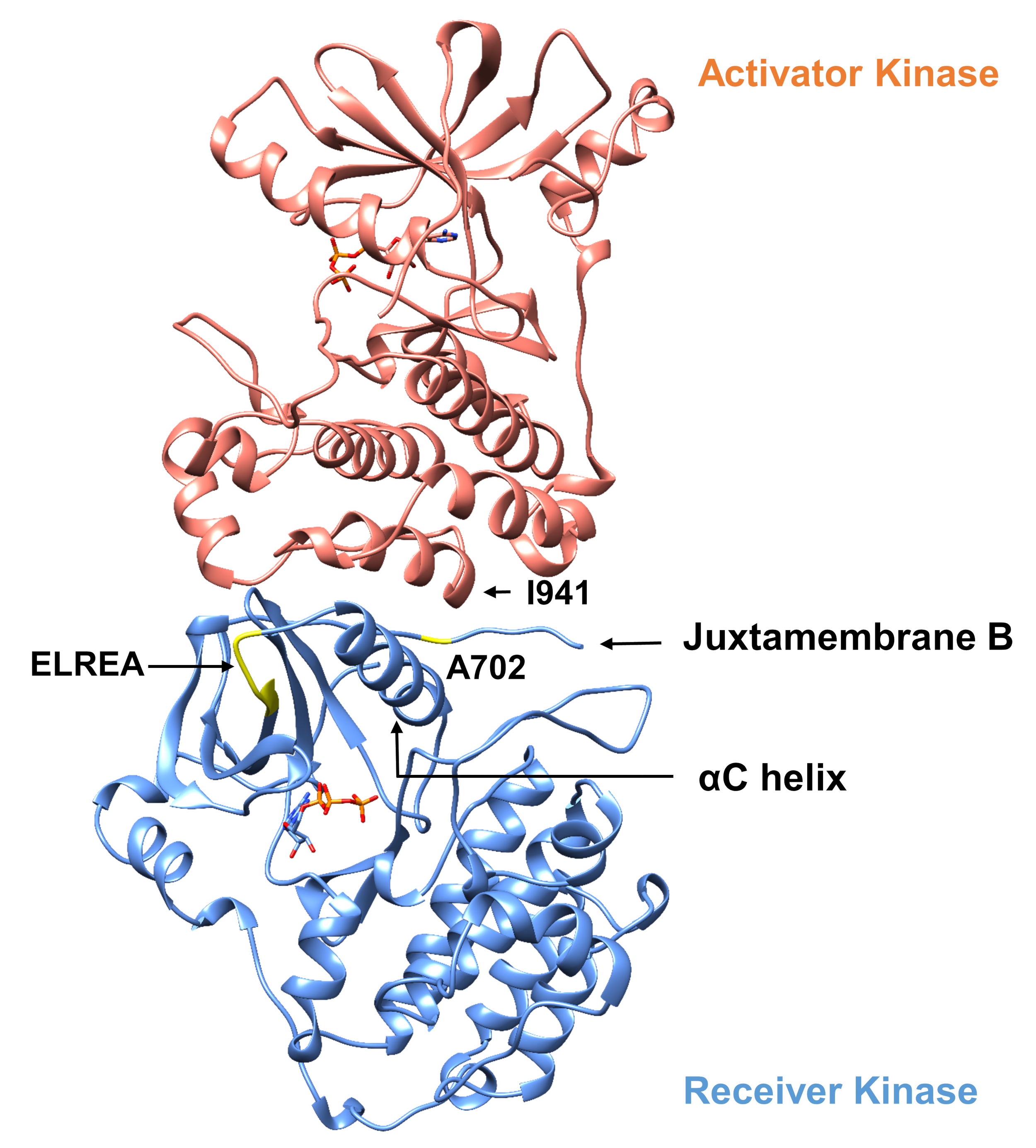
Per quanto riguarda il protocollo descritto di seguito si considerano due mutazioni di attivazione all'interno del dominio della chinasi tirosina (Figura 3). Entrambe le mutazioni si trovano all'interno del dominio della chinasi in luoghi che sperimentano cambiamenti conformazionali locali che determinano se la chinasi è attiva o meno, e quindi le simulazioni MD sono state applicate in entrambi i casi. Nel primo caso, consideriamo le modifiche che influenzano direttamente il sito di rilegatura ATP e i macchinari catalitici del dominio della chinasi del ricevitore EGFR, esaminando specificamente le conseguenze di una mutazione di eliminazione exon 19 che è ampiamente implicata in NSCLC4,7. La mutazione ELREA750 di746, che riduce la lunghezza del loop β3-C che precede l'elica di C - l'elica che si muove verso il sito di legame/attivo sull'attivazione della chinasi e partecipa a formare l'interazione elettrostatica critica tra E762 dell'elica e K745 posizionando l'interazione lisina per l'attivazione con ATP - predispone il dominio per l'attivazione12. Nel secondo caso, consideriamo la mutazione A702V di EGFR, che si è dimostrata una nuova mutazione attivante di guadagno di funzione rivelata dalla piattaforma iScream9 e identificata in un paziente NSCLC22. Alanine-702 sul dominio chinasi ricevitore si trova sul segmento B juxtamembrane all'interfaccia del ricevitore e i domini chinasi attivatore, in cui questo asimmetrico chinasi dimer complesso e chinasi modifiche conformazionali sono necessari per l'attivazione9.

Figura 3: Il dimer di dominio della chinasi asimmetrica di EGFR. La mutazione A702V si trovava nell'interfaccia critica dei domini dell'attivatore e della chinasi del ricevitore, adiacente all'elica di C e vicino all'isolaucine 941 della chinasi dell'attivatore. I cambiamenti conformazionali indotti dalla formazione del dimer asimmetrico portano all'attivazione della chinasi. Il β3-C che contiene la sequenza ELREA precede direttamente l'elica di C; durante l'attivazione, l'elica C si sposta verso l'interno verso il sito di binding ATP. Si prega di fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
Protocollo
NOTA: Le misure dettagliate adottate per esaminare gli effetti della mutazione di EGFR e A702V sulla struttura EGFR utilizzando simulazioni MD sono discusse come segue:
1. Preparazione della struttura
NOTA: Al fine di studiare gli impatti strutturali della mutazione di ELREA, le forme di tipo selvatico e mutante di strutture monomero EGFR attive, associate all'ATP e apo inattive sono preparate come segue.
- Aprire il programma di visualizzazione Chimera23 (https://www.cgl.ucsf.edu/chimera/) per preparare la struttura di chinasi EGFR apo attiva di tipo selvaggio. Nel menu File fare clic sull'opzione Recupera per ID e selezionare il database Protein Data Bank (PDB24) e specificare il codice PDB 2GS225 (risoluzione 2.8). Il PDB è un deposito di strutture 3D risolto da diverse tecniche sperimentali, tra cui la cristallografia a raggi X, la spettroscopia a risonanza magnetica nucleare, la microscopia crioelettrica e la diffrazione dei neutroni.
- Costruire gli elementi strutturali mancanti di 2GS2 prendendo questi segmenti dalle strutture EGFR PDB 1M1426 (2.6 ) e 3W2S27 (1.9. A tale scopo, aprire 1M14 e 3W2S e sovrapporli su 2GS2 utilizzando l'opzione MatchMaker nel menu → confronto Struttura.
- Ritaglia i segmenti da aggiungere da 1M14 e 3W2S. Selezionare gli atomi terminali del residuo prima delle lacune in 2GS2 e gli atomi da aggiungere da 1M14 e 3W2S (per informazioni dettagliate sui segmenti aggiunti, vedere la tabella 1). Nella riga di comando digitare bond sel e premere enter. Questa struttura è la struttura del modello.
- Utilizzare la struttura del modello dal passaggio 1.2 per costruire la forma mutante di eliminazione di EGFR del dominio chinasi EGFR. Produrre la sequenza di formato FASTA di EGFR di EGFR salvando la sequenza della struttura del modello (Favorite → Sequence → File → Save as) e quindi eliminando la sequenza ELREA in corrispondenza di numeri residui 746-750.
- Aprite la sequenza EGFR in Chimera e allineatela con la sequenza della struttura del modello 2GS2 utilizzando il menu Sequenza. Nella finestra di allineamento selezionare l'opzione → Modeller (omologia)28.
- Nella finestra popup specificare la struttura composita 2GS2 come modello e la sequenza mutante come query da modellare. Quindi premere OK. Selezionare un modello mutante tra i modelli risultanti, in base al punteggio zDOPE (in genere il punteggio più basso) e all'ispezione visiva.
- Per preparare la struttura di chinasi EGFR di tipo selvaggio legata all'ATP, utilizzare la struttura PDB2ITX 29 (2,98) come struttura principale. Costruire segmenti mancanti (vedere la tabella 1) utilizzando le strutture 2GS625 (2.6 ) e 3W2S seguendo la procedura descritta nel passaggio 1.2. Convertire il ligando ANP nella struttura risultante in ATP aprendo il file PDB in un editor di testo e cambiando l'atomo di azoto N3B di ANP in un atomo di ossigeno.
- Aprire la struttura nel passaggio 1.4 a Chimera come indicato nel passaggio 1.1. Aggiungere uno ione di magnesio a questa struttura della struttura PDB2ITN 29 (2.47 s) al fine di ottenere un posizionamento simile per lo ione Mg2.
- Con la struttura risultante dal passaggio 1.5, modellare la forma mutante di ZELREA seguendo il passaggio 1.3.
| Apo attivo EGFR | Apo inattivo EGFR | EGFR attivo legato all'ATP | |
| Struttura principale | 2GS2 | 2GS7 | 2ITX |
| Strutture utilizzate per costruire cicli mancanti | 1M14 (723-725) | 3W2S (958-984) | 2GS6 (862-865) |
| 3W2S (967-981) | 4HJO (848-850) | 3W2S (990-1001) |
Tabella 1: strutture utilizzate per creare modelli compositi di strutture attive apo attive, apo inactive e associate ad ATP. Le regioni mancanti (intervallo di amminoacidi tra parentesi) nella struttura principale sono state costruite dalle strutture elencate.
- Per preparare la struttura di chinasi EGFR apoattiva di tipo selvaggio, aprire la struttura PDB2GS7 25 (2.6) come nel punto 1.1 ed eliminare i ligando legati e le acque cristalline. Aggiungere i segmenti mancanti in 2GS7 (vedere la tabella 1) dalle strutture 3W2S e 4HJO30 (2.75) utilizzando la procedura descritta nel passaggio 1.2. In base alla struttura EGFR inattiva finale, preparare il modello mutante utilizzando la procedura descritta al punto 1.3.
NOTA: per lo studio della mutazione A702V, la struttura dimera asimmetrica EGFR viene studiata poiché la mutazione si trova nel segmento juxtamembrane B del dominio chinasi che forma gran parte dell'interfaccia dimer. Le strutture EGFR mutanti di tipo selvaggio e A702V sono preparate come segue: - La struttura dimer asimmetrica di tipo selvaggio è costruita dalla struttura PDB 2GS2, che inizialmente viene visualizzata nella forma monometica. Per eseguire la conversione nell'assieme biologico che contiene le chinasi dell'attivatore e del ricevitore nella disposizione asimmetrica, aprire 2GS2 in Chimera come in (1.1) ed eseguire calcoli di simmetria facendo clic su Strumenti → Struttura di ordine superiore → Cella unità. Selezionare la struttura 2GS2 e immettere Crea copie. Infine, selezionare e salvare un singolo dimer asimmetrico dalle copie multiple del dimer risultante dalle operazioni di simmetria.
- Utilizzando la struttura EGFR asimmetrica di tipo selvaggio del passaggio 1.8, costruire il mutante A702V sostituendo alanine 702 con valine utilizzando l'opzione Strumenti → Struttura di modifica → Rotamers in Chimera.
NOTA: Collettivamente, sei strutture EGFR monometiche e due dimeriche sono preparate rispettivamente per gli studi di mutazione di EGFR e A702V. Ogni struttura viene successivamente elaborata per la simulazione utilizzando la preparazione guidata delle proteine nel programma Maestro31 e le simulazioni MD sono fatte con il programma Amber32. - Aprire la struttura in Maestro utilizzando l'opzione → di importazione del file. Quindi fare clic sul pulsante della preparazione delle proteine e selezionare quanto segue: aggiungere atomi di idrogeno, costruire atomi di catena laterale mancanti, determinare gli stati di protolazione dei residui ionizable al pH 7.0 utilizzando PROPKA, ottimizzare l'orientamento dei residui di asparagine, glutammina e istidina per l'incollaggio dell'idrogeno e infine ridurre al minimo la struttura.
2. Configurazione del sistema
- Aprire il programma saltato incluso nel pacchetto software Amber. Importare il campo di forza ff14SB33 (source leaprc.protein.ff14SB) e LE molecole d'acqua TIP3P34 (source leaprc.water.tip3p). Per i sistemi associati ad ATP importare anche i parametri per ATP35 (loadamberparams frcmod.phosphate, loadamberprep ATP.prep). Quindi, caricare la struttura (mol - loadpdb structure.pdb).
- Risolvi la struttura in una scatola di ottaedrali con molecole d'acqua TIP3P esplicite che si estendono di 10 z in tutte le direzioni dagli atomi superficiali della proteina (solvateoct mol TIP3PBOX 10.0).
- Controllare il sistema costruito (Controllare mol) e neutralizzarlo aggiungendo ioni necessari (addions mol Na 0). Per modellare sufficientemente i sistemi biomolecolari, aggiungere altri atomiNa- /Cl- alla scatola di simulazione per portare la concentrazione di sale del sistema a 0,15 M(addions mol Na x, addions mol Cl- X), dove X viene sostituito dal risultato di: concentrazione di sale desiderata - numero di molecole d'acqua - volume per molecola d'acqua - numero di Avogadro.
- Generare e salvare i file di topologia e coordinate del sistema, che fungono da input per la successiva simulazione di produzione (saveamberparm mol X.prmtop X.inpcrd).
3. Simulazione di dinamica molecolare
- Utilizzando Amber, inizialmente sottopone il sistema di simulazione a 5000 cicli di discesa più ripida e coniugare la minimizzazione dell'energia del gradiente per aggirare le configurazioni sfavorevoli. Eseguire la minimizzazione in più fasi, abbassando gradualmente il sistema di ritenuta applicato sugli atomi solute da 25 kcal mol-1 - -2 a 0 kcal mol-1 - -2.
- Nel file di input di minimizzazione, min.in, regolare la variabile maxcyc per il ciclo di minimizzazione totale (maxcyc - 5000) e ncyc per indicare il numero di cicli per l'algoritmo di discesa più ripido. Utilizzare la variabile restraint_wt per applicare la forza di ritenuta sugli atomi di soluto specificati dal parametro restraintmask. Quindi eseguire la minimizzazione come segue:
$AMBERHOME/bin/sander -O -i min.in -o min.out -p X.prmtop -c X.inpcrd -r min.rst -ref X.inpcrd
NOTA: la strategia e i parametri effettivi utilizzati possono variare in base alle proprie preferenze. I dettagli e le indicazioni sono disponibili nel manuale e nel sito web di Amber (https://ambermd.org/index.php)
- Nel file di input di minimizzazione, min.in, regolare la variabile maxcyc per il ciclo di minimizzazione totale (maxcyc - 5000) e ncyc per indicare il numero di cicli per l'algoritmo di discesa più ripido. Utilizzare la variabile restraint_wt per applicare la forza di ritenuta sugli atomi di soluto specificati dal parametro restraintmask. Quindi eseguire la minimizzazione come segue:
- Riscaldare il sistema per 100 ps da 0 K a 300 K impostando un mol 10 kcalmol -1 - -2 di restrizione sugli atomi solute. A tale scopo, impostare 0,0 , temp0 , dt , nstlim e restraint_wt nel file di input heat.in. Eseguire il riscaldamento con il seguente comando:
$AMBERHOME/bin/sander -O -i heat.in -o heat.out -p X.prmtop -c min.rst -r heat.rst -x heat.mdcrd -ref min.rst - Equilibrare il sistema per 900 ps sotto un insieme NPT; numero costante di atomi, temperatura (temp0 - 300,0) e pressione (ntp 1), controllandola con il metodo Berendsen (ntt - 1). Impostare un taglio di distanza di 9 z (taglio : 9,0) per le interazioni elettrostatiche a lungo raggio. Abbassare gradualmente il sistema di ritenuta dell'atomo soluto a 0,1 kcal mol-1 - -2 (restraint_wt - 0,1). Eseguire il file di input di equil.in che descrive i parametri precedenti come segue:
$AMBERHOME/bin/sander -O -i equil.in -o equil.out -p X.prmtop -c heat.rst -r equil.rst -x equil.mdcrd -ref heat.rst - Finalizzare l'equilibrio con una simulazione non vincolata di 5 ns (set dt : 0,002 ps, ntslim e 2500000).
$AMBERHOME/bin/sander -O -i equil_final.in -o equil_final.out -p X.prmtop -c equil.rst -r equil_final.rst -x equil_final.mdcrd -ref equil.rst - Verificare che il sistema sia stato equilibrato esaminando i valori di temperatura, pressione, densità ed energia.
$AMBERHOME/bin/process_mdout.perl heat.out equil.out equil_final.out
riepilogo xmgrace. TEMP/DENSITY/ETOT/EPTOT/EKTOT - Eseguire la simulazione di produzione per 100 ns (set dt : 0.002 ps, ntslim , 50000000 in prod.in) e salvare le conformazioni ogni 10 ps (ntwx - 5000).
$AMBERHOME/bin/sander -O -i prod.in -o prod.out -p X.prmtop -c equil_final.rst -r prod.rst -x prod.mdrcd -ref equil_final.rst
4. Analisi
- Ispezione visiva
- Visualizzare le conformazioni campio da un'immagine e verso l'alto aprendo i file della topologia ambrata X.prmtop e i file di traiettoria prod.mdcrd corrispondenti in VMD36. Utilizzando comode rappresentazioni di struttura secondaria, analizzare la dinamica strutturale complessiva delle proteine dalla traiettoria registrata. Visualizza interazioni specifiche tra atomi/residui di interesse, come il ponte di sale catalisticamente essenziale K745 - E762.
- In alternativa, salvare più conformazioni campio da un'immagine durante la simulazione in formato PDB e aprirle utilizzando il programma Chimera. Sovrapponi le strutture sulla struttura iniziale o mediana utilizzando l'opzione MatchMaker. Visualizzare la struttura iniziale/mediana in solido e il resto delle strutture allineate in bianco sbiadito. Questo approccio permette di visualizzare i movimenti strutturali registrati con maggiore chiarezza.
NOTA: i suggerimenti per le rappresentazioni efficaci e l'elaborazione di insiemi conformazionali da MDS sono disponibili in Melvin et al.37.
- Analisi RMSD e RMSF
- Calcolare i calcoli della deviazione quadrata (RMSD) e della fluttuazione quadrata (RMSF) con il programma Cpptraj 38 per analizzare la stabilità globale delle proteine ed esaminare la flessibilità delle diverse unità strutturali. Nei file di input rmsd.in e rmsf.in, indicare gli atomi di backbone (per RMSD) e gli atomi C (per RMSF) della struttura iniziale come riferimento per il raccordo RMS. Nei file rmsd/rmsf.in importare i file della topologia ambra (parm X.promtop) e i file di traiettoria corrispondenti (trajin prod.mdcrd). Eseguire quindi il comando Cpptraj -i rmsd/rmsf.in. Tracciare i dati di output per l'analisi.
- In alternativa, allineare gli insiemi conformazionali e colorare ogni residuo in base all'atomo di C. Per farlo, apri le conformazioni in Chimera e allineale con l'opzione Matchmaker.
- Passare a Strumenti → rappresentazione → rendering per attributo. Selezionare Residui dell'insieme conformale e C,RMSD come attributi, quindi fare clic su OK. La traccia a catena delle conformazioni sarà quindi colorata dal blu → rosso →, riflettendo rispettivamente regioni di alta, media e bassa stabilità strutturale.
- Analisi delle obbligazioni a idrogeno
- Analizzare l'interazione tra i legami di idrogeno tra ATP e EGFR di tipo selvaggio. Preparare uno script Cpptraj, hbond.in, per eseguire questa attività. Definire un legame di idrogeno con una distanza donatore-accettante inferiore o uguale a 3,5 e un angolo di legame maggiore o uguale a 135. Specificare l'analisi solo per i legami di idrogeno intermolecolari con la variabile nointramol, ad esempio le obbligazioni di idrogeno tra ATP ed EGFR (hbond All nointramol dist 3.5 out nhb.agr avgout avghb.dat). Eseguire lo script come Cpptraj -i hbond.in.
- Utilizzare questo script per valutare le interazioni intramolecolari, ad esempio tra i residui K745 ed E762, che sono residui chiave per l'attività della chinasi EGFR. A tale scopo, specificare K745 come donatore di obbligazioni di idrogeno ed E762 come accettatore di obbligazioni di idrogeno in hbond.in eseguire lo script di conseguenza.
- Monitoraggio della distanza tra gli atomi
- Misurare la distanza tra K745 e E762 aprendo le traiettorie di tipo selvaggio e apo EGFRs in VMD. Selezionare il segno di più di Glu762 e Nz di Lys745 facendo clic su Mouse → etichetta → bond. Monitorare la distanza durante la simulazione tracciando un grafico con grafici → etichette → legame → grafico.
- Calcoli di energia libera
- Per calcolare le energie libere di legame stimate tra ATP e wild-type/EGFRs, e tra l'attivatore e le chinasi ricevitore di tipo selvaggio/A702V EGFRs, utilizzare il modulo39 della meccanica molecolare generalizzata Born surface (MM-GBSA) disponibile nel pacchetto AMBER. Impostare ATP come ligando e EGFR come recettore nello studio di .ELREA. Nello studio A702V, specificare la chinasi del ricevitore come ligando e la chinasi dell'attivatore come recettore.
- Preparare innanzitutto i file PDB complessi ligando, recettore e legante-recettore separatamente nel programma intercalare impostando il valore PBRadii su mbondi2. Per i file PDB, salvare i file di topologia ambrata della fase gasso (.prmtop) e dei file di coordinate (.inpcrd).
- Quindi, nel file di input mmgbsa, mmgbsa.in, impostare igb , saltcon e 0,1. Eseguire calcoli di energia vincolanti utilizzando le traiettorie delle simulazioni, i file di ambra recettore/ligando preparati e i parametri nel mmgbsa.in con lo script MMPBSA.py disponibile in Ambra come segue:
$AMBERHOME/bin/MMPBSA.py -O -i mmgbsa.in -o mmgbsa.dat -sp X.prmtop -cp complex.prmtop -rp recettore.prmtop -lp ligand.prmtop -y prod.mdcrd -eo output.csv - Analizzare i dati di output, output.csv, tracciando i grafici.
Risultati
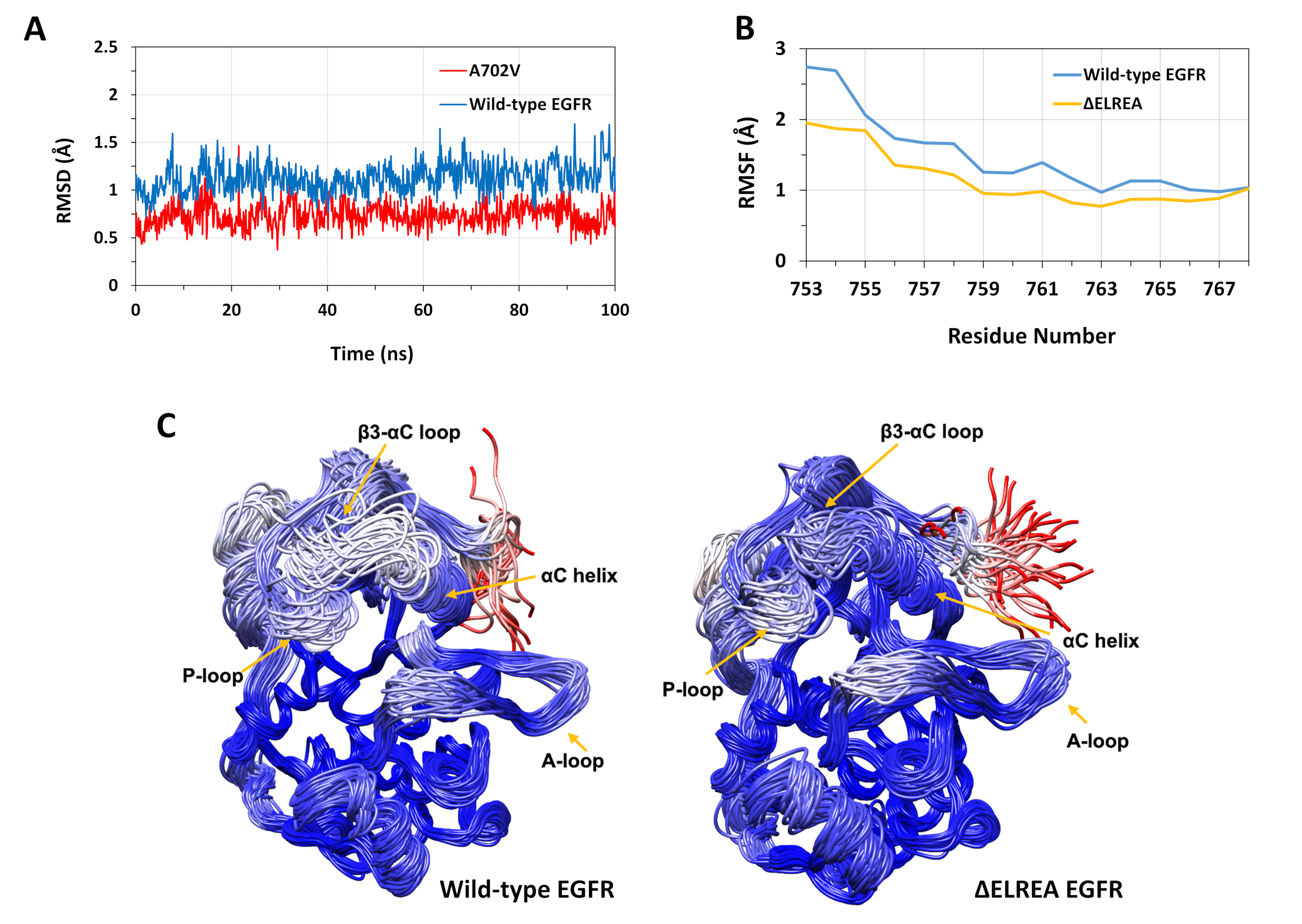
Il protocollo descritto è stato utilizzato per studiare gli effetti strutturali delle mutazioni di EELREA e A702V sulla struttura della chinasi EGFR. Un'applicazione del protocollo consisteva nello studiare l'effetto delle mutazioni sulla stabilità strutturale/conformale locale calcolando i valori RMSD e RMSF delle simulazioni MD. Poiché la mutazione A702V si trova nel segmento juxtamemembrane B, il RMSD di questo segmento della chinasi del ricevitore rispetto alla struttura iniziale è stato calcolato sia per il tipo selvaggio che per gli EGFR A702V. Il risultato (Figura 4A) ha rivelato che il segmento juxtramembrane B del mutante ha aumentato la stabilità conformazionale durante la simulazione di 100 ns (media RMSD 0,7 - 95% intervallo di confidenza (CI) 0,009) rispetto al dominio EGFR chinaase di tipo selvaggio (media RMSD 1.1 - 95% CI 0.01). Questo è molto probabilmente il risultato delle più strette interazioni idrofobiche all'interfaccia dimer a causa della sostituzione dell'alanina 702 (catena laterale del gruppo metile) con un residuo idrofobico più ingombrante, valine (catena laterale del gruppo isopropile), che porta ad un aumento delle interazioni idrofobiche del V702 sul dominio della chinasi del ricevitore con 941 del dominio della chinasi dell'attivatore.
La mutazione di .ELREA si trova β3 loop di tipo -C, adiacente all'elica di C, critica dal punto di vista funzionale; la conformazione dell'elica C è la chiave per spostamenti tra gli stati attivi e inattivi della chinasi EGFR. La stabilità conformazionale dell'elica di C nello stato attivo è stata valutata esaminando il RMSF sugli atomiFigure 4Bdi residui di C . ci sono fluttuazioni più basse nel mutante (media RMSF 1.1 - 95% CI 0.4) rispetto al tipo selvatico (media RMSF 1.5 - 95% CI 0.57); con la più alta differenza di fluttuazioni registrata per i residui N-terminali. Le conformazioni campiore, rispettivamente, sovrapposte alla struttura mediana del dominio della chinasi di tipo selvatico e del dominio chinasi di tipo s'ELEREA supportano anche questi risultati (Figura 4C): sia i domini di chinasi di tipo selvaggio che quelli della chinasi di tipo "EL-EL" hanno una stabilità generale simile per le conformazioni sovrapposte, ad eccezione del loop β3-C e dell'elica di C, che sono chiaramente più stabili in EG. Questi risultati indicano che la cancellazione della sequenza ELREA frena il movimento dell'elica dello stato attivo, limitando e stabilizzando così la conformazione attiva. Inoltre, dal momento che l'elica di C fa parte dell'interfaccia asimmetrica del dimer, i vincoli sull'elica di C nel mutante stabilizzerebbero molto probabilmente il dimer asimmetrico, prolungando la durata dello stato attivato.
Un'altra applicazione del protocollo è studiare il comportamento delle interazioni chiave intra e intermolecolari che si verificano durante la simulazione. Così, l'interazione tra K745 ed E762, che è fondamentale per l'attività ezimatica EGFR, è stato analizzato sia per la forma attiva wild-type che per la chinasi EGFR di forma attiva misurando la percentuale di occupazione dei legami di idrogeno formati tra gli atomi polari della catena laterale dei due residui durante le simulazioni MD (Figura 5A): questa interazione elettrostatica chiave si è formata più spesso nel dominio della chinasi di tipo .ELREA rispetto al dominio di chinasi di tipo selvatico, a causa della più stabile elica. Durante la simulazione sono state valutate anche le interazioni tra i domini di chinasi mg2-ATP e i domini di chinasi di tipo selvaggio e EGFR(Figura 5B) (figura5C): il numero di obbligazioni a idrogeno è stato maggiore per il valore medio di 4,0 - 95% CI 0,03) rispetto al tipo selvaggio EGFR (valore medio 3,2 - 95% CI 0,04). Un'ulteriore analisi dei legami di idrogeno ha rivelato che K745 interagisce più frequentemente con i gruppi di fosfati dell'ATP in EGFR, che è collegato all'interazione più stabile K745-E762 osservata nella simulazione del dominio mutante EGFR kinase.
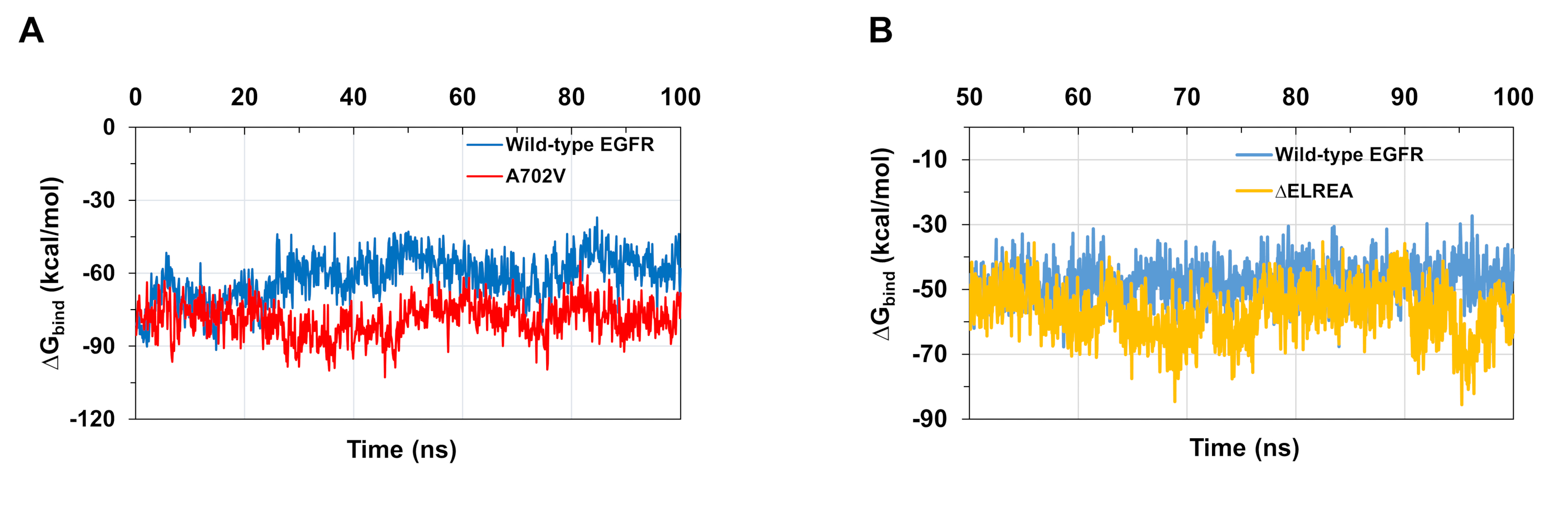
Le simulazioni MD descritte nel protocollo sono utili anche per valutare l'energia relativa libera del legame per le interazioni proteina-proteina e proteina-ligando. Le energie vincolanti tra l'attivatore e i domini di chinasi del ricevitore di tipo selvaggio e A702V EGFR, e tra ATP e i domini di chinasi EGFR mutanti di tipo selvaggio e di tipo zELREA, sono stati calcolati da calcoli di superficie generalizzata della meccanica molecolare (MMGBSA)(Figura 6A): il mutante A702Vbind ha prodotto un valore medio di legame medio inferiore (media dibind legame di ZG - -76 kcal/mol - 9 5% CI 0,47), che rappresenta interazioni di dimer più favorevoli, a differenza del dominio EGFR di tipo selvaggio (media di legame con il GG - -61 kcal/mol - 95% CI 0,61). Questa osservazione è coerente con il segmento juxtamembrane B più stabile e l'interfaccia dimer più stretta a causa delle maggiori interazioni idrofobiche osservate per il dominio della chinasi A702V EGFR. Nel caso dell'associazione ATP ai domini di chinasi EGFR di tipo selvaggio e di EGFR (Figura 6B), i calcoli di MMGBSA prevedono un'associazione ATP più forte con il mutante di tipo zELREA(associazione media di G -57) kcal/mol - 95% CI 0,43) rispetto all'EGFR di tipo selvatico (media dilegame di G -48 kcal/mol - 95% CI 0,33). Questo risultato è in linea con il maggior numero di obbligazioni a idrogeno registrate tra ATP e EGFR(Figura 5C)rispetto al dominio di tipo selvaggio.
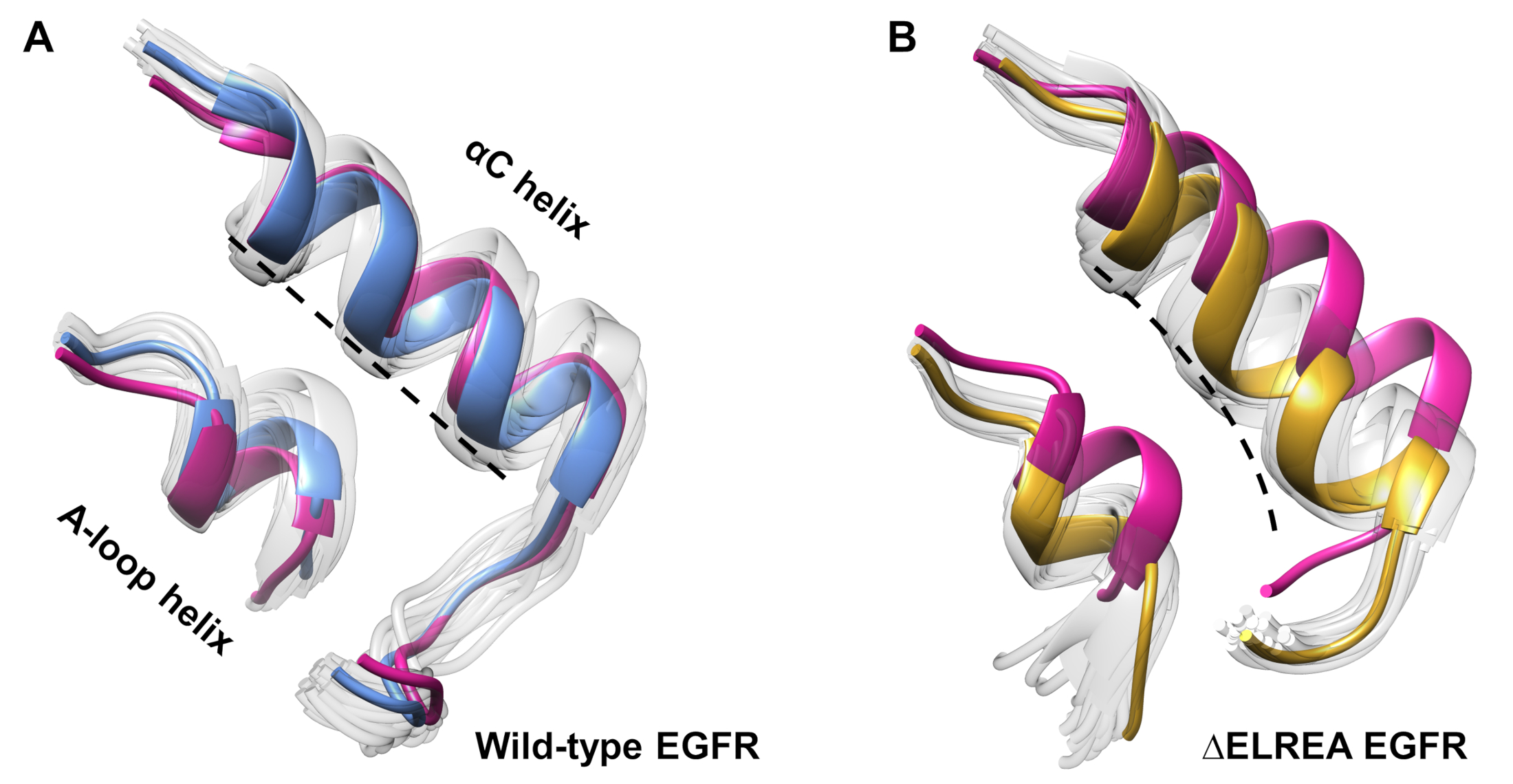
Il protocollo può anche essere utilizzato per studiare i cambiamenti conformazionali osservati durante una simulazione. Nello studio attuale, gli effetti della mutazione di EGFR inattiva sono stati studiati mediante l'ispezione visiva e la sovrapposizione delle conformazioni campiore dalla simulazione. L'analisi ha rivelato un movimento verso l'interno dell'elica di C nel dominio della chinasi EGFR(Figura 7A),un cambiamento strutturale previsto durante la transizione allo stato attivo. Al contrario, l'elica di tipo selvaggio EGFR inattiva ha mantenuto la sua conformazione iniziale (Figura 7B). Così, le simulazioni MD sostengono la proposta che la mutazione di delezione, mostrata sperimentalmente per aumentare l'attività di chinasi40,41, promuove uno spostamento conformazionale dalla chinasi inattiva allo stato attivo.

Figura 4: stabilità conformazionale di tipo selvaggio e mutante del dominio della chinasi EGFR attivo durante le simulazioni MD. (A) RMSD (atomi backbone) sul segmento juxtamembrane B del dominio di chinasi del ricevitore di tipo selvaggio (blu) e A702V (rosso). (B) RMSF (atomi di C) sui residui dell'elica di C: tipo selvatico (blu) e zELREA (oro). (C) Conformazioni campiore sovrapposte di tipo selvaggio (a sinistra) e di dominio chinasi EGFR (a destra) di tipo selvatico (a sinistra); tracce a catena colorate in base agli atomi DI RMSD (C) di ciascun residuo rispetto alla struttura mediana. La colorazione varia dal blu al bianco al rosso, rappresentando regioni di stabilità conformazionale alta a bassa. Si noti che le regioni "libere" N-terminal del dominio di chinasi isolato, di colore rosso, non mostrerebbero questo livello di mobilità nella struttura EGFR intatta. Figure adattate da Chakroborty et al.9 (Figura 4A riprodotta con il permesso del Journal of Biological Chemistry) e Tamirat et al.12. Si prega di fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 5: Caratteristiche principali osservate nella chinasi del ricevitore attivo durante le simulazioni MD: il ponte di sale K745-E762, l'elica di C e le interazioni con ATP. (A) Occupazione percentuale dell'interazione K745-E762 durante la simulazione dei domini di chinasi EGFR di tipo selvaggio (blu) e EGFR (oro). (B) Residui del tipo selvaggio e del mutante di tipo zELREA che interagiscono con ATP (bastone). Le coordinate Mg2 (verde) con ATP e D855. (C) Il numero di obbligazioni a idrogeno formate dall'ATP con entrambi i domini di chinasi di tipo selvaggio e di EGFR durante le simulazioni MD. Figura di Tamirat et al.12. Si prega di fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 6: Durante le simulazioni si osservano energie libere relative inferiori per i domini mutanti della chinasi. (A) Energie di legame calcolate per l'interazione tra l'attivatore e i domini di chinasi del ricevitore del tipo selvaggio (blu) e A702V (rosso) EGFR. (B) Legarel'ATP ai domini di chinasi EGFR di tipo selvaggio (blu) e EGFR (oro). Figure adattate da Chakroborty et al.9 (Figura 6A riprodotta con il permesso del Journal of Biological Chemistry) e Tamirat et al.12. Si prega di fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 7: conformazioni sovrapposte dal dominio di chinasi EGFR inattivo di tipo selvaggio e .ELREA. Conforme dell'elica di tipo ariato(A)e dell'elica A-loop di ( A ) wild-type (strutturamedianain blu) e ( B ) EGRS (oro). Altre conformazioni campiore, bianco sbiadito; strutture iniziali prima delle simulazioni MD, rosa. Figura di Tamirat et al.12. Si prega di fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
Discussione
Il protocollo descritto in questo studio si concentra sull'utilizzo di simulazioni di dinamiche molecolari per studiare le alterazioni strutturali locali e globali che derivano dall'attivazione di mutazioni somatiche del dominio della chinasi EGFR. Sebbene le strutture cristalline a raggi X di tipo selvaggio e mutanti EGFR forniscano informazioni strutturali inestimabili, rappresentano una o poche rappresentazioni statiche. Tuttavia, inerente alla funzione biologica degli ErbB sono le transizioni necessarie tra la chinasi tirosina ezimaticamente inattiva e attiva, invocando cambiamenti dinamici sia nella struttura che nelle interazioni intramolecolari tra i monomeri della chinasi. Sono state quindi effettuate simulazioni MD per sondare la natura dinamica del dominio della tirosina della tirosina EGFR, tra cui la struttura di tipo selvaggio, la mutazione di delezione introdotta da EELREA e la mutazione A702V. Queste simulazioni sono riuscite a chiarire il probabile ruolo di queste mutazioni nelle strutture e come i loro effetti sulla conformazione del dominio della chinasi della tirosina avrebbero portato agli aumenti osservati sperimentalmente nell'attività della chinasi EGFR.
Un passo cruciale in questo protocollo è l'uso di una struttura rilevante per valutare l'impatto della mutazione. Un modo per selezionare una struttura di input di simulazione pertinente consiste nel visualizzare la posizione della mutazione nella struttura statica 3D ed esaminarne il possibile impatto rispetto agli amminoacidi e alle unità strutturali adiacenti. In questo studio, ad esempio, poiché la mutazione A702V EGFR si trova nel segmento juxtamembrane B che forma l'interfaccia dimer asimmetrica, l'uso della struttura dimera per la simulazione in contrapposizione al monomero è fondamentale. L'uso di una struttura monomerica avrebbe esposto il segmento juxtamembrane B della chinasi del ricevente al solvente, privarlo dalle interazioni stabilizzanti, arricchito dalla mutazione a un residuo idrofobico più grande e dalle interazioni con l'isolaucina 941 dai residui del lobo C della chinasi dell'attivatore. Inoltre, è interessante notare che la struttura 3D rappresentata dalle coordinate in un file PDB non corrisponde necessariamente alla struttura biologicamente rilevante che dovrebbe essere utilizzata per lo studio. Ad esempio, con la struttura di ErbB4, codice PDB 3BCE, le coordinate PDB corrispondono a un trimer, ma ciò è dovuto ai contatti di cristallo (pochi contatti tra i monomeri si vedono quando si visualizza questa struttura). Matrici all'interno del file PDB possono essere utilizzate (ad esempio, all'interno di Chimera) per ricostruire le strutture cristallograficamente correlate, che possono essere visualizzate per identificare le catene che corrispondono alla struttura 3D biologicamente rilevante come riportato nella pubblicazioneoriginale 42. Un altro passo essenziale del protocollo è quello di preparare correttamente la struttura di input di simulazione, come la costruzione di aminoacidi mancanti in diverse regioni del ciclo, e soprattutto dove si trova nelle vicinanze della mutazione. Sebbene nel PDB esistano numerose strutture EGFR di tipo selvaggio, sono disponibili solo un numero limitato di strutture EGFR mutanti. Di conseguenza, anche le strutture mutanti devono essere modellate; per una singola mutazione dei residui come A702V, la chimera è stata utilizzata per mutare il residuo; mentre, per la mutazione di delezione di EELREA, è stato utilizzato Modeller.
I vari parametri utilizzati nei file di input di simulazione - ad esempio, il numero di cicli di minimizzazione, il riscaldamento del sistema alla temperatura desiderata in una sola volta o invece il riscaldamento lentamente attraverso diverse temperature intermedie, il periodo di tempo per l'equilibrio e per le simulazioni di produzione - possono essere modificati in base alla molecola di studio, allo scopo del lavoro e alle proprie preferenze. Durante l'esecuzione di simulazioni MD, è anche comune imbattersi in errori che possono derivare dai file di input, problemi relativi al software di simulazione in uso o anche un errore dell'utente. Pertanto, è molto importante comprendere l'origine degli errori esaminando attentamente eventuali messaggi di errore. La maggior parte dei programmi di simulazione dispone di una mailing list in cui gli utenti possono porre domande agli sviluppatori di software e ad altri utenti con cui la maggior parte dei problemi può essere risolta. Inoltre, i manuali utente forniscono assistenza significativa per comprendere i dettagli del protocollo di simulazione, inclusi presupposti e limitazioni. Anche se la simulazione MD è uno strumento importante per esplorare le proprietà dinamiche delle molecole, ricordate che i risultati computazionali devono essere valutati attentamente in combinazione con altre fonti di informazioni per valutarne la validità. Quando possibile, collaborare con ricercatori esperti sulle proteine in esame, in particolare dove vengono effettuati studi sperimentali sul laboratorio umido pertinenti, che servono a fornire risultati per l'interpretazione strutturale e a suggerire esperimenti che possono essere fatti sulla base di osservazioni strutturali per testare ipotesi.
In questo studio, il protocollo è stato efficace nell'esaminare gli impatti strutturali dinamici delle mutazioni di EELREA e A702V sulle strutture della chinasi EGFR. Le simulazioni hanno rivelato che la zeELREA limita l'elica di C funzionalmente essenziale e promuove un passaggio conformazionale dalla chinasi inattiva a una chinasi attiva stabilizzata. I risultati della simulazione sono supportati in modo indipendente dai dati di risposta ai farmaci che hanno dimostrato gli effetti degli inibitori della chinasi della tirosina sulle linee cellulari del cancro del polmone con la mutazione della delezione di EGFR e LGFR di tipo selvaggio, dove è stata segnalata una maggiore inibizione da parte dei farmaci che riconoscono la conformazione della chinasi attiva per l'EGFR12. Con la mutazione A702V, le simulazioni MD indicano, rispetto al tipo selvaggio, una maggiore stabilizzazione dell'interfaccia chinasi attivatore-ricevitore, nonché una maggiore affinità dell'attivatore e della chinasi del ricevitore l'uno per l'altro, sostenendo insieme il mantenimento della conformazione attivata della kinase EGFR. La mutazione A702V, situata sul segmento juxtamembrane B della chinasi del ricevitore, aumenterebbe le interazioni idrofobiche con la chinasi dell'attivatore, funzionando per prolungare la durata dello stato attivato. La mutazione A702V supporta la sopravvivenza cellulare in assenza di fattore di crescita ed è stata identificata in uno screening in vitro per le mutazioni EGFR9.
Divulgazioni
Gli autori non hanno nulla da rivelare.
Riconoscimenti
Questa ricerca è finanziata da sovvenzioni a M.S.J dell'Accademia di Finlandia (308317, 320005), Sigrid Juselius Foundation e Tor, Joe e Pentti Borg memorial fund, e a K.E. dell'Accademia di Finlandia (274728, 316796), della Cancer Foundation of Finland e del Turku University Central Hospital. Il M.E.T. è finanziato dalla rete di biologia instrusa e strutturale dell'Akademi. Ringraziamo il CSC IT Center for Science per le risorse informatiche e il Dr. Jukka Lehtonen per il supporto IT nell'ambito della rete bioinformatica Biocenter Finland; e Biocenter Finlandia rete di infrastrutture di biologia strutturale.
Materiali
| Name | Company | Catalog Number | Comments |
| Amber software | University of California, San Francisco | Version 2018 | Executable |
| Chimera program | Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco | Version 1.13.1 | Executable |
| EGFR struture files | The Protein Data Bank | 3D coordinates of EGFR structures | |
| Maestro | Schrödinger LLC | Version 2018-3 | Executable |
| Modeller program | The Andrej Šali Lab, Departments of Biopharmaceutical Sciences and Pharmaceutical Chemistry, University of California San Francisco | Included in the Chimera program | |
| VMD software | Theoretical and Computational Biophysics Group, University of Illinois at Urbana-Champaign | Version 1.9.3 | Executable |
Riferimenti
- Yarden, Y., Sliwkowski, M. X. Untangling the ErbB signalling network. Nature Reviews Molecular Cell Biology. 2, 127-137 (2001).
- Lemmon, M. A., Schlessinger, J., Ferguson, K. M. The EGFR family: not so prototypical receptor tyrosine kinases. Cold Spring Harbor Perspectives in Biology. 6, a020768 (2014).
- Arteaga, C. L., Engelman, J. A. ERBB receptors: From oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell. 2, 282-303 (2014).
- Mishra, R., Hanker, A. B., Garrett, J. T. Genomic alterations of ERBB receptors in cancer: Clinical implications. Oncotarget. 8, 114371-114392 (2017).
- . cBioPortal for Cancer Genomics Available from: https://www.cbioportal.org (2020)
- Lynch, T. J., et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. New England Journal of Medicine. 350 (21), 2129-2139 (2004).
- Paez, J. G., et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 304 (5676), 1497-1500 (2004).
- Pao, W., et al. EGF receptor gene mutations are common in lung cancers from "never smokers" and are associated with sensitivity of tumors to gefitinib and erlotinib. Proceedings of the National Academy of Sciences U.S.A. 101 (36), 13306-13311 (2004).
- Chakroborty, D., et al. Unbiased in vitro screen for activating EGFR mutations. Journal of Biological Chemistry. 294 (24), 9377-9389 (2019).
- Leahy, D. J. Structure and Function of the Epidermal Growth Factor (EGF/ErbB) Family of Receptors. Advances in Protein Chemistry. 68, 1-27 (2004).
- Roskoski, R. ErbB/HER protein-tyrosine kinases: Structures and small molecule inhibitors. Pharmacological Research. 87, 42-59 (2014).
- Tamirat, M. Z., Koivu, M., Elenius, K., Johnson, M. S. Structural characterization of EGFR exon 19 deletion mutation using molecular dynamics simulation. PLoS ONE. 14 (9), e0222814 (2019).
- Ding, L., et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 455, 1069-1075 (2008).
- Kurppa, K. J., Denessiouk, K., Johnson, M. S., Elenius, K. Activating somatic ERBB4 mutations in non small-cell lung cancer. Oncogene. 35 (10), 1283-1291 (2016).
- Soung, Y. H., et al. Somatic mutations of the ERBB4 kinase domain in human cancers. International Journal of Cancer. 118, 1426-1429 (2006).
- Tvorogov, D., et al. Somatic mutations of ERBB4: selective loss-of-function phenotype affecting signal transduction pathways in cancer. Journal of Biological Chemistry. 284, 5582-5591 (2009).
- Hubbard, S. R., Till, J. H. Protein tyrosine kinase structure and function. Annual Review of Biochemistry. 69 (1), 373-398 (2000).
- Huse, M., Kuriyan, J. The conformational plasticity of protein kinases. Cell. 109 (3), 275-282 (2002).
- Jura, N., et al. Catalytic control in the EGF receptor and its connection to general kinase regulatory mechanisms. Molecular Cell. 42, 9-22 (2011).
- Karplus, M., Kuriyan, M., J, Molecular dynamics and protein function. Proceedings of the National Academy of Sciences U.S.A. 102 (19), 6679-6685 (2005).
- Shan, Y., Arkhipov, A., Kim, E. T., Pan, A. C., Shaw, D. E. Transitions to catalytically inactive conformations in EGFR kinase. Proceedings of the National Academy of Sciences U.S.A. 110 (18), 7270-7275 (2013).
- Reckamp, K. L., et al. A phase I trial to determine the optimal biological dose of celecoxib when combined with erlotinib in advanced non-small cell lung cancer. Clinical Cancer Research. 12 (11 Pt 1), 3381-3388 (2006).
- Pettersen, E. F., et al. UCSF Chimera-a visualization system for exploratory research and analysis. Journal of Computational Chemistry. 25 (13), 1605-1612 (2004).
- Berman, H. M., et al. The Protein Data Bank. Nucleic Acids Research. 28 (1), 235-242 (2000).
- Zhang, X., Gureasko, J., Shen, K., Cole, P. A., Kuriyan, J. An Allosteric Mechanism for Activation of the Kinase Domain of Epidermal Growth Factor Receptor. Cell. 125 (6), 1137-1149 (2006).
- Stamos, J., Sliwkowski, M. X., Eigenbrot, C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. Journal of Biological Chemistry. 277 (48), 46265-46272 (2002).
- Sogabe, S., et al. Structure-Based Approach for the Discovery of Pyrrolo[3,2-d]pyrimidine-Based EGFR T790M/L858R Mutant Inhibitors. ACS Medicinal Chemistry Letters. 4 (2), 201-205 (2013).
- Sali, A., Blundell, T. L. Comparative protein modelling by satisfaction of spatial restraints. Journal of Molecular Biology. 234 (3), 779-815 (1993).
- Yun, C. H., et al. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell. 11 (3), 217-227 (2007).
- Park, J. H., Liu, Y., Lemmon, M. A., Radhakrishnan, R. Erlotinib binds both inactive and active conformations of the EGFR tyrosine kinase domain. Biochemical Journal. 448 (3), 417-423 (2012).
- . Release 2018-3: Maestro Available from: https://www.schrodinger.com/maestro (2018)
- Case, D. A., et al. . AMBER 2018. , (2018).
- Maier, J. A., Martinez, C., Kasavajhala, K., Wickstrom, L., Hauser, K. E., Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. Journal of Chemical Theory and Computation. 11 (8), 3696-3713 (2015).
- Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W., Klein, M. L. Comparison of simple potential functions for simulating liquid water. Journal of Chemical Physics. 79 (2), 926-935 (1983).
- Meagher, K. L., Redman, L. T., Carlson, H. A. Development of polyphosphate parameters for use with the AMBER force field. Journal of Computational Chemistry. 24 (9), 1016-1025 (2003).
- Humphrey, W., Dalke, A., Schulten, K. VMD: Visual molecular dynamics. Journal of Molecular Graphics. 14 (1), 33-38 (1996).
- Melvin, R. L., Salsbury, F. R. Visualizing ensembles in structural biology. Journal of Molecular Graphics and Modelling. 67, 44-53 (2016).
- Roe, D. R., Cheatham, T. E. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. Journal of Chemical Theory and Computation. 9 (7), 3084-3095 (2013).
- Miller, B. R., et al. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. Journal of Chemical Theory and Computation. 8 (9), 3314-3321 (2012).
- Guha, U., et al. Comparisons of tyrosine phosphorylated proteins in cells expressing lung cancer-specific alleles of EGFR and KRAS. Proceedings of the National Academy of Sciences U.S.A. 105 (37), 14112-14117 (2008).
- Furuyama, K., et al. Sensitivity and kinase activity of epidermal growth factor receptor (EGFR) exon 19 and others to EGFR-tyrosine kinase inhibitors. Cancer Science. 104 (5), 584-589 (2013).
- Qiu, C., et al. Mechanism of Activation and Inhibition of the HER4/ErbB4 Kinase. Structure. 6 (3), 460-467 (2008).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati