
Method Article
Entschlüsselung der strukturellen Auswirkungen der Aktivierung von EGFR-Somatischen Mutationen mit Molekulardynamik-Simulation
In diesem Artikel
Zusammenfassung
Ziel dieses Protokolls ist es, mithilfe molekularer Dynamiksimulationen die dynamischen Strukturveränderungen zu untersuchen, die durch aktivierende Mutationen des EGFR-Kinaseproteins auftreten.
Zusammenfassung
Zahlreiche somatische Mutationen, die in der epidermalen Wachstumsfaktor-Rezeptor-Familie (EGFR) (ErbB) von Rezeptor-Tyrosin-Kinasen (RTK) auftreten, wurden von Krebspatienten berichtet, obwohl relativ wenige getestet wurden und nachweislich funktionelle Veränderungen bei ErbBs verursachen. Die ErbB-Rezeptoren werden bei Ligandenbindung verdorren und aktiviert, und dynamische Konformationsänderungen der Rezeptoren sind für die Induktion der nachgeschalteten Signalisierung inhärent. Für zwei Experimentell gezeigte Mutationen zur Veränderung der EGFR-Funktion, A702V und der Löschmutation746ELREA750, veranschaulichen wir im folgenden Protokoll, wie molekulare Dynamik (MD)-Simulationen die (1) konforme Stabilität der mutierten Tyrosinkinasestruktur im Vergleich zu Wildtyp EGFR untersuchen können; (2) strukturelle Folgen und konforme Übergänge und ihr Verhältnis zu beobachteten funktionellen Veränderungen; (3) Auswirkungen von Mutationen auf die Stärke der Bindung atP sowie auf Bindung zwischen den Kinase-Domänen im aktivierten asymmetrischen Dimer; und (4) Auswirkungen der Mutationen auf schlüsselhafte Wechselwirkungen innerhalb der EGFR-Bindungsstelle, die mit dem aktivierten Enzym verbunden sind. Das Protokoll bietet ein detailliertes Schritt-für-Schritt-Verfahren sowie Anleitungen, die generell für die Untersuchung von Proteinstrukturen mit Hilfe von MD-Simulationen als Mittel zur Untersuchung der Strukturdynamik und des Zusammenhangs mit biologischer Funktion nützlich sein können.
Einleitung
Die Humane epidermale Wachstumsfaktor-Rezeptor (EGFR) Familie (ErbB) von Rezeptor-Tyrosin-Kinasen (RTKs) umfasst vier Mitglieder - EGFR/ErbB1/HER1, ErbB2/HER2, ErbB3/HER3 und ErbB4/HER4. Die ErbB-Rezeptoren regulieren grundlegende zelluläre Prozesse wie Zellwachstum und -proliferation, Differenzierung, Migration und Überleben1,2, und sind somit potente Proto-Onkogene. Aberrante Aktivität der ErbB-Rezeptoren, insbesondere EGFR und ErbB2, wurde häufig mit menschlichen Krebserkrankungen in Verbindung gebracht, was ErbB-Rezeptoren zu zentralen Zielen für Krebstherapeutikamacht 2,3.
Mehrere somatische Veränderungen von ERBB-Genen wurden durch menschliche Malignitäten3,4,5berichtet. Die am besten charakterisierten Beispiele sind die wiederkehrenden, aktivierenden Punktmutationen und kurzen In-Frame-Deletionen in der EGFR-Kinase-Domäne bei nicht-kleinzelligem Lungenkrebs (NSCLC). Diese EGFR-Mutationen sind die wichtigsten Treiber für das Krebswachstum und sagen die Empfindlichkeit gegenüber EGFR voraus, die auf Krebsmedikamenteabzielt 6,7,8. Bei den meisten Krebsarten treten jedoch somatische Mutationen in EGFR außerhalb dieser wiederkehrenden "Hotspots" auf und verteilen sich über die gesamte 1210-Rückstandsspanne des Rezeptors. Tatsächlich wurden die meisten Rückstände entlang der EGFR-Primärsequenz bei menschlichem Krebs mutiert9. Abgesehen von den wenigen Hotspots bleibt jedoch die funktionelle Bedeutung der überwiegenden Mehrheit der krebsassoziierten EGFR-Mutationen unbekannt.
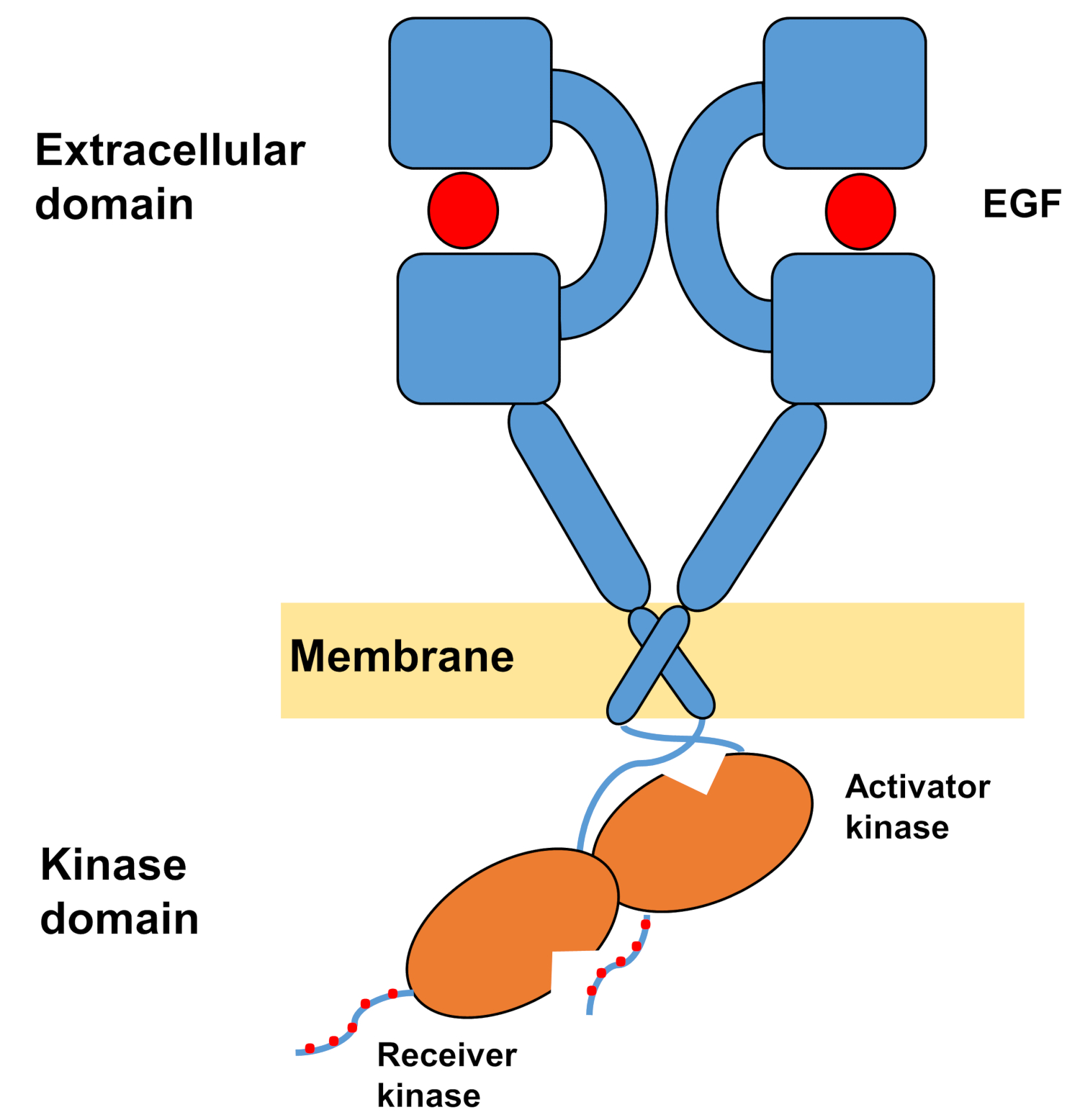
Die monomere Struktur von ErbBs besteht aus einer großen extrazellulären Aminoterminaldomäne, gefolgt von einer einzigen Transmembranhelix, die zur intrazellulären Tyrosinkinase-Domäne und zur C-terminalen Schwanzregion führt, die Docking-Sites für intrazelluläre Signalproteine enthält. Ligandbindung löst eine dramatische Konformationsänderung in der extrazellulären Domäne aus, die die Bildung von Rezeptordimermern erleichtert, indem sie die Dimerisierungsarme freilegt, die symmetrisch übereinander kreuzen und mit ihren aromatischen/hydrophoben Oberflächen interagieren. Bei der Bildung des Rezeptor-Dimers kommen die Tyrosinkinase-Domänen asymmetrisch in Kontakt(Abbildung 1), was zur Aktivierung der Kinasen führt, die die C-Terminalschwänze der Rezeptormonomere phosphorylieren, und anschließend bei der Aktivierung der nachgeschalteten Signalisierung10,11.

Abbildung 1: Struktur des EGFR-Dimers. EGFR dimerisiert, wenn die extrazellulären Domänen den Wachstumsfaktor binden (EGF, epidermaler Wachstumsfaktor). Die Empfängerkinase-Domäne wird dann durch asymmetrische Interaktion mit der Aktivatorkinase-Domäne aktiviert, und die C-Terminalschwänze werden bei Tyrosinrückständen autophosphoryliert (Modifiziert von Tamirat et al.12). Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Aufgrund der dynamischen strukturellen Umlagerungen, die während der  Monomer-Dimer-Übergänge auftreten, zusammen mit der Kinase-Aktivierung, die mit der Bildung eines asymmetrischen Dimers verbunden ist, können Mutationen entlang der gesamten Länge der Rezeptorstruktur potenziell einen Einfluss auf die Rezeptorfunktion haben. Hier beschreiben wir einige Beispiele aus unseren früheren Studien, in denen die Modellierung der Mutation und Visualisierung ausreichten, um die Folgen für die Funktion zu erklären.
Monomer-Dimer-Übergänge auftreten, zusammen mit der Kinase-Aktivierung, die mit der Bildung eines asymmetrischen Dimers verbunden ist, können Mutationen entlang der gesamten Länge der Rezeptorstruktur potenziell einen Einfluss auf die Rezeptorfunktion haben. Hier beschreiben wir einige Beispiele aus unseren früheren Studien, in denen die Modellierung der Mutation und Visualisierung ausreichten, um die Folgen für die Funktion zu erklären.
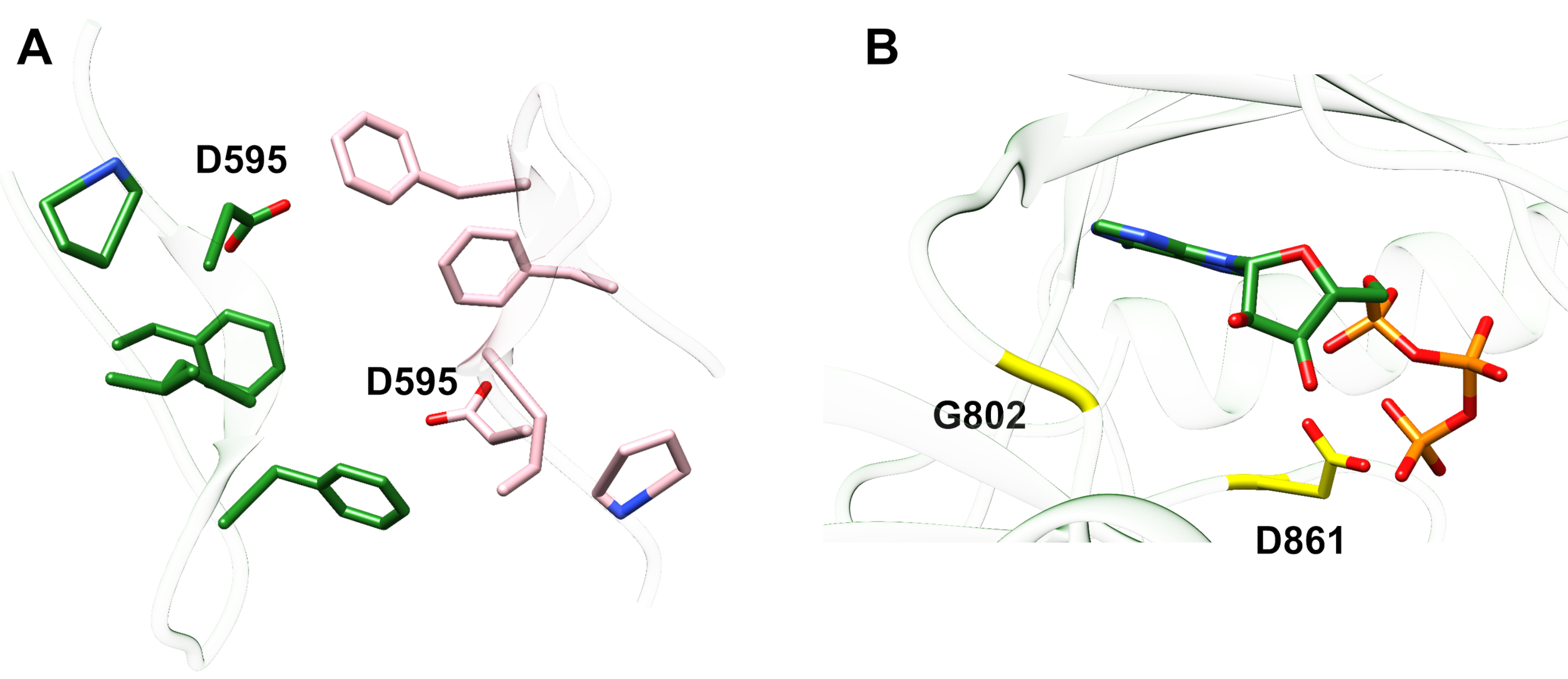
Beispiel 1: Eine gemeldete Mutation, D595V in ErbB413, führte zu erhöhter ErbB4-Dimerisierung und Phosphorylierung14. Die Visualisierung der Position der Mutation war ein entscheidender Faktor für das Verständnis der beobachteten funktionellen Effekte: D595V trat an der symmetrischen Kreuzung der dimerischen Arme der Ectodomain auf (Abbildung 2A). Die Arme sind weitgehend aromatisch und hydrophob, und der Ersatz von polarer Aspartinsäure durch Valin würde erwartet, die "klebrigen" hydrophoben Wechselwirkungen zu erhöhen, den Dimer zu stabilisieren und damit die Zeitdauer zu erhöhen, wenn die Phosphorylierung stattfindet14. Es war zunächst eine Überraschung, aspartat in jedem Arm zu finden, aber im Nachhinein könnte man es als einen Timing-Mechanismus für die Aktivität betrachten, wo die polaren Säure-Seitenketten die Affinität und Lebensdauer des intakten Dimers reduzieren und somit kinasevermittelte Phosphorylierung und Signalisierung begrenzen. Ein Ersatz durch Valine würde diese Sicherung dann durch eine weitere Stabilisierung des ErbB4-Dimers beseitigen.

Abbildung 2: Position einer ErbB4-aktivierenden Mutation und Mutationen, die Kinase-totes ErbB4 produzieren. (A) D595 (Aktivierung der D595V-Mutation) befindet sich auf den aromatischen/hydrophoben dimerischen Armen des ErbB4-Ectodomain-Modells; die Arme assoziiert auf Wachstumsfaktor Bindung; (in der Nähe rückstände werden als Stöcke angezeigt). (B) In ErbB4 hilft G802 (inaktivierende G802dup-Mutation) die Bindungstasche um den Adeninring von ATP und katalytischem D861 (inaktivierende D861Y-Mutation) bindet sowohl Mg2+ (nicht gezeigt) als auch die γ-Phosphat-Gruppe von ATP. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Beispiel 2: Man könnte erwarten, dass somatische Mutationen, die auf die ATP-bindende Stelle der Kinase-Domäne abzielen, enzymatische Aktivität verändern oder eliminieren würden, was zu einem beeinträchtigten oder kinase-toten Rezeptor führen würde, der nicht signalfähig ist. Von neun gemeldeten Mutationen von Patienten mit Brust-, Magen-, Dickdarm- oder NSCLC15hatten zwei der neun getesteten Mutationen eine stark verminderte Phosphorylierungsaktivität16:G802dup (G → GG) und D861Y. Beide inaktivierenden somatischen Mutationen wurden innerhalb der ATP-Bindungsstelle der Tyrosinkinase-Domänenstruktur gefunden (Abbildung 2B): flexibles Glycin, dupliziert, würde die Adenin-Ring-Stelle verändern und kleine Assigsäure, die durch das sperrige Tyrosin in der Nähe der Terminalphosphate ersetzt wird, würde Mg2+-ATP physikalisch an der Bindung hindern. Jedoch ist die da ErbB4 mit ErbB2 einen Heterodimer bilden kann - ErbB2 bindet keinen Wachstumsfaktor und ist auf die Assoziation mit einem ErbB angewiesen, der dies tut, um heterodimerisieren zu können - der ErbB2 (aktiv)-ErbB4(kinase-dead) Heterodimer würde die Zellproliferation über den Erk/Akt-Signalweg stimulieren, aber Zellen würden sich aufgrund von Kinase-toter ErbB4 und fehlender STAT5-Signalsignalaktivierung16nicht unterscheiden.
In neueren Studien wurde deutlich, dass die dynamischen Bewegungen von ErbBs relevant waren, um die Auswirkungen einiger Mutanten auf die ErbB-Funktion zu verstehen, insbesondere Mutationen, die innerhalb der Tyrosinkinase-Domäne auftreten. Die Tyrosinkinase-Domäne besteht aus einem N-Lappen (hauptsächlich β-Blätter) und C-Lappen (weitgehend alpha-helical), die durch die katalytische Stelle getrennt sind, an der ATP bindet. Der N-Lappen enthält die C-Helix und die P-Schleife, während die Aktivierungs- (A-Schleife) und die katalytischen Schleifen in der C-Lappen17,18,19vorhanden sind. Kristallstrukturen der Tyrosinkinase-Domäne zeigten zwei inaktive Konformationen, die Mehrheit der Strukturen hat den Src-ähnlichen inaktiven Zustand. In der aktiven Konformation zeigt das katalytische Aspartat des A-Loops auf die ATP-Bindungsstelle und die 'C-Helix ist auf die ATP-Bindungstasche ausgerichtet( "C-in"-Konformation), wodurch eine starke Glutamat-Lysin-Ionen-Paar-Wechselwirkung gebildet wird.
Da ErbBs und die Komponente Kinase-Domäne hochdynamische Entitäten sind, und insbesondere in Fällen, in denen die Auswirkungen von Mutationen auf Funktion und biologische Aktivität wahrscheinlich eng mit den Konformationszuständen der ErbBs verbunden sind, ist es wichtig, Mutationen in Bezug auf den Bereich der dynamischen Veränderungen zu bewerten, die sie erleben würden. Röntgenkristallstrukturen von ErbBs liefern statische Momentaufnahmen der 3D-Struktur, die für das Verständnis der dynamischen Folgen einer Mutation relevant sein können oder auch nicht. Um den Bereich der dynamischen Veränderungen zu untersuchen, die der "Energielandschaft" entsprechen, die einer dreidimensionalen (3D) Struktur zur Verfügung steht, werden molekulare Dynamik(MD) Simulationen weit verbreitet20verwendet. Bei Mutationen, die zu lokalen Konformationsänderungen innerhalb der Tyrosinkinase-Domäne oder zur Stabilisierung eines Komplexes führen würden, können Simulationen in der Größenordnung von 100 ns ausreichen. Größere konforme Veränderungen (z. B. Übergänge zwischen den aktiven und inaktiven Konformationen der Kinase-Domäne) erfordern jedoch eine längere Simulationszeit - in der Größenordnung von Mikrosekunden21.
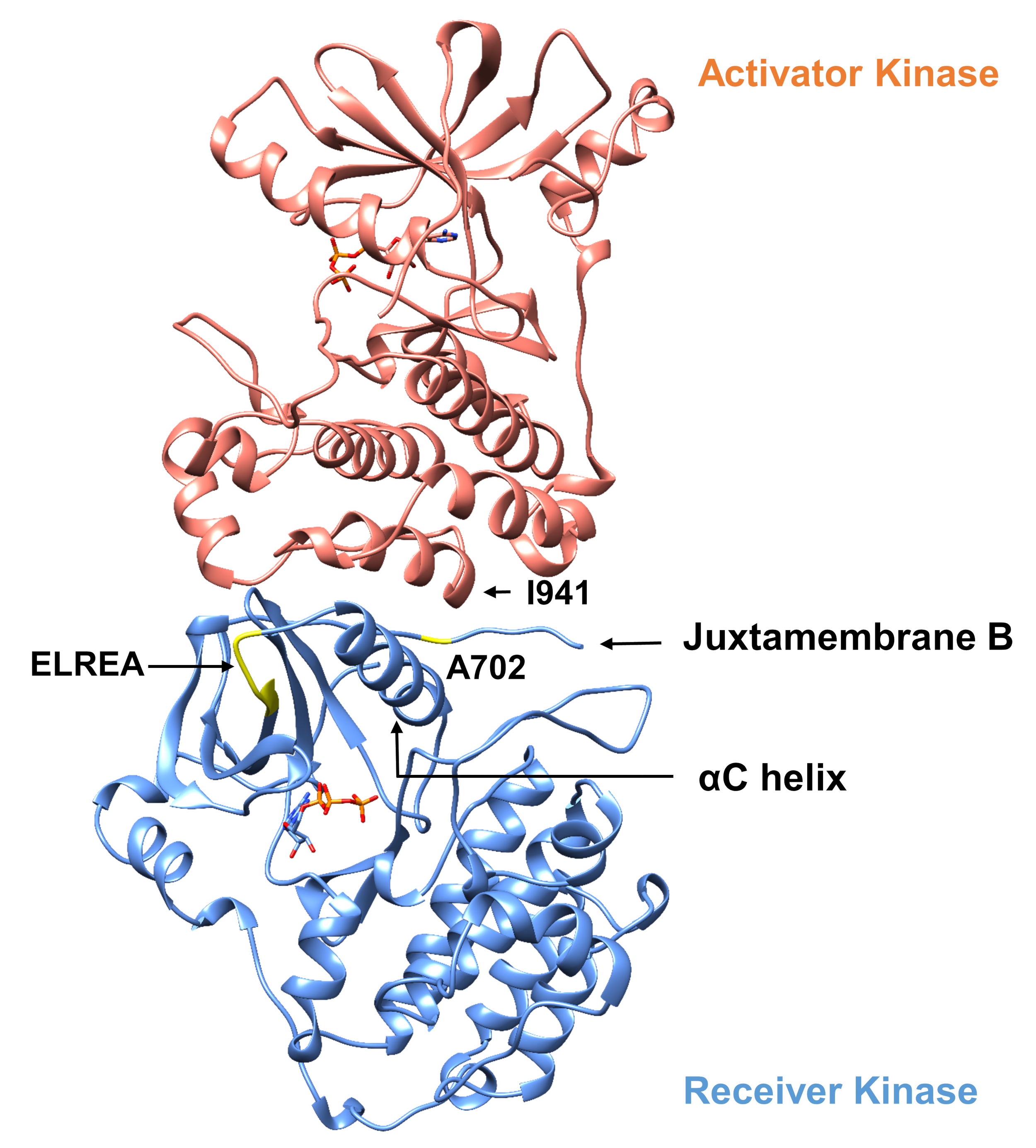
In Bezug auf das unten beschriebene Protokoll betrachten wir zwei aktivierende Mutationen innerhalb der Tyrosinkinase-Domäne (Abbildung 3). Beide Mutationen befinden sich innerhalb der Kinase-Domäne an Orten, an denen lokale Konformationsänderungen auftreten, die bestimmen, ob die Kinase aktiv ist oder nicht, und daher wurden MD-Simulationen in beiden Fällen angewendet. Im ersten Fall betrachten wir Änderungen, die sich direkt auf die ATP-Bindungsstelle und die katalytische Maschinerie der EGFR-Empfängerkinase-Domäne auswirken, und untersuchen insbesondere die Folgen einer Exon-19-Deletionsmutation, die in NSCLC4,7weithin involviert ist. Die Mutation von746ELREA750, die die Länge der Vorlaufzeit von '3-C vor der 'C-Helix - der Helix, die sich zur Bindung/aktiven Stelle der Kinase-Aktivierung bewegt und an der Bildung der kritischen elektrostatischen Wechselwirkung zwischen E762 der Helix und K745 beteiligt ist, durch Positionierung des Lysins zur Interaktion mit ATP - prädisponiert die Domäne für die Aktivierung12. Im zweiten Fall betrachten wir die A702V-Mutation von EGFR, die sich als eine neuartige Funktionsverstärkungs-Aktivierungsmutation herausstellte, die von der iScream-Plattform9 aufgedeckt und bei einem NSCLC-Patienten identifiziert wurde22. Alanin-702 auf der Empfängerkinase-Domäne befindet sich auf dem Nebenmembransegment B an der Schnittstelle des Empfängers und der Aktivatorkinase-Domänen, in denen dieser asymmetrische Kinase-Dimer-Komplex und Kinase-Konformationsänderungen für die Aktivierung9erforderlich sind.

Abbildung 3: Der asymmetrische Kinase-Domänendimer von EGFR. Die A702V-Mutation würde sich an der kritischen Schnittstelle der Aktivator- und Empfängerkinase-Domänen befinden, die an die C-Helix angrenzen und in der Nähe von Isoleucin 941 der Aktivatorkinase liegen. Konformationsveränderungen, die durch die Bildung des asymmetrischen Dimers induziert werden, führen zur Kinaseaktivierung. Die Schleife von 3 -C, die die ELREA-Sequenz enthält, geht direkt der C-Helix voraus; während der Aktivierung bewegt sich die C-Helix nach innen zur ATP-Bindungsstelle. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Protokoll
ANMERKUNG: Die detaillierten Schritte zur Untersuchung der Auswirkungen der Mutation von ELREA und A702V auf die EGFR-Struktur mithilfe von MD-Simulationen werden wie folgt erörtert:
1. Strukturvorbereitung
ANMERKUNG: Um die strukturellen Auswirkungen der ElREA-Mutation zu untersuchen, werden wildtypartige und mutierte Formen apoaktiver, ATP-gebundener aktiver und apoinaktiver EGFR-Monomerstrukturen wie folgt vorbereitet.
- Öffnen Sie das Visualisierungsprogramm Chimera23 (https://www.cgl.ucsf.edu/chimera/), um die wildtypische apo aktive EGFR-Kinasestruktur vorzubereiten. Klicken Sie im Menü Datei auf die Option Nach ID abrufen und wählen Sie die Protein Data Bank (PDB24) Datenbank aus und geben Sie den PDB-Code 2GS225 (Auflösung 2,8 ) an. Die PDB ist ein Endlager für 3D-Strukturen, die durch verschiedene experimentelle Techniken gelöst werden, einschließlich Röntgenkristallographie, Kernspinresonanzspektroskopie, Kryo-Elektronenmikroskopie und Neutronenbeugung.
- Bauen Sie die fehlenden Strukturelemente von 2GS2, indem Sie diese Segmente aus den EGFR PDB-Strukturen 1M1426 (2,6 ) und 3W2S27 (1,9 %) übernehmen. Öffnen Sie dazu 1M14 und 3W2S und überlagern Sie sie auf 2GS2 mit der MatchMaker-Option im Vergleichsmenü Tools → Structure.
- Schneiden Sie die hinzuzufügenden Segmente von 1M14 und 3W2S heraus. Wählen Sie die Endatome des Rückstands vor den Lücken in 2GS2 und die zu zuzubereitenden Atome aus 1M14 und 3W2S (detaillierte Informationen zu den hinzugefügten Segmenten finden Sie in Tabelle 1). Geben Sie in der Befehlszeile bond sel ein und drücken Sie enter. Diese Struktur ist die Vorlagenstruktur.
- Verwenden Sie die Vorlagenstruktur aus Schritt 1.2, um die Mutantform der EGFR-Kinase-Domäne zu erstellen. Erzeugen Sie die FASTA-Formatsequenz von "ELREA EGFR", indem Sie die Sequenz der Vorlagenstruktur (Favorite → Sequence → File → speichern unter) und dann die ELREA-Sequenz bei den Rückstandsnummern 746-750 löschen.
- Öffnen Sie die Sequenz "ELREA EGFR" in Chimera und richten Sie sie über das Menü Sequenz an der Reihenfolge der Vorlagestruktur 2GS2 aus. Wählen Sie im Ausrichtungsfenster die Option Struktur → Modeller (Homologie)28 aus.
- Geben Sie im Popupfenster die 2GS2-Verbundstruktur als Vorlage und die Mutantensequenz als zu modellierende Abfrage an. Drücken Sie dann OK. Wählen Sie ein mutiertes Modell unter den resultierenden Modellen aus, basierend auf der zDOPE-Punktzahl (in der Regel die niedrigste Punktzahl) und der visuellen Inspektion.
- Um die ATP-gebundene Wildtyp-EGFR-Kinasestruktur vorzubereiten, verwenden Sie die PDB-Struktur 2ITX29 (2,98 ) als Hauptstruktur. Erstellen Sie fehlende Segmente (siehe Tabelle 1) mit den Strukturen 2GS625 (2,6 ) und 3W2S nach dem Verfahren in Schritt 1.2. Konvertieren Sie den Liganden ANP in der resultierenden Struktur in ATP, indem Sie die PDB-Datei in einem Texteditor öffnen und das N3B Stickstoffatom von ANP in ein Sauerstoffatom ändern.
- Öffnen Sie die Struktur in Schritt 1.4 in Chimera, wie in Schritt 1.1 angegeben. Fügen Sie dieser Struktur aus der PDB-Struktur 2ITN29 (2,47 ° ) ein Magnesium-Ion hinzu, um eine ähnliche Positionierung für das Mg2+-Ionen zu erreichen.
- Mit der Struktur, die sich aus Schritt 1.5 ergibt, modellieren Sie die Mutantenform der ELREA nach Schritt 1.3.
| Apo aktiv EGFR | Apo inaktive EGFR | ATP-gebundenaktive EGFR | |
| Hauptstruktur | 2GS2 | 2GS7 | 2ITX |
| Strukturen, die zum Erstellen fehlender Schleifen verwendet werden | 1M14 (723-725) | 3W2S (958-984) | 2GS6 (862-865) |
| 3W2S (967-981) | 4HJO (848-850) | 3W2S (990-1001) |
Tabelle 1: Strukturen, die zum Erstellen von zusammengesetzten Modellen von apoaktiven, apo inaktiven und ATP-gebundenen aktiven Strukturen verwendet werden. Fehlende Regionen (Aminosäurebereich in Klammern) in der Hauptstruktur wurden aus den aufgeführten Strukturen aufgebaut.
- Um die wildtypische apo inaktive EGFR-Kinasestruktur vorzubereiten, öffnen Sie die PDB-Struktur 2GS725 (2,6 °C) wie in Schritt 1.1 und löschen Sie die gebundenen Liganden und kristallographischen Gewässer. Fügen Sie die fehlenden Segmente in 2GS7 (siehe Tabelle 1) aus den Strukturen 3W2S und 4HJO30 (2,75 ) nach dem Verfahren in Schritt 1.2 hinzu. Erstellen Sie das Mutantenmodell auf der Grundlage der endgültigen inaktiven EGFR-Struktur nach dem Verfahren in Schritt 1.3.
HINWEIS: Für die A702V-Mutationsstudie wird die asymmetrische Dimerstruktur EGFR untersucht, da sich die Mutation im Vsxtamembran-B-Segment der Kinase-Domäne befindet, die einen großen Teil der Dimer-Schnittstelle bildet. Die wild-type und A702V mutierten EGFR Strukturen werden wie folgt vorbereitet: - Die wildartige asymmetrische Dimerstruktur besteht aus der PDB-Struktur 2GS2, die zunächst in monomerer Form dargestellt wird. Um in die biologische Baugruppe umzuwandeln, die die Aktivator- und Empfängerkiinasen in der asymmetrischen Anordnung enthält, öffnen Sie 2GS2 in Chimera wie in (1.1) und führen Sie Symmetrieberechnungen durch, indem Sie auf das Menü Werkzeuge → Struktur → Einheitszellen klicken. Wählen Sie die 2GS2-Struktur aus und geben Sie Kopien erstellenein. Wählen Sie schließlich einen einzelnen asymmetrischen Dimer aus den mehreren Kopien des Dimers aus, die sich aus den Symmetrievorgängen ergeben, und speichern Sie ihn.
- Mit der wilden asymmetrischen EGFR-Struktur aus Schritt 1.8, bauen Sie die A702V Mutante, indem Sie Alanin 702 durch Valin mit der Option Tools → Structure Edit → Rotamers in Chimera ersetzen.
ANMERKUNG: Insgesamt werden sechs monomerische und zwei dimerische EGFR-Strukturen für die Mutationsstudien "ELREA" bzw. "A702V" vorbereitet. Jede Struktur wird anschließend zur Simulation mit dem Proteinaufbereitungsassistenten im Maestro-Programm31 verarbeitet und MD-Simulationen werden mit dem Bernsteinprogramm32gemacht. - Öffnen Sie die Struktur in Maestro, indem Sie die Option Datei → Importstruktur verwenden. Klicken Sie dann auf den Button des Proteinzubereitungsassistenten und wählen Sie Folgendes aus: Wasserstoffatome hinzufügen, fehlende Seitenkettenatome bauen, Protonationszustände von ionisierbaren Rückständen bei pH 7,0 mit PROPKA bestimmen, die Ausrichtung von Asparagin-, Glutamin- und Histidinrückständen für die Wasserstoffbindung optimieren und schließlich die Struktur minimieren.
2. Systemeinrichtung
- Öffnen Sie das im Amber-Softwarepaket enthaltene Sprungprogramm. Importieren Sie das ff14SB Kraftfeld33 (Quelle leaprc.protein.ff14SB) und TIP3P Wassermoleküle34 (Quelle leaprc.water.tip3p). Für die ATP-gebundenen Systeme importieren sie auch Parameter für ATP35 (loadamberparams frcmod.phosphate, loadamberprep ATP.prep). Dann laden Sie die Struktur (mol = loadpdb structure.pdb).
- Solvate die Struktur in einer Oktaeder-Box mit expliziten TIP3P-Wassermolekülen, die sich 10 A in alle Richtungen von den Oberflächenatomen des Proteins (solvateoct mol TIP3PBOX 10.0) erstreckt.
- Überprüfen Sie das gebaute System (Check mol) und neutralisieren Sie es durch Hinzufügen der notwendigen Ionen (Addionen mol Na+ 0). Um biomolekulare Systeme ausreichend zu modellieren, fügen Sie der Simulationsbox zusätzliche Na+/Cl-Atome hinzu, um die Salzkonzentration des Systems auf 0,15 M zu erhöhen(Zugänge mol Na+ X, Addions mol Cl- X), wobei X durch das Ergebnis ersetzt wird: gewünschte Salzkonzentration * Anzahl der Wassermoleküle * Volumen pro Wassermolekül * Avogadro-Zahl.
- Generieren und speichern Sie die Topologie- und Koordinatendateien des Systems, die als Eingaben für die nachfolgende Produktionssimulation dienen (saveamberparm mol X.prmtop X.inpcrd).
3. Molekulare Dynamiksimulation
- Mit Amber unterwerfen Sie das Simulationssystem zunächst 5000 Zyklen steilster Abstiegs- und Konjugatenergieminimierung, um ungünstige Konfigurationen zu umgehen. Führen Sie die Minimierung in mehreren Schritten durch, und senken Sie schrittweise die auf gelöste Atome aufgetragene Rückhalteregelung von 25 kcal mol-1 -2 bis 0 kcal mol-1 - -2.
- In der Minimierungseingabedatei min.indie variable maxcyc für den gesamten Minimierungszyklus (maxcyc = 5000) und ncyc so an, dass die Anzahl der Zyklen für den steilsten Abstiegsalgorithmus angezeigt wird. Verwenden Sie die restraint_wt Variable, um die durch den Parameter Rückhaltemaske angegebene Rückhaltekraft auf gelöste Atome anzuwenden. Führen Sie dann die Minimierung wie folgt aus:
$AMBERHOME/bin/sander -O -i min.in -o min.out -p X.prmtop -c X.inpcrd -r min.rst -ref X.inpcrd
HINWEIS: Die verwendete Strategie und die tatsächlichen Parameter können je nach den eigenen Vorlieben variieren. Details und Anleitungen finden Sie im Amber-Handbuch und auf der Website (https://ambermd.org/index.php)
- In der Minimierungseingabedatei min.indie variable maxcyc für den gesamten Minimierungszyklus (maxcyc = 5000) und ncyc so an, dass die Anzahl der Zyklen für den steilsten Abstiegsalgorithmus angezeigt wird. Verwenden Sie die restraint_wt Variable, um die durch den Parameter Rückhaltemaske angegebene Rückhaltekraft auf gelöste Atome anzuwenden. Führen Sie dann die Minimierung wie folgt aus:
- Erhitzen Sie das System für 100 ps von 0 K bis 300 K und stellen Sie eine 10 kcal mol-1 x-2-Rückhalteeinrichtung auf gelösten Atomen ein. Setzen Sie dazu tempi = 0.0, temp0 = 300.0, dt = 0.002 ps, nstlim = 50000 und restraint_wt = 10 in der heat.in Eingabedatei. Führen Sie die Heizung mit folgendem Befehl durch:
$AMBERHOME/bin/sander -O -i heat.in -o heat.out -p X.prmtop -c min.rst -r heat.rst -x heat.mdcrd -ref min.rst - Das System für 900 ps unter einem NPT-Ensemble ausdemieren; konstante Anzahl von Atomen, Temperatur (temp0 = 300.0) und Druck (ntp = 1), steuerung mit der Berendsen-Methode (ntt = 1). Stellen Sie für elektrostatische Wechselwirkungen mit großer Reichweite einen Abstandsabstand von 9 °(Schnitt = 9,0) ein. Nach und nach die solute Atom-Rückhalteeinrichtung auf 0,1 kcal mol-1 --2 (restraint_wt = 0,1) senken. Führen Sie die Ausgleichseingabedatei equil.in aus, die die oben genannten Parameter wie folgt beschreibt:
$AMBERHOME/bin/sander -O -i equil.in -o equil.out -p X.prmtop -c heat.rst -r equil.rst -x equil.mdcrd -ref heat.rst - Abschluss des Gleichgewichts mit einer hemmungslosen 5 ns Simulation (Satz dt = 0,002 ps, ntslim = 2500000).
$AMBERHOME/bin/sander -O -i equil_final.in -o equil_final.out -p X.prmtop -c equil.rst -r equil_final.rst -x equil_final.mdcrd -ref equil.rst - Überprüfen Sie, ob das System ausgeglichen wurde, indem Sie temperatur-, Druck-, Dichte- und Energiewerte untersuchen.
$AMBERHOME/bin/process_mdout.perl heat.out equil.out equil_final.out
xmgrace Zusammenfassung. TEMP/DENSITY/ETOT/EPTOT/EKTOT - Führen Sie die Produktionssimulation für 100 ns (set dt = 0.002 ps, ntslim = 50000000 in prod.in) und speichern Sie konformationen alle 10 ps (ntwx = 5000).
$AMBERHOME/bin/sander -O -i prod.in -o prod.out -p X.prmtop -c equil_final.rst -r prod.rst -x prod.mdrcd -ref equil_final.rst
4. Analyse
- Visuelle Inspektion
- Visualisieren Sie die Während der Wildtyp- und Mutanten-EGFR-Kinase-Simulationen abgetasteten Konformationen, indem Sie die X.prmtop Bernsteintopologiedateien und die entsprechenden prod.mdcrd-Trajektoriendateien in VMD36öffnen. Analysieren Sie anhand bequemer Sekundärstrukturdarstellungen die gesamte strukturelle Dynamik der Proteine aus der aufgezeichneten Flugbahn. Zeigen Sie spezifische Wechselwirkungen zwischen Atomen/Rückständen von Interesse an, wie z. B. die katalytisch essentielle Salzbrücke K745 - E762.
- Alternativ können Sie mehrere Konformationen speichern, die während der Simulation im PDB-Format abgetastet wurden, und öffnen Sie sie mit dem Chimera-Programm. Überlagern Sie die Strukturen in der Anfangs- oder Medianstruktur mit der Option MatchMaker. Zeigen Sie die Anfangs-/Medianstruktur in Volumenkörper und den Rest der ausgerichteten Strukturen in verblasstem Weiß an. Dieser Ansatz ermöglicht es, die aufgezeichneten Strukturbewegungen klarer zu visualisieren.
HINWEIS: Vorschläge für effektive Darstellungen und Dieverarbeitung von Konformationsensembles von MDS finden Sie in Melvin et al.37.
- RMSD- und RMSF-Analyse
- Berechnen Sie wurzelmittlere quadratische Abweichung (RMSD) und Wurzel-mittlere quadratische Fluktuation (RMSF) Berechnungen mit dem Cpptraj-Programm 38, um die globale Stabilität der Proteine zu analysieren und die Flexibilität der verschiedenen Struktureinheiten zu untersuchen. Geben Sie in den rmsd.in und rmsf.in Eingabedateien die Backbone-Atome (für RMSD) und die C-Atome (für RMSF) der Anfangsstruktur als Referenz für RMS-Fitting an. In den Dateien rmsd/rmsf.in importieren sie die Bernsteintopologiedateien (parm X.promtop) und die entsprechenden Trajektoriendateien (trajin prod.mdcrd). Führen Sie dann den Befehl Cpptraj -i rmsd/rmsf.inaus. Plotten Sie die Ausgabedaten für die Analyse.
- Alternativ können Sie konforme Ensembles ausrichten und jeden Rückstand auf der Grundlage des C-Atoms RMSD färben. Öffnen Sie dazu die Konformitäten in Chimera, und richten Sie sie an der Matchmaker-Option aus.
- Wechseln Sie zu Tools → Darstellung → Rendern nach Attribut. Wählen Sie Residues des Konformationsensembles und Cé RMSD als Attribute aus, und klicken Sie auf OK. Die Kettenspur der Konformationen wird dann von blau → weiß → rot gefärbt, die jeweils Bereiche mit hoher, mittlerer und niedriger struktureller Stabilität widerspiegeln.
- Wasserstoffbindungsanalyse
- Analysieren Sie die Wasserstoff-Bindungsinteraktion zwischen ATP und Wildtyp/ELREA EGFRs. Bereiten Sie ein Cpptraj-Skript hbond.invor, um diese Aufgabe auszuführen. Definieren Sie eine Wasserstoffbindung mit einem Spender-Akzeptanz-Abstand von weniger oder gleich 3,5 ° und einem Bindungswinkel von mehr als oder gleich 135°. Analyse nur für intermolekulare Wasserstoffbindungen mit der Nointramol-Variablen, d.h. Wasserstoffbindungen zwischen ATP und EGFR (hbond All nointramol dist 3.5 out nhb.agr avgout avghb.dat). Führen Sie das Skript als Cpptraj -i hbond.inaus.
- Verwenden Sie dieses Skript, um intramolekulare Wechselwirkungen zu bewerten, z. B. zwischen den Rückständen K745 und E762, die Schlüsselrückstände für die EGFR-Kinaseaktivität sind. Geben Sie dazu K745 als Wasserstoffbondspender und E762 als Wasserstoffbindungsakzeptor in hbond.in an und führen Sie das Skript entsprechend aus.
- Überwachung des Abstands zwischen Atomen
- Messen Sie den Abstand zwischen K745 und E762, indem Sie die Flugbahnen von Wildtyp- und ELREA-APO-EGFRs in VMDöffnen. Wählen Sie die C-Werte von Glu762 und Nz von Lys745 aus, indem Sie auf maus→ Etikett → Bondklicken. Überwachen Sie den Abstand während der Simulation, indem Sie ein Diagramm mit Grafiken → Beschriftungen → → Diagrammskalonen.
- Freie Energieberechnungen
- Um die geschätzten bindungsfreien Energien zwischen ATP und Wildtyp/ELREA EGFRs sowie zwischen aktivator- und Empfängerkinasen von Wildtyp/A702V EGFRs zu berechnen, verwenden Sie das im AMBER-Paket verfügbare molekularmechanik-generalisierte Born-Oberfläche (MM-GBSA)-Modul39. Stellen Sie ATP als Liganden und EGFR als Rezeptor in der Studie "ELREA" ein. Geben Sie in der A702V-Studie die Empfängerkinase als Liganden und die Aktivatorkinase als Rezeptor an.
- Zuerst bereiten Sie die Liganden-, Rezeptor- und Ligandenrezeptor-Komplexen PDB-Dateien getrennt im Schaltprogramm vor, indem Sie den PBRadii-Wert auf mbondi2einstellen. Speichern Sie für die PDB-Dateien die Gasphase Bernsteintopologie (.prmtop) und koordinieren Sie (.inpcrd) Dateien.
- Dann, in der mmgbsa-Eingabedatei, mmgbsa.in, setzen Sie igb = 2, saltcon = 0.1. Führen Sie Bindungsenergieberechnungen mit den Flugbahnen der Simulationen, den vorbereiteten Rezeptor-/Ligand-Bernsteindateien und den Parametern im mmgbsa.in mit dem in Bernstein verfügbaren MMPBSA.py Skript wie folgt aus:
$AMBERHOME/bin/MMPBSA.py -O -i mmgbsa.in -o mmgbsa.dat -sp X.prmtop -cp complex.prmtop -rp receptor.prmtop -lp ligand.prmtop -y prod.mdcrd -eo output.csv - Analysieren Sie die Ausgabedaten, output.csv, indem Sie Diagramme zeichnen.
Ergebnisse
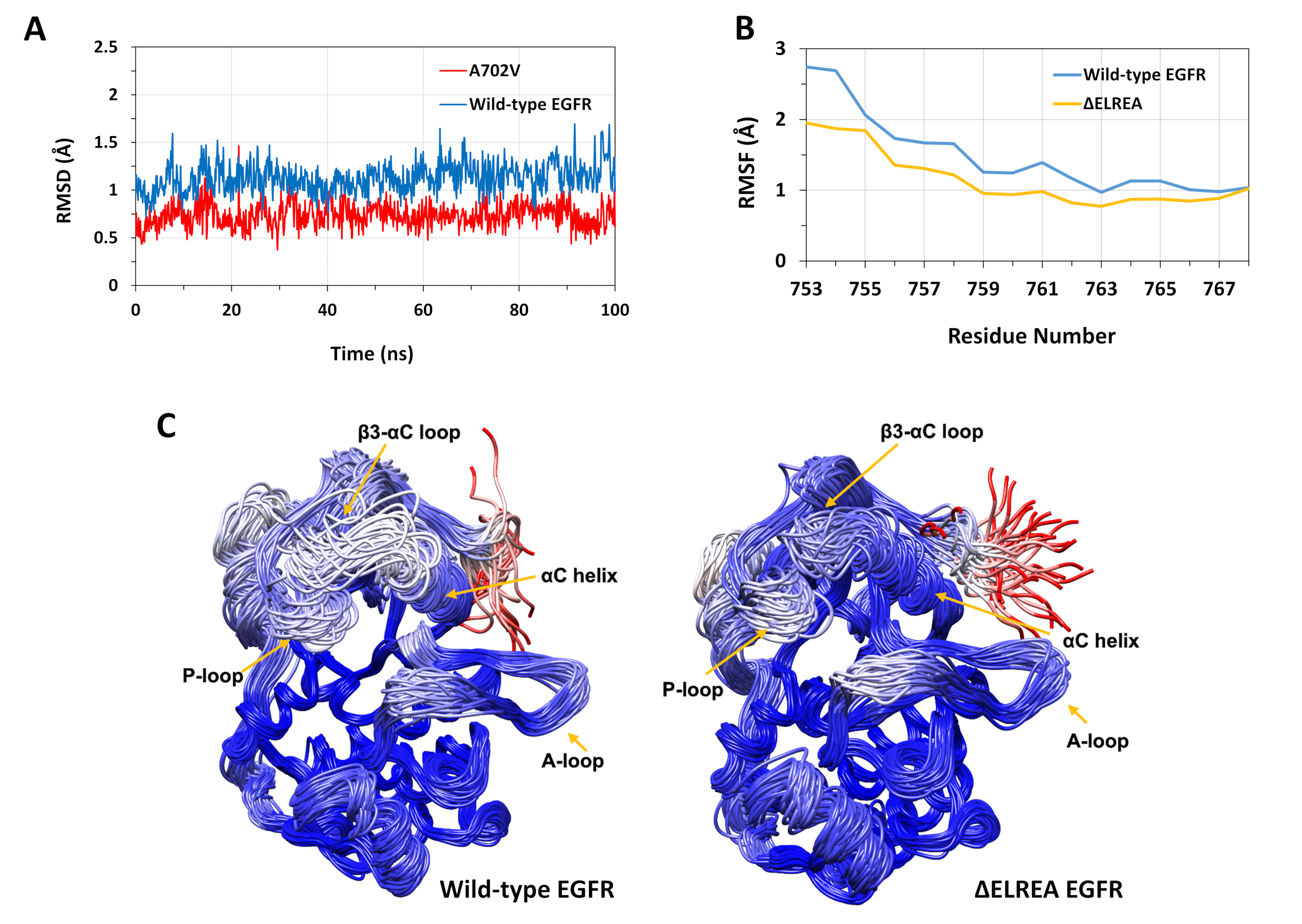
Das beschriebene Protokoll wurde verwendet, um die strukturellen Auswirkungen der Mutationen von ELREA und A702V auf die EGFR-Kinasestruktur zu untersuchen. Eine Anwendung des Protokolls bestand darin, die Auswirkungen der Mutationen auf die lokale strukturelle/konforme Stabilität zu untersuchen, indem RMSD- und RMSF-Werte aus den MD-Simulationen berechnet wurden. Da sich die A702V-Mutation im Segment der Juxtamemembrane B befindet, wurde die RMSD dieses Segments der Empfängerkinase relativ zur Ausgangsstruktur sowohl für die Wildtyp- als auch für die A702V-EGFRs berechnet. Das Ergebnis (Abbildung 4A) zeigte, dass das Juxtramembran-B-Segment des Mutanten während der 100 ns-Simulation die Konformationsstabilität erhöht hat (durchschnittliche RMSD 0,7 - 95% Konfidenzintervall (CI) 0,009) im Vergleich zur Wildtyp-EGFR-Kinase-Domäne (durchschnittliche RMSD 1,1 - 95% CI 0,01). Dies ist sehr wahrscheinlich das Ergebnis der engeren hydrophoben Wechselwirkungen an der Dimer-Schnittstelle durch den Ersatz von Alanin 702 (Methylgruppen-Seitenkette) durch einen sperrigeren hydrophoben Rückstand, Valin (Isopropylgruppe Seitenkette), was zu erhöhten hydrophoben Wechselwirkungen von V702 auf der Empfängerkinase-Domäne mit Isoleucin 941 der Aktivatorkinase-Domäne führt.
Die ElREA-Mutation befindet sich an der Schleife von 3 -C, die an die funktional-kritische C-Helix angrenzt; Die Konformation der C-Helix ist der Schlüssel zu Verschiebungen zwischen den aktiven und inaktiven Zuständen der EGFR-Kinase. Die Konformationsstabilität der C-Helix im aktiven Zustand wurde bewertet, indem der RMSF über die C-Atome von Rückständen innerhalb der Spirale während MD-Simulationen untersucht wurde (Abbildung 4B): Insgesamt gibt es geringere Schwankungen im Mutanten (durchschnittliche RMSF 1,1 - 95% CI 0,4) im Vergleich zum Wildtyp (durchschnittlicher RMSF 1,5 - 95% CI 0,57); mit dem höchsten Unterschied in den Schwankungen, die bei den N-Terminalrückständen gemessen wurden. Die abgetasteten Konformationen, die jeweils auf die Medianstruktur der Wildtyp-Kinase-Domäne und der Kinase-Domäne elREA überlagert werden, unterstützen ebenfalls diese Ergebnisse(Abbildung 4C):Sowohl die Wildtyp- als auch die ELREA-Kinase-Domänen weisen insgesamt eine ähnliche Stabilität für die überlagerten Konkonformationen mit Ausnahme der -3-C-Schleife und der 'C-Helix, die in der 'ELREA EGFR' deutlich stabiler sind. Diese Ergebnisse deuten darauf hin, dass das Löschen der ELREA-Sequenz die Bewegung der aktiven Zustands-C-Helix bremst, wodurch die aktive Konformation zurückhält und somit stabilisiert wird. Da die C-Helix Teil der asymmetrischen Dimer-Schnittstelle ist, würden die Beschränkungen der C-Helix in der Mutante sehr wahrscheinlich den asymmetrischen Dimer stabilisieren und die Dauer des aktivierten Zustands verlängern.
Eine weitere Anwendung des Protokolls besteht darin, das Verhalten wichtiger intra- und intermolekularer Wechselwirkungen zu untersuchen, die während der Simulation stattfinden. Somit ist die Wechselwirkung zwischen K745 und E762, die für die enzymatische Tätigkeit von EGFR von grundlegender Bedeutung ist, wurde sowohl für die aktive Form des Wildtyps als auch für die EGFR-Kinase analysiert, indem die prozentuale Belegung von Wasserstoffbindungen gemessen wurde, die zwischen den seitlichen Polaratomen der beiden Rückstände der beiden Rückstände während der MD-Simulationen gebildet wurden (Abbildung 5A): Diese schlüsselelektrische Wechselwirkung wurde im Bereich der Kinase von ELREA im Vergleich zur Wildtyp-Kinase häufiger gebildet, aufgrund der stabileren C-Helix, die E762 aufnimmt. Die Wechselwirkungen zwischen Mg2+-ATP und den Wildtyp- und ELREA-EGFR-Kinase-Domänen (Abbildung 5B) während der Simulation wurden ebenfalls bewertet (Abbildung 5C): Die Anzahl der Wasserstoffbindungen war für ELREA (Durchschnittswert 4,0 - 95% KI 0,03) größer als für den Wildtyp EGFR (Durchschnittswert 3,2 - 95% CI 0,04). Eine weitere Analyse der Wasserstoffbindungen ergab, dass K745 häufiger mit den Phosphatgruppen von ATP in der 'ELREA EGFR' interagiert, was mit der stabileren K745-E762-Wechselwirkung verbunden ist, die in der Simulation der egFR-Kinase-Domäne von ELREA festgestellt wurde.
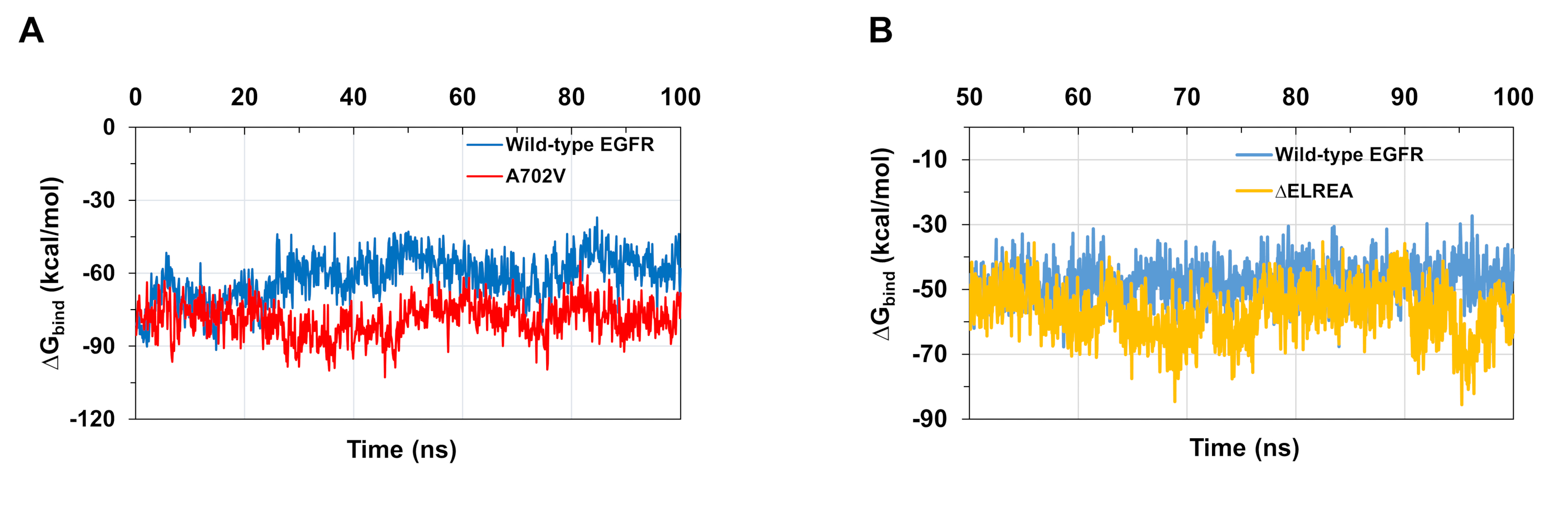
MD-Simulationen, wie im Protokoll beschrieben, sind auch nützlich bei der Beurteilung der relativen freien Bindungsenergie für Protein-Protein- und Protein-Ligand-Wechselwirkungen. Die Bindungsenergien zwischen den Aktivator- und Empfängerkinase-Domänen von Wildtyp- und A702V-EGFRs sowie zwischen ATP und den Wildtyp- und ELREA-mutierten EGFR-Kinase-Domänen, wurden aus molekularmechanischen generalisierten Born-Oberflächen-Berechnungen (MMGBSA) berechnet (Abbildung 6A): Der A702V-Mutant erzeugte einen niedrigeren durchschnittlichenG-Bindungswert (durchschnittliche G-Bindung = -76 kcal/mol - 95% CI 0,47), was im Gegensatz zur Wild-Typ-EGFR-Domäne (durchschnittliche G-Bindung = -61 kcal/mol - 95% CI 0,61) günstigere Dimer-Wechselwirkungen darstellt.bind bind Diese Beobachtung stimmt mit dem stabileren Vsadamembrane B-Segment und der engeren Dimer-Schnittstelle aufgrund erhöhter hydrophober Wechselwirkungen für die A702V EGFR-Kinase-Domäne überein. Im Falle der ATP-Bindung an die Wildtyp- und ELREA-EGFR-Kinase-Domänen (Abbildung 6B) prognostizieren MMGBSA-Berechnungen eine stärkere ATP-Bindung mit dem Mutant von 'ELREA (durchschnittlicheG-Bindung -57 kcal/mol - 95% CI 0.43) im Vergleich zu Wildtyp EGFR (durchschnittlicheG-Bindung -48 kcal/mol - 95% CI 0,33). Dieses Ergebnis steht im Einklang mit der größeren Anzahl von Wasserstoffbindungen, die zwischen ATP und ELREA EGFR (Abbildung 5C) im Vergleich zur Wildtyp-Domäne registriert wurden.
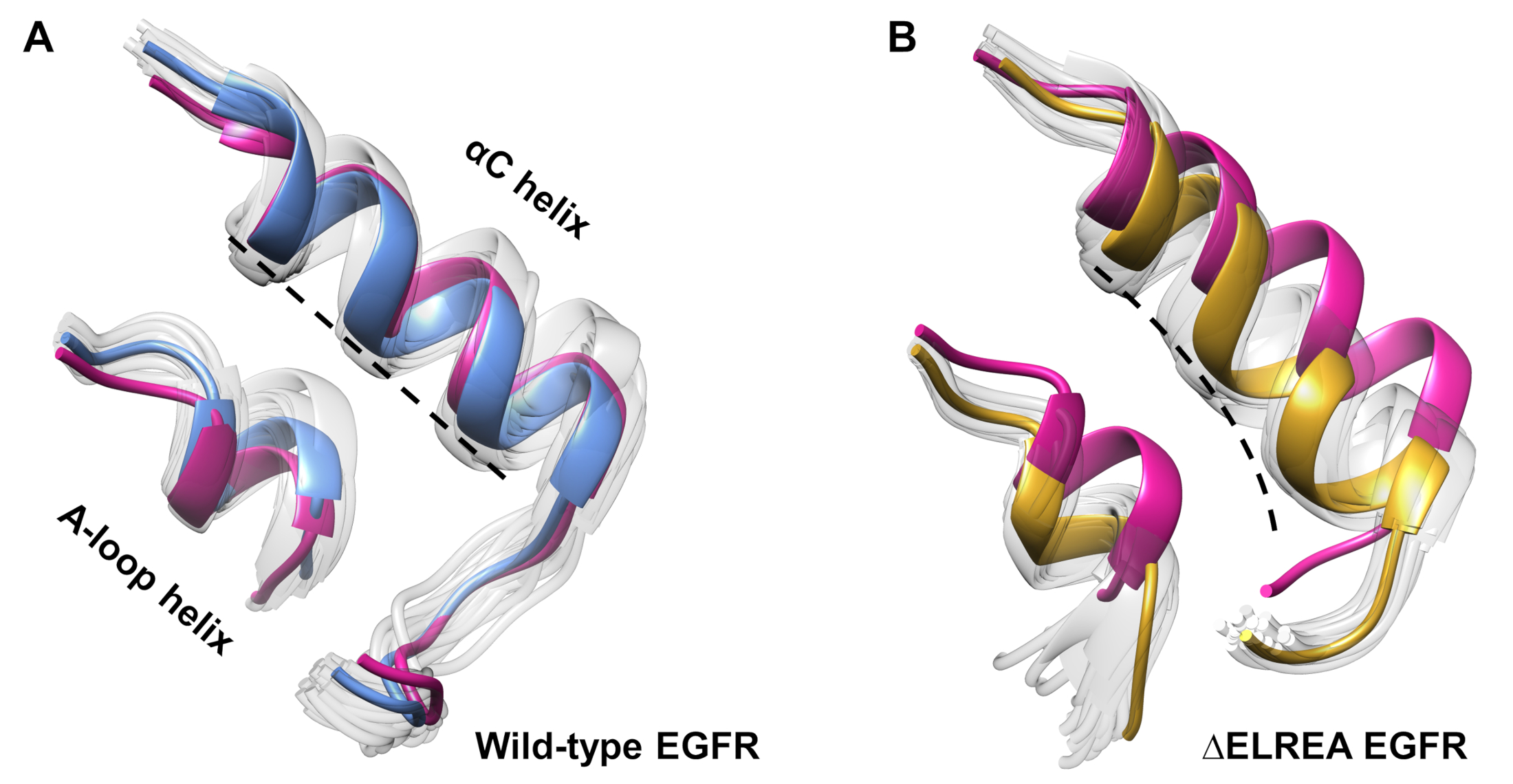
Das Protokoll kann auch verwendet werden, um konforme Veränderungen zu untersuchen, die während einer Simulation beobachtet wurden. In der aktuellen Studie wurden die Auswirkungen der ElREA-Mutation auf die inaktive EGFR-Konformation durch visuelle Inspektion und Überlagerung der beprobten Konformationen aus der Simulation untersucht. Die Analyse deckte eine innere Bewegung der C-Helix in der Domäne der EGFR-Kinase(Abbildung 7A) auf, eine strukturelle Veränderung, die während des Übergangs zum aktiven Zustand erwartet wird. Im Gegensatz dazu behielt die C-Helix des inaktiven Wildtyps EGFR ihre Anfangskonformation bei (Abbildung 7B). So unterstützen die MD-Simulationen den Vorschlag, dass die Extiermutation, die experimentell zur Erhöhung der Kinaseaktivität40,41gezeigt wird, eine konformale Verschiebung von der inaktiven Kinase in den aktiven Zustand fördert.

Abbildung 4: Wildtyp- und mutierte Konformationsstabilität der aktiven EGFR-Kinase-Domäne während MD-Simulationen. (A) RMSD (Backbone-Atome) über das Vsxtamembran-B-Segment der Wildtyp-Empfängerkinasedomäne (Blau) und A702V (rot). (B) RMSF (C-Atome) über Rückstände der C-Helix: Wildtyp (blau) und ELREA (Gold). (C) Überlagerte stichprobenartige Übereinstimmungen von Wildtyp (links) und EGFR-Kinase-Domäne von EGFR; Kettenspuren, die auf rmSD-Atomen (C-Atome) der einzelnen Rückstände in Bezug auf die Medianstruktur gefärbt sind. Die Färbung reicht von blau über weiß bis rot und stellt Regionen mit hoher bis niedriger Konformationsstabilität dar. Beachten Sie, dass die "freien" N-Terminal-Regionen der isolierten Kinase-Domäne, rot gefärbt, dieses Maß an Mobilität in der intakten EGFR-Struktur nicht aufweisen würden. Figuren nach Chakroborty et al.9 (Abbildung 4A reproduziert mit Genehmigung des Journal of Biological Chemistry) und Tamirat et al.12. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 5: Die wichtigsten Merkmale, die in der aktiven Empfängerkinase während MD-Simulationen zu sehen sind: die Salzbrücke K745-E762, die C-Helix und Wechselwirkungen mit ATP. (A) Prozentuale Belegung der K745-E762-Wechselwirkung während der Simulation der Wildtyp-(blau) und der EGFR-Kinasendomänen "ELREA" (Gold). (B) Rückstände des Wildtyps und der Mutanten, die mit ATP (Sticks) interagieren. Mg2+ (grün) koordiniert mit ATP und D855. (C) Die Anzahl der Wasserstoffbindungen, die von ATP bei MD-Simulationen sowohl mit den Wildtyp- als auch mit den EGFR-Kinase-Domänen von ELREA gebildet werden. Abbildung aus Tamirat et al.12. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 6: Für die mutierten Kinase-Domänen werden während der Simulationen niedrigere relative freie Bindungsenergien beobachtet. (A) Bindungsenergien berechnet für die Wechselwirkung zwischen den Aktivator- und Empfängerkinase-Domänen der Wildtyp-EGFRs (blau) und A702V (rot). (B)G-Bindung von ATP an Wildtyp (blau) und ELREA (Gold) EGFR-Kinase-Domänen. Figuren nach Chakroborty et al.9 (Abbildung 6A reproduziert mit Genehmigung des Journal of Biological Chemistry) und Tamirat et al.12. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}

Abbildung 7: Überlagerte Übereinstimmungen aus der inaktiven EGFR-Kinase-Domäne des Wildtyps und der ELREA. Konformation der C-Helix und der A-Loop-Helix von (A) Wildtyp (Medianstruktur in blau) und (B) ELREA EGFRs (Gold). Andere abgetastete Konformationen, verblasst weiß; Anfangsstrukturen vor den MD-Simulationen, rosa. Abbildung aus Tamirat et al.12. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Diskussion
Das in dieser Studie beschriebene Protokoll konzentriert sich auf die Verwendung molekularer Dynamiksimulationen zur Untersuchung lokaler und globaler struktureller Veränderungen, die sich aus der Aktivierung somatischer Mutationen der EGFR-Kinase-Domäne ergeben. Obwohl Röntgenkristallstrukturen von wilden und mutierten EGFRs unschätzbare strukturelle Erkenntnisse liefern, zeigen sie eine oder wenige statische Darstellungen. Der biologischen Funktion von ErbBs sind jedoch die notwendigen Übergänge zwischen der enzymatisch inaktiven und aktiven Tyrosinkinase, die dynamische Veränderungen sowohl in der Struktur als auch in intramolekularen Wechselwirkungen zwischen Kinasemonomeren hervorruft. Md-Simulationen wurden daher durchgeführt, um die Dynamik der EGFR-Tyrosinkinase-Domäne zu untersuchen, einschließlich der Wildtypstruktur, der eingeführten ElREA-Deletionsmutation und der A702V-Mutation. Diese Simulationen konnten die wahrscheinliche Rolle dieser Mutationen in den Strukturen und die Frage, wie ihre Auswirkungen auf die Konformation der Tyrosinkinase-Domäne zu den experimentell beobachteten Anstiegen der EGFR-Kinase-Aktivität führen würden, aufklären.
Ein entscheidender Schritt in diesem Protokoll ist die Verwendung einer relevanten Struktur, um die Auswirkungen der Mutation zu bewerten. Eine Möglichkeit, eine relevante Simulationseingabestruktur auszuwählen, besteht darin, die Position der Mutation in der statischen 3D-Struktur zu visualisieren und deren mögliche Auswirkungen auf die benachbarten Aminosäuren und Struktureinheiten zu untersuchen. Da sich in dieser Studie beispielsweise die A702V EGFR-Mutation am Vsadamembrane-B-Segment befindet, das die asymmetrische Dimer-Schnittstelle bildet, ist die Verwendung der Dimerstruktur für die Simulation im Gegensatz zum Monomer entscheidend. Die Verwendung einer monomeren Struktur hätte das Nebeneinander-B-Segment der Empfängerkinase dem Lösungsmittel ausgesetzt und es den stabilisierenden Wechselwirkungen entzogen, die durch die Mutation zu einem größeren hydrophoben Rückstand und Wechselwirkungen mit Isoleucin 941 aus den C-Lappenrückständen der Aktivatorkinase verstärkt wurden. Darüber hinaus ist es bemerkenswert, dass die 3D-Struktur, die durch die Koordinaten in einer HVE-Datei dargestellt wird, nicht notwendigerweise der biologisch relevanten Struktur entspricht, die für die Untersuchung verwendet werden sollte. Mit der Struktur von ErbB4, PDB-Code 3BCE, entsprechen die PDB-Koordinaten einem Trimmer, aber dies ist auf Kristallkontakte zurückzuführen (bei der Visualisierung dieser Struktur sind nur wenige Kontakte zwischen den Monomeren zu sehen). Matrizen innerhalb der PDB-Datei können (z.B. innerhalb von Chimera) verwendet werden, um die kristallographisch verwandten Strukturen zu rekonstruieren, die visualisiert werden können, um Ketten zu identifizieren, die der biologisch relevanten 3D-Struktur entsprechen, wie in der ursprünglichen Publikation42berichtet. Ein weiterer wesentlicher Schritt des Protokolls ist die richtige Vorbereitung der Simulationseingabestruktur, wie z. B. der Aufbau fehlender Aminosäuren in verschiedenen Schleifenregionen, insbesondere dort, wo sie sich in der Nähe der Mutation befinden. Obwohl im PDB zahlreiche Wildtyp-EGFR-Strukturen vorhanden sind, steht nur eine begrenzte Anzahl mutierter EGFR-Strukturen zur Verfügung. Folglich müssen auch die mutierten Strukturen modelliert werden; für eine einzelne Rückstandsmutation wie A702V wurde Chimera verwendet, um den Rückstand zu mutieren; in der Erwägung, dass für die Löschmutation "ELREA" Modeller verwendet wurde.
Die verschiedenen Parameter, die in den Simulationseingabedateien verwendet werden - zum Beispiel die Anzahl der Minimierungszyklen, das Erhitzen des Systems auf die gewünschte Temperatur in einem Rutsch oder das langsame Erhitzen durch mehrere Zwischentemperaturen, der Zeitraum für den Ausgleich und für die Produktionssimulationen - können anhand des Untersuchungsmoleküls, des Arbeitsziels und der eigenen Vorlieben modifiziert werden. Bei der Durchführung von MD-Simulationen ist es auch üblich, auf Fehler zu stoßen, die aus den Eingabedateien entstehen können, Probleme im Zusammenhang mit der in Gebrauch sein den Simulationssoftware oder sogar einen Benutzerfehler. Daher ist es sehr wichtig, die Ursache der Fehler zu verstehen, indem Sie alle Fehlermeldungen sorgfältig prüfen. Die meisten Simulationsprogramme verfügen über eine Mailingliste, in der Benutzer Fragen an die Softwareentwickler und andere Benutzer stellen können, mit denen die meisten Probleme gelöst werden können. Darüber hinaus bieten Benutzerhandbücher eine wichtige Hilfe beim Verständnis der Details des Simulationsprotokolls, einschließlich Annahmen und Einschränkungen. Obwohl MD-Simulation ein wichtiges Werkzeug ist, um die dynamischen Eigenschaften von Molekülen zu erforschen, denken Sie daran, dass Rechenergebnisse sorgfältig in Verbindung mit anderen Informationsquellen ausgewertet werden müssen, um ihre Gültigkeit zu bewerten. Arbeiten Sie nach Möglichkeit mit Forschern zusammen, die Experten für die untersuchten Proteine sind, insbesondere wenn relevante Experimente im Nasslabor durchgeführt werden, die dazu dienen, Ergebnisse für die strukturelle Interpretation zu liefern und Experimente vorzuschlagen, die auf strukturellen Beobachtungen basieren, um Hypothesen zu testen.
In dieser Studie untersuchte das Protokoll die dynamischen strukturellen Auswirkungen der Mutationen von ELREA und A702V auf die EGFR-Kinasestrukturen. Die Simulationen ergaben, dass die funktional essentielle C-Helix von der Funktion die Funktion -ELREA zurückhält und eine konformale Verschiebung von der inaktiven Kinase zu einer stabilisierten aktiven Kinase fördert. Die Simulationsergebnisse werden unabhängig von Medikamentenreaktionsdaten unterstützt, die die Auswirkungen von Tyrosinkinase-Inhibitoren auf Lungenkrebszelllinien mit der Löschmutation elREA und dem Wildtyp EGFR zeigten, bei denen eine größere Hemmung durch Medikamente, die die aktive Kinase-Konformation erkannten, für DIE ElREA berichtet wurde als für den Wildtyp EGFR12. Mit der A702V-Mutation weisen die MD-Simulationen im Vergleich zum Wildtyp auf eine erhöhte Stabilisierung der Aktivator-Empfänger-Kinase-Schnittstelle sowie eine höhere Affinität des Aktivators und der Empfängerkinase füreinander hin, zusammen unterstützen sie die Aufrechterhaltung der aktivierten Konformation der EGFR-Kinase. Die A702V-Mutation, die sich auf dem Adxtamembran-B-Segment der Empfängerkinase befindet, würde die hydrophoben Wechselwirkungen mit der Aktivatorkinase erhöhen und die Dauer des aktivierten Zustands verlängern. Die A702V-Mutation unterstützt das Zellüberleben ohne Wachstumsfaktor und wurde in einem In-vitro-Screening auf EGFR-Mutationen9identifiziert.
Offenlegungen
Die Autoren haben nichts zu verraten.
Danksagungen
Diese Forschung wird durch Stipendien an M.S.J von der Akademie Finnlands (308317, 320005), Sigrid Juselius Foundation und Tor, Joe und Pentti Borg Memorial Fund sowie an K.E. von der Akademie finnlands (274728, 316796), der Cancer Foundation of Finland und des Turku University Central Hospital finanziert. M.Z.T. wird vom Doktorandennetzwerk der Informations- und Strukturbiologie finanziert. Wir danken dem CSC IT Center for Science für die Rechenressourcen und Dr. Jukka Lehtonen für die IT-Unterstützung im Rahmen des Biocenter Finland Bioinformatics Netzwerks; und Biocenter Finland strukturelle Biologie Infrastrukturnetzwerk.
Materialien
| Name | Company | Catalog Number | Comments |
| Amber software | University of California, San Francisco | Version 2018 | Executable |
| Chimera program | Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco | Version 1.13.1 | Executable |
| EGFR struture files | The Protein Data Bank | 3D coordinates of EGFR structures | |
| Maestro | Schrödinger LLC | Version 2018-3 | Executable |
| Modeller program | The Andrej Šali Lab, Departments of Biopharmaceutical Sciences and Pharmaceutical Chemistry, University of California San Francisco | Included in the Chimera program | |
| VMD software | Theoretical and Computational Biophysics Group, University of Illinois at Urbana-Champaign | Version 1.9.3 | Executable |
Referenzen
- Yarden, Y., Sliwkowski, M. X. Untangling the ErbB signalling network. Nature Reviews Molecular Cell Biology. 2, 127-137 (2001).
- Lemmon, M. A., Schlessinger, J., Ferguson, K. M. The EGFR family: not so prototypical receptor tyrosine kinases. Cold Spring Harbor Perspectives in Biology. 6, a020768 (2014).
- Arteaga, C. L., Engelman, J. A. ERBB receptors: From oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell. 2, 282-303 (2014).
- Mishra, R., Hanker, A. B., Garrett, J. T. Genomic alterations of ERBB receptors in cancer: Clinical implications. Oncotarget. 8, 114371-114392 (2017).
- . cBioPortal for Cancer Genomics Available from: https://www.cbioportal.org (2020)
- Lynch, T. J., et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. New England Journal of Medicine. 350 (21), 2129-2139 (2004).
- Paez, J. G., et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 304 (5676), 1497-1500 (2004).
- Pao, W., et al. EGF receptor gene mutations are common in lung cancers from "never smokers" and are associated with sensitivity of tumors to gefitinib and erlotinib. Proceedings of the National Academy of Sciences U.S.A. 101 (36), 13306-13311 (2004).
- Chakroborty, D., et al. Unbiased in vitro screen for activating EGFR mutations. Journal of Biological Chemistry. 294 (24), 9377-9389 (2019).
- Leahy, D. J. Structure and Function of the Epidermal Growth Factor (EGF/ErbB) Family of Receptors. Advances in Protein Chemistry. 68, 1-27 (2004).
- Roskoski, R. ErbB/HER protein-tyrosine kinases: Structures and small molecule inhibitors. Pharmacological Research. 87, 42-59 (2014).
- Tamirat, M. Z., Koivu, M., Elenius, K., Johnson, M. S. Structural characterization of EGFR exon 19 deletion mutation using molecular dynamics simulation. PLoS ONE. 14 (9), e0222814 (2019).
- Ding, L., et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 455, 1069-1075 (2008).
- Kurppa, K. J., Denessiouk, K., Johnson, M. S., Elenius, K. Activating somatic ERBB4 mutations in non small-cell lung cancer. Oncogene. 35 (10), 1283-1291 (2016).
- Soung, Y. H., et al. Somatic mutations of the ERBB4 kinase domain in human cancers. International Journal of Cancer. 118, 1426-1429 (2006).
- Tvorogov, D., et al. Somatic mutations of ERBB4: selective loss-of-function phenotype affecting signal transduction pathways in cancer. Journal of Biological Chemistry. 284, 5582-5591 (2009).
- Hubbard, S. R., Till, J. H. Protein tyrosine kinase structure and function. Annual Review of Biochemistry. 69 (1), 373-398 (2000).
- Huse, M., Kuriyan, J. The conformational plasticity of protein kinases. Cell. 109 (3), 275-282 (2002).
- Jura, N., et al. Catalytic control in the EGF receptor and its connection to general kinase regulatory mechanisms. Molecular Cell. 42, 9-22 (2011).
- Karplus, M., Kuriyan, M., J, Molecular dynamics and protein function. Proceedings of the National Academy of Sciences U.S.A. 102 (19), 6679-6685 (2005).
- Shan, Y., Arkhipov, A., Kim, E. T., Pan, A. C., Shaw, D. E. Transitions to catalytically inactive conformations in EGFR kinase. Proceedings of the National Academy of Sciences U.S.A. 110 (18), 7270-7275 (2013).
- Reckamp, K. L., et al. A phase I trial to determine the optimal biological dose of celecoxib when combined with erlotinib in advanced non-small cell lung cancer. Clinical Cancer Research. 12 (11 Pt 1), 3381-3388 (2006).
- Pettersen, E. F., et al. UCSF Chimera-a visualization system for exploratory research and analysis. Journal of Computational Chemistry. 25 (13), 1605-1612 (2004).
- Berman, H. M., et al. The Protein Data Bank. Nucleic Acids Research. 28 (1), 235-242 (2000).
- Zhang, X., Gureasko, J., Shen, K., Cole, P. A., Kuriyan, J. An Allosteric Mechanism for Activation of the Kinase Domain of Epidermal Growth Factor Receptor. Cell. 125 (6), 1137-1149 (2006).
- Stamos, J., Sliwkowski, M. X., Eigenbrot, C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. Journal of Biological Chemistry. 277 (48), 46265-46272 (2002).
- Sogabe, S., et al. Structure-Based Approach for the Discovery of Pyrrolo[3,2-d]pyrimidine-Based EGFR T790M/L858R Mutant Inhibitors. ACS Medicinal Chemistry Letters. 4 (2), 201-205 (2013).
- Sali, A., Blundell, T. L. Comparative protein modelling by satisfaction of spatial restraints. Journal of Molecular Biology. 234 (3), 779-815 (1993).
- Yun, C. H., et al. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell. 11 (3), 217-227 (2007).
- Park, J. H., Liu, Y., Lemmon, M. A., Radhakrishnan, R. Erlotinib binds both inactive and active conformations of the EGFR tyrosine kinase domain. Biochemical Journal. 448 (3), 417-423 (2012).
- . Release 2018-3: Maestro Available from: https://www.schrodinger.com/maestro (2018)
- Case, D. A., et al. . AMBER 2018. , (2018).
- Maier, J. A., Martinez, C., Kasavajhala, K., Wickstrom, L., Hauser, K. E., Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. Journal of Chemical Theory and Computation. 11 (8), 3696-3713 (2015).
- Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W., Klein, M. L. Comparison of simple potential functions for simulating liquid water. Journal of Chemical Physics. 79 (2), 926-935 (1983).
- Meagher, K. L., Redman, L. T., Carlson, H. A. Development of polyphosphate parameters for use with the AMBER force field. Journal of Computational Chemistry. 24 (9), 1016-1025 (2003).
- Humphrey, W., Dalke, A., Schulten, K. VMD: Visual molecular dynamics. Journal of Molecular Graphics. 14 (1), 33-38 (1996).
- Melvin, R. L., Salsbury, F. R. Visualizing ensembles in structural biology. Journal of Molecular Graphics and Modelling. 67, 44-53 (2016).
- Roe, D. R., Cheatham, T. E. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. Journal of Chemical Theory and Computation. 9 (7), 3084-3095 (2013).
- Miller, B. R., et al. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. Journal of Chemical Theory and Computation. 8 (9), 3314-3321 (2012).
- Guha, U., et al. Comparisons of tyrosine phosphorylated proteins in cells expressing lung cancer-specific alleles of EGFR and KRAS. Proceedings of the National Academy of Sciences U.S.A. 105 (37), 14112-14117 (2008).
- Furuyama, K., et al. Sensitivity and kinase activity of epidermal growth factor receptor (EGFR) exon 19 and others to EGFR-tyrosine kinase inhibitors. Cancer Science. 104 (5), 584-589 (2013).
- Qiu, C., et al. Mechanism of Activation and Inhibition of the HER4/ErbB4 Kinase. Structure. 6 (3), 460-467 (2008).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten