
Se requiere una suscripción a JoVE para ver este contenido. Inicie sesión o comience su prueba gratuita.
Method Article
Imágenes de células vivas de matrices unicelulares (LISCA) - una técnica versátil para cuantificar la cinética celular
En este artículo
Resumen
Presentamos un método para la adquisición de cursos de tiempo reportero de fluorescencia de células individuales utilizando matrices micropatterned. El protocolo describe la preparación de matrices unicelulares, la configuración y el funcionamiento de la microscopía de lapso de tiempo de barrido de células vivas y una herramienta de análisis de imágenes de código abierto para la preselección automatizada, el control visual y el seguimiento de los cursos de tiempo de fluorescencia integrados en células por sitio de adhesión.
Resumen
Imágenes de células vivas de matrices unicelulares (LISCA) es un método versátil para recopilar cursos de tiempo de señales de fluorescencia de células individuales en alto rendimiento. En general, la adquisición de cursos de tiempo unicelulares de células cultivadas se ve obstaculizada por la motilidad celular y la diversidad de formas celulares. Las micro-matrices adhesivas estandarizan las condiciones unicelulares y facilitan el análisis de imágenes. LISCA combina microarrays unicelulares con microscopía de lapso de tiempo de barrido y procesamiento automatizado de imágenes. Aquí, se describen los pasos experimentales de tomar cursos de tiempo de fluorescencia unicelular en un formato LISCA. Transfectamos las células adherentes a una matriz micropatterned utilizando la codificación de ARNm para la proteína fluorescente verde mejorada (eGFP) y supervisar la cinética de expresión de eGFP de cientos de células en paralelo a través de microscopía de lapso de tiempo de barrido. Las pilas de datos de imagen son procesadas automáticamente por un software recientemente desarrollado que integra la intensidad de fluorescencia sobre los contornos celulares seleccionados para generar cursos de tiempo de fluorescencia unicelulares. Demostramos que los cursos de tiempo de expresión de eGFP después de la transfección de ARNm están bien descritos por un modelo de traducción cinética simple que revela la expresión y las tasas de degradación de ARNm. Otros usos de LISCA para las correlaciones del tiempo del acontecimiento de marcadores múltiples en el contexto del apoptosis de la señalización se discuten.
Introducción
En los últimos años, la importancia de los experimentos unicelulares se ha hecho evidente. Los datos de células individuales permiten la investigación de la variabilidad célula a célula, la resolución de correlaciones de parámetros intracelulares y la detección de cinéticas celulares que permanecen ocultas en las mediciones deconjuntos 1,2,3. Con el fin de investigar la cinética celular de miles de células individuales en paralelo, se necesitan nuevos enfoques que permitan el monitoreo de las células en condiciones estandarizadas durante un período de tiempo de varias horas hasta varios días seguido de un análisis cuantitativo de datos 4. Aquí, presentamos imágenes de células vivas de matrices unicelulares (LISCA), que combina el uso de matrices microestructuradas con microscopía de lapso de tiempo y análisis de imágenes automatizado.
Se han establecido y publicado en la literatura5,6. Aquí, describimos brevemente microescala plasma-iniciado patrón de proteínas (μPIPP). Un protocolo detallado de la fabricación de matrices unicelulares utilizando μPIPP también se encuentra en la referencia7. El uso de matrices unicelulares permite la alineación de miles de células en puntos de adhesión estandarizados que presentan microambientes definidos para cada célula y, por lo tanto, reduce una fuente de variabilidad experimental (Figura 1A). Las matrices unicelulares se utilizan para monitorear los cursos de tiempo de los marcadores fluorescentes con el propósito de indicar una variedad de procesos celulares. La microscopía a largo plazo en el modo de lapso de tiempo de barrido permite monitorear una gran área de las matrices unicelulares y, por lo tanto, tomar muestras de datos unicelulares en alto rendimiento durante un tiempo de observación de varias horas o incluso días. Esto genera pilas de línea de tiempo de imágenes de cada posición de la matriz (Figura 1B). Con el fin de reducir la gran cantidad de datos de imagen y extraer los cursos de tiempo de fluorescencia unicelular deseados de una manera eficiente, se requiere un procesamiento automatizado de imágenes que aproveche el posicionamiento de las células (Figura 1C).
El reto de LISCA es adaptar los protocolos experimentales y las herramientas computacionales para formar un ensayo de alto rendimiento que genere datos cuantitativos y reproducibles de cinética celular. En este artículo proporcionamos una descripción paso a paso de los métodos individuales y cómo se combinan en un ensayo LISCA. Como ejemplo, discutimos el curso del tiempo de la expresión mejorada de la proteína fluorescente verde (eGFP) después de la entrega artificial del mRNA. La expresión del eGFP que sigue entrega del mRNA es descrita por las ecuaciones de la tarifa de reacción que modelan la traducción y la degradación del mRNA. Ajustar la función del modelo para el curso temporal de la concentración de eGFP a la lectura LISCA de la intensidad de fluorescencia para cada célula individual a lo largo del tiempo produce las mejores estimaciones de los parámetros del modelo, como la tasa de degradación del ARNm. Como resultado representativo discutimos la eficacia de la entrega del mRNA de dos diversos agentes lípido-basados de la transfección y cómo sus distribuciones del parámetro diferencian.

Figura 1:Representación del flujo de trabajo lisca combinando (A) micro-patrones de matrices unicelulares (B) microscopía de lapso de tiempo de barrido y (C) análisis automatizado de imágenes de series de imágenes grabadas. Las matrices unicelulares consisten en un patrón bidimensional de cuadrados adhesivos celulares con un interespacio repelente celular que conduce a una disposición de las células en el micropatrón, como se puede ver en la imagen de contraste de fase, así como la imagen de fluorescencia de las células que expresan eGFP(A). Toda el área microestructurada se toma una imagen en un modo de lapso de tiempo de escaneo tomando repetidamente imágenes en una secuencia de posiciones (B). Las series de imágenes grabadas se procesan para leer la intensidad de fluorescencia por célula a lo largo del tiempo(C). Barras de escala: 500 μm (A), 200 μm (C). Haga clic aquí para ver una versión más amplia de esta figura.
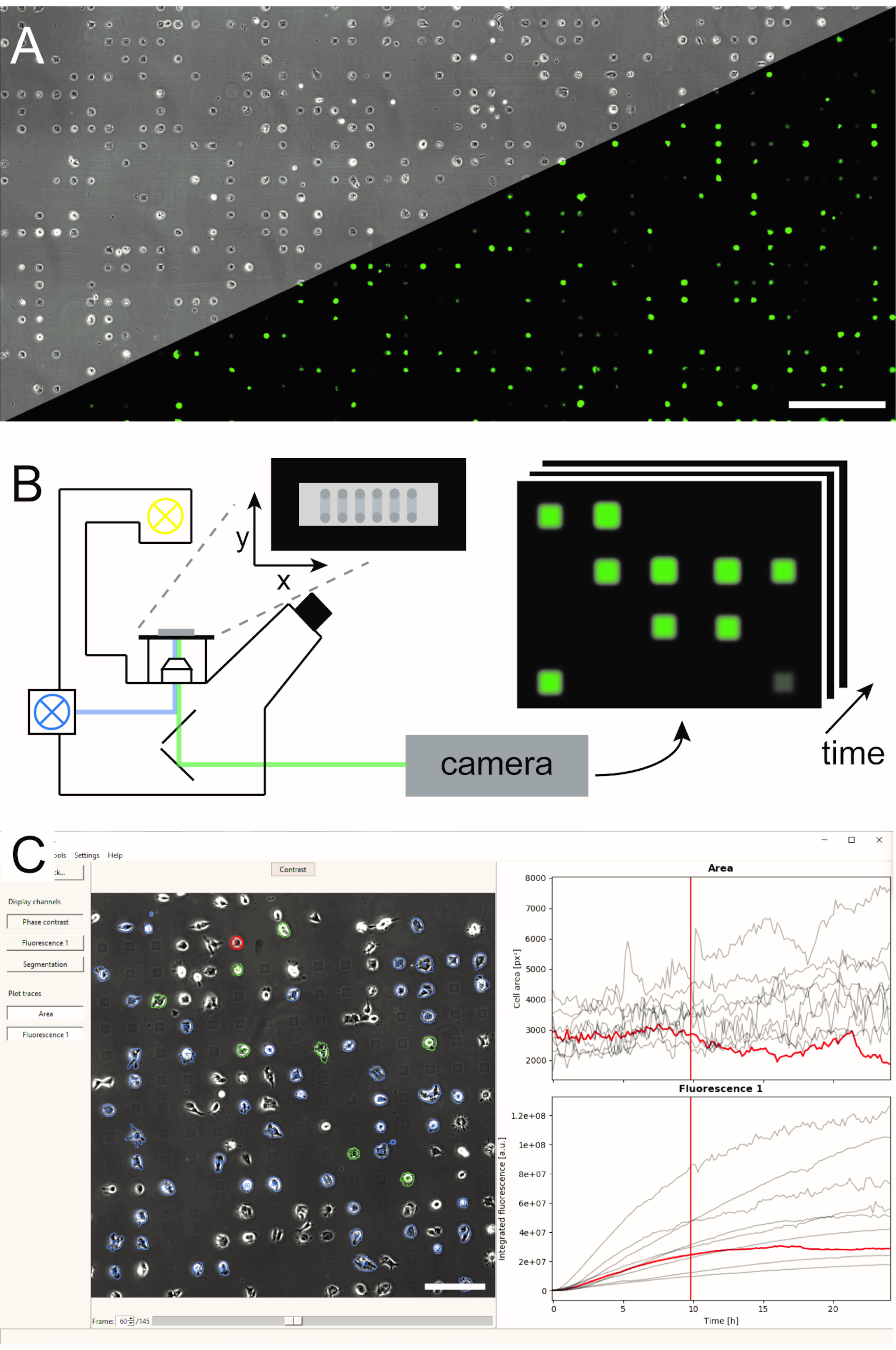{kind=link}
Protocolo

Figura 2:Adquisición de datos que combina microarrays unicelulares (A) con microscopía de lapso de tiempo de barrido (B). Como preparación del experimento time-lapse, se prepara una matriz unicelular con un micropatrón 2D de cuadrados de adhesión (1), seguido de la siembra celular y la alineación de las células en el micropatrón (2), así como la conexión de un sistema de perfusión al portaobjetos de seis canales, que permite el manejo de líquidos durante la medición de lapso de tiempo (3). Se establece un experimento de lapso de tiempo de barrido (4) y las células se transfectan en el microscopio inyectando una solución de lipoplex de ARNm a través del sistema de perfusión durante el experimento de lapso de tiempo (5). Barras de escala: 200 μm. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
1. Fabricación de matrices unicelulares microestructuradas (Figura 2A)
- Prepare los materiales necesarios para la fabricación de matrices μPIPP.
- Preparar solución salina tamponada con fosfato estéril (PBS) a pH 7.4.
- Prepare agua ultrapura estéril con una resistividad de al menos 18 MΩcm a 25 °C.
- Prepare la solución de trabajo PLL(20 kDa)-g[3.5] PEG(2 kDa) (PLL-PEG) con una concentración de 2 mg/mL de PLL-PEG en agua ultrapura que contenga 150 mM de NaCl y 10 mM de ácido 4-(2-hidroxietil)-1-piperazinatanosulfónico (HEPES).
- Preparar una solución de proteína de matriz extracelular para recubrimiento superficial: 1 mg/mL de fibronectina (FN) en PBS.
- Preparar una oblea de silicio con un micropatrón fabricado por fotolitografía8 que funciona como un maestro reutilizable. El micropatrón consiste en cuadrados con una longitud de borde de 30 μm, una profundidad de 12 μm y una distancia inter-cuadrada de 60 μm, dispuestos en seis franjas cada una con un ancho de 6 mm y una altura de 18 mm.
- Mezcle un monómero de polidimetilsiloxano (PDMS) con un reticulador al 9% (masa %) usando un kit de elastómero de silicona y desgasificarlo durante aproximadamente 30 minutos hasta que esté libre de burbujas usando un desecador. Funda la oblea de silicio con una capa pdms de aproximadamente 3-5 mm de espesor y desgasificarla de nuevo durante unos 30 minutos hasta que esté libre de burbujas.
- Coloque la oblea de silicio con el PDMS en un horno de hornear a 50 °C para curar el PDMS durante al menos 4 h.
- Corte los sellos pdms.
- Utilice un bisturí y cortar fuera de la capa PDMS una obra maestra PDMS que contiene las seis rayas micropattern.
- Coloque la obra maestra de PDMS en un banco con el micropatrón mirando hacia arriba.
- Corte cada una de las seis rayas de micropatrón de la obra maestra de PDMS con una navaja de afeitar en un sello de PDMS. Tenga cuidado de que los bordes de los sellos PDMS estén abiertos cortando parte del área estampada.
- Coloque los sellos PDMS en una cubierta de una diapositiva de seis canales (Figura 3-1).
- Utilice un cubrebocas sin recubrimiento y marque las posiciones de los canales de la corredera de seis canales rayando cuidadosamente la lámina de protección de la cubierta. Luego coloque el cubrebocas en el banco con la lámina de protección mirando hacia abajo.
- Coloque los sellos PDMS con el micropatrón mirando hacia abajo en el cubrebocas en las posiciones marcadas del canal usando pinzas.
- Compruebe la fijación de los sellos PDMS bajo un microscopio. Si un sello PDMS está completamente unido a la cubierta, los cuadrados en contacto aparecen más oscuros que el interespacio. La fijación del sello PDMS al cubrebocas es crucial para la calidad del micropatrón.
- Coloque el cubrebocas con los seis sellos pdms en él en un limpiador de plasma y tratarlo con plasma de oxígeno (presión 0.2 mbar, ~ 40 W durante 3 min) para hacer las superficies entre los sellos PDMS y el cubrebocas hidrofílico (Figura 3-2).
- Lleve a cabo todos los pasos adicionales de la fabricación de micropatrones en un gabinete de bioseguridad. Utilice 15 μL de la solución PLL-PEG y pipetee una gota de ella junto a cada sello PDMS para que la solución PLL-PEG se absorba en el patrón hidrofílico del sello PDMS (Figura 3-3). Deje que el PLL-PEG incube durante 20 min a temperatura ambiente.
- Enjuague 1 mL de agua ultrapura sobre el cubrebocas con los sellos PDMS y retire los sellos PDMS usando pinzas (Figura 3-4). Luego enjuague el cubrebocas una segunda vez con 1 mL de agua ultrapura y déjelo secar.
- Cuando la cubierta se haya secado por completo, pegue una diapositiva pegajosa de seis canales a la cubierta (Figura 3-4). Tenga cuidado de que las áreas micropatterned se alineen con la parte inferior de los canales.
- Funcionalizar los cuadrados de adhesión con FN.
- Llene 40 μL de PBS en cada canal.
- Prepare una solución de FN de 100 μg/mL en PBS.
- Añadir 40 μL de la solución FN a cada canal (Figura 3-5). Mezcle la solución de FN con el PBS en el canal a fondo eliminando 40 μL de un depósito y agregándolo al depósito opuesto del mismo canal durante 3 veces para generar una solución homogénea. Incubar la solución de FN durante 45 min a temperatura ambiente.
- Lavar cada canal tres veces con 120 μL de PBS (Figura 3-6).
- Para marcar para saber si hay calidad del patrón, utilice un FN fluorescente etiquetado en el paso 9.2. (Figura 4A).
NOTA: Recomendamos preparar la matriz μPIPP no más de un día antes de la siembra celular, ya que PLL-PEG y FN no están unidos covalentemente al sustrato y la calidad del patrón puede disminuir con el tiempo. Guarde la matriz μPIPP preparada en la nevera.

Figura 3:Fabricación de microarrays unicelulares por μPIPP. (1) Los sellos PDMS con una estructura de micropatrón tridimensional en la superficie están dispuestos en una cubierta de una diapositiva de seis canales. (2) El cubrebocas con los sellos pdms en él se trata con plasma de oxígeno para hacer las superficies hidrofílicas. (3) Se añade PLL-PEG. Es absorbido en la microestructura por las fuerzas capilares y hace que las superficies no cubiertas por el sello PDMS repelente celular. (4) El cubrebocas se enjuaga con agua para eliminar el PLL-PEG restante. Luego, se quitan los sellos PDMS y se pega una diapositiva a pegajosa de seis canales en la barra de cubierta. (5) Fibronectin, una proteína de la matriz extracelular, se agrega para hacer las áreas sin el célula-adhesivo de PLL-PEG. (6) El portaobjetos de seis canales se lava con solución salina tamponada con fosfato. Haga clic aquí para ver una versión más amplia de esta figura.
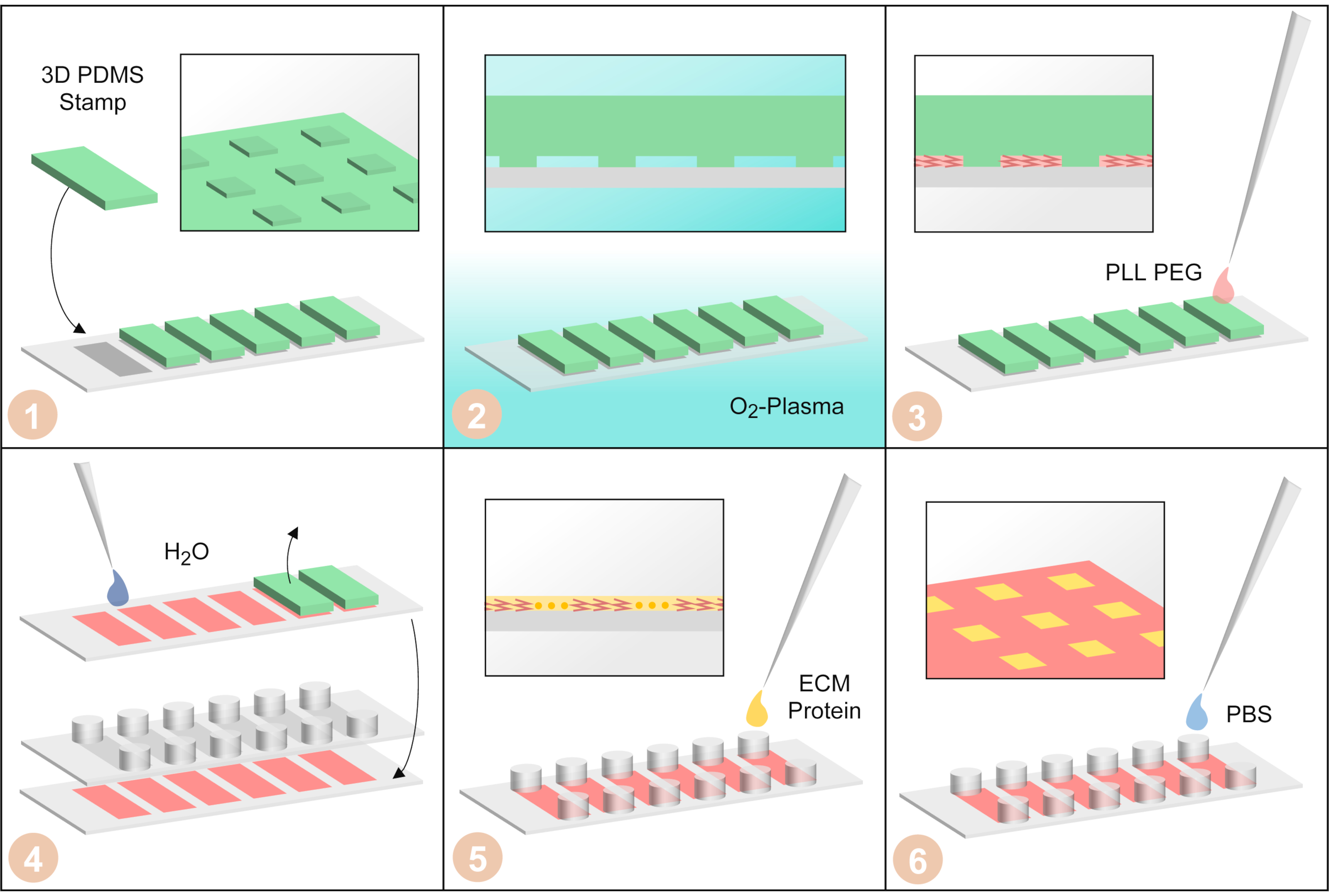{kind=link}
2. Siembra celular (Figura 2A)
NOTA: Para los siguientes pasos de lavado, agregue el líquido respectivo a un depósito y luego retire un volumen igual de líquido del depósito opuesto de un canal.
- Lave cada canal con 120 μL de 37 °C totalmente suplementados con el medio de crecimiento celular. Antes de añadir la suspensión de la célula, asegúrese de que sólo los canales están llenos de medio, pero no los depósitos.
- Desconecte las células HuH7 de un matraz de cultivo celular siguiendo su protocolo estándar para el paso de células y ajuste la concentración de suspensión celular a 4 x 105 células/mL.
- Añadir 40 μL de suspensión celular y mezclar el medio de crecimiento celular con la suspensión celular retirando 40 μL de un depósito y añadiéndolo al reservorio opuesto del mismo canal durante 3 veces para alcanzar una distribución celular homogénea (Figura 4B).
- Retire 40 μL de suspensión del canal para que solo el canal esté lleno de suspensión celular.
- Coloque el portaobjetos en una incubadora y compruebe la adhesión celular 1 h después de la siembra con un microscopio de contraste de fase.
- Añadir 120 μL de medio de crecimiento celular caliente a 37 °C.
- Pero la diapositiva hacia atrás en la incubadora durante más 3 h para permitir la autoorganización celular en el micropatrón (Figura 4C).

Figura 4:Autoorganización celular y control de calidad de la matriz μPIPP. (A) La superficie microestructurada consiste en puntos de adhesión recubiertos de FN al cuadrado que se muestran en rojo rodeados por un polímero repelente celular. (B)Después de la siembra celular, las células HuH7 se distribuyen aleatoriamente y(C)se adhieren principalmente en los puntos de adhesión durante un período de tiempo de 4 h. Reimpreso con permiso 7. Barras de escala: 200 μm. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
3. Sistema de perfusión (Figura 2A)
NOTA: El uso de un sistema de perfusión sólo es necesario si es necesario añadir reactivos o marcadores fluorescentes durante el transcurso de la medición del lapso de tiempo. Dependiendo de sus necesidades, puede conectar cada canal a un sistema de perfusión separado o conectar varios canales en serie al mismo sistema de perfusión. El número de sistemas de perfusión corresponde al número de condiciones experimentales independientes. Conecte los tubos en condiciones estériles en un gabinete de bioseguridad y evite la inclusión de burbujas de aire en el sistema de perfusión. Si no se utiliza ningún sistema de perfusión, agregue los reactivos/marcadores en un gabinete de bioseguridad antes de la medición de lapso de tiempo. El sistema de perfusión es fabricado internamente, el material utilizado se enumera en la Tabla de Materiales. El montaje del sistema de perfusión ha sido descrito previamente9.
- Use una jeringa de 1 ml (con cuchara de reemplazo) y llene la jeringa con 1 mL de medio de crecimiento celular de 37 °C.
- Conecte la jeringa al tubo de entrada usando la válvula y llene el tubo con medio.
- Conecte el tubo de entrada a un depósito de un canal y asegúrese de que no haya burbujas de aire atrapadas.
- Para conectar otro canal en serie a este sistema de perfusión, conecte un conector serie al depósito opuesto al tubo de entrada del canal actual. Continúe con el siguiente canal y conecte el extremo libre del conector serie a uno de sus depósitos.
- Repita los pasos anteriores hasta que el número necesario de canales estén conectados en serie.
- Conecte el tubo de salida directamente al depósito libre del canal actual. Llene el tubo conectado con el medio para comprobar que el sistema de la perfusión no se escapa.
- Repita los pasos anteriores hasta que los seis canales de la diapositiva estén conectados a un sistema de perfusión.
- Coloque el portaobjetos con el sistema o sistemas de perfusión conectados de nuevo en la incubadora hasta su uso posterior o colóquelo directamente en la cámara de calentamiento del microscopio precalentado a 37 °C para la medición del lapso de tiempo.
4. Microscopía de lapso de tiempo (Figura 2B)
NOTA: Para mediciones a largo plazo, mantenga una temperatura estable de 37 °C y un nivel estable de CO2. Como alternativa al medio de crecimiento celular dependiente delCO2,utilice el medio L15 para el que no se requiere ningún sistema de incubación de gases.
NOTA: Para obtener imágenes cuantitativas, utilice el medio de crecimiento celular sin rojo fenol durante la medición de lapso de tiempo para reducir la fluorescencia de fondo y utilice los mismos ajustes del protocolo de lapso de tiempo, así como el mismo microscopio para réplicas técnicas.
- Configure un protocolo de lapso de tiempo para grabar una imagen de contraste de fase y una imagen de fluorescencia con tiempos de exposición de 750 ms (dependiendo de la cámara), un intervalo de tiempo de 10 minutos entre bucles consecutivos a través de la lista de posición y un tiempo de observación de 30 h, utilizando un objetivo de 10x y filtros de fluorescencia apropiados.
- Coloque la corredera de seis canales con las celdas en las matrices unicelulares en el soporte de la muestra de la cámara de calentamiento caliente de 37 °C. Si los sistemas de perfusión están conectados a la corredera de seis canales, fije los tubos al escenario con un poco de cinta para asegurarse de que la corredera de seis canales no se mueva durante el intercambio de líquido. Inserte los extremos libres de los tubos de salida a través de un orificio de un tubo de reacción de 15 mL para recoger los residuos líquidos.
- Establezca la lista de posiciones para la medición de lapso de tiempo de escaneo. Asegúrese de que el número de posiciones se puede escanear dentro del intervalo de tiempo definido entre bucles consecutivos a través de la lista de posiciones. Con un objetivo de 10x, se pueden configurar 10-30 posiciones por canal para escanear el área total del micropatrón dependiendo del tamaño del chip de la cámara.
- Inicie la medición de lapso de tiempo. Para una mejor calidad de imagen de las mediciones a largo plazo, utilice un sistema automatizado de corrección de enfoque.
5. Marcador fluorescente - transfección de ARNm (Figura 2B)
NOTA: Para una transfección en dos canales conectados por un sistema de tubería, se necesita un volumen total de 600 μL de mezcla de transfección (300 μL para un canal). Los volúmenes indicados se refieren a una transfección en dos canales conectados.
- Prepare una solución de agente de transfección diluyendo 1 μL de agente de transfección en 200 μL de medio reducido en suero y deje que la solución se incube durante 5 min a temperatura ambiente.
- Prepare una solución de ARNm diluyendo 300 ng de codificación de ARNm para eGFP en 150 μL de medio reducido en suero.
- Prepare la mezcla de transfección añadiendo 150 μL de la solución del agente de transfección a la solución de ARNm y mézclela bien. Dejar que la mezcla de transfección se incube durante 20 min a temperatura ambiente.
- Enjuague el sistema de tubos con 1 mL de PBS caliente de 37 °C utilizando una jeringa durante la incubación de la mezcla de transfección. Al enjuagar los tubos, asegúrese de que la etapa del microscopio no se mueva. Pausar la medición de lapso de tiempo si es necesario.
- Diluya la mezcla de transfección a la concentración final de ARNm de 0,5 ng/μL añadiendo un medio reducido en suero de 300 μL.
- Enjuague el sistema de tubos con la mezcla de transfección usando una jeringa y deje que los lipoplexos de ARNm se incuben durante 1 h (pausar la medición de lapso de tiempo si es necesario).
- Detenga la incubación de transfección y enjuague los lipoplexos de ARNm no unidos lavándolos con 1 mL de 37 °C de medio de crecimiento celular totalmente suplementado con una jeringa (pausar la medición de lapso de tiempo si es necesario).
6. Análisis de imágenes y lectura de fluorescencia
- Al ejecutar el análisis de imágenes por primera vez, instale la versión 0.1.6 del software de código abierto "Automated Microstructure Analysis in Python" (PyAMA) desde la ubicación10 citada de acuerdo con las instrucciones proporcionadas allí.
- Asegúrese de que los canales de imagen (contraste de fase y fluorescencia) estén disponibles como archivos TIFF de 16 bits de varias imágenes. Si es necesario, conviértalos en consecuencia.
- Inicie PyAMA y haga clic en Abrir pila ... para abrir imágenes para su análisis.
- Para cada archivo TIFF de varias imágenes que se abra, haga clic en Abrir y seleccione el archivo para que se muestre en la lista de archivos cargados en el lado izquierdo del cuadro de diálogo (Figura 5-1).
- Marque los canales a incluir en el análisis. Para cada canal, realice los pasos siguientes.
- Seleccione en la lista de archivos cargados el archivo TIFF que contiene el canal.
- En la sección Agregar nuevo canal, seleccione el índice del canal en el archivo TIFF. La indización se basa en cero; el primer canal tiene índice 0, el segundo canal tiene índice 1 y así sucesivamente.
- Seleccione el tipo de canal. Seleccione Contraste de fase o Fluorescencia para los canales de imagen correspondientes y Segmentación para un canal binario que indique los contornos de celda.
- Opcionalmente, introduzca una etiqueta del canal para distinguir diferentes canales de fluorescencia: eGFP y DAPI.
- Después de configurar el canal, haga clic en Agregar.
- Cuando todos los canales agregados se muestren en la lista de canales del lado derecho del cuadro de diálogo, haga clic en Aceptar para cargar la pila.
- Para realizar la segmentación utilizando el algoritmo de segmentación incorporado de PyAMA para el reconocimiento celular basado en las imágenes de contraste de fase (Figura 5-2), vaya a Herramientas | Binarize... e introduzca un nombre de archivo para el archivo NumPy con el canal binarized.
Nota : en la versión actual, cargar el canal binarized requiere volver a cargar todos los canales. - Para realizar una corrección de fondo11 en un canal de fluorescencia (Figura 5-3), asegúrese de que el canal de fluorescencia y un canal de segmentación están cargados. Si no se carga ningún canal de segmentación, asegúrese de que se carga un canal de contraste de fase para la segmentación automática. Vaya a "Herramientas > corrección en segundo plano..." y seleccione un nombre de archivo para el archivo TIFF resultante con el canal de fluorescencia corregido.
Nota : en la versión actual, cargar el canal corregido en segundo plano requiere volver a cargar todos los canales. - Inspeccione las celdas preseleccionadas(Figura 5-4)y su señal de fluorescencia integrada(Figura 5-5)desplazándose por los marcos de tiempo, viendo los canales enumerados en el menú del canal en el lado izquierdo y haciendo clic en las celdas para resaltar sus cursos de tiempo de fluorescencia(Figura 1C). Utilice la selección de celdas para excluir de un análisis posterior las células que no son viables, que no están confinadas a un punto de adhesión o que están unidas a otra célula. Para alternar la selección de celdas para su lectura, presione Mayús y haga clic en la celda, o resalte la celda y presione Entrar.
- Guarde los cursos de tiempo unicelulares para el área celular y la fluorescencia integrada (Figura 5-6) haciendo clic en Archivo | Guarde y seleccione un directorio en el que guardar.

Figura 5:Procesamiento automatizado de imágenes de lapso de tiempo utilizando PyAMA. (1) Se importan series de imágenes de contraste de fase y fluorescencia para cada posición de imagen. (2) Los contornos de celda están determinados por la segmentación en la pila de imágenes de contraste de fase. (3) Se aplica una corrección de fondo a las imágenes de fluorescencia. (4) Los contornos de celda se rastrean a lo largo del tiempo y se preseleccionados para la exportación. (5) La intensidad de fluorescencia se integra en función de los contornos celulares rastreados. (6) Se evalúan las zonas unicelulares y las intensidades de fluorescencia integradas y se exportan los cursos de tiempo para cada célula. Barras de escala: 100 μm. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
- Para analizar la cinética de traslación después de la transfección por ARNm, ajuste un modelo de traslación basado en ecuaciones de velocidad bioquímica a cada ciclo de tiempo unicelular como se describió anteriormente por Reiser et al.12. Los datos y el código utilizados en dicho estudio están a disposición del público13.
- Para cada curso de tiempo unicelular, recupere los parámetros de ajuste estimados del modelo de traducción que representan la tasa de degradación del ARNm y el punto de tiempo de inicio de la traducción. Un conjunto de datos de ejemplo se describe en la sección de resultados representativos.
- Realice análisis adicionales sobre las distribuciones de las mejores estimaciones de los parámetros para condiciones experimentales variadas para investigar la variabilidad de célula a célula dentro de las poblaciones celulares.
Resultados
El enfoque LISCA permite recoger de manera eficiente cursos de tiempo de fluorescencia de células individuales. Como ejemplo representativo, describimos cómo se aplica el método LISCA para medir la expresión de eGFP unicelular después de la transfección. Los datos del experimento LISCA se utilizan para evaluar la cinética de administración de ARNm, que es importante para el desarrollo de fármacos eficientes de ARNm.
En particular, demostramos el impacto diferente de dos sistemas de ad...
Discusión
Aquí describimos LISCA como una técnica versátil para seguir la cinética celular de las etiquetas fluorescentes intracelulares a nivel unicelular. Para realizar un experimento LISCA exitoso, cada uno de los pasos descritos de la sección de protocolo debe establecerse individualmente y, a continuación, se deben combinar todos los pasos. Cada uno de los tres aspectos principales de LISCA cuenta con pasos cruciales.
Fabricación de microarrays unicelulares
La calidad del microar...
Divulgaciones
Los autores declaran que no tienen intereses financieros contrapuestos.
Agradecimientos
Este trabajo fue apoyado por subvenciones de la Fundación Alemana de la Ciencia (DFG) al Centro de Investigación Colaborativa (SFB) 1032. El apoyo del Ministerio Federal alemán de Educación, Investigación y Tecnología (BMBF) en el marco del proyecto cooperativo 05K2018-2017-06716 Medisoft, así como una subvención de la Bayerische Forschungsstiftung son agradecidos. Anita Reiser recibió el apoyo de una beca DFG a través de la Graduate School of Quantitative Biosciences Munich (QBM).
Materiales
| Name | Company | Catalog Number | Comments |
| Adtech Polymer Engineering PTFE Microtubing | Fisher Scientific | 10178071 | |
| baking oven | Binder | 9010-0190 | |
| CFI Plan Fluor DL 10x | Nikon | MRH20100 | |
| Desiccator | Roth | NX07.1 | |
| Eclipse Ti-E | Nikon | ||
| eGFP mRNA | Trilink | L-7601 | |
| Female Luer to Tube Connector | MEDNET | FTL210-6005 | |
| Fetal bovine serum | Thermo Fisher | 10270106 | |
| Fibronectin | Yo Proteins | 663 | |
| Filter set eGFP | AHF | F46-002 | |
| Fisherbrand Translucent Platinum-Cured Silicone Tubing | Fisher Scientific | 11768088 | |
| HEPES (1 M) | Thermo Fisher | 15630080 | |
| Incubation Box | Okolab | OKO-H201 | |
| incubator | Binder | 9040-0012 | |
| L-15 without phenol red | Thermo Fisher | 21083027 | |
| Lipofectamine 2000 | Thermo Fisher | 11668027 | |
| Male Luer | in-house fabricated consisting of teflon | ||
| Male Luer to Tube Connector | MEDNET | MTLS210-6005 | alternative to in-house fabricated male luers |
| NaCl (5 M) | Thermo Fisher | AM9760G | |
| Needleless Valve to Male Luer Connector | MEDNET | NVFMLLPC | |
| NIS Elements | Nikon | Imaging software Version 5.02.00 | |
| NOA81 | Thorlabs | NOA81 | Fast Curing Optical Adhesive for tube system assembly |
| Opti-MEM | Thermo Fisher | 31985062 | |
| PCO edge 4.2 M-USB-HQ-PCO | pco | ||
| Phosphate buffered saline (PBS) | in-house prepared | ||
| Plasma Cleaner | Diener Femto | Pico-BRS | |
| PLL(20 kDa)-g[3.5]-PEG(2 kDa) | SuSoS AG | ||
| silicon wafer mit mircorstructures | in-house fabricated | ||
| Sola Light Engine | Lumencor | ||
| sticky slide VI 0.4 | ibidi | 80608 | |
| Sylgard 184 Silicone Elastomer Kit | Dow Corning | 1673921 | |
| Tango 2 | Märzhäuser | 00-24-626-0000 | |
| Ultrapure water | in-house prepared | ||
| uncoated coverslips | ibidi | 10813 | |
| Injekt-F Solo, 1 mL | Omilab | 9166017V | with replacement sporn |
Referencias
- Altschuler, S. J., Wu, L. F. Cellular heterogeneity: do differences make a difference. Cell. 141 (4), 559-563 (2010).
- Locke, J. C., Elowitz, M. B. Using movies to analyse gene circuit dynamics in single cells. Nature Reviews Microbiology. 7 (5), 383-392 (2009).
- Spencer, S. L., Gaudet, S., Albeck, J. G., Burke, J. M., Sorger, P. K. Non-genetic origins of cell-to-cell variability in TRAIL-induced apoptosis. Nature. 459 (7245), 428-432 (2009).
- El-Ali, J., Sorger, P. K., Jensen, K. F. Cells on chips. Nature. 442 (7101), 403-411 (2006).
- Segerer, F. J., et al. Versatile method to generate multiple types of micropatterns. Biointerphases. 11 (1), 011005 (2016).
- Piel, M., Théry, M. . Micropatterning in cell biology, part A/B/C. , (2014).
- Reiser, A., Zorn, M. L., Murschhauser, A., Rädler, J. O., Ertl, P., Rothbauer, M. . Cell-Based Microarrays: Methods and Protocols. , 41-54 (2018).
- Picone, R., Baum, B., McKendry, R. . Methods in cell biology. 119, 73-90 (2014).
- Reiser, A. . Single-cell time courses of mRNA transport and translation kinetics. , (2019).
- . Softmatter LMU-Raedler Group Available from: https://github.com/SoftmatterLMU-RaedlerGroup/pyama (2020)
- . MIAAB, 29262 Available from: https://github.com/SoftmatterLMU-RaedlerGroup/pyama (2020)
- Reiser, A., et al. Correlation of mRNA delivery timing and protein expression in lipid-based transfection. Integrative Biology. 11 (9), 362-371 (2019).
- Reiser, A., Woschée, D., Mehrotra, N., Krzysztoń, R. S., Strey, H. H., Rädler, J. O. Supplementing data and code for: "Correlation of mRNA delivery timing and protein expression in lipid-based transfection". Zenodo. , (2019).
- Ferizi, M., et al. Stability analysis of chemically modified mRNA using micropattern-based single-cell arrays. Lab on a Chip. 15 (17), 3561-3571 (2015).
- Fröhlich, F., et al. Multi-experiment nonlinear mixed effect modeling of single-cell translation kinetics after transfection. NPJ systems biology and applications. 4 (1), 1-12 (2018).
- Krzysztoń, R., et al. Single-cell kinetics of siRNA-mediated mRNA degradation. Nanomedicine: Nanotechnology, Biology and Medicine. 21, 102077 (2019).
- Röttgermann, P. J., Alberola, A. P., Rädler, J. O. Cellular self-organization on micro-structured surfaces. Soft Matter. 10 (14), 2397-2404 (2014).
- Röttgermann, P. J., Dawson, K. A., Rädler, J. O. Time-resolved study of nanoparticle induced apoptosis using microfabricated single cell arrays. Microarrays. 5 (2), 8 (2016).
- Murschhauser, A., et al. A high-throughput microscopy method for single-cell analysis of event-time correlations in nanoparticle-induced cell death. Communications Biology. 2 (1), 1-11 (2019).
- Chatzopoulou, E. I., et al. Chip-based platform for dynamic analysis of NK cell cytolysis mediated by a triplebody. Analyst. 141 (7), 2284-2295 (2016).
- Sztilkovics, M., et al. Single-cell adhesion force kinetics of cell populations from combined label-free optical biosensor and robotic fluidic force microscopy. Scientific Reports. 10 (1), 1-13 (2020).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoThis article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados