
Для просмотра этого контента требуется подписка на Jove Войдите в систему или начните бесплатную пробную версию.
Method Article
Визуализация живых клеток одноклеточных массивов (LISCA) - универсальный метод количественной оценки клеточной кинетики
В этой статье
Резюме
Представлен метод получения флуоресцентных репортерных временных курсов из одиночных клеток с использованием микроструктурированных массивов. Протокол описывает подготовку одноэлементных массивов, настройку и эксплуатацию покадровой микроскопии сканирования живых клеток и инструмент анализа изображений с открытым исходным кодом для автоматизированного предварительного выбора, визуального контроля и отслеживания интегрированных в клетку курсов времени флуоресценции на сайт адгезии.
Аннотация
Live-cell Imaging of Single-Cell Arrays (LISCA) является универсальным методом сбора временных курсов флуоресцентных сигналов от отдельных клеток с высокой пропускной способностью. В целом, приобретение одноклеточных временных курсов из культивируемых клеток затруднено подвижностью клеток и разнообразием форм клеток. Клеевые микрорешетки стандартизируют одноэлементные условия и облегчают анализ изображений. LISCA сочетает в себе одноэлементные микрочипы со сканирующей покадровой микроскопией и автоматизированной обработкой изображений. Здесь мы описываем экспериментальные этапы прохождения курсов времени флуоресценции с одной клеткой в формате LISCA. Мы трансфектируем клетки, прилипающие к микроструктурированному массиву, используя кодирование мРНК для усиленного зеленого флуоресцентного белка (eGFP), и отслеживаем кинетику экспрессии eGFP сотен клеток параллельно с помощью сканирующей покадровой микроскопии. Стеки данных изображения автоматически обрабатываются недавно разработанным программным обеспечением, которое интегрирует интенсивность флуоресценции по выбранным контурам клеток для создания одноэлементных курсов флуоресценции. Показано, что временные курсы экспрессии eGFP после трансфекции мРНК хорошо описываются простой кинетической моделью трансляции, которая выявляет скорость экспрессии и деградации мРНК. Обсуждаются дальнейшие применения LISCA для корреляций времени событий множественных маркеров в контексте сигнального апоптоза.
Введение
В последние годы важность одноклеточных экспериментов стала очевидной. Данные от отдельных клеток позволяют проводить исследование межклеточной изменчивости, разрешение корреляций внутриклеточных параметров и обнаружение клеточной кинетики, которые остаются скрытыми в ансамблевых измерениях1,2,3. Для параллельного исследования клеточной кинетики тысяч одиночных клеток необходимы новые подходы, позволяющие контролировать клетки в стандартизированных условиях в течение периода времени от нескольких часов до нескольких дней с последующим количественным анализом данных 4. Здесь мы представляем Live-cell Imaging of Single-Cell Arrays (LISCA), которая сочетает в себе использование микроструктурированных массивов с покадровой микроскопией и автоматизированным анализом изображений.
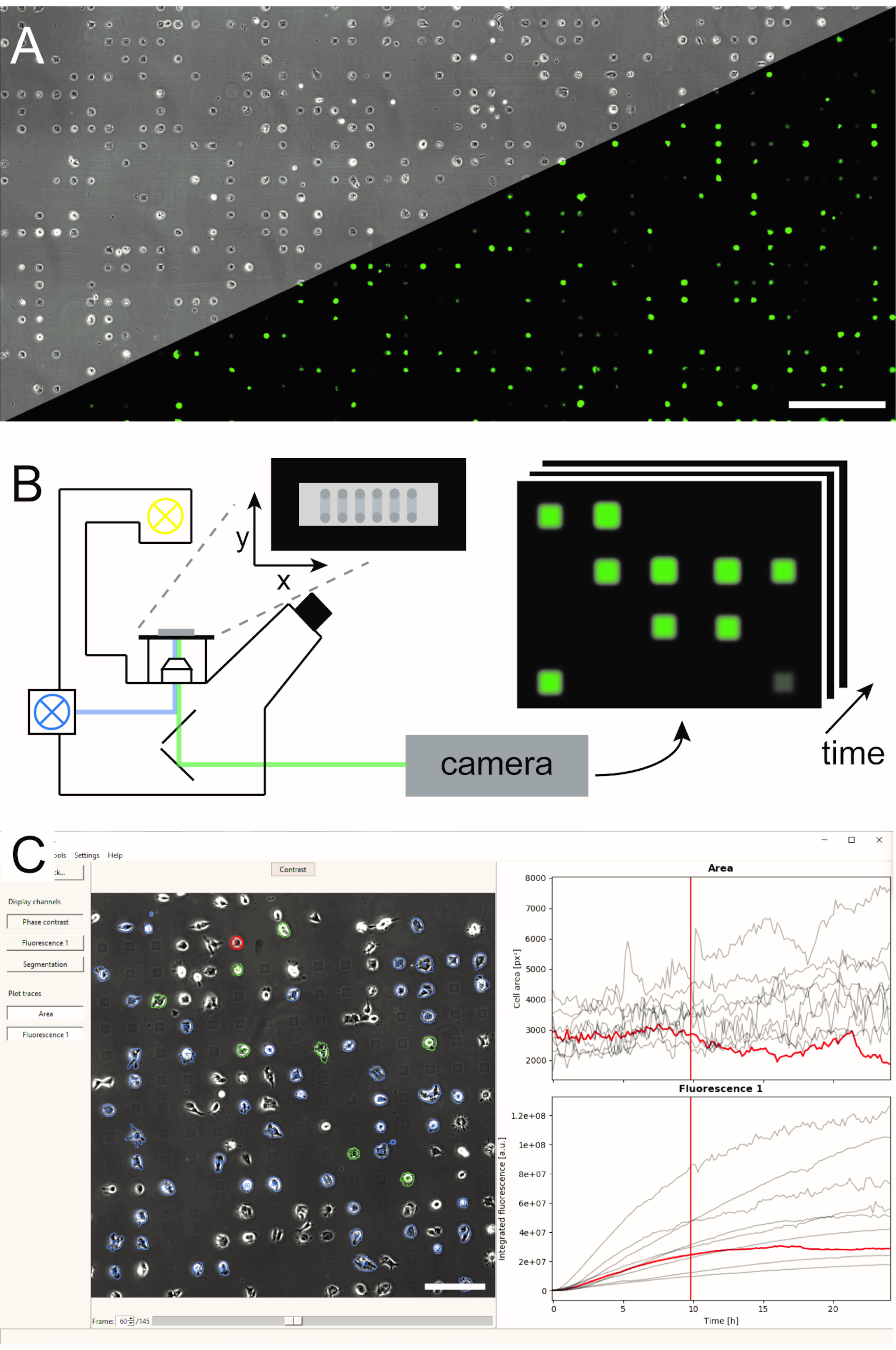
Несколько методов генерации микроструктурированных одноклеточных массивов были установлены и опубликованы влитературе5,6. Здесь мы кратко опишем микромасштабное плазменно-инициированное белковое паттернирование (μPIPP). Подробный протокол изготовления одноэлеческого массива с использованием μPIPP также приведен в ссылке7. Использование одноклеточных массивов позволяет выровнять тысячи клеток по стандартизированным пятнам адгезии, представляющим определенные микросреды для каждой клетки, и, таким образом, уменьшает источник экспериментальной изменчивости(рисунок 1A). Одноклеточные массивы используются для мониторинга временных ходов флуоресцентных маркеров, направленных на указание на различные клеточные процессы. Долгосрочная микроскопия в режиме замедленной съемки сканирования позволяет контролировать большую площадь одноклеточных массивов и, следовательно, отбирать одноклеточные данные с высокой пропускной способностью в течение нескольких часов или даже дней. При этом генерируются стеки изображений из каждой позиции массива(рисунок 1B). Чтобы уменьшить большой объем данных изображения и эффективно извлечь желаемые одноэлементные курсы времени флуоресценции, требуется автоматизированная обработка изображений, которая использует преимущества позиционирования ячеек(рисунок 1C).
Задача LISCA состоит в том, чтобы адаптировать экспериментальные протоколы и вычислительные инструменты для формирования высокопроизводительного анализа, который генерирует количественные и воспроизводимые данные клеточной кинетики. В этой статье мы предоставляем пошаговое описание отдельных методов и того, как они сочетаются в анализе LISCA. В качестве примера мы обсуждаем временной ход экспрессии усиленного зеленого флуоресцентного белка (eGFP) после искусственной доставки мРНК. Экспрессия eGFP после доставки мРНК описывается уравнениями скорости реакции, моделируя трансляцию и деградацию мРНК. Подгонка функции модели для временного хода концентрации eGFP к показаниям LISCA интенсивности флуоресценции для каждой отдельной ячейки с течением времени дает наилучшие оценки параметров модели, таких как скорость деградации мРНК. В качестве репрезентативного результата мы обсуждаем эффективность доставки мРНК двух различных трансфекционных агентов на основе липидов и то, как различаются их распределения параметров.

Рисунок 1:Представление рабочего процесса LISCA, сочетающего (A) микроструктурные одноклеточные массивы (B) сканирующей покадровой микроскопии и (C) автоматизированный анализ изображений записанных серий изображений. Одноклеточные массивы состоят из двумерного рисунка клеточно-адгезивных квадратов с клеточно-репеллентным межпространством, ведущим к расположению ячеек на микроструктуре, как видно на фазоконтрастном изображении, а также на флуоресцентном изображении эГФП-экспрессирующих клеток(А). Вся микроструктурированная область визуализируется в режиме сканирования покадровой съемки многократно, делая снимки в последовательности позиций(B). Записанные серии изображений обрабатываются для считывания интенсивности флуоресценции на ячейку с течением времени(C). Шкала стержней: 500 мкм(A),200 мкм(C). Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}
протокол

Рисунок 2:Сбор данных, сочетающий одноклеточные микрочипы (А) со сканирующей покадровой микроскопией (В). В качестве подготовки покадрового эксперимента готовят одноэлементную матрицу с 2D-микрошаблоном квадратов адгезии (1), за которой следует посев клеток и выравнивание ячеек на микрошаблоне (2), а также подключение перфузионной системы к шестиканальному слайду, что позволяет обрабатывать жидкость во время покадрового измерения (3). Проводится сканирующий эксперимент с замедленной съемкой (4), и клетки трансфектируются на микроскопе путем введения раствора липоплекса мРНК через перфузионную систему во время покадрового эксперимента (5). Шкала: 200 мкм. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}
1. Изготовление микроструктурированного одноэлементного массива(рисунок 2A)
- Подготовьте материалы, необходимые для изготовления массива μPIPP.
- Готовят стерильный фосфатно-буферный физиологический раствор (PBS) при рН 7,4.
- Приготовьте стерильную сверхчистую воду с удельным сопротивлением не менее 18 МОм см при 25 °C.
- Готовят PLL(20 кДа)-г[3.5] PEG(2 кДа) (PLL-PEG) рабочий раствор с концентрацией 2 мг/мл PLL-PEG в сверхчистой воде, содержащей 150 мМ NaCl и 10 мМ 4-(2-гидроксиэтил)-1-пиперазинэтансульфоновой кислоты (HEPES).
- Приготовьте раствор белка внеклеточного матрикса для покрытия поверхности: 1 мг/мл фибронектина (FN) в PBS.
- Подготовьте кремниевую пластину с микрошаблоном, изготовленную с помощью фотолитографии8, которая функционирует как многоразовый мастер. Микрошаблон состоит из квадратов с длиной кромки 30 мкм, глубиной 12 мкм и межквартовым расстоянием 60 мкм, расположенных в шесть полос, каждая из которых имеет ширину 6 мм и высоту 18 мм.
- Смешайте полидиметилсилоксан (PDMS) мономер с 9% сшивателем (масс.%) используя силиконовый эластомерный набор и дегазуйте его в течение примерно 30 минут, пока он не освободится от пузырьков с помощью осушителя. Отлить кремниевую пластину слоем PDMS толщиной примерно 3-5 мм и снова дегазируйте ее в течение примерно 30 минут, пока она не освободится от пузырьков.
- Поместите кремниевую с PDMS в печь для выпечки при 50 °C, чтобы вылечить PDMS в течение не менее 4 ч.
- Вырежьте штампы PDMS.
- Используйте скальпель и вырежьте из слоя PDMS один шедевр PDMS, который содержит шесть микрошаблонных полос.
- Поместите шедевр PDMS на скамейку с микрошаблоном вверх.
- Вырежьте каждую из шести микрошаблонных полос шедевра PDMS бритвенным лезвием на штамп PDMS. Позаботьтесь о том, чтобы края штампов PDMS были открыты, отрезав часть узорчатой области.
- Поместите штампы PDMS на крышку шестиканального слайда(рисунок 3-1).
- Используйте крышку без покрытия и отметьте положения канала шестиканального слайда, тщательно поцарапав защитную пленку крышки. Затем поместите крышку на скамейку с защитной фольгой, обращенной вниз.
- Поместите штампы PDMS с микрошаблоном вниз на крышку в отмеченных положениях канала с помощью пинцетам.
- Проверьте прикрепление штампов PDMS под микроскопом. Если штамп PDMS полностью прикреплен к крышке, квадраты в контакте кажутся темнее, чем промежуточное пространство. Прикрепление штампа PDMS к крышке имеет решающее значение для качества микроструктуры.
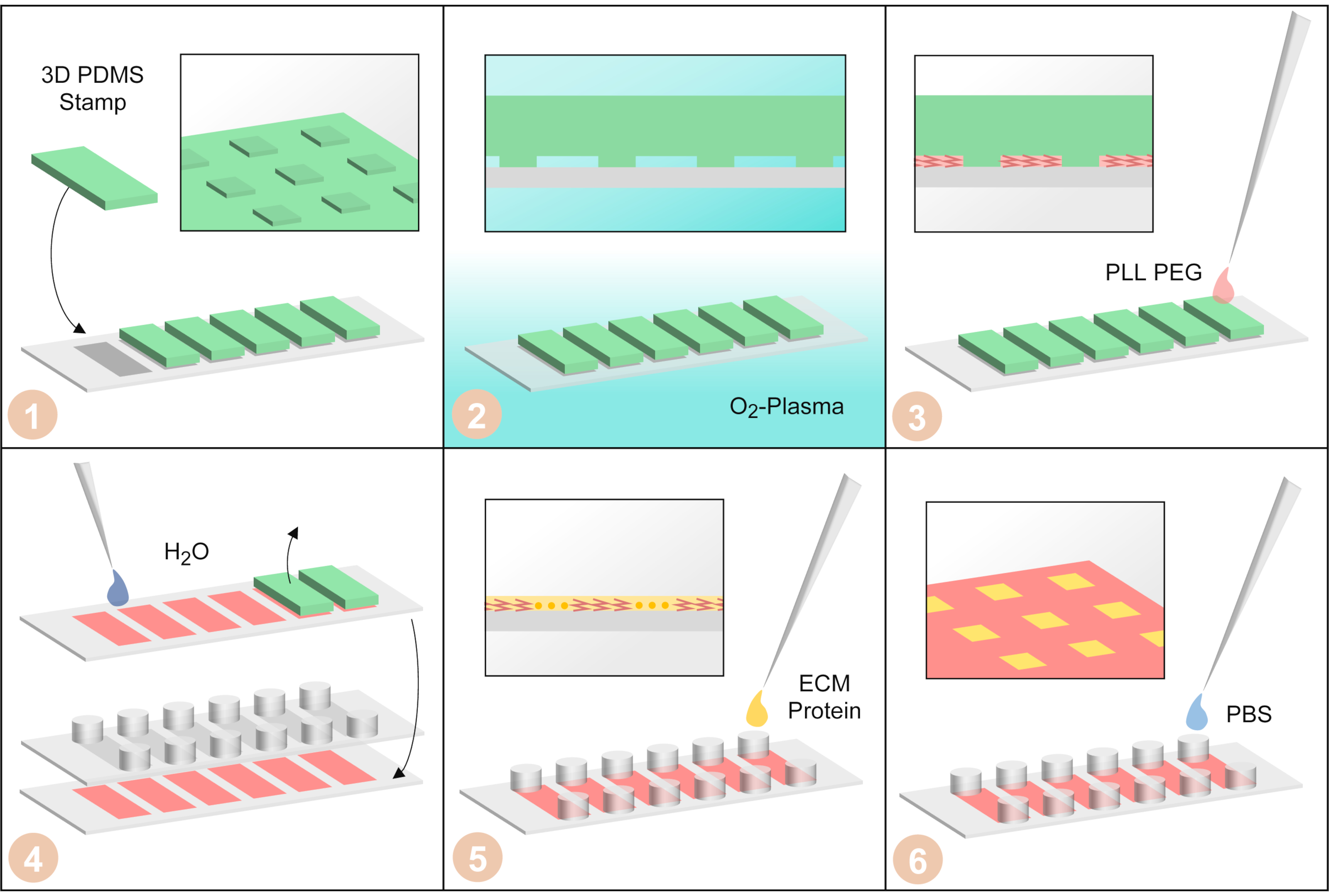
- Поместите крышку с шестью штампами PDMS на ней в плазмоочиститель и обработайте ее кислородной плазмой (давление 0,2 мбар, ~40 Вт в течение 3 мин), чтобы сделать поверхности между штампами PDMS и крышкой гидрофильными(рисунок 3-2).
- Провести все дальнейшие этапы изготовления микроструктуры в шкафу биобезопасности. Используйте 15 мкл раствора PLL-PEG и пипетку одной капли рядом с каждой маркой PDMS, чтобы раствор PLL-PEG впитывался в гидрофильный рисунок штампа PDMS(рисунок 3-3). Дайте PLL-PEG инкубировать в течение 20 мин при комнатной температуре.
- Промойте 1 мл сверхчистой воды поверх крышки со штампами PDMS на нем и удалите штампы PDMS с помощью пинцета(рисунок 3-4). Затем промойте крышку второй раз 1 мл сверхчистой воды и дайте ей высохнуть.
- Когда крышка полностью высохнет, приклеите шестиканальный липкий слайд к крышке(рисунок 3-4). Позаботьтесь о том, чтобы микроструктурированные участки выровняли нижнюю часть каналов.
- Функционализируйте квадраты адгезии с FN.
- Заполните 40 мкл PBS в каждый канал.
- Приготовьте раствор FN 100 мкг/мл в PBS.
- Добавьте 40 мкл раствора FN к каждому каналу(рисунок 3-5). Тщательно перемешайте раствор FN с PBS в канале, удалив 40 мкл из одного резервуара и добавив его в противоположный резервуар того же канала в течение 3 раз, чтобы получить однородный раствор. Инкубировать раствор FN в течение 45 мин при комнатной температуре.
- Промывайте каждый канал три раза 120 мкл PBS(рисунок 3-6).
- Чтобы проверить качество рисунка, используйте флуоресцентно маркированный FN на шаге 9.2. (Рисунок 4А).
ПРИМЕЧАНИЕ: Мы рекомендуем готовить матрицу μPIPP не более чем за один день до посева клеток, поскольку ФАПХ-ПЭГ и FN не связаны ковалентно с субстратом, и качество рисунка может со временем снизиться. Храните подготовленный массив μPIPP в холодильнике.

Рисунок 3:Изготовление одноэлементных микрочипов μPIPP. (1) Штампы PDMS с трехмерной микроструктурой на поверхности расположены на крышке шестиканального слайда. (2) Крышка со штампами PDMS обрабатывается кислородной плазмой, чтобы сделать поверхности гидрофильными. (3) Добавлена БЛХ-ПЭГ. Он поглощается в микроструктуру капиллярными силами и делает поверхности не покрытыми отталкивая клетки pdms. (4) Крышка промывается водой для удаления оставшихся PLL-PEG. Затем штампы PDMS удаляются, и шестиканальный липкий слайд приклеивается к крышке. (5) Фибронектин, белок внеклеточного матрикса, добавляют, чтобы сделать области без клеточного адгезивного ФЛАЛ-ПЭГ. (6) Шестиканальный затвор промывают фосфатно-буферным физиологическим раствором. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}
2. Посев клеток(рисунок 2А)
ПРИМЕЧАНИЕ: Для следующих этапов промывки добавьте соответствующую жидкость в один резервуар, а затем извлеките равный объем жидкости из противоположного резервуара канала.
- Промыть каждый канал 120 мкл 37 °C полностью дополненной средой для роста клеток. Перед добавлением клеточной суспензии убедитесь, что только каналы заполнены средой, но не резервуарами.
- Отсоедините клетки HuH7 от колбы клеточной культуры в соответствии со стандартным протоколом для прохождения клеток и отрегулируйте концентрацию клеточной суспензии до 4 x10 5 клеток / мл.
- Добавьте 40 мкл клеточной суспензии и смешайте среду роста клеток с клеточной суспензией, удалив 40 мкл из одного резервуара и добавив его в противоположный резервуар того же канала в течение 3 раз, чтобы достичь однородного распределения клеток(рисунок 4B).
- Извлеките 40 мкл суспензии из канала так, чтобы только канал был заполнен клеточной суспензией.
- Поместите затвор в инкубатор и проверьте адгезию клеток через 1 ч после посева с помощью фазоконтрастного микроскопа.
- Добавьте 120 мкл теплой клеточной питательной среды с 37 °C.
- Но скольжение обратно в инкубатор еще на 3 ч для обеспечения клеточной самоорганизации на микроструктуре(рисунок 4С).

Рисунок 4:Клеточная самоорганизация и контроль качества массива μPIPP. (A) Микроструктурированная поверхность состоит из квадратных адгезионных пятен с FN-покрытием, показанных красным цветом, окруженных клеточно-репеллентным полимером. (B) После посева клеток клетки HuH7 распределяются случайным образом и(C)прилипают в основном к пятнам адгезии в течение периода времени 4 ч. Перепечатано с разрешения 7. Шкала: 200 мкм. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}
3. Перфузионные системы(рисунок 2А)
ПРИМЕЧАНИЕ: Использование перфузионной системы требуется только в том случае, если реагенты или флуоресцентные маркеры должны быть добавлены в ходе покадрового измерения. В зависимости от ваших потребностей вы можете подключить каждый канал к отдельной перфузионной системе или подключить несколько каналов последовательно к одной и той же перфузионной системе. Количество перфузионных систем соответствует числу независимых экспериментальных условий. Соедините трубки в стерильных условиях в шкафу биобезопасности и избегайте включения пузырьков воздуха в перфузионную систему. Если перфузионные системы не используются, добавьте реагенты/маркеры в шкаф биобезопасности перед покадровым измерением. Перфузионная система изготавливается на месте, используемый материал указан в Таблице материалов. Сборка перфузионной системы была описанаранее 9.
- Используйте шприц 1 мл (с заменяющим спором) и заполните шприц 1 мл 37 °C клеточной питательной среды.
- Подключите шприц к впускной трубке с помощью клапана и заполните трубку средой.
- Подключите впускную трубку к резервуару канала и убедитесь, что пузырьки воздуха не задерживаются.
- Чтобы подключить другой канал последовательно к этой перфузионной системе, подключите последовательный разъем к резервуару, противоположному впускной трубке текущего канала. Перейдите к следующему каналу и подключите свободный конец последовательного разъема к одному из его резервуаров.
- Повторяйте предыдущие шаги до тех пор, пока необходимое количество каналов не будет подключено последовательно.
- Подключите выпускную трубу непосредственно к свободному резервуару текущего канала. Заполните подключенную трубку средой, чтобы убедиться, что перфузионный режим не протекает.
- Повторяйте предыдущие шаги до тех пор, пока все шесть каналов слайда не будут подключены к перфузионной системе.
- Поместите затвор с подключенной перфузионной системой (системами) обратно в инкубатор до дальнейшего использования или поместите его непосредственно в нагревательную камеру микроскопа, предварительно нагретого до 37 °C для измерения замедленной съемки.
4. Покадровая микроскопия(рисунок 2B)
ПРИМЕЧАНИЕ: Для долгосрочных измерений поддерживайте стабильную температуру 37 °C и стабильный уровень CO2. В качестве альтернативыCO2-зависимойсреде для роста клеток используют среду L15, для которой не требуется система инкубации газа.
ПРИМЕЧАНИЕ: Для количественной визуализации используйте среду роста клеток без фенол-красного цвета во время измерения замедленной съемки для уменьшения фоновой флуоресценции и используйте те же настройки протокола покадровой съемки, а также тот же микроскоп для технических реплик.
- Настройте протокол замедленной съемки для записи фазоконтрастного изображения и флуоресцентного изображения со временем экспозиции 750 мс (в зависимости от камеры), 10-минутным временным интервалом между последовательными циклами через список позиций и временем наблюдения 30 ч, используя 10-кратный объектив и соответствующие флуоресцентные фильтры.
- Поместите шестиканальный затвор с ячейками на одноэлементные массивы в держатель образца теплой нагревательной камеры с 37 °C. Если перфузионные системы подключены к шестиканальному затвору, закрепите трубки на сцене с помощью какой-либо ленты, чтобы гарантировать, что шестиканальный слайд не перемещается во время обмена жидкости. Вставьте свободные концы выпускных трубок через отверстие реакционной трубки объемом 15 мл для сбора жидких отходов.
- Задайте список позиций для измерения интервала сканирования. Убедитесь, что количество позиций может быть проверено в течение определенного интервала времени между последовательными циклами через список позиций. При 10-кратном объективе можно установить 10-30 позиций на канал для сканирования общей площади микрошаблонного изображения в зависимости от размера чипа камеры.
- Запустите покадровое измерение. Для лучшего качества изображения при длительных измерениях используйте автоматизированную систему коррекции фокусировки.
5. Флуоресцентный маркер - трансфекция мРНК(рисунок 2В)
ПРИМЕЧАНИЕ: Для трансфекции в двух каналах, соединенных системой трубок, необходим общий объем трансфекционной смеси 600 мкл (300 мкл на один канал). Указанные объемы относятся к трансфекции в двух соединенных каналах.
- Готовят раствор трансфекционного агента, разбавляя 1 мкл трансфектного агента в 200 мкл сывороточно-восстановленной среды и дайте раствору инкубироваться в течение 5 мин при комнатной температуре.
- Готовят раствор мРНК путем разбавления 300 нг мРНК, кодирующей eGFP, в 150 мкл сывороточно-восстановленной среды.
- Приготовьте трансфекционную смесь, добавив 150 мкл раствора трансфекционного агента в раствор мРНК и хорошо перемешайте его. Дайте трансфекционной смеси инкубировать в течение 20 мин при комнатной температуре.
- Промывайте систему труб 1 мл теплого PBS при 37 °C с помощью шприца во время инкубации трансфекционной смеси. При промывке трубок следите за тем, чтобы ступень микроскопа не двигался. При необходимости приостановите покадровое измерение.
- Разбавляют трансфекционную смесь до конечной концентрации мРНК 0,5 нг/мкл путем добавления 300 мкл сывороточно-восстановленной среды.
- Промыть систему труб трансфекционной смесью с помощью шприца и дать липоплексам мРНК инкубироваться в течение 1 ч (при необходимости приостановите покадровое измерение).
- Прекратить трансфекционную инкубацию и вымыть несвязанные липоплексы мРНК путем промывки 1 мл теплой полностью дополненной питательной среды клеток с помощью шприца (при необходимости приостановите покадровое измерение).
6. Анализ изображений и считывание флуоресценции
- При первом запуске анализа изображений установите версию 0.1.6 программного обеспечения с открытым исходным кодом «Автоматизированный анализ микроструктуры в Python» (PyAMA) из указанного местоположения10 в соответствии с инструкциями, приведенными там.
- Убедитесь, что каналы изображения (фазовая контрастность и флуоресценция) доступны в виде 16-разрядных файлов TIFF с несколькими изображениями. При необходимости конвертируйте их соответствующим образом.
- Запустите PyAMA и нажмите на Открыть стек..., чтобы открыть изображения для анализа.
- Для каждого файла TIFF с несколькими изображениями нажмите кнопку Открыть и выберите файл так, чтобы он отображался в списке загруженных файлов в левой части диалогового окна(рисунок 5-1).
- Отметьте каналы для включения в анализ. Для каждого канала выполните следующие действия.
- Выберите в списке загруженных файлов TIFF-файл, содержащий канал.
- В разделе Добавить новый каналвыберите индекс канала в TIFF-файле. Индексирование основано на нуле; первый канал имеет индекс 0, второй канал имеет индекс 1 и так далее.
- Выберите тип канала. Выберите Фазовый контраст или Флуоресценция для соответствующих каналов изображения и Сегментация для двоичного канала, указывающего контуры ячеек.
- При желании введите метку канала для различения различных каналов флуоресценции: eGFP и DAPI.
- После настройки канала нажмите кнопку Добавить.
- Когда все добавленные каналы отобразятся в списке каналов в правой части диалогового окна, нажмите кнопку ОК, чтобы загрузить стек.
- Чтобы выполнить сегментацию с помощью встроенного алгоритма сегментации PyAMA для распознавания ячеек на основе фазово-контрастных изображений(рисунок 5-2),перейдите в Tools | Бинаризовать... и введите имя файла NumPy с бинаризованным каналом.
ПРИМЕЧАНИЕ: В текущей версии загрузка бинаризованного канала требует перезагрузки всех каналов. - Для выполнения фоновой коррекции11 на флуоресцентномканале (фиг.5-3)убедитесь, что канал флуоресценции и канал сегментации загружены. Если канал сегментации не загружен, убедитесь, что для автоматической сегментации загружен канал фазовой контрастности. Перейдите в «Инструменты > коррекции фона...» и выберите имя результирующего TIFF-файла с исправленным каналом флуоресценции.
ПРИМЕЧАНИЕ: В текущей версии загрузка канала с коррекцией фона требует перезагрузки всех каналов. - Осмотрите предварительно выбранные ячейки(рисунок 5-4)и их интегрированный флуоресцентный сигнал(рисунок 5-5),прокрутив таймфреймы, просмотрев каналы, перечисленные в меню каналов с левой стороны и нажав на ячейки, чтобы выделить их курсы времени флуоресценции(рисунок 1С). Используйте выбор клеток, чтобы исключить клетки, которые не являются жизнеспособными, не ограничены местом адгезии или прикреплены к другой клетке из дальнейшего анализа. Переключите выделение ячеек для считывания, нажав клавишу «Shift» и щелкнув ячейку, или выделив ячейку и нажав клавишу ВВОД.
- Сохраните одноклеточные курсы времени для области ячейки и встроенной флуоресценции(рисунок 5-6),нажав на Файл | Сохранить и выбрать каталог для сохранения.

Рисунок 5:Автоматизированная обработка изображений покадровых серий изображений с использованием PyAMA. (1) Импортируются фазоконтрастные и флуоресцентные серии изображений для каждой позиции изображения. (2) Контуры клеток определяются сегментацией на стеке фазоконтрастных изображений. (3) Фоновая коррекция применяется к флуоресцентным изображениям. (4) Контуры ячеек отслеживаются с течением времени и предварительно выбираются для экспорта. (5) Интенсивность флуоресценции интегрируется на основе отслеживаемых контуров клеток. (6) Оцениваются площади одноклеточных клеток и интегральная интенсивность флуоресценции и экспортируются временные курсы для каждой клетки. Шкала: 100 мкм. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}
- Чтобы проанализировать кинетику трансляции после трансфекции мРНК, подогнать модель трансляции, основанную на биохимических уравнениях скорости, к каждому одноклеточной временной куче, как описано ранее Reiser et al.12. Данные и код, использованные в этом исследовании, являются общедоступными13.
- Для каждого одноклеточного временного курса извлеките расчетные параметры подгонки модели трансляции, которые представляют скорость деградации мРНК и временную точку начала трансляции. Пример набора данных обсуждается в разделе репрезентативных результатов.
- Провести дальнейший анализ распределения наилучших оценок параметров для различных экспериментальных условий для исследования изменчивости между клетками в клеточных популяциях.
Результаты
Подход LISCA позволяет эффективно собирать курсы флуоресценции из отдельных клеток. В качестве репрезентативного примера мы описываем, как метод LISCA применяется для измерения одноклеточной экспрессии eGFP после трансфекции. Данные эксперимента LISCA используются для оценки кинетики достав...
Обсуждение
Здесь мы описали LISCA как универсальный метод для отслеживания клеточной кинетики внутриклеточных флуоресцентных меток на одноклеточном уровне. Для того, чтобы провести успешный эксперимент LISCA, каждый из описанных этапов раздела протокола должен быть установлен индивидуально, а зате?...
Раскрытие информации
Авторы заявляют, что у них нет конкурирующих финансовых интересов.
Благодарности
Эта работа была поддержана грантами Немецкого научного фонда (DFG) Центру совместных исследований (SFB) 1032. Мы с благодарностью отмечаем поддержку Федерального министерства образования, исследований и технологий Германии (BMBF) в рамках совместного проекта 05K2018-2017-06716 Medisoft, а также грант от Bayerische Forschungsstiftung. Анита Рейзер была поддержана стипендией DFG через Высшую школу количественных биологических наук Мюнхена (QBM).
Материалы
| Name | Company | Catalog Number | Comments |
| Adtech Polymer Engineering PTFE Microtubing | Fisher Scientific | 10178071 | |
| baking oven | Binder | 9010-0190 | |
| CFI Plan Fluor DL 10x | Nikon | MRH20100 | |
| Desiccator | Roth | NX07.1 | |
| Eclipse Ti-E | Nikon | ||
| eGFP mRNA | Trilink | L-7601 | |
| Female Luer to Tube Connector | MEDNET | FTL210-6005 | |
| Fetal bovine serum | Thermo Fisher | 10270106 | |
| Fibronectin | Yo Proteins | 663 | |
| Filter set eGFP | AHF | F46-002 | |
| Fisherbrand Translucent Platinum-Cured Silicone Tubing | Fisher Scientific | 11768088 | |
| HEPES (1 M) | Thermo Fisher | 15630080 | |
| Incubation Box | Okolab | OKO-H201 | |
| incubator | Binder | 9040-0012 | |
| L-15 without phenol red | Thermo Fisher | 21083027 | |
| Lipofectamine 2000 | Thermo Fisher | 11668027 | |
| Male Luer | in-house fabricated consisting of teflon | ||
| Male Luer to Tube Connector | MEDNET | MTLS210-6005 | alternative to in-house fabricated male luers |
| NaCl (5 M) | Thermo Fisher | AM9760G | |
| Needleless Valve to Male Luer Connector | MEDNET | NVFMLLPC | |
| NIS Elements | Nikon | Imaging software Version 5.02.00 | |
| NOA81 | Thorlabs | NOA81 | Fast Curing Optical Adhesive for tube system assembly |
| Opti-MEM | Thermo Fisher | 31985062 | |
| PCO edge 4.2 M-USB-HQ-PCO | pco | ||
| Phosphate buffered saline (PBS) | in-house prepared | ||
| Plasma Cleaner | Diener Femto | Pico-BRS | |
| PLL(20 kDa)-g[3.5]-PEG(2 kDa) | SuSoS AG | ||
| silicon wafer mit mircorstructures | in-house fabricated | ||
| Sola Light Engine | Lumencor | ||
| sticky slide VI 0.4 | ibidi | 80608 | |
| Sylgard 184 Silicone Elastomer Kit | Dow Corning | 1673921 | |
| Tango 2 | Märzhäuser | 00-24-626-0000 | |
| Ultrapure water | in-house prepared | ||
| uncoated coverslips | ibidi | 10813 | |
| Injekt-F Solo, 1 mL | Omilab | 9166017V | with replacement sporn |
Ссылки
- Altschuler, S. J., Wu, L. F. Cellular heterogeneity: do differences make a difference. Cell. 141 (4), 559-563 (2010).
- Locke, J. C., Elowitz, M. B. Using movies to analyse gene circuit dynamics in single cells. Nature Reviews Microbiology. 7 (5), 383-392 (2009).
- Spencer, S. L., Gaudet, S., Albeck, J. G., Burke, J. M., Sorger, P. K. Non-genetic origins of cell-to-cell variability in TRAIL-induced apoptosis. Nature. 459 (7245), 428-432 (2009).
- El-Ali, J., Sorger, P. K., Jensen, K. F. Cells on chips. Nature. 442 (7101), 403-411 (2006).
- Segerer, F. J., et al. Versatile method to generate multiple types of micropatterns. Biointerphases. 11 (1), 011005 (2016).
- Piel, M., Théry, M. . Micropatterning in cell biology, part A/B/C. , (2014).
- Reiser, A., Zorn, M. L., Murschhauser, A., Rädler, J. O., Ertl, P., Rothbauer, M. . Cell-Based Microarrays: Methods and Protocols. , 41-54 (2018).
- Picone, R., Baum, B., McKendry, R. . Methods in cell biology. 119, 73-90 (2014).
- Reiser, A. . Single-cell time courses of mRNA transport and translation kinetics. , (2019).
- . Softmatter LMU-Raedler Group Available from: https://github.com/SoftmatterLMU-RaedlerGroup/pyama (2020)
- . MIAAB, 29262 Available from: https://github.com/SoftmatterLMU-RaedlerGroup/pyama (2020)
- Reiser, A., et al. Correlation of mRNA delivery timing and protein expression in lipid-based transfection. Integrative Biology. 11 (9), 362-371 (2019).
- Reiser, A., Woschée, D., Mehrotra, N., Krzysztoń, R. S., Strey, H. H., Rädler, J. O. Supplementing data and code for: "Correlation of mRNA delivery timing and protein expression in lipid-based transfection". Zenodo. , (2019).
- Ferizi, M., et al. Stability analysis of chemically modified mRNA using micropattern-based single-cell arrays. Lab on a Chip. 15 (17), 3561-3571 (2015).
- Fröhlich, F., et al. Multi-experiment nonlinear mixed effect modeling of single-cell translation kinetics after transfection. NPJ systems biology and applications. 4 (1), 1-12 (2018).
- Krzysztoń, R., et al. Single-cell kinetics of siRNA-mediated mRNA degradation. Nanomedicine: Nanotechnology, Biology and Medicine. 21, 102077 (2019).
- Röttgermann, P. J., Alberola, A. P., Rädler, J. O. Cellular self-organization on micro-structured surfaces. Soft Matter. 10 (14), 2397-2404 (2014).
- Röttgermann, P. J., Dawson, K. A., Rädler, J. O. Time-resolved study of nanoparticle induced apoptosis using microfabricated single cell arrays. Microarrays. 5 (2), 8 (2016).
- Murschhauser, A., et al. A high-throughput microscopy method for single-cell analysis of event-time correlations in nanoparticle-induced cell death. Communications Biology. 2 (1), 1-11 (2019).
- Chatzopoulou, E. I., et al. Chip-based platform for dynamic analysis of NK cell cytolysis mediated by a triplebody. Analyst. 141 (7), 2284-2295 (2016).
- Sztilkovics, M., et al. Single-cell adhesion force kinetics of cell populations from combined label-free optical biosensor and robotic fluidic force microscopy. Scientific Reports. 10 (1), 1-13 (2020).
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеСмотреть дополнительные статьи
This article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены