
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Live-Cell Imaging of Single-Cell Arrays (LISCA) - eine vielseitige Technik zur Quantifizierung der zellulären Kinetik
In diesem Artikel
Zusammenfassung
Wir stellen eine Methode zur Erfassung von Fluoreszenz-Reporter-Zeitkursen aus einzelnen Zellen mit mikrostrukturierten Arrays vor. Das Protokoll beschreibt die Vorbereitung von Einzelzellarrays, den Aufbau und Betrieb der Live-Cell-Scanning-Zeitraffermikroskopie und ein Open-Source-Bildanalyse-Tool zur automatisierten Vorauswahl, visuellen Kontrolle und Verfolgung von zellintegrierten Fluoreszenzzeitverläufen pro Adhäsionsstelle.
Zusammenfassung
Live-cell Imaging of Single-Cell Arrays (LISCA) ist eine vielseitige Methode, um Zeitverläufe von Fluoreszenzsignalen einzelner Zellen bei hohem Durchsatz zu sammeln. Im Allgemeinen wird der Erwerb von Einzelzellzeitverläufen aus kultivierten Zellen durch die Zellmotilität und die Vielfalt der Zellformen behindert. Adhäsive Mikroarrays standardisieren Einzelzellbedingungen und erleichtern die Bildanalyse. LISCA kombiniert Einzelzell-Microarrays mit Rasterzeitraffermikroskopie und automatisierter Bildverarbeitung. Hier beschreiben wir die experimentellen Schritte der Teilnahme an einzelligen Fluoreszenzzeitkursen im LISCA-Format. Wir transfizieren Zellen, die an einem mikrostrukturierten Array haften, indem wir mRNA verwenden, das für ein verbessertes grün fluoreszierendes Protein (eGFP) kodiert, und überwachen die eGFP-Expressionskinetik von Hunderten von Zellen parallel mittels Rasterzeitraffermikroskopie. Die Bilddatenstapel werden automatisch von einer neu entwickelten Software verarbeitet, die die Fluoreszenzintensität über ausgewählte Zellkonturen integriert, um Einzelzell-Fluoreszenzzeitverläufe zu erzeugen. Wir zeigen, dass eGFP-Expressionszeitkurse nach der mRNA-Transfektion durch ein einfaches kinetisches Translationsmodell gut beschrieben werden, das Expressions- und Abbauraten von mRNA aufdeckt. Weitere Anwendungen von LISCA für Ereigniszeitkorrelationen multipler Marker im Kontext der Signalapoptose werden diskutiert.
Einleitung
In den letzten Jahren hat sich die Bedeutung von Einzelzellexperimenten gezeigt. Daten aus einzelnen Zellen ermöglichen die Untersuchung der Zell-zu-Zell-Variabilität, die Auflösung intrazellulärer Parameterkorrelationen und die Detektion zellulärer Kinetik, die in Ensemblemessungen verborgen bleiben1,2,3. Um die zelluläre Kinetik von Tausenden von Einzelzellen parallel zu untersuchen, bedarf es neuer Ansätze, die es ermöglichen, die Zellen unter standardisierten Bedingungen über einen Zeitraum von mehreren Stunden bis zu mehreren Tagen zu überwachen, gefolgt von einer quantitativen Datenanalyse 4. Hier stellen wir Live-cell Imaging of Single-Cell Arrays (LISCA) vor, das den Einsatz mikrostrukturierter Arrays mit Zeitraffermikroskopie und automatisierter Bildanalyse kombiniert.
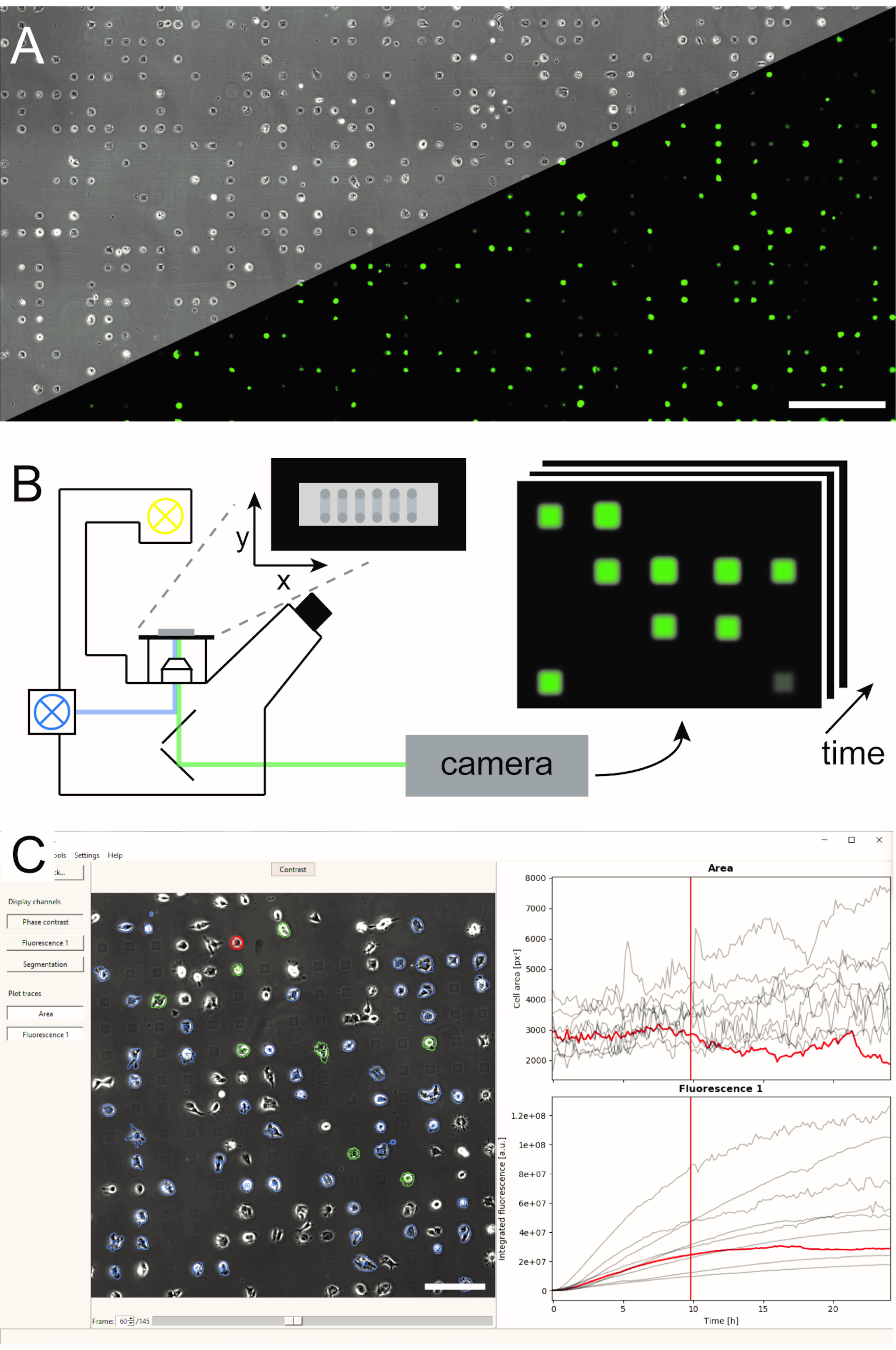
Mehrere Methoden zur Erzeugung mikrostrukturierter Einzelzellarrays wurden etabliert und in literatur5,6veröffentlicht. Hier beschreiben wir kurz das mikroskalige plasmainitiierte Proteinmuster (μPIPP). Ein detailliertes Protokoll der Einzelzellarray-Herstellung mit μPIPP finden Sie auch in Referenz7. Die Verwendung von Einzelzellarrays ermöglicht die Ausrichtung von Tausenden von Zellen auf standardisierten Adhäsionsstellen, die definierte Mikroumgebungen für jede Zelle darstellen, und reduziert so eine Quelle experimenteller Variabilität (Abbildung 1A). Einzelzellarrays werden verwendet, um die Zeitverläufe von fluoreszierenden Markern zu überwachen, die eine Vielzahl von zellulären Prozessen anzeigen sollen. Die Langzeitmikroskopie im Rasterzeitraffermodus ermöglicht die Überwachung eines großen Bereichs der Einzelzellarrays und damit die Abtastung von Einzelzelldaten in hohem Durchsatz über eine Beobachtungszeit von mehreren Stunden oder sogar Tagen. Dadurch werden Zeitlinienstapel von Bildern aus jeder Position des Arrays generiert (Abbildung 1B). Um die große Menge an Bilddaten zu reduzieren und die gewünschten Einzelzellfluoreszenzzeitverläufe effizient zu extrahieren, ist eine automatisierte Bildverarbeitung erforderlich, die sich die Positionierung der Zellen zunutze macht (Bild 1C).
Die Herausforderung von LISCA besteht darin, die experimentellen Protokolle und Berechnungswerkzeuge anzupassen, um einen Hochdurchsatz-Assay zu bilden, der quantitative und reproduzierbare Daten der zellulären Kinetik generiert. In diesem Artikel beschreiben wir Schritt für Schritt die einzelnen Methoden und wie sie in einem LISCA-Assay kombiniert werden. Als Beispiel diskutieren wir den zeitlichen Verlauf der Expression von enhanced green fluorescent protein (eGFP) nach künstlicher mRNA-Abgabe. Die eGFP-Expression nach mRNA-Abgabe wird durch Reaktionsratengleichungen beschrieben, die die Translation und den Abbau von mRNA modellieren. Die Anpassung der Modellfunktion für den zeitlichen Verlauf der eGFP-Konzentration an die LISCA-Ablesung der Fluoreszenzintensität für jede einzelne Zelle im Zeitverlauf liefert beste Schätzungen von Modellparametern wie der mRNA-Abbaurate. Als repräsentatives Ergebnis diskutieren wir die mRNA-Abgabeeffizienz von zwei verschiedenen lipidbasierten Transfektionsmitteln und wie sich ihre Parameterverteilungen unterscheiden.

Abbildung 1: Darstellung des LISCA-Workflows, der (A) mikrogemusterte Einzelzellarrays (B), Rasterzeitraffermikroskopie und (C) automatisierte Bildanalyse von aufgezeichneten Bildserien kombiniert. Die Einzelzellarrays bestehen aus einem zweidimensionalen Muster von zellklebenden Quadraten mit einem zellabweisenden Zwischenraum, der zu einer Anordnung der Zellen auf dem Mikromuster führt, wie im Phasenkontrastbild sowie im Fluoreszenzbild von eGFP-exprimierenden Zellen (A) zu sehen ist. Der gesamte mikrostrukturierte Bereich wird im Scan-Zeitraffermodus abgebildet, wobei wiederholt Bilder an einer Sequenz von Positionen (B) aufnahmen. Aufgezeichnete Bildserien werden verarbeitet, um die Fluoreszenzintensität pro Zelle über die Zeit auszulesen (C). Maßstabsstäbe: 500 μm (A), 200 μm (C). Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
Protokoll

Abbildung 2: Datenerfassung bei der Einzelzell-Microarrays (A) mit Rasterzeitraffermikroskopie (B) kombiniert werden. Zur Vorbereitung des Zeitrafferexperiments wird ein Einzelzellarray mit einem 2D-Mikromuster aus Adhäsionsquadrats vorbereitet (1), gefolgt von der Zellaussaat und der Ausrichtung der Zellen auf dem Mikromuster (2) sowie dem Anschluss eines Perfusionssystems an den Sechskanal-Objektträger, der ein Liquid Handling während der Zeitraffermessung ermöglicht (3). Ein Rasterzeitrafferexperiment wird aufgebaut (4) und die Zellen werden am Mikroskop transfiziert, indem während des Zeitrafferexperiments eine mRNA-Lipoplexlösung durch das Perfusionssystem injiziert wird (5). Maßstabsbalken: 200 μm. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
1. Mikrostrukturierte Einzelzell-Array-Fertigung (Abbildung 2A)
- Bereiten Sie die Materialien vor, die für die Herstellung von μPIPP-Arrays benötigt werden.
- Bereiten Sie sterile phosphatgepufferte Kochsalzlösung (PBS) bei pH 7,4 vor.
- Steriles Reinstwasser mit einem Widerstand von mindestens 18 MΩcm bei 25 °C vorbereiten.
- PLL(20 kDa)-g[3.5] PEG(2 kDa) (PLL-PEG) Arbeitslösung mit einer Konzentration von 2 mg/ml PLL-PEG in Reinstwasser mit 150 mM NaCl und 10 mM 4-(2-Hydroxyethyl)-1-piperazinethansulfonsäure (HEPES) vorbereiten.
- Bereiten Sie eine extrazelluläre Matrixproteinlösung für die Oberflächenbeschichtung vor: 1 mg / ml Fibronektin (FN) in PBS.
- Bereiten Sie einen Siliziumwafer mit einem durch Photolithographie hergestellten Mikromuster8 vor, das als wiederverwendbarer Master fungiert. Das Mikromuster besteht aus Quadraten mit einer Kantenlänge von 30 μm, einer Tiefe von 12 μm und einem Interquadratabstand von 60 μm, die in sechs Streifen mit einer Breite von jeweils 6 mm und einer Höhe von 18 mm angeordnet sind.
- Mischen Sie ein Polydimethylsiloxan (PDMS) Monomer mit 9% Vernetzer (Masse %) verwenden Sie ein Silikonelastomer-Kit und entgasen Sie es für ca. 30 minuten, bis es mit einem Trockenmittel blasenfrei ist. Den Siliziumwafer mit einer ca. 3-5 mm dicken PDMS-Schicht gießen und für ca. 30 min wieder entgasen, bis er blasenfrei ist.
- Legen Sie die Siliziumwaffel mit dem PDMS in einen Backofen bei 50 °C, um das PDMS für mindestens 4 h auszuhärten.
- Schneiden Sie die PDMS-Stempel aus.
- Verwenden Sie ein Skalpell und schneiden Sie aus der PDMS-Schicht ein PDMS-Meisterwerk aus, das die sechs Mikromusterstreifen enthält.
- Stellen Sie das PDMS-Meisterwerk auf eine Bank mit dem Mikromuster nach oben.
- Schneiden Sie jeden der sechs Mikromusterstreifen des PDMS-Meisterwerks mit einer Rasierklinge in einen PDMS-Stempel. Achten Sie darauf, dass die Kanten der PDMS-Stempel offen sind, indem Sie einen Teil des gemusterten Bereichs abschneiden.
- Platzieren Sie die PDMS-Stempel auf einem Abdeckstrich eines sechskanaligen Dias (Abbildung 3-1).
- Verwenden Sie einen unbeschichteten Deckrutsch und markieren Sie die Kanalpositionen des Sechskanalträgers, indem Sie die Schutzfolie des Deckstücks vorsichtig zerkratzen. Legen Sie dann den Abdeckklip mit der Schutzfolie nach unten auf die Bank.
- Platzieren Sie die PDMS-Stempel mit dem mikromuster nach unten gerichteten Mikromuster auf dem Abdeckbügel an den markierten Kanalpositionen mit einer Pinzette.
- Überprüfen Sie die Befestigung der PDMS-Stempel unter einem Mikroskop. Wenn ein PDMS-Stempel vollständig am Coverlip befestigt ist, erscheinen die in Kontakt tretenden Quadrate dunkler als der Zwischenraum. Die Befestigung des PDMS-Stempels am Coverlip ist entscheidend für die Mikromusterqualität.
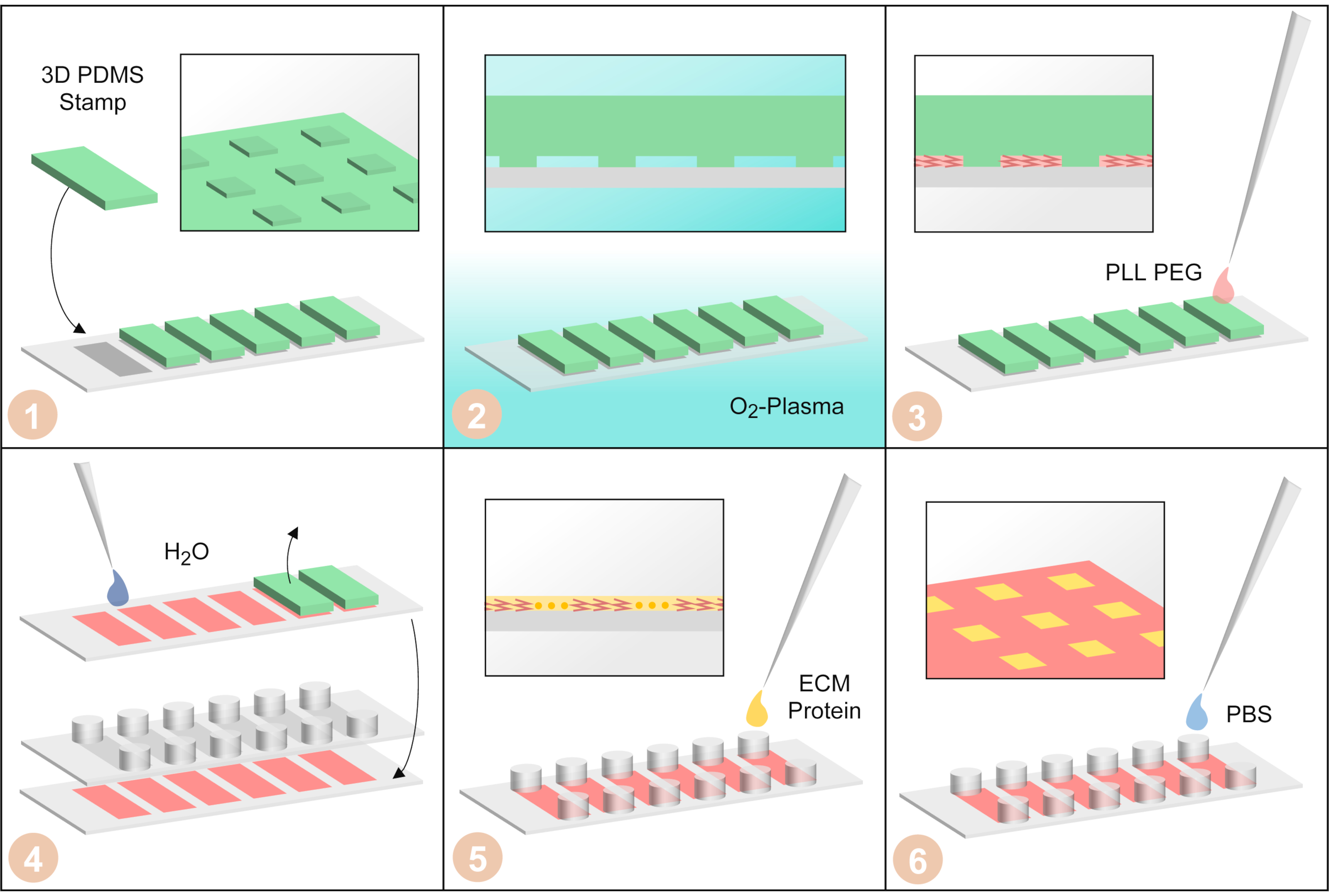
- Legen Sie den Abdeckklip mit den sechs PDMS-Stempeln in einen Plasmareiniger und behandeln Sie ihn mit Sauerstoffplasma (Druck 0,2 mbar, ~40 W für 3 min), um die Oberflächen zwischen den PDMS-Stempeln und dem Abdeckr hydrophil zu machen (Abbildung 3-2).
- Führen Sie alle weiteren Schritte der Mikromusterherstellung in einer Biosicherheitswerkbank durch. Verwenden Sie 15 μL der PLL-PEG-Lösung und pipetieren Sie einen Tropfen davon neben jedem PDMS-Stempel, so dass die PLL-PEG-Lösung in das hydrophile Muster des PDMS-Stempels aufgenommen wird (Abbildung 3-3). Lassen Sie den PLL-PEG 20 min bei Raumtemperatur inkubieren.
- Spülen Sie 1 ml Reinstwasser mit den PDMS-Stempeln über den Abdeckr und entfernen Sie die PDMS-Stempel mit einer Pinzette (Abbildung 3-4). Anschließend den Abdeckt ein zweites Mal mit 1 ml Reinstwasser abspülen und trocknen lassen.
- Wenn der Abdeckklip vollständig getrocknet ist, kleben Sie einen sechskanaligen klebrigen Schlitten auf den Coverslip (Abbildung 3-4). Achten Sie darauf, dass die mikrostrukturierten Bereiche mit der Unterseite der Kanäle ausgerichtet sind.
- Funktionalisieren Sie die Adhäsionsquadrate mit FN.
- Füllen Sie 40 μL PBS in jeden Kanal.
- Bereiten Sie eine 100 μg/ml FN-Lösung in PBS vor.
- Zu jedem Kanal werden 40 μL der FN-Lösung hinzugefügt(Abbildung 3-5). Mischen Sie die FN-Lösung gründlich mit dem PBS im Kanal, indem Sie 40 μL aus einem Reservoir entfernen und es 3 Mal in das gegenüberliegende Reservoir desselben Kanals geben, um eine homogene Lösung zu erzeugen. Inkubieren Sie die FN-Lösung für 45 min bei Raumtemperatur.
- Waschen Sie jeden Kanal dreimal mit 120 μL PBS (Abbildung 3-6).
- Um die Musterqualität zu überprüfen, verwenden Sie ein fluoreszierend markiertes FN in Schritt 9.2. (Abbildung 4A).
HINWEIS: Wir empfehlen, das μPIPP-Array nicht mehr als einen Tag vor der Zellaussaat vorzubereiten, da PLL-PEG und FN nicht kovalent an das Substrat gebunden sind und die Qualität des Musters im Laufe der Zeit abnehmen kann. Bewahren Sie das vorbereitete μPIPP-Array im Kühlschrank auf.

Abbildung 3: Einzelzellige Microarray-Herstellung durch μPIPP. (1) PDMS-Stempel mit dreidimensionaler Mikrostruktur auf der Oberfläche sind auf einem Deckslip eines sechskanaligen Objektträgers angeordnet. (2) Der Abdeckklip mit den PDMS-Stempeln darauf wird mit Sauerstoffplasma behandelt, um die Oberflächen hydrophil zu machen. (3) PLL-PEG wird hinzugefügt. Es wird durch Kapillarkräfte in die Mikrostruktur aufgenommen und macht die Oberflächen, die nicht vom PDMS-Stempel bedeckt sind, zellabweisend. (4) Der Abdeckr wird mit Wasser gespült, um das verbleibende PLL-PEG zu entfernen. Dann werden die PDMS-Stempel entfernt und ein sechskanaliger klebriger Schieber wird auf den Coverlip geklebt. (5) Fibronektin, ein Protein der extrazellulären Matrix, wird zugegeben, um die Bereiche ohne PLL-PEG-Zellkleber herzustellen. (6) Der sechskanalige Schlitten wird mit phosphatgepufferter Kochsalzlösung gewaschen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
2. Zellaussaat (Abbildung 2A)
HINWEIS: Für die folgenden Waschschritte fügen Sie die jeweilige Flüssigkeit in ein Reservoir und entfernen Sie dann ein gleiches Flüssigkeitsvolumen aus dem gegenüberliegenden Reservoir eines Kanals.
- Waschen Sie jeden Kanal mit 120 μL 37 °C vollständig ergänztem Zellwachstumsmedium. Stellen Sie vor der Zugabe der Zellsuspension sicher, dass nur die Kanäle mit Medium gefüllt sind, nicht aber die Reservoirs.
- Lösen Sie HuH7-Zellen aus einem Zellkulturkolben gemäß Ihrem Standardprotokoll für die Zellpassage und stellen Sie die Zellsuspensionskonzentration auf 4 x 105 Zellen/ml ein.
- Fügen Sie 40 μL Zellsuspension hinzu und mischen Sie das Zellwachstumsmedium mit der Zellsuspension, indem Sie 40 μL aus einem Reservoir entfernen und es 3 Mal in das gegenüberliegende Reservoir desselben Kanals geben, um eine homogene Zellverteilung zu erreichen (Abbildung 4B).
- Entfernen Sie 40 μL Suspension aus dem Kanal, so dass nur der Kanal mit Zellsuspension gefüllt ist.
- Legen Sie den Objektträger in einen Inkubator und überprüfen Sie die Zellhaftung 1 h nach der Aussaat mit einem Phasenkontrastmikroskop.
- Fügen Sie 120 μL warmes Zellwachstumsmedium mit 37 °C hinzu.
- Aber das Rutschen zurück in den Inkubator für weitere 3 h, um zelluläre Selbstorganisation auf dem Mikromuster zu ermöglichen (Abbildung 4C).

Abbildung 4: Zelluläre Selbstorganisation und Qualitätskontrolle des μPIPP-Arrays. (A) Die mikrostrukturierte Oberfläche besteht aus quadratischen FN-beschichteten Adhäsionsflecken, die rot dargestellt sind und von einem zellabweisenden Polymer umgeben sind. (B) Nach der Zellaussaat werden die HuH7-Zellen zufällig verteilt und (C) haften hauptsächlich an den Adhäsionsstellen über einen Zeitraum von 4 h. Nachdruck mit Genehmigung 7. Maßstabsbalken: 200 μm. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
3. Perfusionssystem (Abbildung 2A)
HINWEIS: Die Verwendung eines Perfusionssystems ist nur erforderlich, wenn im Laufe der Zeitraffermessung Reagenzien oder fluoreszierende Marker hinzugefügt werden müssen. Je nach Bedarf können Sie jeden Kanal an ein separates Perfusionssystem anschließen oder mehrere Kanäle in Reihe mit demselben Perfusionssystem verbinden. Die Anzahl der Perfusionssysteme entspricht der Anzahl der unabhängigen Versuchsbedingungen. Verbinden Sie die Röhrchen unter sterilen Bedingungen in einer Biosicherheitswerkbank und vermeiden Sie die Aufnahme von Luftblasen in das Perfusionssystem. Wenn kein Perfusionssystem verwendet wird, fügen Sie die Reagenzien/Marker vor der Zeitraffermessung in eine Biosicherheitswerkbank ein. Das Perfusionssystem wird selbst hergestellt, das verwendete Material ist in der Materialtabelleaufgeführt. Der Aufbau des Perfusionssystems wurde zuvor beschrieben9.
- Verwenden Sie eine 1-ml-Spritze (mit Ersatzsporn) und füllen Sie die Spritze mit 1 ml 37 °C Zellwachstumsmedium.
- Verbinden Sie die Spritze mit dem Einlassrohr über das Ventil und füllen Sie das Röhrchen mit Medium.
- Schließen Sie das Einlassrohr an ein Reservoir eines Kanals an und stellen Sie sicher, dass keine Luftblasen eingeschlossen sind.
- Um einen weiteren Kanal in Reihe mit diesem Perfusionssystem zu verbinden, schließen Sie einen seriellen Stecker an das Reservoir gegenüber dem Einlassrohr des Stromkanals an. Fahren Sie mit dem nächsten Kanal fort und verbinden Sie das freie Ende des seriellen Anschlusses mit einem seiner Behälter.
- Wiederholen Sie die vorherigen Schritte, bis die erforderliche Anzahl von Kanälen in Reihe geschaltet ist.
- Schließen Sie das Auslassrohr direkt an das freie Reservoir des Stromkanals an. Füllen Sie das angeschlossene Röhrchen mit Medium, um zu überprüfen, ob das Perfusionssystem nicht ausläuft.
- Wiederholen Sie die vorherigen Schritte, bis alle sechs Kanäle des Objektträgers mit einem Perfusionssystem verbunden sind.
- Legen Sie den Objektträger mit dem/den angeschlossenen Perfusionssystem(en) bis zur weiteren Verwendung wieder in den Inkubator oder legen Sie ihn direkt in die Heizkammer des auf 37 °C vorgewärmten Mikroskops zur Zeitraffermessung.
4. Zeitraffermikroskopie (Abbildung 2B)
HINWEIS: Halten Sie für Langzeitmessungen eine stabile Temperatur von 37 °C und einen stabilen CO2-Gehalt aufrecht. Als Alternative zum CO2-abhängigenZellwachstumsmedium verwenden Sie L15-Medium, für das kein Gasinkubationssystem erforderlich ist.
HINWEIS: Verwenden Sie für die quantitative Bildgebung während der Zeitraffermessung ein Zellwachstumsmedium ohne Phenolrot, um die Hintergrundfluoreszenz zu reduzieren, und verwenden Sie die gleichen Einstellungen des Zeitrafferprotokolls sowie das gleiche Mikroskop für technische Replikate.
- Richten Sie ein Zeitrafferprotokoll für die Aufnahme eines Phasenkontrastbildes und eines Fluoreszenzbildes mit Belichtungszeiten von 750 ms (abhängig von der Kamera), 10-minütigem Zeitintervall zwischen aufeinanderfolgenden Schleifen durch die Positionsliste und einer Beobachtungszeit von 30 h mit einem 10-fachen Objektiv und geeigneten Fluoreszenzfiltern ein.
- Legen Sie den sechskanaligen Objektträger mit den Zellen auf die Einzelzellenarrays in den Probenhalter der 37 °C warmen Heizkammer. Wenn Perfusionssysteme mit dem sechskanaligen Schlitten verbunden sind, befestigen Sie die Rohre mit etwas Klebeband an der Bühne, um sicherzustellen, dass der sechskanalige Schlitten während des Flüssigkeitsaustauschs nicht bewegt wird. Führen Sie die freien Enden der Auslassrohre durch ein Loch eines 15-ml-Reaktionsrohrs ein, um den flüssigen Abfall zu sammeln.
- Legen Sie die Positionsliste für die Scanzeitraffermessung fest. Stellen Sie sicher, dass die Anzahl der Positionen innerhalb des definierten Zeitintervalls zwischen aufeinanderfolgenden Schleifen durch die Positionsliste gescannt werden kann. Mit einem 10-fachen Objektiv können 10-30 Positionen pro Kanal eingestellt werden, um den gesamten Mikromusterbereich abhängig von der Größe des Kamerachips zu scannen.
- Starten Sie die Zeitraffermessung. Für eine bessere Bildqualität von Langzeitmessungen verwenden Sie ein automatisiertes Fokuskorrektursystem.
5. Fluoreszierender Marker - mRNA-Transfektion (Abbildung 2B)
HINWEIS: Für eine Transfektion in zwei Kanälen, die durch ein Schlauchsystem verbunden sind, wird ein Gesamtvolumen von 600 μL Transfektionsmischung benötigt (300 μL für einen Kanal). Die angegebenen Volumina beziehen sich auf eine Transfektion in zwei miteinander verbundenen Kanälen.
- Bereiten Sie eine Transfektionsmittellösung vor, indem Sie 1 μL Transfektionsmittel in 200 μL serumreduziertem Medium verdünnen und die Lösung 5 min bei Raumtemperatur inkubieren lassen.
- Bereiten Sie eine mRNA-Lösung vor, indem Sie 300 ng mRNA, die für eGFP kodiert, in 150 μL serumreduziertes Medium verdünnen.
- Bereiten Sie die Transfektionsmischung vor, indem Sie 150 μL der Transfektionsmittellösung in die mRNA-Lösung geben und gut mischen. Lassen Sie die Transfektionsmischung 20 minuten bei Raumtemperatur inkubieren.
- Spülen Sie das Schlauchsystem während der Inkubation der Transfektionsmischung mit 1 mL 37 °C warmem PBS mit einer Spritze. Achten Sie beim Spülen der Röhrchen darauf, dass sich der Mikroskopstand nicht bewegt. Pausieren Sie die Zeitraffermessung bei Bedarf.
- Verdünnen Sie die Transfektionsmischung auf die endgültige mRNA-Konzentration von 0,5 ng/μL, indem Sie 300 μL serumreduziertes Medium hinzufügen.
- Spülen Sie das Schlauchsystem mit der Transfektionsmischung mit einer Spritze und lassen Sie die mRNA-Lipoplexe 1 h inkubieren (pausieren Sie die Zeitraffermessung, falls erforderlich).
- Stoppen Sie die Transfektionsinkubation und spülen Sie die ungebundenen mRNA-Lipoplexe aus, indem Sie mit 1 ml 37 °C warmes, vollständig ergänztes Zellwachstumsmedium mit einer Spritze waschen (pause die Zeitraffermessung falls erforderlich).
6. Bildanalyse und Fluoreszenzanzeige
- Wenn Sie die Bildanalyse zum ersten Mal ausführen, installieren Sie die Version 0.1.6 der Open-Source-Software "Automated Microstructure Analysis in Python" (PyAMA) von der zitierten Stelle10 gemäß den dort bereitgestellten Anweisungen.
- Stellen Sie sicher, dass die Bildkanäle (Phasenkontrast und Fluoreszenz) als 16-Bit-TIFF-Dateien mit mehreren Bildern verfügbar sind. Wenn nötig, konvertieren Sie sie entsprechend.
- Starten Sie PyAMA und klicken Sie auf Open stack..., um Bilder zur Analyse zu öffnen.
- Klicken Sie für jede zu öffnende TIFF-Datei mit mehreren Bildern auf Öffnen und wählen Sie die Datei so aus, dass sie in der Liste der geladenen Dateien auf der linken Seite des Dialogfelds angezeigt wird (Abbildung 5-1).
- Markieren Sie die Kanäle, die in die Analyse aufgenommen werden sollen. Führen Sie für jeden Kanal die folgenden Schritte aus.
- Wählen Sie in der Liste der geladenen Dateien die TIFF-Datei aus, die den Kanal enthält.
- Wählen Sie im Abschnitt Neuen Kanal hinzufügenden Index des Kanals in der TIFF-Datei aus. Die Indizierung erfolgt nullbasiert. Der erste Kanal hat Index 0, der zweite Kanal hat Index 1 und so weiter.
- Wählen Sie den Kanaltyp aus. Wählen Sie Phasenkontrast oder Fluoreszenz für die entsprechenden Bildkanäle und Segmentierung für einen Binärkanal, der die Zellkonturen anzeigt.
- Geben Sie optional eine Beschriftung des Kanals ein, um verschiedene Fluoreszenzkanäle zu unterscheiden: eGFP und DAPI.
- Klicken Sie nach dem Konfigurieren des Kanals auf Hinzufügen.
- Wenn alle hinzugefügten Kanäle in der Kanalliste auf der rechten Seite des Dialogfelds angezeigt werden, klicken Sie auf OK, um den Stack zu laden.
- Um die Segmentierung mit dem integrierten Segmentierungsalgorithmus von PyAMA für die Zellerkennung basierend auf den Phasenkontrastbildern durchzuführen (Abbildung 5-2), gehen Sie zu Tools | Binarisieren... und geben Sie einen Dateinamen für die NumPy-Datei mit dem binarisierten Kanal ein.
HINWEIS: In der aktuellen Version erfordert das Laden des binarisierten Kanals das Erneutladen aller Kanäle. - Um eine Hintergrundkorrektur11 auf einem Fluoreszenzkanal durchzuführen (Abbildung 5-3), stellen Sie sicher, dass der Fluoreszenzkanal und ein Segmentierungskanal geladen sind. Wenn kein Segmentierungskanal geladen ist, stellen Sie sicher, dass ein Phasenkontrastkanal für die automatische Segmentierung geladen wird. Gehen Sie zu "Tools > Hintergrundkorrektur..." und wählen Sie einen Dateinamen für die resultierende TIFF-Datei mit dem korrigierten Fluoreszenzkanal.
HINWEIS: In der aktuellen Version erfordert das Laden des hintergrundkorrigierten Kanals das Erneutladen aller Kanäle. - Untersuchen Sie die vorausgewählten Zellen (Abbildung 5-4) und ihr integriertes Fluoreszenzsignal (Abbildung 5-5), indem Sie durch die Zeitrahmen scrollen, die im Kanalmenü auf der linken Seite aufgeführten Kanäle anzeigen und auf Zellen klicken, um ihre Fluoreszenzzeitverläufe hervorzuheben (Abbildung 1C). Verwenden Sie die Zellauswahl, um Zellen auszuschließen, die nicht lebensfähig sind, nicht auf einen Adhäsionsfleck beschränkt oder an eine andere Zelle gebunden sind, von der weiteren Analyse. Schalten Sie die Auswahl der Zellen zum Auslesen um, indem Sie die Umschalttaste drücken und auf die Zelle klicken oder die Zelle markieren und die Eingabetastedrücken.
- Speichern Sie die Einzelzellzeitverläufe für den Zellbereich und die integrierte Fluoreszenz (Abbildung 5-6), indem Sie auf Datei | Speichern und auswählen Sie ein Verzeichnis, in dem Gespeichert werden soll.

Abbildung 5: Automatisierte Bildverarbeitung von Zeitraffer-Bildserien mit PyAMA. (1) Phasenkontrast- und Fluoreszenzbildserien für jede Bildposition werden importiert. (2) Zellkonturen werden durch Segmentierung auf dem Phasenkontrast-Bildstapel bestimmt. (3) Auf die Fluoreszenzbilder wird eine Hintergrundkorrektur angewendet. (4) Die Zellkonturen werden über die Zeit verfolgt und für den Export vorausgewählt. (5) Die Fluoreszenzintensität wird anhand der verfolgten Zellkonturen integriert. (6) Einzelzellbereiche und integrierte Fluoreszenzdicken werden ausgewertet und Zeitverläufe für jede Zelle exportiert. Maßstabsbalken: 100 μm. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
{kind=link}
- Um die Translationskinetik nach der mRNA-Transfektion zu analysieren, passen Sie ein Translationsmodell basierend auf biochemischen Geschwindigkeitsgleichungen an jeden Einzelzellzeitverlauf an, wie zuvor von Reiser et al.12beschrieben. Die in dieser Studie verwendeten Daten und Codes sind öffentlich zugänglich13.
- Rufen Sie für jeden Einzelzellzeitverlauf die geschätzten Anpassungsparameter des Translationsmodells ab, die die mRNA-Abbaurate und den Zeitpunkt des Translationsbeginns darstellen. Ein Beispieldatensatz wird im Abschnitt "Repräsentative Ergebnisse" erläutert.
- Führen Sie weitere Analysen der Verteilung der besten Schätzungen der Parameter für verschiedene experimentelle Bedingungen durch, um die Zell-zu-Zell-Variabilität innerhalb der Zellpopulationen zu untersuchen.
Ergebnisse
Der LISCA-Ansatz ermöglicht es, Fluoreszenzzeitkurse von einzelnen Zellen effizient zu erfassen. Als repräsentatives Beispiel skizzieren wir, wie die LISCA-Methode zur Messung der einzelligen eGFP-Expression nach der Transfektion angewendet wird. Die Daten des LISCA-Experiments werden verwendet, um die mRNA-Abgabekinetik zu bewerten, die für die Entwicklung effizienter mRNA-Medikamente wichtig ist.
Insbesondere zeigen wir die unterschiedlichen Auswirkungen zweier lipidbasierter mRNA-Abgabes...
Diskussion
Hier beschrieben wir LISCA als eine vielseitige Technik, um die zelluläre Kinetik intrazellulärer fluoreszierender Markierungen auf Einzelzellebene zu verfolgen. Um ein erfolgreiches LISCA-Experiment durchführen zu können, muss jeder der beschriebenen Schritte des Protokollabschnitts einzeln festgelegt und dann alle Schritte kombiniert werden. Jeder der drei Hauptaspekte von LISCA zeichnet sich durch entscheidende Schritte aus.
Einzelzell-Microarray-Herstellung
Die Qualität de...
Offenlegungen
Die Autoren erklären, dass sie keine konkurrierenden finanziellen Interessen haben.
Danksagungen
Diese Arbeit wurde durch Zuschüsse der Deutschen Forschungsgemeinschaft (DFG) zum Sonderforschungsbereich (SFB) 1032 unterstützt. Die Unterstützung durch das Bundesministerium für Bildung, Forschung und Technologie (BMBF) im Rahmen des Kooperationsprojekts 05K2018-2017-06716 Medisoft sowie eine Förderung durch die Bayerische Forschungsstiftung werden dankbar gewürdigt. Anita Reiser wurde durch ein DFG-Fellowship der Graduate School of Quantitative Biosciences Munich (QBM) gefördert.
Materialien
| Name | Company | Catalog Number | Comments |
| Adtech Polymer Engineering PTFE Microtubing | Fisher Scientific | 10178071 | |
| baking oven | Binder | 9010-0190 | |
| CFI Plan Fluor DL 10x | Nikon | MRH20100 | |
| Desiccator | Roth | NX07.1 | |
| Eclipse Ti-E | Nikon | ||
| eGFP mRNA | Trilink | L-7601 | |
| Female Luer to Tube Connector | MEDNET | FTL210-6005 | |
| Fetal bovine serum | Thermo Fisher | 10270106 | |
| Fibronectin | Yo Proteins | 663 | |
| Filter set eGFP | AHF | F46-002 | |
| Fisherbrand Translucent Platinum-Cured Silicone Tubing | Fisher Scientific | 11768088 | |
| HEPES (1 M) | Thermo Fisher | 15630080 | |
| Incubation Box | Okolab | OKO-H201 | |
| incubator | Binder | 9040-0012 | |
| L-15 without phenol red | Thermo Fisher | 21083027 | |
| Lipofectamine 2000 | Thermo Fisher | 11668027 | |
| Male Luer | in-house fabricated consisting of teflon | ||
| Male Luer to Tube Connector | MEDNET | MTLS210-6005 | alternative to in-house fabricated male luers |
| NaCl (5 M) | Thermo Fisher | AM9760G | |
| Needleless Valve to Male Luer Connector | MEDNET | NVFMLLPC | |
| NIS Elements | Nikon | Imaging software Version 5.02.00 | |
| NOA81 | Thorlabs | NOA81 | Fast Curing Optical Adhesive for tube system assembly |
| Opti-MEM | Thermo Fisher | 31985062 | |
| PCO edge 4.2 M-USB-HQ-PCO | pco | ||
| Phosphate buffered saline (PBS) | in-house prepared | ||
| Plasma Cleaner | Diener Femto | Pico-BRS | |
| PLL(20 kDa)-g[3.5]-PEG(2 kDa) | SuSoS AG | ||
| silicon wafer mit mircorstructures | in-house fabricated | ||
| Sola Light Engine | Lumencor | ||
| sticky slide VI 0.4 | ibidi | 80608 | |
| Sylgard 184 Silicone Elastomer Kit | Dow Corning | 1673921 | |
| Tango 2 | Märzhäuser | 00-24-626-0000 | |
| Ultrapure water | in-house prepared | ||
| uncoated coverslips | ibidi | 10813 | |
| Injekt-F Solo, 1 mL | Omilab | 9166017V | with replacement sporn |
Referenzen
- Altschuler, S. J., Wu, L. F. Cellular heterogeneity: do differences make a difference. Cell. 141 (4), 559-563 (2010).
- Locke, J. C., Elowitz, M. B. Using movies to analyse gene circuit dynamics in single cells. Nature Reviews Microbiology. 7 (5), 383-392 (2009).
- Spencer, S. L., Gaudet, S., Albeck, J. G., Burke, J. M., Sorger, P. K. Non-genetic origins of cell-to-cell variability in TRAIL-induced apoptosis. Nature. 459 (7245), 428-432 (2009).
- El-Ali, J., Sorger, P. K., Jensen, K. F. Cells on chips. Nature. 442 (7101), 403-411 (2006).
- Segerer, F. J., et al. Versatile method to generate multiple types of micropatterns. Biointerphases. 11 (1), 011005 (2016).
- Piel, M., Théry, M. . Micropatterning in cell biology, part A/B/C. , (2014).
- Reiser, A., Zorn, M. L., Murschhauser, A., Rädler, J. O., Ertl, P., Rothbauer, M. . Cell-Based Microarrays: Methods and Protocols. , 41-54 (2018).
- Picone, R., Baum, B., McKendry, R. . Methods in cell biology. 119, 73-90 (2014).
- Reiser, A. . Single-cell time courses of mRNA transport and translation kinetics. , (2019).
- . Softmatter LMU-Raedler Group Available from: https://github.com/SoftmatterLMU-RaedlerGroup/pyama (2020)
- . MIAAB, 29262 Available from: https://github.com/SoftmatterLMU-RaedlerGroup/pyama (2020)
- Reiser, A., et al. Correlation of mRNA delivery timing and protein expression in lipid-based transfection. Integrative Biology. 11 (9), 362-371 (2019).
- Reiser, A., Woschée, D., Mehrotra, N., Krzysztoń, R. S., Strey, H. H., Rädler, J. O. Supplementing data and code for: "Correlation of mRNA delivery timing and protein expression in lipid-based transfection". Zenodo. , (2019).
- Ferizi, M., et al. Stability analysis of chemically modified mRNA using micropattern-based single-cell arrays. Lab on a Chip. 15 (17), 3561-3571 (2015).
- Fröhlich, F., et al. Multi-experiment nonlinear mixed effect modeling of single-cell translation kinetics after transfection. NPJ systems biology and applications. 4 (1), 1-12 (2018).
- Krzysztoń, R., et al. Single-cell kinetics of siRNA-mediated mRNA degradation. Nanomedicine: Nanotechnology, Biology and Medicine. 21, 102077 (2019).
- Röttgermann, P. J., Alberola, A. P., Rädler, J. O. Cellular self-organization on micro-structured surfaces. Soft Matter. 10 (14), 2397-2404 (2014).
- Röttgermann, P. J., Dawson, K. A., Rädler, J. O. Time-resolved study of nanoparticle induced apoptosis using microfabricated single cell arrays. Microarrays. 5 (2), 8 (2016).
- Murschhauser, A., et al. A high-throughput microscopy method for single-cell analysis of event-time correlations in nanoparticle-induced cell death. Communications Biology. 2 (1), 1-11 (2019).
- Chatzopoulou, E. I., et al. Chip-based platform for dynamic analysis of NK cell cytolysis mediated by a triplebody. Analyst. 141 (7), 2284-2295 (2016).
- Sztilkovics, M., et al. Single-cell adhesion force kinetics of cell populations from combined label-free optical biosensor and robotic fluidic force microscopy. Scientific Reports. 10 (1), 1-13 (2020).
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenWeitere Artikel entdecken
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten