
Se requiere una suscripción a JoVE para ver este contenido. Inicie sesión o comience su prueba gratuita.
Method Article
Visualización de la proximidad inducida por caspasas inflamatorias en macrófagos derivados de monocitos humanos
En este artículo
Resumen
Este protocolo describe el flujo de trabajo para obtener macrófagos derivados de monocitos (MDM) a partir de muestras de sangre humana, un método simple para introducir eficientemente reporteros de complementación de fluorescencia bimolecular de caspasa inflamatoria (BiFC) en MDM humanos sin comprometer la viabilidad y el comportamiento celular, y un enfoque basado en imágenes para medir la activación de la caspasa inflamatoria en células vivas.
Resumen
Las caspasas inflamatorias incluyen caspasa-1, -4, -5, -11 y -12 y pertenecen al subgrupo de caspasas iniciadoras. La caspasa-1 es necesaria para garantizar la correcta regulación de la señalización inflamatoria y se activa por dimerización inducida por proximidad después del reclutamiento a inflamasomas. La caspasa-1 es abundante en el linaje celular monocítico e induce la maduración de las citoquinas proinflamatorias interleucina (IL)-1β e IL-18 a moléculas secretadas activas. Las otras caspasas inflamatorias, caspasa-4 y -5 (y su homólogo murino caspasa-11) promueven la liberación de IL-1β al inducir piroptosis. La complementación de fluorescencia bimolecular de caspasa (BiFC) es una herramienta utilizada para medir la proximidad inflamatoria inducida por caspasa como una lectura de la activación de la caspasa. El prodominio caspasa-1, -4 o -5, que contiene la región que se une al inflamasoma, se fusiona con fragmentos no fluorescentes de la proteína fluorescente amarilla Venus (Venus-N [VN] o Venus-C [VC]) que se asocian para reformar el complejo fluorescente de Venus cuando las caspasas experimentan proximidad inducida. Este protocolo describe cómo introducir a estos reporteros en macrófagos primarios derivados de monocitos humanos (MDM) utilizando nucleofection, tratar las células para inducir la activación de la caspasa inflamatoria y medir la activación de la caspasa mediante fluorescencia y microscopía confocal. La ventaja de este enfoque es que se puede utilizar para identificar los componentes, los requisitos y la localización del complejo de activación de la caspasa inflamatoria en las células vivas. Sin embargo, se deben considerar controles cuidadosos para evitar comprometer la viabilidad y el comportamiento de las células. Esta técnica es una herramienta poderosa para el análisis de interacciones dinámicas de caspasa a nivel de inflamasoma, así como para el interrogatorio de las cascadas de señalización inflamatoria en MDM vivos y monocitos derivados de muestras de sangre humana.
Introducción
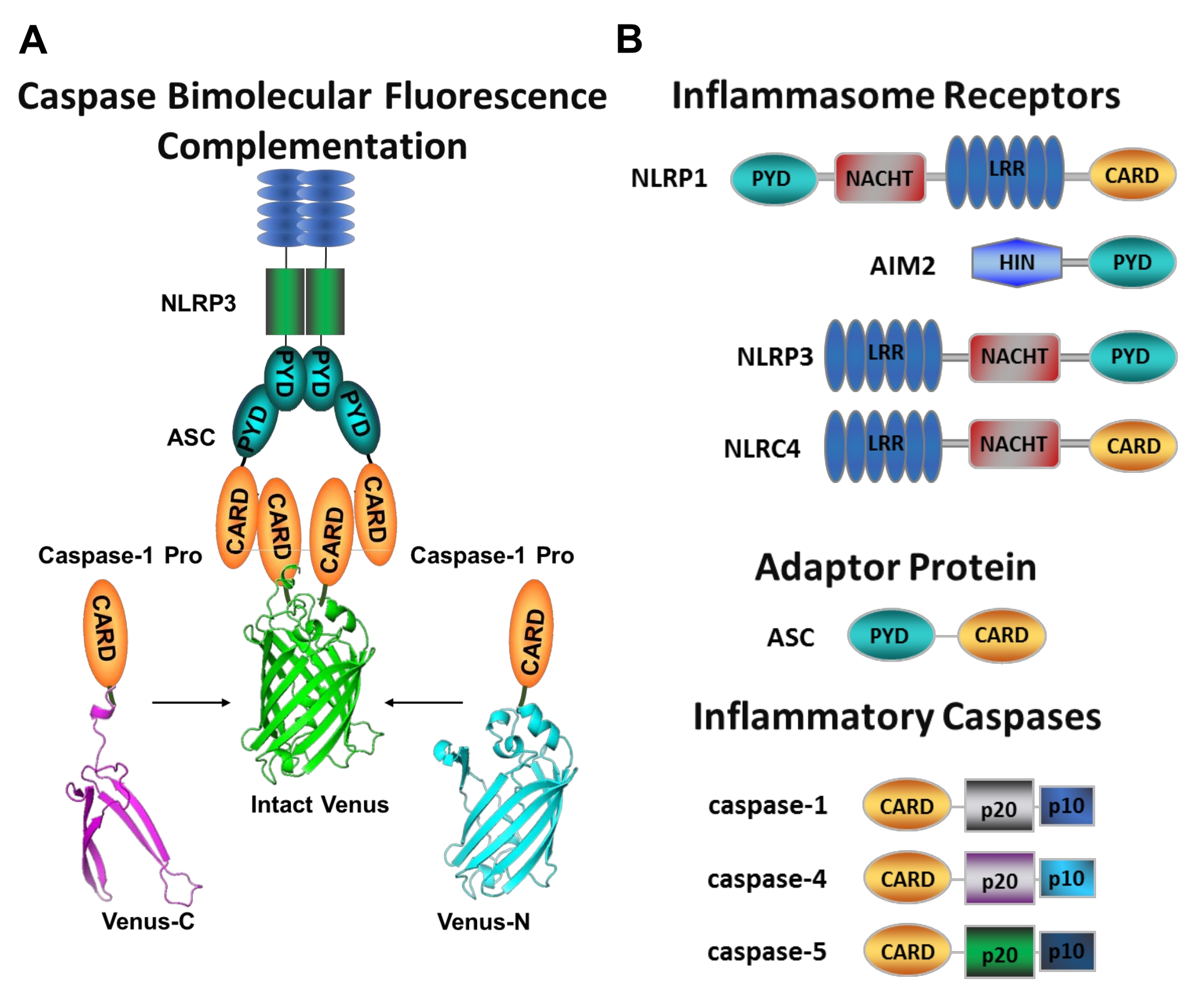
Las caspasas son una familia de proteasas de cisteína aspartato que se pueden agrupar en caspasas iniciadoras y caspasas verdugo. Las caspasas verdugosas comprenden caspasa-3, -6 y -7. Se encuentran naturalmente en las células como dímeros y son escindidos por las caspasas iniciadoras para ejecutar la apoptosis1. Las caspasas iniciadoras incluyen caspasa humana-1, -2, -4, -5, -8, -9, -10 y -12. Se encuentran como zimógenos inactivos (pro-caspasas) que se activan por dimerización inducida por proximidad y se estabilizan mediante escisión autoprotolítica 2,3. Las caspasas inflamatorias son un subconjunto de las caspasas iniciadoras2 y abarcan caspasas-1, -4, -5 y -12 en humanos, y caspasa-1, -11 y -12 enratones 4,5. En lugar de un papel apoptótico, juegan un papel central en la inflamación. Median el procesamiento proteolítico y la secreción de pro-interleucina (IL)-1β y pro-IL-18 6,7, que son las primeras citoquinas que se liberan en respuesta a invasores patógenos 8,9. Caspasa-1 se activa al reclutar en su plataforma de activación; un complejo proteico de gran peso molecular denominado inflamasoma (Figura 1A)10. La dimerización de la caspasa-4, -5 y -11 ocurre independientemente de estas plataformas a través de una vía inflamasómica no canónica 11,12.
Los inflamasomas canónicos son complejos de proteínas multiméricas citosólicas que consisten en una proteína sensora de inflamasoma, la proteína adaptadora ASC (proteína similar a una mota asociada a la apoptosis que contiene una CARD) y la proteína efectora caspasa-110. Los inflamasomas canónicos más estudiados son la familia de receptores similares a NOD que contienen un dominio de pirina (NLRP), NLRP1 y NLRP3, la familia NLR que contiene una CARD (NLRC), NLRC4 y la ausente en el melanoma 2 (AIM2). Cada uno de ellos contiene un dominio de pirina, un CARD o ambos dominios. El dominio CARD media la interacción entre las caspasas que contienen CARD y sus activadores ascendentes. Por lo tanto, la molécula de andamio ASC, que se compone de un dominio de pirina N-terminal (PYD) y un motivo CARD C-terminal 13,14, es necesaria para el reclutamiento de caspasa-1 a los inflamasomas NLRP1 10, NLRP315 y AIM216.
Cada inflamasoma lleva el nombre de su proteína sensora única que reconoce distintos estímulos proinflamatorios (Figura 1B). Los activadores de esta vía se denominan estímulos canónicos. Los inflamasomas sirven como sensores para los componentes microbianos y el estrés tisular, y se ensamblan para desencadenar una respuesta inflamatoria robusta a través de la activación de las caspasas inflamatorias17. El ensamblaje del inflamasoma inicia la activación de la caspasa-1 para mediar la maduración y secreción de sus principales sustratos pro-IL-1β y pro-IL-18. Este proceso se produce a través de un mecanismo de dos pasos. En primer lugar, un estímulo de cebado regula al alza la expresión de ciertas proteínas inflamasomas y pro-IL-1β a través de la activación de la vía NF-κB. En segundo lugar, un estímulo intracelular (canónico) induce el ensamblaje del inflamasoma y el reclutamiento de procaspasa-1 6,7.
Caspasa-4 y caspasa-5 son los ortólogos humanos de la caspasa murina-1111. Se activan de manera independiente del inflamasoma por el lipopolisacárido intracelular (LPS), una molécula que se encuentra en la membrana externa de la bacteria Gram-negativa18,19,20, y por el hemo extracelular, un producto de la hemólisis de glóbulos rojos21. Se ha propuesto que el LPS se une directamente al motivo CARD de estas proteínas e induce su oligomerización20. La activación de la caspasa-4 o caspasa-5 promueve la liberación de IL-1β al inducir una forma inflamatoria de muerte celular llamada piroptosis a través de la escisión de la proteína formadora de poros gasdermin D (GSDMD)18,19. Además, el eflujo de iones potásicos resultantes de la caspasa-4 y la muerte piroptótica mediada por GSDMD induce la activación del inflamasoma NLRP3 y la posterior activación de la caspasa-1 22,23. Por lo tanto, la caspasa-4, -5 y -11 se consideran sensores intracelulares para LPS que son capaces de inducir la activación de la piroptosis y la caspasa-1 en respuesta a estímulos específicos11,24.

Figura 1: Ensayo de caspasas inflamatorias y complementación de fluorescencia caspasa-bimolecular (BiFC). (A) Diagrama que muestra el sistema caspasa-BiFC, donde dos prodominios de caspasa-1 (C1-pro) unidos a cada fragmento no fluorescente de Venus (Venus-C o Venus-N) se reclutan para la plataforma de activación NLRP3, obligando a Venus a replegarse y fluorescenciar. Este complejo aparece como una mancha verde bajo el microscopio y sirve como una lectura para la proximidad inflamatoria inducida por la caspasa, que es el primer paso en la activación de la caspasa iniciadora. (B) Esquema que muestra la organización del dominio de los componentes del inflamasoma y las caspasas inflamatorias. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Medir la activación específica de las caspasas iniciadoras es difícil, y no hay muchos métodos disponibles para hacerlo mediante enfoques de imágenes. La complementación de fluorescencia bimolecular de caspasa (BiFC) se puede utilizar para visualizar la activación de la caspasa inflamatoria directamente en células vivas (Figura 1A)25. Esta técnica ha sido recientemente adaptada para su uso en macrófagos derivados de monocitos humanos (MDM)21. Caspasa BiFC mide el primer paso en la activación de la caspasa inflamatoria, proximidad inducida para facilitar la dimerización. Se utiliza la expresión de plásmidos que codifican el prodominio caspasa que contiene CARD fusionado con fragmentos no fluorescentes de la proteína fluorescente amarilla fotostable Venus (Venus-C [VC]) y Venus-N [VN]). Cuando los dos prodominios de caspasa se reclutan en su plataforma de activación o experimentan proximidad inducida, las dos mitades de Venus se acercan y se ven obligadas a replegarse y fluorescenciar (ver Figura 1A, B). Esto proporciona una lectura en tiempo real de la activación específica de la caspasa inflamatoria.
Los MDM humanos expresan abundantemente genes inflamasomas y receptores de reconocimiento de patrones que identifican señales de peligro y productos patógenos. Esto proporciona un tipo de célula ideal para el interrogatorio de las vías inflamatorias de la caspasa. Además, pueden derivarse de sangre periférica e incluso de muestras de pacientes para evaluar la activación de la caspasa inflamatoria en un estado de enfermedad específico. Este protocolo describe cómo introducir a los reporteros de caspasa BiFC en MDM utilizando nucleofection, un método de transfección basado en electroporación, cómo tratar las células para inducir la activación inflamatoria de la caspasa y cómo visualizar los complejos activos de caspasa utilizando enfoques de microscopía. Además, esta metodología se puede adaptar para determinar la composición molecular de estos complejos, la localización subcelular, la cinética y el tamaño de estas estructuras altamente ordenadas 25,26,27.
Protocolo
Este protocolo sigue las directrices del comité de ética de investigación humana de Baylor College of Medicine para la manipulación de muestras humanas. Las muestras de sangre se manejan siguiendo las pautas de seguridad institucionales para muestras humanas. Las muestras de sangre se obtienen en un banco de sangre regional, donde se recogen con solución de citrato fosfato dextrosa (CPD). Sin embargo, la sangre recolectada con otros anticoagulantes como la heparina sódica, la heparina de litio o el EDTA también se puede utilizar para este protocolo28,29.
1. Aislamiento de monocitos humanos y diferenciación en macrófagos
- Obtenga sangre anticoagulada de individuos sanos no identificados en un banco de sangre regional y aísle las células mononucleares de sangre periférica (PBMC) como se indica a continuación.
NOTA: Realice todos los pasos en una campana de flujo laminar de cultivo de tejidos. Use solo tubos estériles y use guantes. Agregue lejía al 10% a todos los productos relacionados con la sangre cuando se desechen. PBS estéril (1x) o DPBS (sin Ca2+ y Mg2+) se puede utilizar indistintamente.- Preparar el tampón de dilución: Suplementar 1x PBS estéril con 2% de FBS y 0,5 mM de EDTA.
- Preparar el medio de cultivo: Suplementar el medio RPMI-1640 con FBS (10% (v/v)), glutamax (2 mM) y Penicilina/Estreptomicina (50 I.U./50 μg/mL)
- Preenfríe el búfer de funcionamiento (Tabla de materiales) de acuerdo con el protocolo del fabricante.
- Diluir la sangre entera con dos volúmenes de tampón de dilución. Usando una pipeta serológica, transfiera 15 ml de la sangre anticoagulada a un tubo de 50 ml que contenga 30 ml del tampón de dilución. Mezclar suavemente por inversión.
- Por cada 10 ml de sangre total o 30 ml de sangre diluida, agregue 15 ml del medio de gradiente de densidad a un tubo vacío de 50 ml.
- Cubra el medio de gradiente de densidad del paso 1.1.5 con 30 ml de sangre diluida lenta y constantemente utilizando una pipeta serológica de 25 ml. Mantenga la punta de la pipeta contra la pared del tubo y el tubo en un ángulo inclinado.
- Transfiera cuidadosamente los tubos a una centrífuga de cuchara oscilante. Evite perturbar las dos fases. Centrifugar los tubos a 400 x g a temperatura ambiente (RT) durante 25 min con aceleración y desaceleración ajustadas al valor mínimo.
- Retire con cuidado la capa de plasma superior (transparente) con una pipeta de 10 ml y deséchela en un recipiente con lejía (10%).
- Recoger la capa interfase (blanca) de células mononucleares de sangre periférica (PBMC, Figura 2) con una pipeta de 10 ml y transferirla a un tubo fresco de 50 ml. Combine la capa blanca de diferentes tubos del mismo donante en un tubo de 50 ml hasta 30 ml.
- Llevar cada tubo a un volumen total de 50 ml con el tampón de dilución del paso 1.1.1 y centrifugar a 300 x g y 4 °C durante 10 min. Retire el sobrenadante con una pipeta de 10 ml y deséchelo en un recipiente con lejía (10%).
- Resuspender cada gránulo de celda en 1 ml del búfer de funcionamiento preenfriado del paso 1.1.3 utilizando una micropipeta p1000. Combine las suspensiones celulares del mismo donante en un nuevo tubo de 15 ml. Lleve el volumen de cada tubo a 15 ml con tampón de funcionamiento preenfriado y mezcle bien por inversión.
- Tome una alícuota de 20 μL de la suspensión celular del paso 1.1.11 y prepare una dilución de 1:100 usando 1x PBS estéril. Determine el número de células usando un hemocitómetro.
- Centrifugar la suspensión celular del paso 1.1.11 a 300 x g y 4 °C durante 10 min y retirar el sobrenadante con una pipeta de 10 ml. Si es necesario, use una micropipeta p200 para eliminar el sobrenadante por completo.
- Resuspender los PBMC aislados en 80 μL de macS preenfriado ejecutando buffer por cada 1 x 107 celdas, sumando un máximo de 800 μL del buffer.
- Agregue 20 μL de microperlas CD14 antihumanas por cada 1 x 107 células o hasta 100 μL por muestra de sangre (~ 100 ml de sangre sin diluir). Mezclar bien por inversión y colocar en un rotador de tubo durante 20 min con mezcla continua a 4 °C.
- Retire las muestras del rotador del tubo, agregue 10 ml de tampón de funcionamiento preenfriado a cada tubo y centrífuga a 300 x g (aceleración = 5, desaceleración = 5) y 4 ° C durante 10 min.
- Retire el sobrenadante con una pipeta de 10 ml y vuelva a suspender hasta 1 x 108 celdas en 500 μL de tampón de funcionamiento preenfriado (2 x 108/ml).
- Realizar el aislamiento de células CD14 positivas mediante clasificación de células magnéticas utilizando un sistema manual o automatizado (Tabla de Materiales) según las instrucciones del fabricante.
- Tome una alícuota de 20 μL de la suspensión celular del paso 1.1.18 después de la selección cd14 positiva y prepare una dilución de 1:100 utilizando 1x PBS estéril. Determine el número de células contando las células en un hemocitómetro.
- Centrifugar las células CD14 positivas a 300 x g y RT durante 10 min. Retire el sobrenadante con una pipeta de 10 ml o un sistema de vacío.
- Resuspendir el pellet celular desde el paso 1.1.20 en medio de cultivo precalentado desde el paso 1.1.2 hasta una densidad celular final de 1 x 107 células/ml.
- Sembrar los monocitos CD14 positivos aislados a una densidad celular de 5 x 106 células.
- En una placa de cultivo de tejidos de 10 cm, agregue 10 ml de medio de cultivo del paso 1.1.2 suplementado con 50 ng / ml de factor estimulante de colonias de granulocitos y macrófagos (GM-CSF).
- Añadir 0,5 ml de la suspensión celular del paso 1.1.21 al medio de cultivo gota a gota y girar suavemente la placa. Incubar las células en una incubadora de cultivo de tejidos humidificados (37 °C, 5% CO2) durante la noche.
- Al día siguiente, aspire el medio utilizando un sistema de vacío para eliminar las células que no se unieron durante la noche. Añadir 10 ml de medio de cultivo fresco suplementado con GM-CSF (50 ng/mL) e incubar células en una incubadora de cultivo de tejidos humidificados (37 °C, 5% CO2) durante 7 días para permitir una diferenciación completa (ver Figura 3A para la aparición de monocitos CD14+ en varias etapas de diferenciación en GM-CSF). Intercambie el medio de cultivo cada 2-3 días y complemente con GM-CSF fresco (50 ng / ml) cada vez.

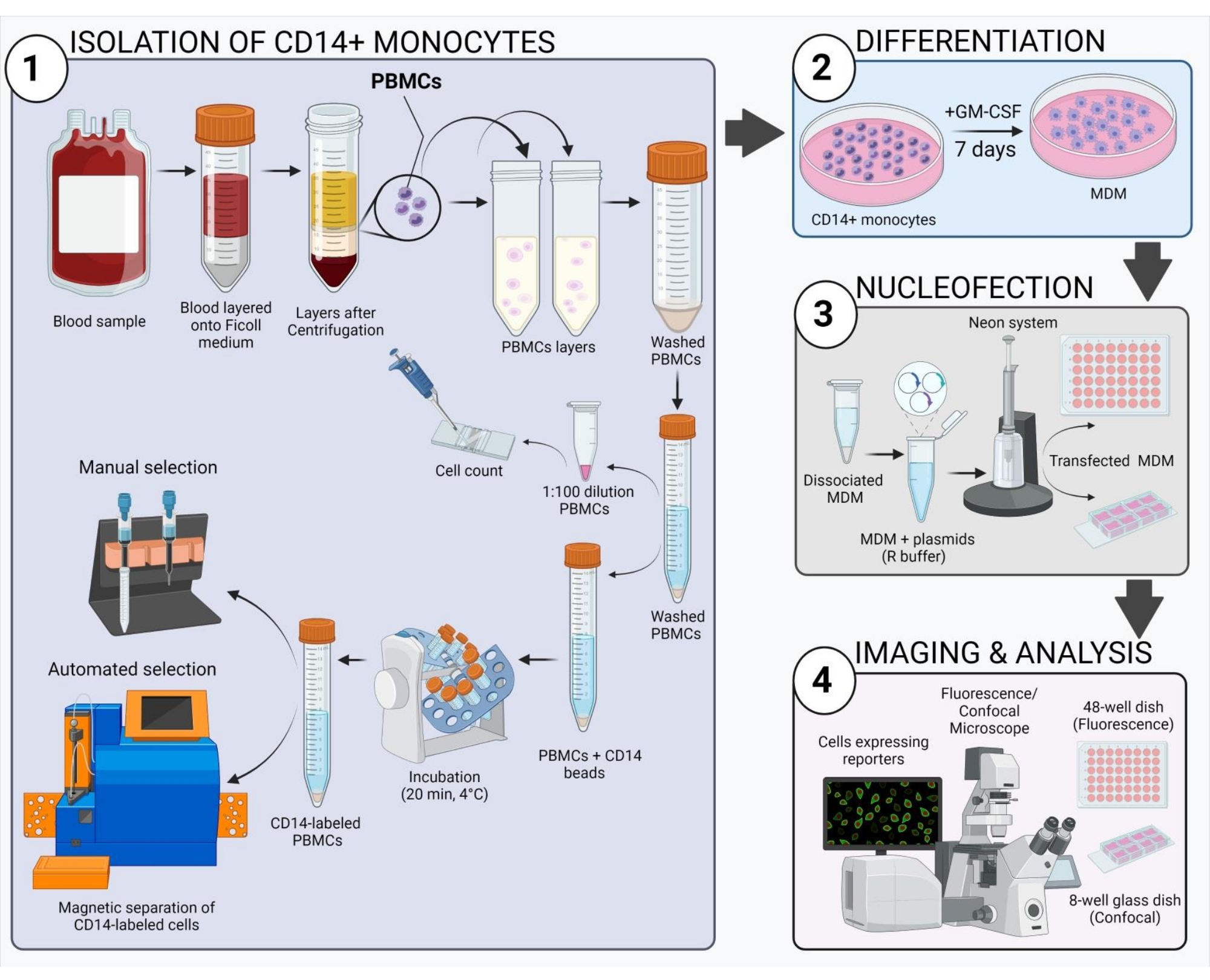
Figura 2: Descripción general esquemática del flujo de trabajo experimental. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
2. Preparación de componentes de electroporación
NOTA: Este protocolo está diseñado para una punta de neón de 10 μL (Tabla de materiales). Para cada transfección, use 1-2 x 105 células. Se recomienda sembrar células transfectadas en una placa de 48 pocillos o en una placa con cámara de 8 pocillos (10 células transfectadas por μL por pozo). Se puede usar 1x DPBS estéril (sin Ca2+ y Mg2+) en lugar de PBS.
- El día 7, prepare el medio libre de antibióticos complementando el medio RPMI-1640 con FBS (10 % (v / v)) y glutamax (2 mM).
- Coloque el medio RPMI-1640 sin suero, la solución de tripsina-EDTA (0,25%), 1 PBS estéril (sin Ca2+ y Mg2+) y el medio de cultivo completo a partir del paso 1.1.2 en un baño de agua a 37 °C.
- Si usa platos con fondo de vidrio (para microscopía confocal), cubra los platos con hidrobromuro de poli-D-lisina.
- Cubra un plato de 8 platos bien recamarados con 200 μL de hidrobromuro de poli-D-lisina (0,1 mg/ml en 1x PBS estéril) e incube durante 5 min a RT.
- Aspire la solución de poli-D-lisina y lave el vidrio una vez con 1 PBS estéril. Aspire el PBS y continúe con el paso 2.4.
- Añadir 200 μL de medio libre de antibióticos por pocillo de la placa de 48 pocillos u 8 platos bien recalibrados y preincubar en una incubadora de cultivo de tejidos humidificados (37 °C, 5% de CO2) hasta que esté listo para colocar las células transfectadas.
3. Preparación de celdas para electroporación
NOTA: El rendimiento de MDM de un plato de 10 cm al final del período de diferenciación de 7 días es de aproximadamente 1,5 x 106 células. Se puede usar 1x DPBS estéril (sin Ca2+ y Mg2+) en lugar de PBS. Este protocolo se optimizó para que la mayoría de los macrófagos se separen de la placa con el mantenimiento de la viabilidad e integridad celular. Los MDM son difíciles de separar de las placas de cultivo celular. Por lo tanto, puede ser necesario realizar los pasos 3.2 y 3.3 dos veces para disociar las células. Asegúrese de que cada tiempo de incubación con tripsina-EDTA (0,25%) no exceda de 5 min.
- Aspire el medio de macrófagos totalmente diferenciados en platos de 10 cm y lave la monocapa celular con un medio RPMI-1640 sin suero caliente. Asegúrese de quitar completamente el medio.
- Cosechar las células añadiendo 2 ml de solución tibia de tripsina-EDTA (0,25%) por plato de 10 cm e incubar en una incubadora de cultivo de tejido humidificado (37 °C, 5% CO2) durante 5 min.
- Complete el desprendimiento celular canalizando suavemente la solución de tripsina-EDTA (0,25%) hacia arriba y hacia abajo sobre toda el área del plato usando una micropipeta p1000. Transfiera la suspensión celular a un tubo cónico de 15 ml que contenga 5 ml de medio de cultivo completo caliente a partir del paso 1.1.2.
- Lleve el plato a un microscopio de campo brillante y verifique si hay desprendimiento celular en varios campos de visión. Si todavía hay una cantidad considerable de celdas unidas, repita los pasos 3.2-3.3.
- Centrifugar la suspensión celular a 250 x g durante 5 min a RT.
- Aspire el medio y resuspenda las células en 10 ml de 1x PBS estéril precalentado a 37 °C. Tome una alícuota de 20 μL para determinar el número de células utilizando un hemocitómetro.
- Tome 1-2 x 105 células por transfección prevista y colóquelas en un tubo de 15 ml. Llevar a un volumen final de 15 ml con PBS estéril precalentado 1x. Centrifugar a 250 x g durante 5 min a RT.
- Aspirar el PBS y centrifugar una vez más durante 1 min a 250 x g. Retire cualquier PBS residual del pellet celular usando una micropipeta p200.
4. Nucleofection de componentes de caspasa BiFC en macrófagos derivados de monocitos humanos
NOTA: Esta sección del protocolo se realiza utilizando el Sistema de Transfección de Neón (Tabla de Materiales). Este protocolo describe los pasos para transfectar 1-2 x 105 celdas utilizando una punta de neón de 10 μL (Tabla de materiales). Si usa una punta de neón de 100 μL, amplíe la escala en consecuencia. Evite exponer las células al tampón de resuspensión R durante más de 15 minutos, ya que esto puede disminuir la viabilidad celular y la eficiencia de la transfección.
- Diluya el plásmido reportero (es decir, mCherry o dsRedmito a 100 ng / μL) en agua libre de nucleasa o 0.5x TE buffer para visualizar las células transfectadas.
- Diluir los plásmidos BiFC de caspasa a una concentración adecuada en agua libre de nucleasa o 0,5x tampón TE, de modo que el volumen total de plásmidos no exceda el 30% del volumen total de transfección de 10 μL (es decir, 300 ng/μL de C1 Pro-VC y 300 ng/μL de C1 Pro-VN).
- Preparar un microtubo estéril de 1,5 ml por transfección prevista y añadir la cantidad adecuada de plásmido informador (es decir, 50 ng o 0,5 μL) y plásmidos BiFC caspasa (es decir, 300 ng o 1 μL de C1 Pro-VC y 300 ng o 1 μL de fragmento C1 Pro-VN). Mantenga los microtubos en el capó en todo momento.
- Coloque la estación de pipeta, el dispositivo, las puntas, los tubos de electroporación y la pipeta en una campana de flujo laminar estéril.
NOTA: La estación de pipeta, el dispositivo, las puntas, los tubos de electroporación y la pipeta están incluidos en el sistema de transfección de neón. - Conecte el conector de alto voltaje y sensor de la estación de pipeta a los puertos traseros del dispositivo según las instrucciones del fabricante. Mantenga la estación de pipeta cerca del dispositivo.
- Conecte el cable de alimentación a la entrada de CA trasera y proceda a conectar el dispositivo a la toma de corriente. Presione el interruptor de encendido para encender el dispositivo.
- Introduzca los parámetros de transfección en la pantalla de inicio que se muestra cuando el dispositivo está encendido y conectado adecuadamente. Presione Voltaje, ingrese 1000 y presione Listo para establecer el voltaje a 1000 V. Presione Ancho, ingrese 40 y presione Listo para establecer la duración del pulso en 40 ms. Por último, presione # Pulsos, ingrese 2 y presione Listo para establecer el número de pulsos eléctricos en 2.
- Tome uno de los tubos de electroporación (provistos en el kit) y llénelo con 3 ml de tampón electrolítico E (para puntas de 10 μL y provisto en el kit) en RT. Inserte el tubo de electroporación en el soporte de pipeta en la estación de pipeta. Asegúrese de que el electrodo en el lado del tubo esté orientado hacia adentro y que se escuche un sonido de clic cuando se inserta el tubo.
- Tome el pellet celular del paso 3.8 y agregue 10 μL de tampón R de resuspensión R precalentado (proporcionado en el kit) por cada 1-2 x 105 celdas. Mezclar suavemente con una micropipeta p20. Añadir 10 μL de la suspensión celular a cada tubo configurado en el paso 4.3 y mezclar suavemente con una micropipeta p20.
- Tome la pipeta e inserte una punta presionando el botón Pulsador en la segunda parada. Asegúrese de que la abrazadera recoja completamente el vástago de montaje del pistón en la punta y que no se observe ningún espacio en la parte superior de la pipeta.
- Para aspirar la muestra, presione el botón Pulsador en la pipeta hasta la primera parada y sumérjase en el primer tubo que contiene la mezcla de ADN celular/plásmido. Aspire lentamente la mezcla en la punta de la pipeta.
NOTA: Evite las burbujas de aire, ya que pueden causar arcos durante la electroporación y, si el dispositivo las detecta, pueden evitar la entrega del pulso eléctrico. Si se observan burbujas de aire, suelte el contenido en el tubo e intente aspirar de nuevo. - Inserte la pipeta con la muestra con mucho cuidado en el soporte de la pipeta. Asegúrese de que la pipeta haga clic y que esté colocada correctamente.
- Presione Inicio en la pantalla táctil y espere hasta que se entreguen los pulsos eléctricos. Un mensaje en la pantalla indicará la finalización.
- Retire lentamente la pipeta de la estación e inmediatamente agregue la suspensión celular transfectada en el pozo correspondiente con un medio libre de antibióticos precalentado del paso 2.4 presionando lentamente el botón hasta la primera parada.
NOTA: Esta punta se puede reutilizar hasta tres veces para el mismo plásmido; de lo contrario, deséchelo en un contenedor de residuos de riesgo biológico presionando el botón Pulsador hasta la segunda parada. - Repita los pasos 4.10-4.14 para cada tubo que contenga una mezcla de ADN celular/plásmido.
- Balancee suavemente la placa con células transfectadas e incube durante 1-3 h en una incubadora de cultivo de tejido humidificado (37 °C, 5% CO2).
- Añadir a cada pocillo 200 μL de medio de cultivo precalentado (medio completo) a partir del paso 1.1.2. Coloque el plato en la incubadora de cultivo de tejidos humidificados (37 °C, 5% CO2) nuevamente. Permita al menos 24 h para la expresión génica.
- Al día siguiente, inspeccione la viabilidad celular y la eficiencia de la transfección utilizando un microscopio de epifluorescencia.
- Encienda el microscopio de epifluorescencia y la caja de la fuente de luz fluorescente según las instrucciones del fabricante y coloque la placa de cultivo en el escenario del microscopio.
- Seleccione el objetivo 10x o 20x y el filtro de 568 nm (RFP).
- Para estimar la viabilidad de la celda, pulse el botón LED de luz transmitida (TL) para visualizar todas las celdas del campo seleccionado. Mientras mira el ocular del microscopio, gire la perilla de enfoque hasta que se observen las células y verifique la unión de la célula en el campo seleccionado.
NOTA: Las células totalmente unidas representan las células viables, mientras que las células flotantes representan las células no viables. Si la confluencia del pozo es alta, la presencia de células no unidas podría ser el resultado de una sobreestimación del número de células y no el resultado de una baja viabilidad. Sin embargo, la baja confluencia acompañada de un alto contenido de células flotantes significa una baja viabilidad que puede resultar del arco durante la electroporación, la toxicidad de los plásmidos o la sobreexposición al tampón R de resuspensión. No utilice pozos que muestren este último comportamiento. - Para estimar la eficiencia de la transfección, concéntrese en las células en el campo seleccionado bajo la luz transmitida como se describió anteriormente. Cuente el número total de celdas en el campo seleccionado. Con el LED de luz transmitida (TL) apagado, presione el botón LED de luz reflejada (RL) para encenderlo.
- Enfoque fino en la fluorescencia del gen reportero (glóbulos rojos) y contar el número total de células fluorescentes rojas. Repita estos pasos (4.18.4-4.18.5) para al menos dos campos más por pozo.
5. Tratamiento de la adquisición de datos transfectados de MDM y caspasa BiFC
NOTA: Si planea obtener imágenes de las células utilizando un microscopio de epifluorescencia o confocal, se recomienda el tratamiento con qVD-OPh (20 μM) durante 1 h previo tratamiento con el estímulo elegido para prevenir la muerte celular dependiente de caspasa (predominantemente apoptosis). Esto se utiliza en imágenes para evitar que las células se despeguen debido a la apoptosis, lo que las hace muy difíciles de visualizar a medida que se mueven fuera del plano focal. Tenga en cuenta que el reclutamiento de caspasa a la plataforma de activación y la caspasa BiFC asociada no depende de la actividad catalítica de la caspasa y, en consecuencia, la inhibición de la caspasa no afectará este paso.
- Tratar con el estímulo elegido aproximadamente 24 h después de la transfección e incubar durante el tiempo que sea necesario para cada fármaco.
- Prepare el medio de imagen complementando el medio de cultivo del paso 1.1.2 con Hepes (20 mM, pH 7.2-7.5) y 2-mercaptoetanol (55 μM).
- Agregue la concentración deseada de estímulo al medio de imagen precalentado y mezcle suavemente.
- Retire los medios de las células con cuidado con una micropipeta p1000 y agregue 500 μL de la solución de estímulo desde el paso 5.1.2 hacia el lado del pozo.
- Para ejecutar pozos de control no tratados, agregue un medio de imágenes sin el estímulo.
- Incubar las células en una incubadora de cultivo de tejidos humidificados (37 °C, 5% CO2) durante el tiempo indicado para cada tratamiento.
- Visualice las células usando un microscopio de epifluorescencia o confocal.
- Encienda el microscopio y la fuente de luz fluorescente, siguiendo las instrucciones del fabricante.
- Seleccione el objetivo 10x o 20x y coloque la placa de cultivo en el escenario del microscopio.
- Usando el ocular del microscopio, encuentre células bajo el filtro de 568 nm y concéntrese en las células que expresan el reportero dsRedmito/mCherry (glóbulos rojos).
- Cuente todos los glóbulos rojos en el campo visual y registre el número.
- Mientras esté en el mismo campo visual, cambie al filtro 488 o 512 (GFP o YFP), proceda a contar el número de glóbulos rojos que también son verdes (Venus positivo o BiFC positivo) y registre el número.
- Cuente al menos 100 dsRedmito/mCherry -células positivas de un mínimo de tres campos visuales individuales.
- Calcule el porcentaje de células transfectadas positivas para Venus por campo visual y promedie los porcentajes resultantes para cada tratamiento (pozo) para obtener la desviación estándar.
- Imagen de las células usando un microscopio de epifluorescencia o confocal
NOTA: Para adquirir imágenes confocales utilizando un objetivo de 20x o un aumento mayor, las células deben estar chapadas en placas de vidrio a menos que el microscopio esté equipado con un objetivo de paso largo.- Siga los pasos 5.2.1-5.2.3. Si utiliza un microscopio confocal con el objetivo de aceite 40x, 60x o 63x, coloque una gota de aceite sobre el objetivo.
- Visualice la imagen en vivo de las celdas en la pantalla de la computadora según lo adquirido por la cámara. Utilice la fuente de luz de epifluorescencia para imágenes fluorescentes o cambie la fuente de luz a los láseres para imágenes confocales.
- Ajuste el enfoque y la posición de las celdas utilizando el control del joystick y la rueda de enfoque.
- Establezca el porcentaje de potencia láser y el tiempo de exposición para los láseres de 512 nm o 488 nm (YFP o GFP) y 568 nm (RFP) para que la señal en la imagen se vea bien y no alcance la saturación.
- Encienda la captura en vivo y examine la imagen resultante. Asegúrese de que se vea un pico distinto para cada flúor en los histogramas de visualización para ambos canales.
- Ajuste la potencia del láser y el tiempo de exposición según sea necesario. Mantenga estos valores lo más bajos posible mientras puede detectar ambas señales fluorescentes (RFP y GFP / YFP).
- Mientras visualiza la imagen en vivo de las celdas, tome múltiples imágenes representativas de un campo que contenga una o más celdas que expresen el reportero mCherry/dsRedmito para cada pozo de la placa y guarde los datos.
Resultados
El esquema que se muestra en la Figura 2 ofrece una visión general de cómo obtener, transfectar e imagenr la MDM humana. Después de la incubación de los monocitos CD14+ seleccionados con GM-CSF durante 7 días, la morfología celular cambia en el transcurso del período de diferenciación (Figura 3A), pasando de células de suspensión esférica a células de suspensión esférica a delgadas y completamente unidas (días 3 y 4), y, por último, a células m...
Discusión
Este protocolo describe el flujo de trabajo para obtener macrófagos a partir de monocitos aislados de muestras de sangre humana y un método para introducir eficientemente los reporteros biFC de caspasa inflamatoria en MDM humano sin comprometer la viabilidad y el comportamiento celular.
Este protocolo aprovecha la técnica BiFC35 para etiquetar las caspasas inflamatorias en el dominio de reclutamiento de caspasa (CARD) con fragmentos no fluorescentes de la proteína f...
Divulgaciones
Los autores declaran que no tienen intereses financieros contrapuestos.
Agradecimientos
Agradecemos a los miembros del laboratorio de LBH pasados y presentes que contribuyeron al desarrollo de esta técnica. Este laboratorio cuenta con el apoyo de NIH/NIDDK T32DK060445 (BEB), NIH/NIDDK F32DK121479 (BEB), NIH/NIGMS R01GM121389 (LBH). La figura 2 se dibujó utilizando el software Biorender.
Materiales
| Name | Company | Catalog Number | Comments |
| 48 well tissue culture2:34 plates | Genesee Scientific | 25-108 | |
| 10 cm Tissue Culture Dishes | VWR | 25382-166 | |
| 2 Mercaptoethanol 1000x | Thermo Fisher Scientific | 21985023 | |
| 8 well chambered coverglass with 1.5 HP coverglass | Cellvis | c8-1.5H-N | |
| AutoMACS columns | Miltenyi (Biotec) | 130-021-101 | For automated separation using AutoMACS Pro Separator only |
| AutoMACS Pro Separator | Miltenyi (Biotec) | 130-092-545 | For automated separation using AutoMACS Pro Separator only |
| AutoMACS Pro Washing Solution | Miltenyi (Biotec) | 130-092-987 | For automated separation using AutoMACS Pro Separator only |
| AutoMACS Rinsing Solution | Miltenyi (Biotec) | 130-091-222 | For automated separation using AutoMACS Pro Separator only |
| AutoMacs running buffer | Miltenyi (Biotec) | 130-091-221 | For manual or automated separation using QuadroMACS or AutoMACS pro Separator |
| Axio Observer Z1 motorized inverted microscope equipped with a CSU-X1A 5000 spinning disk unit | Zeiss | Any confocal microscope equipped with a laser module fitted with laser lines of 568 nm (RFP) and 488 or 512 nm (GFP or YFP) wavelengths can be used | |
| AxioObserver A1, Research Grade Inverted Microscope | Zeiss | Any epifluorescence microscope with fluorescence filters capable of exciting 568 nm (RFP) and 488 or 512 nm (GFP or YFP) wavelengths can be used | |
| CD14+ MICROBEADS | Miltenyi (Biotec) | 130-050-201 | For manual or automated separation using QuadroMACS or AutoMACS pro Separator |
| DPBS without calcium chloride and magnesium chloride | Sigma | D8537-6x500ML | |
| DsRed mito plasmid | Clontech | 632421 | Similar plasmids that can be used as fluorescent reporters can be found on Addgene |
| Fetal Bovine Serum | Thermo Fisher Scientific | 10437028 | |
| Ficoll-Paque PLUS 6 x 100 mL | Sigma | GE17-1440-02 | |
| GlutaMAX Supplement (100x) | Thermo Fisher Scientific | 35050079 | |
| GM-CSF | Thermo Fisher Scientific | PHC2011 | |
| Hemin BioXtra, from Porcine, ≥96.0% (HPLC) | Sigma | 51280-1G | |
| HEPES | Thermo Fisher Scientific | 15630106 | |
| Inflammatory caspase BiFC plasmids | Available by request from LBH lab | ||
| LPS-EB Ultrapure | Invivogen | TLRL-3PELPS | |
| LS Columns | Miltenyi (Biotec) | 130-042-401 | For manual separation using QuadroMACS Separator only |
| MACS 15 mL Tube Rack | Miltenyi (Biotec) | 130-091-052 | For manual separation using QuadroMACS Separator only |
| MACS MultiStand | Miltenyi (Biotec) | 130-042-303 | For manual separation using QuadroMACS Separator only |
| mCherry plasmid | Yungpeng Wang Lab | Similar plasmids that can be used as fluorescent reporters can be found on Addgene | |
| Neon Transfection System | Thermo Fisher Scientific | MPK5000 | Includes Neon electroporation device, pipette and pipette station |
| Neon Transfection System 10 µL Kit | Thermo Fisher Scientific | MPK1096 | Includes resuspension buffer R, resuspension buffer T, electrolytic buffer E, 96 x 10 µL Neon tips and Neon electroporation tubes |
| Nigericin sodium salt, ready made solution | Sigma | SML1779-1ML | |
| Penicillin-Streptomycin (10,000 U/mL) | Thermo Fisher Scientific | 15140122 | |
| Poly-D-Lysine Hydrobromide | Sigma | P7280-5mg | |
| QuadroMACS Separator | Miltenyi (Biotec) | 130-090-976 | For manual separation using QuadroMACS Separator only |
| qVD-OPh | Fisher (ApexBio) | 50-101-3172 | |
| RPMI 1640 Medium | Thermo Fisher Scientific | 11875119 | |
| Trypsin-EDTA (0.25%), phenol red | Thermo Fisher Scientific | 25200072 | |
| UltraPure 0.5 M EDTA, pH 8.0 | Thermo Fisher Scientific | 15575020 | |
| Zeiss Zen 2.6 (blue edition) software | Zeiss | Any software used to operate the confocal microscope of choice |
Referencias
- Boatright, K. M., et al. A unified model for apical caspase activation. Molecular Cell. 11 (2), 529-541 (2003).
- Pop, C., Salvesen, G. S. Human caspases: activation, specificity, and regulation. Journal of Biological Chemistry. 284 (33), 21777-21781 (2009).
- Boice, A., Bouchier-Hayes, L. Targeting apoptotic caspases in cancer. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 1867 (6), 118688 (2020).
- Bolívar, B. E., Vogel, T. P., Bouchier-Hayes, L. Inflammatory caspase regulation: maintaining balance between inflammation and cell death in health and disease. The FEBS Journal. 286 (14), 2628-2644 (2019).
- Martinon, F., Tschopp, J. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell. 117 (5), 561-574 (2004).
- Lamkanfi, M., Vishva, M. D. Mechanisms and functions of inflammasomes. Cell. 157 (5), 1013-1022 (2014).
- Viganò, E., et al. Human caspase-4 and caspase-5 regulate the one-step noncanonical inflammasome activation in monocytes. Nature Communications. 6, 8761 (2015).
- Cerretti, D. P., et al. Molecular cloning of the interleukin-1 beta converting enzyme. Science. 256 (5053), 97 (1992).
- van de Veerdonk, F. L., Netea, M. G., Dinarello, C. A., Joosten, L. A. Inflammasome activation and IL-1β and IL-18 processing during infection. Trends in Immunology. 32 (3), 110-116 (2011).
- Martinon, F., Burns, K., Tschopp, J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Molecular Cell. 10 (2), 417-426 (2002).
- Kayagaki, N., et al. Noncanonical inflammasome activation targets caspase-11. Nature. 479 (7371), 117-121 (2011).
- Kayagaki, N., et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science. 341 (6151), 1246-1249 (2013).
- Masumoto, J., et al. ASC, a novel 22-kDa protein, aggregates during apoptosis of human promyelocytic leukemia HL-60 cells. The Journal of Biological Chemistry. 274 (48), 33835-33838 (1999).
- Bertin, J., DiStefano, P. S. The PYRIN domain: a novel motif found in apoptosis and inflammation proteins. Cell Death and Differentiation. 7 (12), 1273-1274 (2000).
- Agostini, L., et al. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity. 20 (3), 319-325 (2004).
- Bürckstümmer, T., et al. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nature Immunology. 10 (3), 266-272 (2009).
- Latz, E., Xiao, T. S., Stutz, A. Activation and regulation of the inflammasomes. Nature Reviews Immunology. 13 (6), 397-411 (2013).
- Kayagaki, N., et al. Caspase-11 cleaves gasdermin D for noncanonical inflammasome signalling. Nature. 526 (7575), 666-671 (2015).
- Shi, J., et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 526 (7575), 660-665 (2015).
- Shi, J., et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 514 (7521), 187-192 (2014).
- Bolívar, B. E., et al. Noncanonical Roles of Caspase-4 and Caspase-5 in Heme-Driven IL-1β Release and Cell Death. The Journal of Immunology. 206 (8), 1878-1889 (2021).
- Schmid-Burgk, J. L., et al. Caspase-4 mediates noncanonical activation of the NLRP3 inflammasome in human myeloid cells. European Journal of Immunology. 45 (10), 2911-2917 (2015).
- Baker, P. J., et al. NLRP3 inflammasome activation downstream of cytoplasmic LPS recognition by both caspase-4 and caspase-5. European Journal of Immunology. 45 (10), 2918-2926 (2015).
- Cullen, S. P., Kearney, C. J., Clancy, D. M., Martin, S. J. Diverse activators of the NLRP3 inflammasome promote IL-1β secretion by triggering necrosis. Cell Reports. 11 (10), 1535-1548 (2015).
- Sanders, M. G., et al. Single-cell imaging of inflammatory caspase dimerization reveals differential recruitment to inflammasomes. Cell Death & Disease. 6, 1813 (2015).
- Charendoff, C. I., Bouchier-Hayes, L. Lighting up the pathways to caspase activation using bimolecular fluorescence complementation. Journal of Visualized Experiments: JoVE. (133), e57316 (2018).
- Bouchier-Hayes, L., et al. Characterization of cytoplasmic caspase-2 activation by induced proximity. Molecular Cell. 35 (6), 830-840 (2009).
- Riedhammer, C., Halbritter, D., Weissert, R. Peripheral blood mononuclear cells: Isolation, Freezing, thawing, and culture. Methods in Molecular Biology. 1304, 53-61 (2016).
- Betsou, F., Gaignaux, A., Ammerlaan, W., Norris, P. J., Stone, M. Biospecimen science of blood for peripheral blood mononuclear cell (PBMC) functional applications. Current Pathobiology Reports. 7 (2), 17-27 (2019).
- Perregaux, D., et al. IL-1 beta maturation: evidence that mature cytokine formation can be induced specifically by nigericin. The Journal of Immunology. 149 (4), 1294-1303 (1992).
- Cheneval, D., et al. Increased mature interleukin-1β (IL-1β) secretion from THP-1 cells induced by nigericin is a result of activation of p45 IL-1β-converting enzyme processing. Journal of Biological Chemistry. 273 (28), 17846-17851 (1998).
- Fernandes-Alnemri, T., et al. The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death and Differentiation. 14 (9), 1590-1604 (2007).
- Lu, A., et al. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell. 156 (6), 1193-1206 (2014).
- Muller-Eberhard, U., Javid, J., Liem, H. H., Hanstein, A., Hanna, M. Brief report: Plasma concentrations of hemopexin, haptoglobin and heme in patients with various hemolytic diseases. Blood. 32 (5), 811-815 (1968).
- Shyu, Y. J., Liu, H., Deng, X., Hu, C. D. Identification of new fluorescent protein fragments for bimolecular fluorescence complementation analysis under physiological conditions. Biotechniques. 40 (1), 61-66 (2006).
- Thornberry, N. A., et al. A novel heterodimeric cysteine protease is required for interleukin-1βprocessing in monocytes. Nature. 356 (6372), 768-774 (1992).
- Jensen, K., Anderson, J. A., Glass, E. J. Comparison of small interfering RNA (siRNA) delivery into bovine monocyte-derived macrophages by transfection and electroporation. Veterinary Immunology and Immunopathology. 158 (3-4), 224-232 (2014).
- Tada, Y., Sakamoto, M., Fujimura, T. Efficient gene introduction into rice by electroporation and analysis of transgenic plants: use of electroporation buffer lacking chloride ions. Theoretical and Applied Genetics. 80 (4), 475-480 (1990).
- Bhowmik, P., et al. Targeted mutagenesis in wheat microspores using CRISPR/Cas9. Scientific Reports. 8 (1), 6502 (2018).
- Sansom, D. M., Manzotti, C. N., Zheng, Y. What's the difference between CD80 and CD86. Trends in Immunology. 24 (6), 314-319 (2003).
- Kurokawa, M., Kornbluth, S. Caspases and kinases in a death grip. Cell. 138 (5), 838-854 (2009).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoExplorar más artículos
This article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados