
É necessária uma assinatura da JoVE para visualizar este conteúdo. Faça login ou comece sua avaliação gratuita.
Method Article
Visualização de Caspases Inflamatórias Induzidas proximidade em macrófagos derivados de monócitos humanos
Neste Artigo
Resumo
Este protocolo descreve o fluxo de trabalho para obter macrófagos derivados de monócitos (MDM) a partir de amostras de sangue humano, um método simples para introduzir eficientemente os repórteres de complementação de fluorescência bimolecular inflamatória (BiFC) em MDM humano sem comprometer a viabilidade e o comportamento celular, e uma abordagem baseada em imagens para medir a ativação inflamatória de caspase em células vivas.
Resumo
As caspases inflamatórias incluem caspase-1, -4, -5, -11 e -12 e pertencem ao subgrupo de caspases iniciadores. O Caspase-1 é necessário para garantir a correta regulação da sinalização inflamatória e é ativado pela dimerização induzida pela proximidade após o recrutamento para inflamações. Caspase-1 é abundante na linhagem celular monocítica e induz a maturação das citocinas pró-inflamatórias interleucina (IL)-1β e IL-18 a moléculas secretas ativas. As outras caspases inflamatórias, caspase-4 e -5 (e seu caspase-11 homólogo murino) promovem a liberação de IL-1β induzindo a piroptose. Caspase Bimolecular Fluorescence Complementation (BiFC) é uma ferramenta usada para medir a proximidade induzida por caspase inflamatória como uma leitura da ativação do caspase. O caspase-1, -4, ou -5 prodomínio, que contém a região que se liga ao inflamatório, é fundido a fragmentos não fluorescentes da proteína fluorescente amarela Vênus (Venus-N [VN] ou Venus-C [VC]) que associam a reforma do complexo fluorescente de Vênus quando as caspas sofrem proximidade induzida. Este protocolo descreve como introduzir esses repórteres em macrófagos derivados do monócito humano primário (MDM) usando nucleofeipulsão, tratar as células para induzir ativação inflamatória de caspase e medir a ativação de caspase usando fluorescência e microscopia confocal. A vantagem dessa abordagem é que ela pode ser usada para identificar os componentes, requisitos e localização do complexo de ativação inflamatória de caspase em células vivas. No entanto, controles cuidadosos precisam ser considerados para evitar comprometer a viabilidade e o comportamento celular. Esta técnica é uma poderosa ferramenta para a análise de interações dinâmicas de caspase no nível inflamatório, bem como para o interrogatório das cascatas inflamatórias de sinalização em MDM vivos e monócitos derivados de amostras de sangue humano.
Introdução
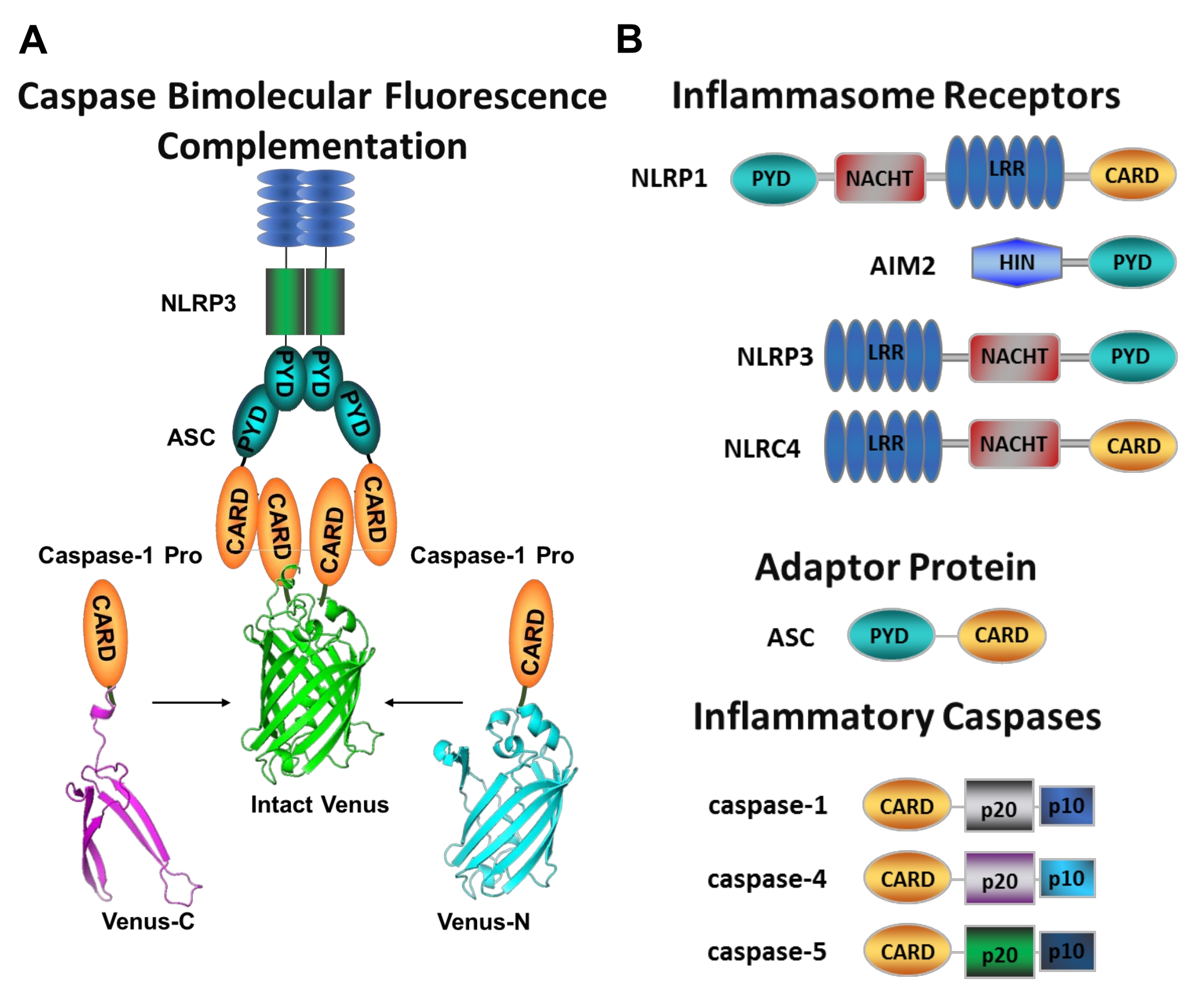
As caspases são uma família de proteases de cisteína aspartate que podem ser agrupadas em caspases iniciadores e caspases carrascos. As caspases carrascos compreendem caspase-3, -6 e -7. Eles são naturalmente encontrados em células como dimers e são cortados pelas caspases iniciadores para executar apoptose1. As caspases iniciais incluem caspase humana-1, -2, -4, -5, -8, -9, -10 e -12. Eles são encontrados como zymogens inativos (pro-caspases) que são ativados pela dimerização induzida pela proximidade e estabilizados pelo decote auto-proteolítico 2,3. As caspases inflamatórias são um subconjunto das caspasesiniciadores 2 e englobam caspase-1, -4, -5 e -12 em humanos, e caspase-1, -11 e -12 no rato 4,5. Em vez de um papel apoptótico, eles desempenham um papel central na inflamação. Eles mediam o processamento proteolítico e a secreção de pró-interleucina (IL)-1β e pró-IL-18 6,7, que são os primeiros citocinas a serem liberados em resposta aos invasores patogênicos 8,9. O Caspase-1 é ativado após o recrutamento para sua plataforma de ativação; um grande complexo de proteína de peso molecular denominado inflamado (Figura 1A)10. A dimerização da caspase-4, -5 e -11 ocorre independentemente dessas plataformas através de uma via inflamamsome nãocanônica11,12.
Os inflamamossscópios canônicos são complexos de proteína multimédica citosolic que consistem em uma proteína sensormasome inflamada, a proteína adaptadora ASC (proteína associada à apoptose contendo um CARD), e o caspase de proteína effectora-110. Os inflamatórios canônicos mais bem estudados são a família receptora semelhante ao NOD contendo um domínio de pirina (NLRP), NLRP1 e NLRP3, a família NLR contendo um CARD (NLRC), NLRC4, e a ausência no melanoma 2 (AIM2). Cada um deles contém um domínio pirin, um CARD ou ambos os domínios. O domínio CARD media a interação entre as caixas contendo cartão e seus ativadores upstream. Portanto, a molécula de andaime ASC, que é composta por um domínio de pirina n-terminal (PYD) e um motivo de CARD terminal C13,14, é necessária para o recrutamento de caspase-1 para o NLRP110, NLRP315 e AIM216 inflamações.
Cada inflamatório é nomeado após sua proteína sensorada única que reconhece distintos estímulos pró-inflamatórios (Figura 1B). Ativadores desta via são denominados estímulos canônicos. Os inflamatórios servem como sensores para componentes microbianos e estresse tecidual, e montam-se para desencadear uma resposta inflamatória robusta através da ativação das caspases inflamatórias17. A montagem inflamada inicia a ativação caspase-1 para mediar a maturação e a secreção de seus substratos principais pró-IL-1β e pró-IL-18. Esse processo ocorre através de um mecanismo de duas etapas. Primeiro, um estímulo de priming regula a expressão de certas proteínas inflamadas e pró-IL-1β através da ativação da via NF-κB. Em segundo lugar, um estímulo intracelular (canônico) induz a montagem inflamada e o recrutamento de procaspase-1 6,7.
Caspase-4 e caspase-5 são os ortopedes humanos da caspase murina-1111. Eles são ativados de forma inflamada e independente por lipopolysacarídeo intracelular (LPS), uma molécula encontrada na membrana externa das bactérias Gram-negativas18,19,20, e por heme extracelular, um produto da hemólise de glóbulos vermelhos21. Foi proposto que o LPS se liga diretamente ao motivo card dessas proteínas e induz sua oligomerização20. A ativação do caspase-4 ou caspase-5 promove a liberação do IL-1β induzindo uma forma inflamatória de morte celular chamada piroptose através do decote da proteína gasdermin D (GSDMD)18,19. Além disso, o efflux de íons de potássio resultantes da morte piropopótica mediada por caspase-4 e GSDMD induz a ativação do inflamador NLRP3 e posterior ativação do caspase-122,23. Portanto, caspase-4, -5 e -11 são considerados sensores intracelulares para LPS que são capazes de induzir piroptose e ativação caspase-1 em resposta a estímulos específicos11,24.

Figura 1: Caspases inflamatórias e ensaio de complementação de fluorescência caspase-bimolecular (BiFC). (A) Diagrama mostrando o sistema caspase-BiFC, onde dois prodomínios caspase-1 (C1-pro) ligados a cada fragmento não fluorescente de Vênus (Venus-C ou Venus-N) são recrutados para a plataforma de ativação NLRP3, forçando Vênus a se repatar e fluorescer. Este complexo aparece como uma mancha verde sob o microscópio e serve como uma leitura para a proximidade inflamatória induzida por caspase, que é o primeiro passo na ativação do caspase iniciador. (B) Esquema mostrando a organização do domínio de componentes inflamatórios e caspases inflamatórias. Clique aqui para ver uma versão maior desta figura.
{kind=link}
Medir a ativação específica de caspases iniciadores é difícil, e não há muitos métodos disponíveis para fazê-lo por abordagens de imagem. A Complementação da Fluorescência Bimolecular Caspase (BiFC) pode ser usada para visualizar a ativação inflamatória de caspase diretamente em células vivas (Figura 1A)25. Esta técnica foi recentemente adaptada para uso em macrófagos derivados de monócitos humanos (MDM)21. Caspase BiFC mede o primeiro passo na ativação inflamatória de caspase, induzida proximidade para facilitar a dimerização. A expressão de plasmídeos que codificam o caspase prodomínio contendo card fundido a fragmentos não fluorescentes da proteína fluorescente amarela fotostável Vênus (Venus-C [VC]) e Venus-N [VN]) são usados. Quando os dois prodomínios caspase são recrutados para sua plataforma de ativação ou sofrem proximidade induzida, as duas metades de Vênus são trazidas de perto e forçadas a se reabastecer e fluoresce (ver Figura 1A,B). Isso fornece uma leitura em tempo real da ativação específica de caspase inflamatória.
O MDM humano expressa abundantemente genes inflamados e receptores de reconhecimento de padrões que identificam sinais de perigo e produtos patógenos. Isso fornece um tipo de célula ideal para o interrogatório de vias inflamatórias caspase. Além disso, podem ser derivados do sangue periférico e até mesmo de amostras de pacientes para avaliar a ativação inflamatória de caspase em um estado específico da doença. Este protocolo descreve como introduzir os repórteres de caspase BiFC no MDM usando nucleofecção, um método de transfecção baseado em eletroporação, como tratar as células para induzir a ativação inflamatória do caspase e como visualizar os complexos ativos de caspase usando abordagens de microscopia. Além disso, essa metodologia pode ser adaptada para determinar a composição molecular desses complexos, localização subcelular, cinética e tamanho dessas estruturas altamente ordenadas 25,26,27.
Access restricted. Please log in or start a trial to view this content.
Protocolo
Este protocolo segue as diretrizes do comitê de ética em pesquisa humana da Baylor College of Medicine para a manipulação de amostras humanas. As amostras de sangue são tratadas seguindo as diretrizes institucionais de segurança para amostras humanas. As amostras de sangue são obtidas em um banco de sangue regional, onde são coletadas com solução de fosfato dextrose citrato (CPD). No entanto, o sangue coletado com outros anticoagulantes como heparina de sódio, heparina de lítio ou EDTA também pode ser usado para este protocolo28,29.
1. Isolamento dos monócitos humanos e diferenciação em macrófagos
- Obtenha sangue anticoagulado de indivíduos saudáveis desidentiados em um banco de sangue regional e isole as células mononucleares periféricas (PBMCs) conforme indicado abaixo.
NOTA: Realize todas as etapas em uma capa de fluxo laminar de cultura de tecido. Use apenas tubos estéreis e use luvas. Adicione 10% de alvejante a todos os produtos relacionados ao sangue ao descartar. PBS estéril (1x) ou DPBS (sem Ca2+ e Mg2+) pode ser usado intercambiavelmente.- Prepare o tampão de diluição: Suplementar 1x PBS estéril com 2% de FBS e 0,5 mM EDTA.
- Prepare o meio de cultura: Suplemento RPMI-1640 médio com FBS (10% (v/v)), glutamax (2 mM) e Penicilina/ Estreptomicina (50 I.U./50 μg/mL)
- Pré-, consulte o buffer de execução (Tabela de Materiais) de acordo com o protocolo do fabricante.
- Diluir o sangue inteiro com dois volumes de tampão de diluição. Utilizando um pipeto sorológico, transfira 15 mL do sangue anticoagulado para um tubo de 50 mL contendo 30 mL do tampão de diluição. Misture suavemente por inversão.
- Para cada 10 mL de sangue inteiro ou 30 mL de sangue diluído, adicione 15 mL do gradiente de densidade médio a um tubo vazio de 50 mL.
- Camada o gradiente de densidade médio a partir da etapa 1.1.5 com 30 mL de sangue diluído lentamente e constantemente usando um tubo sorológico de 25 mL. Mantenha a ponta da tubulação contra a parede do tubo e o tubo em um ângulo inclinado.
- Transfira cuidadosamente os tubos para uma centrífuga de balde balançando. Evite perturbar as duas fases. Centrifugar os tubos a 400 x g à temperatura ambiente (RT) por 25 minutos com aceleração e desaceleração definidas ao valor mínimo.
- Remova cuidadosamente a camada de plasma superior (clara) usando uma tubulação de 10 mL e descarte em um recipiente com alvejante (10%).
- Colete a camada interfase (branca) de células mononucleares de sangue periféricos (PBMCs, Figura 2) com uma tubulação de 10 mL e transfira para um tubo fresco de 50 mL. Combine a camada branca de diferentes tubos do mesmo doador em um tubo de 50 mL até 30 mL.
- Leve cada tubo a um volume total de 50 mL com o tampão de diluição da etapa 1.1.1 e centrífuga a 300 x g e 4 °C por 10 min. Remova o supernatante com uma tubulação de 10 mL e descarte-o em um recipiente com alvejante (10%).
- Resuspenque cada pelota de célula em 1 mL do buffer de corrida pré-resfriado a partir da etapa 1.1.3 usando um micropipette p1000. Combine suspensões celulares do mesmo doador em um novo tubo de 15 mL. Leve o volume de cada tubo para 15 mL com tampão de corrida pré-resfriado e misture bem por inversão.
- Pegue uma alíquota de 20 μL da suspensão celular a partir da etapa 1.1.11 e prepare uma diluição de 1:100 usando PBS 1x estéril. Determine o número da célula usando um hemócito.
- Centrifugar a suspensão celular da etapa 1.1.11 a 300 x g e 4 °C por 10 min e remover o supernatante com uma tubulação de 10 mL. Se necessário, use uma micropipette p200 para remover completamente o sobrenatante.
- Resuspenque os PBMCs isolados em 80 μL de buffer de execução MACS pré-resfriado para cada célula de 1 x 107 , somando um máximo de 800 μL do buffer.
- Adicione 20 μL de MicroEsferas CD14 anti-humanas por cada 1 x 107 células ou até 100 μL por amostra de sangue (~100 mL de sangue não diluído). Misture bem por inversão e coloque em um rotador de tubo por 20 minutos com mistura contínua a 4 °C.
- Remova as amostras do rotador do tubo, adicione 10 mL de tampão de corrida pré-resfriado em cada tubo e centrífugas a 300 x g (aceleração = 5, desaceleração = 5) e 4 °C por 10 min.
- Remova o supernatante com uma tubulação de 10 mL e resuspende até 1 x 108 células em 500 μL de tampão de corrida pré-resfriado (2 x 108/mL).
- Realize o isolamento das células CD14 positivas por meio de um sistema manual ou automatizado (Tabela de Materiais) de acordo com as instruções do fabricante.
- Pegue uma alíquota de 20 μL da suspensão celular a partir da etapa 1.1.18 após a seleção CD14-positiva e prepare uma diluição de 1:100 usando PBS 1x estéril. Determine o número da célula contando as células em um hemótmetro.
- Centrifugar as células CD14-positivas a 300 x g e RT por 10 min. Remova o supernatante usando uma tubulação de 10 mL ou um sistema de vácuo.
- Resuspenque a pelota celular da etapa 1.1.20 em meio de cultura pré-aquecida da etapa 1.1.2 para uma densidade celular final de 1 x 107 células/mL.
- Semente os monócitos isolados CD14-positivos a uma densidade celular de 5 x 106 células.
- Em um prato de cultura tecidual de 10 cm, adicione 10 mL de meio de cultura a partir da etapa 1.1.2 complementada com 50 ng/mL fator estimulante de colônia granulocito-macrófago (GM-CSF).
- Adicione 0,5 mL da suspensão da célula da etapa 1.1.21 ao meio de cultura dropwise e gire suavemente a placa. Incubar células em uma incubadora de cultura tecidificada (37 °C, 5% CO2) durante a noite.
- No dia seguinte, aspire o meio usando um sistema de vácuo para remover células que não se anexaram durante a noite. Adicione 10 mL de cultura fresca de cultura fresca suplementada GM-CSF (50 ng/mL) e incubar células em uma incubadora de cultura tecidificada (37 °C, 5% CO2) por 7 dias para permitir a completa diferenciação (ver Figura 3A para o aparecimento de monócitos CD14+ em vários estágios de diferenciação em GM-CSF ). Troque o meio de cultura a cada 2-3 dias e complemente com GM-CSF fresco (50 ng/mL) cada vez.

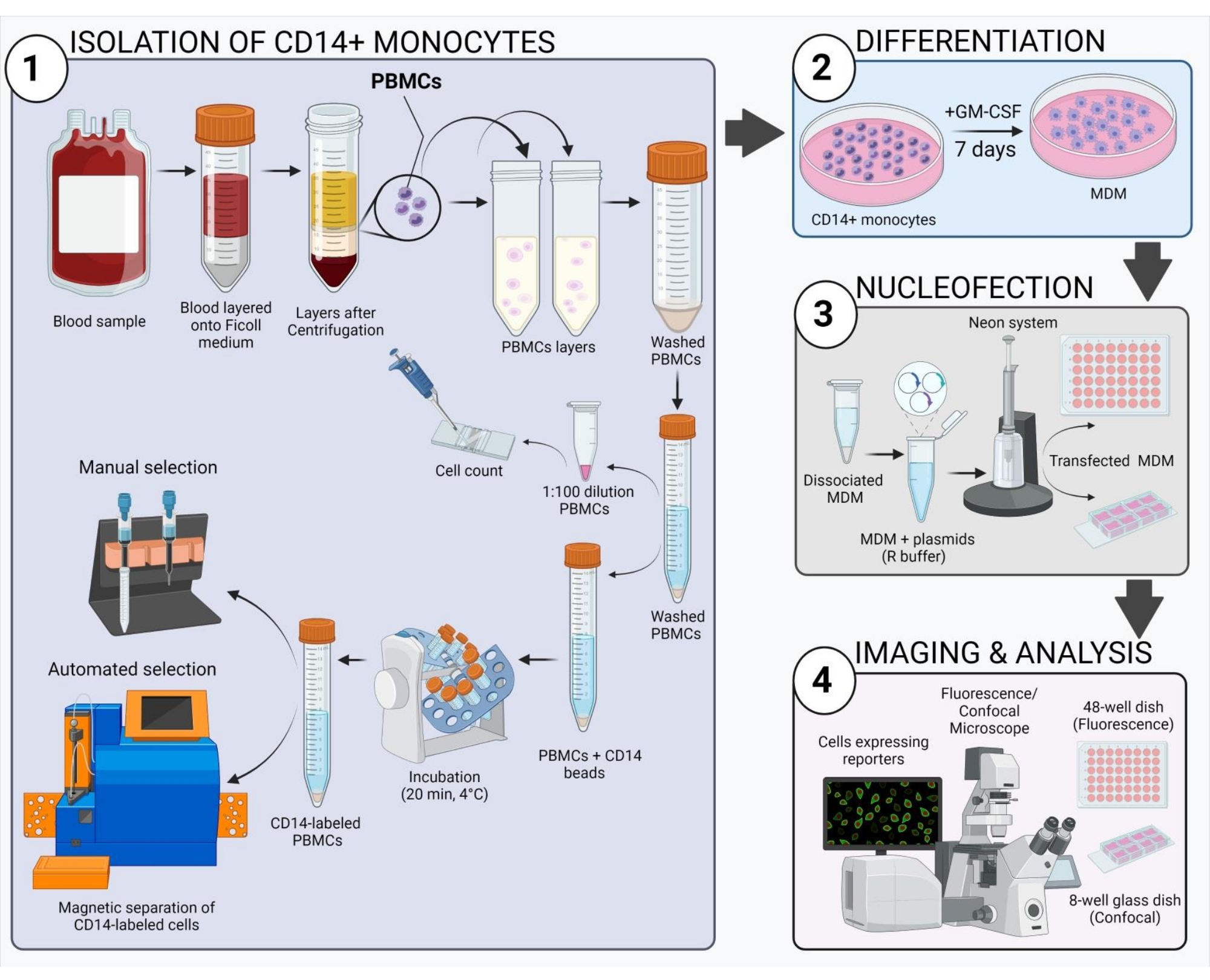
Figura 2: Visão geral do fluxo de trabalho experimental. Clique aqui para ver uma versão maior desta figura.
{kind=link}
2. Preparação de componentes de eletroporação
NOTA: Este protocolo foi projetado para uma dica de 10 μL-Neon (Tabela de Materiais). Para cada transfecção, use células 1-2 x 105 . Recomenda-se a sementes células transfeinadas em uma placa de 48 poços ou 8-bem câmara (10 células transfeinadas μL por poço). 1x DPBS estéreis (sem Ca2+ e Mg2+) podem ser usados no lugar de PBS.
- No dia 7, prepare o meio sem antibióticos suplementando o meio RPMI-1640 com FBS (10 % (v/v)) e glutamax (2 mM).
- Coloque a solução RPMI-1640 sem soro, a solução trypsin-EDTA (0,25%), 1x PBS estéril (sem Ca2+ e Mg2+), e meio cultura completa a partir da etapa 1.1.2 em um banho de água de 37 °C.
- Se usar pratos com fundo de vidro (para microscopia confocal) cubra os pratos com hidrobromida de poli-D-lisina.
- Cubra um prato de 8 câmaras com 200 μL de hidrobromida de poli-D-lysine (0,1 mg/mL em PBS estéril de 1x) e incubar por 5 min na RT.
- Aspire a solução de poli-D-lysine e lave o vidro uma vez com PBS 1x estéril. Aspire o PBS e prossiga com a etapa 2.4.
- Adicione 200 μL de meio livre de antibióticos por poço da placa de 48 poços ou 8 pratos bem-acomodados e pré-incubado em uma incubadora de cultura de tecido umidificado (37 °C, 5% CO2) até estar pronto para emplacar as células transfeinadas.
3. Preparação de células para eletroporação
NOTA: O rendimento de MDM de um prato de 10 cm no final do período de diferenciação de 7 dias é de aproximadamente 1,5 x 106 células. 1x DPBS estéreis (sem Ca2+ e Mg2+) podem ser usados no lugar de PBS. Este protocolo foi otimizado para que a maioria dos macrófagos sejam separados da placa com a manutenção da viabilidade e integridade celular. MDM são difíceis de separar das placas de cultura celular. Portanto, pode ser necessário realizar as etapas 3.2 e 3.3 duas vezes para dissociar as células. Certifique-se de que cada tempo de incubação com trippsin-EDTA (0,25%) não exceda 5 min.
- Aspire a mídia de macrófagos totalmente diferenciados em pratos de 10 cm e lave a monocamada celular com meio RPMI-1640 sem soro quente. Certifique-se de remover completamente o meio.
- Colher as células adicionando 2 mL de solução quente de trippsina-EDTA (0,25%) por prato de 10 cm e incubar em uma incubadora de cultura de tecido umidificado (37 °C, 5% CO2) por 5 min.
- Complete o desprendimento celular, tubondo suavemente a solução trypsin-EDTA (0,25%) para cima e para baixo em toda a área do prato usando uma micropipette p1000. Transfira a suspensão celular para um tubo cônico de 15 mL contendo 5 mL de meio de cultura completa quente a partir da etapa 1.1.2.
- Leve o prato para um microscópio de campo brilhante e verifique se há desprendimento celular em vários campos de visão. Se houver uma quantidade considerável de células ainda anexadas, repita as etapas 3.2-3.3.
- Centrifugar a suspensão celular a 250 x g por 5 min no RT.
- Aspire o médio e resuspenque as células em 10 mL de PBS estéreis de 1x pré-aquecido a 37 °C. Tome uma alíquota de 20 μL para determinar o número da célula usando um hemócito.
- Pegue 1-2 x 105 células por transfecção pretendida e coloque em um tubo de 15 mL. Leve a um volume final de 15 mL com PBS 1x estéril pré-aquecido. Centrifugar a 250 x g por 5 min no RT.
- Aspire o PBS e centrífuga mais uma vez por 1 min a 250 x g. Remova qualquer PBS residual da pelota celular usando uma micropipette p200.
4. Nucleofecção de componentes caspase BiFC em macrófagos derivados de monócitos humanos
NOTA: Esta seção do protocolo é realizada utilizando-se o Sistema de Transfecção neon (Tabela de Materiais). Este protocolo descreve as etapas para transfetar células 1-2 x 105 usando uma ponta de 10 μL neon (Tabela de Materiais). Se usar uma ponta neon de 100 μL, aumente em conformidade. Evite expor células ao tampão de resuspensão R por mais de 15 minutos, pois isso pode diminuir a viabilidade celular e a eficiência da transfecção.
- Diluir o plasmídeo repórter (ou seja, mCherry ou dsRedmito a 100 ng/μL) em água sem nuclease ou tampão te 0,5x para visualizar as células transfeminadas.
- Diluir os plasmídeos Caspase BiFC para uma concentração apropriada em água sem nuclease ou tampão te de 0,5x, de modo que o volume total de plasmídeos não exceda 30% do volume total de transfecção de 10 μL (ou seja, 300 ng/μL de C1 Pro-VC e 300 ng/μL de C1 Pro-VN).
- Prepare um microtubo estéril de 1,5 mL por transfecção pretendida e adicione a quantidade apropriada de plasmid repórter (ou seja, 50 ng ou 0,5 μL) e plasmídeos Caspase BiFC (ou seja, 300 ng ou 1 μL de C1 Pro-VC e 300 ng ou 1 μL de fragmento C1 Pro-VN). Mantenha os microtubos no capô o tempo todo.
- Coloque a estação de pipeta, dispositivo, pontas, tubos de eletroporação e pipeta em uma coifa de fluxo laminar estéril.
NOTA: A estação de pipeta, dispositivo, pontas, tubos de eletroporação e pipeta estão incluídos no Sistema de Transfecção Neon. - Conecte o conector de alta tensão e sensor na estação de pipeta às portas traseiras do dispositivo, de acordo com as instruções do fabricante. Mantenha a estação de pipeta perto do dispositivo.
- Conecte o cabo de alimentação à entrada ca traseira e prossiga para conectar o dispositivo à tomada elétrica. Pressione o interruptor de alimentação para ligar o dispositivo.
- Digite os parâmetros de transfecção na tela de inicialização exibida quando o dispositivo estiver ligado e conectado adequadamente. Pressione a Tensão, digite 1000 e pressione em Done para definir a tensão em 1000 V. Pressione na largura, digite 40 e pressione em Feito para definir a duração do pulso para 40 ms. Por fim, pressione #Pulsos, digite 2 e pressione Em Feito para definir o número de pulsos elétricos para 2.
- Pegue um dos tubos de eletroporação (fornecidos no kit) e encha-o com 3 mL de tampão eletrolítico E (para 10 pontas μL e fornecido no kit) na RT. Insira o tubo de eletroporação no suporte de pipeta na estação pipeta. Certifique-se de que o eletrodo na lateral do tubo está voltado para dentro e que um som de clique seja ouvido quando o tubo estiver inserido.
- Pegue a pelota celular a partir da etapa 3.8 e adicione 10 μL de tampão R de resuspensão pré-aquecida (fornecido no kit) para cada célula de 1-2 x 105 . Misture delicadamente com uma micropipette p20. Adicione 10 μL da suspensão celular a cada tubo configurado na etapa 4.3 e misture suavemente com uma micropipette p20.
- Pegue a pipeta e insira uma ponta pressionando o botão Pressionar até a segunda parada. Certifique-se de que o grampo pegue completamente a haste de montagem do pistão na ponta e que nenhuma lacuna seja observada na parte superior da pipeta.
- Para aspirar a amostra, pressione o botão Pressionar a pipeta até a primeira parada e mergulhe no primeiro tubo contendo mistura de DNA celular/plasmídeo. Aspire lentamente a mistura na ponta da pipeta.
NOTA: Evite bolhas de ar, pois elas podem causar arco durante a eletroporação, e se detectadas pelo dispositivo, podem impedir a entrega do pulso elétrico. Se forem observadas bolhas de ar, solte o conteúdo no tubo e tente aspirar novamente. - Insira a pipeta com a amostra com muito cuidado no suporte da pipeta. Certifique-se de que a pipeta clique e que ela esteja bem colocada.
- Pressione Inicie na tela sensível ao toque e espere até que os pulsos elétricos sejam entregues. Uma mensagem na tela indicará a conclusão.
- Remova lentamente a pipeta da estação e adicione imediatamente a suspensão celular transfeinada no poço correspondente com meio pré-aquecido sem antibióticos a partir do passo 2.4 pressionando lentamente o botão de pressão para a primeira parada.
NOTA: Esta dica pode ser reutilizada até três vezes para o mesmo plasmídeo; caso contrário, descarte-o em um recipiente de resíduos de risco biológico pressionando o botão Apertar para a segunda parada. - Repita as etapas 4.10-4.14 para cada tubo contendo uma mistura de DNA celular/plasmídeo.
- Balance suavemente a placa com células transfeinadas e incubar por 1-3 h em uma incubadora de cultura tecidificada (37 °C, 5% CO2).
- Adicione a cada poço 200 μL de meio de cultura pré-aquecido (meio completo) a partir da etapa 1.1.2. Coloque o prato na incubadora de cultura tecidificada (37 °C, 5% CO2) novamente. Permita pelo menos 24 horas para expressão genética.
- No dia seguinte, inspecione a viabilidade celular e a eficiência da transfecção usando um microscópio de epifluorescência.
- Ligue o microscópio de epifluorescência e a caixa de fonte de luz fluorescente de acordo com as instruções do fabricante e coloque o prato de cultura no estágio do microscópio.
- Selecione o objetivo de 10x ou 20x e o filtro RFP de 568 nm (RFP).
- Para estimar a viabilidade celular, pressione o botão LED de luz transmitida (TL) para visualizar todas as células no campo selecionado. Ao olhar para a ocular do microscópio, gire o botão de foco até que as células sejam observadas e verifique se há apego celular no campo selecionado.
NOTA: As células totalmente conectadas representam as células viáveis, enquanto as células flutuantes representam células não viáveis. Se a confluência do poço for alta, a presença de células não anexadas pode ser o resultado de uma superestimação do número celular e não o resultado da baixa viabilidade. No entanto, a baixa confluência acompanhada de um alto teor de células flutuantes significa baixa viabilidade que pode resultar de arco durante a eletroporação, toxicidade plasmóide ou superexposição para resuspensão R tampão. Não use poços que exibam o último comportamento. - Para estimar a eficiência da transfecção, concentre-se nas células do campo selecionado sob luz transmitida como descrito acima. Conte o número total de células no campo selecionado. Com o LED de luz transmitida (TL) desligado, pressione o botão LED de luz refletida (RL) para ligar.
- Foco fino na fluorescência genética repórter (células vermelhas) e contar o número total de células fluorescentes vermelhas. Repita estes passos (4.18.4-4.18.5) para pelo menos mais dois campos por poço.
5. Tratamento de MDM transfectado e aquisição de dados caspase BiFC
NOTA: Se o planejamento de imagem das células usando um microscópio epifluorescência ou confocal, o tratamento com qVD-OPh (20 μM) para tratamento prévio de 1 h com o estímulo escolhido é aconselhado para prevenir a morte celular dependente de caspase (predominantemente apoptose). Isso é usado em imagens para evitar que as células decolem devido à apoptose, tornando-as muito difíceis de imagem à medida que saem do plano focal. Observe que o recrutamento caspase para a plataforma de ativação e o Caspase BiFC associado não depende da atividade catalítica do caspase e, consequentemente, a inibição de caspase não afetará essa etapa.
- Trate com estímulo escolhido aproximadamente 24 horas após a transfecção e incubar pelo tempo necessário para cada droga.
- Prepare o meio de imagem suplementando o meio cultura da etapa 1.1.2 com Hepes (20 mM, pH 7.2-7.5) e 2-mercaptoetanol (55 μM).
- Adicione a concentração desejada de estímulo ao meio de imagem pré-aquecido e misture suavemente.
- Remova a mídia das células cuidadosamente com uma micropipette p1000 e adicione 500 μL da solução de estímulo da etapa 5.1.2 para baixo na lateral do poço.
- Para executar poços de controle não tratados, adicione o meio de imagem sem o estímulo.
- Incubar as células em uma incubadora de cultura tecidificada (37 °C, 5% DE CO2) pelo tempo indicado para cada tratamento.
- Visualize as células usando uma epifluorescência ou microscópio confocal.
- Ligue o microscópio e a fonte de luz fluorescente, seguindo as instruções do fabricante.
- Selecione o objetivo de 10x ou 20x e coloque o prato de cultura no palco do microscópio.
- Usando a ocular do microscópio, encontre células sob o filtro de 568 nm e foque nas células expressando o repórter dsRedmito/mCherry (células vermelhas).
- Conte todas as células vermelhas no campo visual e regissume o número.
- Enquanto no mesmo campo visual, mude para o filtro 488 ou 512 (GFP ou YFP), proceda à contagem do número de células vermelhas que também são verdes (Venus-positivo ou BiFC-positivo) e registre o número.
- Conte pelo menos 100 células dsRedmito/mCherry -positivas de um mínimo de três campos visuais individuais.
- Calcule a porcentagem de células transfetadas positivas por campo visual e calcule as porcentagens resultantes de cada tratamento (bem) para obter o desvio padrão.
- Imagem das células usando uma epifluorescência ou microscópio confocal
NOTA: Para adquirir imagens confocal usando um objetivo de 20x ou uma ampliação maior, as células devem ser banhadas em pratos de vidro, a menos que o microscópio esteja equipado com um objetivo de passagem longa.- Siga os passos 5.2.1-5.2.3. Se usar um microscópio confocal com o objetivo do óleo 40x, 60x ou 63x, coloque uma gota de óleo no objetivo.
- Visualize a imagem ao vivo das células na tela do computador como adquirido pela câmera. Use a fonte de luz de epifluorescência para imagens fluorescentes ou mude a fonte de luz para os lasers para imagens confocal.
- Ajuste o foco e a posição das células usando o controle do joystick e a roda de foco.
- Defina a porcentagem de potência laser e o tempo de exposição para os lasers de 512 nm ou 488 nm (YFP ou GFP) e 568 nm (RFP) para que o sinal na imagem pareça bom e não atinja a saturação.
- Ligue a captura ao vivo e examine a imagem resultante. Certifique-se de que um pico distinto seja visto para cada fluor no histogramas de exibição para ambos os canais.
- Ajuste a potência do laser e o tempo de exposição conforme necessário. Mantenha esses valores o mais baixos possível enquanto ainda é capaz de detectar ambos os sinais fluorescentes (RFP e GFP/YFP).
- Ao visualizar a imagem ao vivo das células, pegue várias imagens representativas de um campo que contenha uma ou mais células expressando o repórter mCherry/dsRedmito para cada poço da placa e salve os dados.
Access restricted. Please log in or start a trial to view this content.
Resultados
O esquema mostrado na Figura 2 dá uma visão geral de como obter, transfetar e imagem humana MDM. Após a incubação dos monócitos CD14+ selecionados com GM-CSF por 7 dias, a morfologia celular muda ao longo do período de diferenciação (Figura 3A), passando de células de suspensão esféricas para spindly e totalmente anexadas (dias 3 e 4), e, por último, para células mais difundidas quando totalmente diferenciadas (dia 7). As células totalmente difer...
Access restricted. Please log in or start a trial to view this content.
Discussão
Este protocolo descreve o fluxo de trabalho para obter macrófagos de monócitos isolados de amostras de sangue humano e um método para introduzir eficientemente os repórteres de Caspase Inflamatório BiFC em MDM humano sem comprometer a viabilidade e o comportamento celular.
Este protocolo aproveita a técnica BiFC35 para marcar as caspases inflamatórias no domínio de recrutamento caspase (CARD) com fragmentos não fluorescentes da proteína fluorescente dividida V...
Access restricted. Please log in or start a trial to view this content.
Divulgações
Os autores declaram que não têm interesses financeiros concorrentes.
Agradecimentos
Agradecemos aos membros do laboratório passado e presente da LBH que contribuíram para o desenvolvimento dessa técnica. Este laboratório é suportado por NIH/NIDDK T32DK060445 (BEB), NIH/NIDDK F32DK121479 (BEB), NIH/NIGMS R01GM121389 (LBH). A Figura 2 foi desenhada usando o software Biorender.
Access restricted. Please log in or start a trial to view this content.
Materiais
| Name | Company | Catalog Number | Comments |
| 48 well tissue culture2:34 plates | Genesee Scientific | 25-108 | |
| 10 cm Tissue Culture Dishes | VWR | 25382-166 | |
| 2 Mercaptoethanol 1000x | Thermo Fisher Scientific | 21985023 | |
| 8 well chambered coverglass with 1.5 HP coverglass | Cellvis | c8-1.5H-N | |
| AutoMACS columns | Miltenyi (Biotec) | 130-021-101 | For automated separation using AutoMACS Pro Separator only |
| AutoMACS Pro Separator | Miltenyi (Biotec) | 130-092-545 | For automated separation using AutoMACS Pro Separator only |
| AutoMACS Pro Washing Solution | Miltenyi (Biotec) | 130-092-987 | For automated separation using AutoMACS Pro Separator only |
| AutoMACS Rinsing Solution | Miltenyi (Biotec) | 130-091-222 | For automated separation using AutoMACS Pro Separator only |
| AutoMacs running buffer | Miltenyi (Biotec) | 130-091-221 | For manual or automated separation using QuadroMACS or AutoMACS pro Separator |
| Axio Observer Z1 motorized inverted microscope equipped with a CSU-X1A 5000 spinning disk unit | Zeiss | Any confocal microscope equipped with a laser module fitted with laser lines of 568 nm (RFP) and 488 or 512 nm (GFP or YFP) wavelengths can be used | |
| AxioObserver A1, Research Grade Inverted Microscope | Zeiss | Any epifluorescence microscope with fluorescence filters capable of exciting 568 nm (RFP) and 488 or 512 nm (GFP or YFP) wavelengths can be used | |
| CD14+ MICROBEADS | Miltenyi (Biotec) | 130-050-201 | For manual or automated separation using QuadroMACS or AutoMACS pro Separator |
| DPBS without calcium chloride and magnesium chloride | Sigma | D8537-6x500ML | |
| DsRed mito plasmid | Clontech | 632421 | Similar plasmids that can be used as fluorescent reporters can be found on Addgene |
| Fetal Bovine Serum | Thermo Fisher Scientific | 10437028 | |
| Ficoll-Paque PLUS 6 x 100 mL | Sigma | GE17-1440-02 | |
| GlutaMAX Supplement (100x) | Thermo Fisher Scientific | 35050079 | |
| GM-CSF | Thermo Fisher Scientific | PHC2011 | |
| Hemin BioXtra, from Porcine, ≥96.0% (HPLC) | Sigma | 51280-1G | |
| HEPES | Thermo Fisher Scientific | 15630106 | |
| Inflammatory caspase BiFC plasmids | Available by request from LBH lab | ||
| LPS-EB Ultrapure | Invivogen | TLRL-3PELPS | |
| LS Columns | Miltenyi (Biotec) | 130-042-401 | For manual separation using QuadroMACS Separator only |
| MACS 15 mL Tube Rack | Miltenyi (Biotec) | 130-091-052 | For manual separation using QuadroMACS Separator only |
| MACS MultiStand | Miltenyi (Biotec) | 130-042-303 | For manual separation using QuadroMACS Separator only |
| mCherry plasmid | Yungpeng Wang Lab | Similar plasmids that can be used as fluorescent reporters can be found on Addgene | |
| Neon Transfection System | Thermo Fisher Scientific | MPK5000 | Includes Neon electroporation device, pipette and pipette station |
| Neon Transfection System 10 µL Kit | Thermo Fisher Scientific | MPK1096 | Includes resuspension buffer R, resuspension buffer T, electrolytic buffer E, 96 x 10 µL Neon tips and Neon electroporation tubes |
| Nigericin sodium salt, ready made solution | Sigma | SML1779-1ML | |
| Penicillin-Streptomycin (10,000 U/mL) | Thermo Fisher Scientific | 15140122 | |
| Poly-D-Lysine Hydrobromide | Sigma | P7280-5mg | |
| QuadroMACS Separator | Miltenyi (Biotec) | 130-090-976 | For manual separation using QuadroMACS Separator only |
| qVD-OPh | Fisher (ApexBio) | 50-101-3172 | |
| RPMI 1640 Medium | Thermo Fisher Scientific | 11875119 | |
| Trypsin-EDTA (0.25%), phenol red | Thermo Fisher Scientific | 25200072 | |
| UltraPure 0.5 M EDTA, pH 8.0 | Thermo Fisher Scientific | 15575020 | |
| Zeiss Zen 2.6 (blue edition) software | Zeiss | Any software used to operate the confocal microscope of choice |
Referências
- Boatright, K. M., et al. A unified model for apical caspase activation. Molecular Cell. 11 (2), 529-541 (2003).
- Pop, C., Salvesen, G. S. Human caspases: activation, specificity, and regulation. Journal of Biological Chemistry. 284 (33), 21777-21781 (2009).
- Boice, A., Bouchier-Hayes, L. Targeting apoptotic caspases in cancer. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 1867 (6), 118688(2020).
- Bolívar, B. E., Vogel, T. P., Bouchier-Hayes, L. Inflammatory caspase regulation: maintaining balance between inflammation and cell death in health and disease. The FEBS Journal. 286 (14), 2628-2644 (2019).
- Martinon, F., Tschopp, J. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell. 117 (5), 561-574 (2004).
- Lamkanfi, M., Vishva, M. D. Mechanisms and functions of inflammasomes. Cell. 157 (5), 1013-1022 (2014).
- Viganò, E., et al. Human caspase-4 and caspase-5 regulate the one-step noncanonical inflammasome activation in monocytes. Nature Communications. 6, 8761(2015).
- Cerretti, D. P., et al. Molecular cloning of the interleukin-1 beta converting enzyme. Science. 256 (5053), 97(1992).
- van de Veerdonk, F. L., Netea, M. G., Dinarello, C. A., Joosten, L. A. Inflammasome activation and IL-1β and IL-18 processing during infection. Trends in Immunology. 32 (3), 110-116 (2011).
- Martinon, F., Burns, K., Tschopp, J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Molecular Cell. 10 (2), 417-426 (2002).
- Kayagaki, N., et al. Noncanonical inflammasome activation targets caspase-11. Nature. 479 (7371), 117-121 (2011).
- Kayagaki, N., et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science. 341 (6151), 1246-1249 (2013).
- Masumoto, J., et al. ASC, a novel 22-kDa protein, aggregates during apoptosis of human promyelocytic leukemia HL-60 cells. The Journal of Biological Chemistry. 274 (48), 33835-33838 (1999).
- Bertin, J., DiStefano, P. S. The PYRIN domain: a novel motif found in apoptosis and inflammation proteins. Cell Death and Differentiation. 7 (12), 1273-1274 (2000).
- Agostini, L., et al. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity. 20 (3), 319-325 (2004).
- Bürckstümmer, T., et al. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nature Immunology. 10 (3), 266-272 (2009).
- Latz, E., Xiao, T. S., Stutz, A. Activation and regulation of the inflammasomes. Nature Reviews Immunology. 13 (6), 397-411 (2013).
- Kayagaki, N., et al. Caspase-11 cleaves gasdermin D for noncanonical inflammasome signalling. Nature. 526 (7575), 666-671 (2015).
- Shi, J., et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 526 (7575), 660-665 (2015).
- Shi, J., et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 514 (7521), 187-192 (2014).
- Bolívar, B. E., et al. Noncanonical Roles of Caspase-4 and Caspase-5 in Heme-Driven IL-1β Release and Cell Death. The Journal of Immunology. 206 (8), 1878-1889 (2021).
- Schmid-Burgk, J. L., et al. Caspase-4 mediates noncanonical activation of the NLRP3 inflammasome in human myeloid cells. European Journal of Immunology. 45 (10), 2911-2917 (2015).
- Baker, P. J., et al. NLRP3 inflammasome activation downstream of cytoplasmic LPS recognition by both caspase-4 and caspase-5. European Journal of Immunology. 45 (10), 2918-2926 (2015).
- Cullen, S. P., Kearney, C. J., Clancy, D. M., Martin, S. J. Diverse activators of the NLRP3 inflammasome promote IL-1β secretion by triggering necrosis. Cell Reports. 11 (10), 1535-1548 (2015).
- Sanders, M. G., et al. Single-cell imaging of inflammatory caspase dimerization reveals differential recruitment to inflammasomes. Cell Death & Disease. 6, 1813(2015).
- Charendoff, C. I., Bouchier-Hayes, L. Lighting up the pathways to caspase activation using bimolecular fluorescence complementation. Journal of Visualized Experiments: JoVE. (133), e57316(2018).
- Bouchier-Hayes, L., et al. Characterization of cytoplasmic caspase-2 activation by induced proximity. Molecular Cell. 35 (6), 830-840 (2009).
- Riedhammer, C., Halbritter, D., Weissert, R. Peripheral blood mononuclear cells: Isolation, Freezing, thawing, and culture. Methods in Molecular Biology. 1304, 53-61 (2016).
- Betsou, F., Gaignaux, A., Ammerlaan, W., Norris, P. J., Stone, M. Biospecimen science of blood for peripheral blood mononuclear cell (PBMC) functional applications. Current Pathobiology Reports. 7 (2), 17-27 (2019).
- Perregaux, D., et al. IL-1 beta maturation: evidence that mature cytokine formation can be induced specifically by nigericin. The Journal of Immunology. 149 (4), 1294-1303 (1992).
- Cheneval, D., et al. Increased mature interleukin-1β (IL-1β) secretion from THP-1 cells induced by nigericin is a result of activation of p45 IL-1β-converting enzyme processing. Journal of Biological Chemistry. 273 (28), 17846-17851 (1998).
- Fernandes-Alnemri, T., et al. The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death and Differentiation. 14 (9), 1590-1604 (2007).
- Lu, A., et al. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell. 156 (6), 1193-1206 (2014).
- Muller-Eberhard, U., Javid, J., Liem, H. H., Hanstein, A., Hanna, M. Brief report: Plasma concentrations of hemopexin, haptoglobin and heme in patients with various hemolytic diseases. Blood. 32 (5), 811-815 (1968).
- Shyu, Y. J., Liu, H., Deng, X., Hu, C. D. Identification of new fluorescent protein fragments for bimolecular fluorescence complementation analysis under physiological conditions. Biotechniques. 40 (1), 61-66 (2006).
- Thornberry, N. A., et al. A novel heterodimeric cysteine protease is required for interleukin-1βprocessing in monocytes. Nature. 356 (6372), 768-774 (1992).
- Jensen, K., Anderson, J. A., Glass, E. J. Comparison of small interfering RNA (siRNA) delivery into bovine monocyte-derived macrophages by transfection and electroporation. Veterinary Immunology and Immunopathology. 158 (3-4), 224-232 (2014).
- Tada, Y., Sakamoto, M., Fujimura, T. Efficient gene introduction into rice by electroporation and analysis of transgenic plants: use of electroporation buffer lacking chloride ions. Theoretical and Applied Genetics. 80 (4), 475-480 (1990).
- Bhowmik, P., et al. Targeted mutagenesis in wheat microspores using CRISPR/Cas9. Scientific Reports. 8 (1), 6502(2018).
- Sansom, D. M., Manzotti, C. N., Zheng, Y. What's the difference between CD80 and CD86. Trends in Immunology. 24 (6), 314-319 (2003).
- Kurokawa, M., Kornbluth, S. Caspases and kinases in a death grip. Cell. 138 (5), 838-854 (2009).
Access restricted. Please log in or start a trial to view this content.
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoExplore Mais Artigos
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados