
Se requiere una suscripción a JoVE para ver este contenido. Inicie sesión o comience su prueba gratuita.
Method Article
Preparación de aziridinas no activadas enantiopure y síntesis de biemamida B, D y epiallo-isomuscarina
En este artículo
Resumen
En este estudio, preparamos ambos enantiómeros de aziridina-2-carboxilato, que se utilizan en la síntesis asimétrica de alcaloides, incluyendo biemamida B y D, y (-)-epiallo-isomuscarina.
Resumen
Las aziridinas heterociclos que contienen nitrógeno son sintéticamente muy valiosas para la preparación de moléculas azacíclicas y acíclicas. Sin embargo, es muy difícil y laborioso hacer aziridinas en formas ópticamente puras a gran escala para aplicar la síntesis asimétrica de compuestos aza. Afortunadamente, logramos con éxito tanto los enantiómeros (2R)- como (2S)-aziridina-2-carboxilatos con el grupo α-metilbencil donante de electrones en el anillo de nitrógeno como aziridinas no activadas. Estas aziridinas iniciales tienen dos grupos funcionales distintos: anillo de tres miembros altamente reactivo y carboxilato versátil. Son aplicables en la apertura del anillo o la transformación del anillo con aziridina y en la transformación del grupo funcional a otros a partir del carboxilato. Ambos enantiómeros se utilizaron en la preparación de compuestos amino acíclicos y/o aza-heterocíclicos biológicamente importantes de manera asimétrica. Específicamente, este informe describe la primera síntesis asimétrica conveniente de ambos enantiómeros de 5, productos naturales marinos de tipo 6-dihidrouracilo biemamida B y D como posibles inhibidores de TGF-β. Esta síntesis consistió en la reacción de apertura de anillo regio- y estereoselectiva de aziridina-2-carboxilato y la posterior formación de 4-aminoteterahidropirimidina-2,4-diona. Un ejemplo más en este protocolo trató una reacción de Mukaiyama altamente estereoselectiva de aziridina-2-carboxilato y silil enol éter, después de la apertura del anillo intramolecular de aziridina para proporcionar un acceso fácil y fácil a (-)-epiallo-isomuscarina.
Introducción
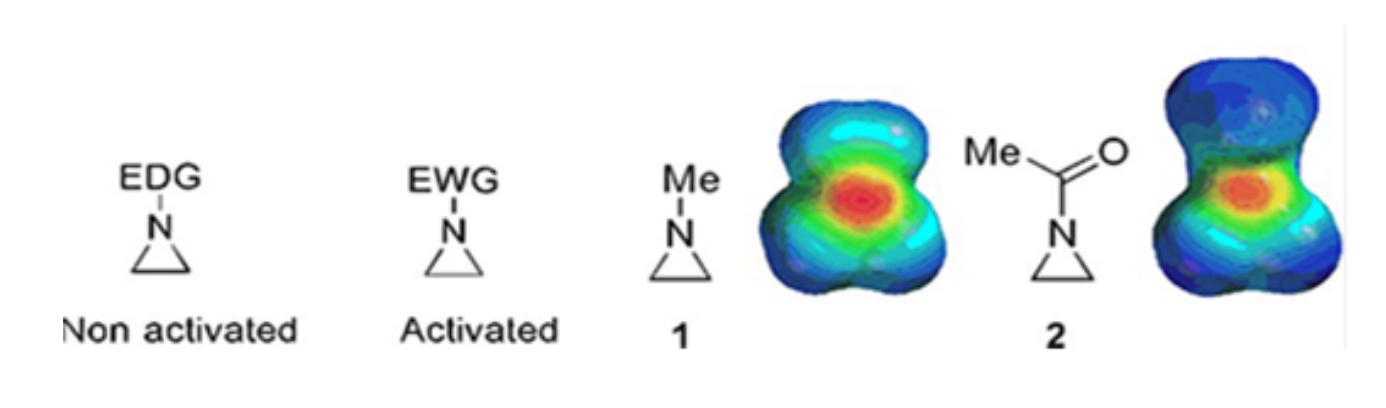
Pequeños anillos que consisten en ciclopropanos, oxiranos y aziridinas se encuentran en varios compuestos como productos naturales y medicamentos 1,2. Se utilizan principalmente como materiales de partida que explotan su cepa de anillo. Entre los compuestos de tres anillos, la aziridina se ha estudiado menos extensamente debido a su inestabilidad y reactividad incontrolable3. Como se muestra en los mapas de potencial electrostático (Figura 1), un grupo unido al anillo de aziridina-nitrógeno, ya sea donante de electrones o atraídor de electrones, hace que la basicidad del nitrógeno sea diferente. Esta diferencia proporciona un contraste sorprendente con la reactividad y selectividad de las aziridinas correspondientes.

Figura 1: Estructuras químicas de aziridinas "activadas" y "no activadas" y mapas de potencial electrostático de sus ejemplos representativos N-metilaziridina y N-acetilaziridina4. Esta cifra ha sido modificada con permiso de Ranjith et al.4. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
Cuando el anillo de nitrógeno tiene un grupo de extracción de electrones, como sulfonato, fosfonato y carbamato, lo llamamos aziridina "activada". Esto es fácilmente reactivo con nucleófilos para compensar su inestabilidad con un alcance limitado de regioquímica. Estas aziridinas activadas se preparan a través de varios métodos catalíticos y se utilizan como material de partida. Gran parte de la química reciente de la aziridina se ha ocupado de estas aziridinas activadas. Sin embargo, las aziridinas activadas sufren ciertas restricciones resultantes de su inestabilidad y alcance de reacción limitado de la apertura del anillo. Por otro lado, las aziridinas que llevan sustituyentes donantes de electrones, como el alquilo o los grupos alquilo sustituidos, en el anillo de nitrógeno llamado "no activado"4, son relativamente estables en la mayoría de las circunstancias y pueden dejarse en el banco durante mucho tiempo sin una descomposición significativa. Las reacciones nucleófilas de apertura de anillo de la aziridina no activada ocurren a través de la formación de iones aziridinio. La mayoría de las reacciones de apertura de anillos de aziridina y transformaciones de anillos proceden de una manera altamente regioquímica. Sin embargo, muy pocos informes de la literatura discuten la preparación de aziridinas no activadas ópticamente puras con sustituyentes en las posiciones C2 o C3 5,6.
Este trabajo muestra la preparación exitosa de α-metilbencil-que contienen aziridina-2-carboxilato quiral como su mezcla diastereomérica, específicamente (-)-mentolilo (-)-feniletilaziridina-2-carboxilatos como su mezcla diastereomérica, a partir de la reacción de 2,3-dibromopropionato y (1R)-feniletilamina. A partir de esta mezcla diastereomérica, se obtuvieron enantiopios (1R)-feniletil-(2R)- y (2S)-aziridina-2-carboxilatos como sus ésteres (-)-mentolilo en formas ópticamente puras mediante recristalización selectiva de MeOH y n-pentano en escalas de varios cien kilos (Figura 1)7. Estos ésteres (-)-mentolílicos se pueden convertir fácilmente en sus ésteres etílicos o metílicos mediante transesterificación en presencia de carbonato de magnesio o potasio7. Estos compuestos también se pueden preparar fácilmente a escala de laboratorio a partir de las reacciones de alquil 2,3-dibromopropionatos o el triflato de vinilo de α-cetoster con 2-feniletilamina quiral seguido de la separación de la mezcla diastereomérica mediante cromatografía simple de columna flash8.
Una vez que tenemos aziridina-2-carboxilato quiral enantiopure, podemos sintetizar varias moléculas diana cíclicas y acíclicas que contienen nitrógeno biológicamente importantes basadas en transformaciones de grupos funcionales de carboxilato y reacciones de apertura de anillos de aziridina altamente regio- y estereoselectivas 6,9,10. La primera síntesis asimétrica conveniente se aplicó tanto para los enantiómeros de 5, 6-dihidrouracilo tipo productos naturales marinos biemamida B y D como potenciales inhibidores de TGF-β11,12. En segundo lugar, la síntesis diastereoselectiva de β-(aziridin-2-yl)-β-hidroxi cetonas se logró mediante la reacción aldol de Mukaiyama de 1-(1-feniletil)-aziridina-2-carboxaldehído ópticamente puro y varios silanos enol en presencia de ZnCl2, en alto rendimiento (>82%) con estereoselectividad casi perfecta (98:2 dr) a través de un estado de transición controlado por quelación. Estos fueron utilizados para la síntesis asimétrica de alcaloides epiallo-isomuscarinos 13,14,15.
Protocolo
1. Síntesis de la mezcla diastereomérica del derivado de aziridina quiral (-)-mentolil éster (1)
- Agregue 2,3-dibromopropano (-)-mentol éster 1a (5,0 g, 13,58 mM, 1,0 equiv) y una barra de agitación magnética en un matraz de fondo redondo de dos cuellos de 250 ml secado al horno bajo atmósfera de nitrógeno (N2).
- Añadir acetonitrilo anhidro (60 ml) al matraz de reacción con una jeringa hermética.
- Luego enfríe la mezcla de reacción a 0 ° C con un baño de hielo y revuelva la mezcla de reacción durante 5 minutos.
- Agregue carbonato de potasio (5.6 g, 40.74 mM, 3.0 equiv) en la mezcla de reacción a la misma temperatura y deje remover durante 30 min.
- Agregue (2R)-feniletilamina (2.0 mL, 16.29 nM, 1.2 equiv) de manera superficial a temperatura ambiente (RT) y permita que la mezcla de reacción se agite durante 12 h.
- Monitorear el progreso de la reacción usando cromatografía de capa delgada usando hexano 9:1 v/v:acetato de etilo (EtOAc; Rf = 0,4) como eluyente.
- Una vez completada la reacción, filtre la mezcla sobre el papel de filtro (tamaño de poro 70 mm).
- Luego, agregue agua (30 ml) en el filtrado orgánico y extraiga la capa orgánica con Et2O (2 x 50 ml) dos veces usando un embudo de separación.
- Secar los extractos orgánicos combinados sobre 7,5 g de Na2SO4 anhidro y concentrarlos en vacío (<15 mbar) utilizando un evaporador rotativo.
NOTA: Ahora se obtiene una mezcla cruda de aziridina quiral diastereomérica que contiene ambos isómeros de (R)-(1R,2 S,5 R)-2-isopropil-5-metilciclohexil1-((R)-1-feniletil)aziridina-2-carboxilato y (S)-(1R,2 S,5 R)-2-isopropil-5-metilciclohexil1-((R)-1-feniletil)aziridina-2-carboxilato (4,1 g, 90%). - Aislamiento de aziridina quiral (R)-(1R,2S,5R)-2-isopropil-5-metilciclohexil 1-((R)-1-feniletil)aziridina-2-carboxilato (2) por un método de cristalización selectiva
- Añadir 8,7 g de la mezcla cruda de aziridina quiral (-)-mentol derivado del éster 1 y disolver en 70 ml de metanol a un matraz de fondo redondo de un solo cuello de 250 ml secado al horno.
- Ahora caliente la mezcla de reacción hasta 70 ° C usando un baño de agua caliente, luego enfríe la mezcla de reacción a -10 ° C hasta que se forme un cristal sólido.
- Filtrar el compuesto sólido sobre un papel de filtro (tamaño de poro 70 mm) para obtener 2,2 g de (R)-(1R,2S,5R)-2-isopropil-5-metilciclohexil 1-((R)-1-feniletil)aziridina-2-carboxilato (2) éster.
- Concentrar la solución filtrada de nuevo en vacuo (<15 mbar) utilizando un evaporador rotativo, disolver 50 mL de etanol en la mezcla de reacción restante y recristalizar a -10 °C para obtener 1,2 g de (R)-(1R,2S,5R)-2-isopropil-5-metilciclohexil 1-((R)-1-feniletil)aziridina-2-carboxilato (2).
- En este momento, use el otro etanol alcohólico de la misma manera que el metanol.
- Después de la recristalización, filtre nuevamente sobre un papel de filtro (tamaño de poro 70 mm), concentre los 5,3 g crudos restantes de solución de filtrado completamente en vacío (<15 mbar) utilizando un evaporador rotativo y agregue 50 ml de un disolvente de hidrocarburo pentano.
- Mantenga la solución de reacción restante a -15 °C.
NOTA: Ahora se obtiene un compuesto sólido de casi 1,9 g de (S)-(1R,2S,5R)-2-isopropil-5-metilciclohexil1-((R)-1-feniletil)aziridina-2-carboxilato éster (3). - Después de obtener los cristales, concentre la solución nuevamente en vacuo (<15 mbar) utilizando un evaporador rotativo y disuelva en 30 ml de disolvente de hidrocarburo de pentano.
- Recristalizar de nuevo a -15 °C para obtener 0,8 g de (S)-(1R,2S,5R)-2-isopropil-5-metilciclohexil1-((R)-1-feniletil)aziridina-2-carboxilato éster (2').
- Obtención (R)-etilo 1-((R)-1-feniletil)aziridina-2-carboxilato (3)
- Añadir (R)-(1R,2 S,5 R)-2-isopropil-5-metilciclohexil1-((R)-1-feniletil)aziridina-2-carboxilato (2) (0,167 g, 0,57 mM) y una barra de agitación magnética en un matraz de fondo redondo de dos cuellos seco al horno de 25 ml bajo atmósfera de nitrógeno (N2).
- Agregue 1,8 ml de etanol al matraz de reacción con una jeringa hermética y revuelva a RT.
- Luego agregue carbonato de potasio (0.40 g, 20.28 mmol, 4.0 equiv) y revuelva a RT durante 2 días.
- Monitorear el progreso de la reacción usando cromatografía de capa delgada (eluente, 8:2 v/v, hexano:acetato de etilo (EtOAc), Rf = 0.6).
- Una vez completada la reacción, filtre la mezcla sobre papel de filtro (tamaño de poro 70 mm), luego agregue agua (5 ml) al filtrado orgánico y extraiga la capa orgánica con CH2Cl2 (2 x 15 ml) dos veces utilizando un embudo de separación.
- Secar los extractos orgánicos combinados sobre 3,0 g de Na2SO4 anhidro y concentrarlos en vacío (<15 mbar) utilizando un evaporador rotativo.
- Purificar el producto crudo mediante cromatografía de columna de fase normal en gel de sílice (malla 70-230) para proporcionar un producto puro (R)-etilo 1-((R)-1-feniletil)aziridina-2-carboxilato (3) (950 mg, 88%). Rf (30% EtOAc/hexano = 0,50).
2. Regio y aziridina estereoselectiva apertura del anillo por nucleófilo de azida para la síntesis total de biemamida B y biemamida D
- Síntesis de (R)-etilo 2-azido-3-(((R)-1-feniletil)amino)propanoato (5)
- Transfiera (500 mg, 2,20 mM, 1,0 equiv) de quiral (S)-etilo 1-((R)-1-feniletil)aziridina-2-carboxilato (4) y una barra de agitación magnética en un matraz de fondo redondo de dos cuellos de 50 ml secado al horno en atmósfera abierta.
- Añadir etanol acuoso al 50% (15 ml) a la mezcla de reacción.
- Enfríe la mezcla de reacción a 0 °C y agregue ácido sulfúrico concentrado (36 N) gota a gota para mantener un pH de casi 4.0, y revuelva durante 5 min.
- Agregue azida de sodio (370 mg, 5.70 mM, 2.5 equiv) a 0 ° C y deje que la mezcla de reacción se revuelva durante 10 minutos a la misma temperatura y luego se caliente a RT.
- Luego, agregue AlCl3∙6H2O (55 mg, 0.22 mM, 0.1 equiv) como catalizador al mismo RT y deje remover durante 3 h adicionales.
- Monitorizar el progreso de la reacción mediante cromatografía en capa fina (eluyente, 6:4 v/v, hexano:acetato de etilo (EtOAc), Rf = 0,2).
- Apague la mezcla de reacción con dos porciones de NaHCO 3 saturado de 20ml.
- Luego filtre la mezcla cruda sobre una almohadilla de celita con etanol (2 x 10 ml).
- Concentrar la mezcla de reacción en vacuo (<15 mbar) utilizando un evaporador rotatorio.
- Extraer la capa orgánica con CH2Cl2 (2 x 50 mL) dos veces utilizando un embudo de separación.
- Luego seque la capa orgánica combinada sobre 5.0 g de Na2SO4 anhidro durante 5 min.
- Concentre la capa orgánica cruda en vacío (<15 mbar) utilizando un evaporador rotativo para obtener un producto de azida cruda.
- Purificar el producto crudo con cromatografía de columna de fase normal sobre gel de sílice (malla 70-230) eluyendo con 40% etOAc/hexano (Rf = 0,20) para permitir 490 mg (90% de rendimiento) de (R)-etilo 2-azido-3-((((R)-1-feniletil)amino)propanoato (5) como líquido viscoso.
- Síntesis de (9H-Fluoren-9-il)metil(3-((R)-3-metil-2,4-dioxo-1-((R)-1 feniletil)hexahidro pirimidina-5-il)amino)-3-oxopropil)carbamato (7)
- Transfiera 150 mg de quiral (R)-5-amino-3-metil-1-((R)-1-feniletil) dihidropirimidina-2,4(1H,3H)-diona (6) (150 mg, 0,60 mM, 1,0 equiv) y una barra de agitación magnética en un matraz de fondo redondo de dos cuellos seco de 25 ml secado al horno bajo atmósfera de N2 .
- Añadir CH2Cl2 seco (15,0 ml) al matraz de reacción con una jeringa hermética.
- Luego enfríe la mezcla de reacción a 0 ° C con un baño de hielo y revuelva la mezcla de reacción durante 5 minutos.
- Añadir Fmoc-beta-alanina (377 mg, 1,20 mM, 2,0 equiv) y DIPEA (0,67 mL, 3,64 mM, 6,0 equiv) a 0 °C y remover durante 5 min.
- Añadir EDCI (347 mg, 1,82 mM, 3,0 equiv) y HOBt (165 mg, 1,21 mM, 2,0 equiv) a la mezcla de reacción a 0 °C y dejar remover durante 10 min a la misma temperatura.
- Mantenga la mezcla de reacción en RT y deje remover durante 8 h adicionales.
- Monitoree el progreso de la reacción utilizando cromatografía de capa delgada utilizando (2:8 v/v, hexano:acetato de etilo (EtOAc), Rf = 0,4) como eluyente.
- Apague la mezcla de reacción con agua (10 ml).
- Lave la capa orgánica combinada con salmuera (15 ml), luego extraiga la capa orgánica con CH2Cl2 (2 x 20 ml) dos veces usando un embudo de separación.
- Luego seque la capa orgánica combinada sobre 5.0 g de Na2SO4 anhidro durante 5 min y concéntrela en vacuo (<15 mbar) usando un evaporador rotativo.
- Purificar el producto crudo con cromatografía en columna de fase normal sobre gel de sílice (malla 70-230) eluyendo con 80% etOAc/hexano (Rf = 0,40) para costear 295 mg de (7) (90% de rendimiento).
3. Reacción estereoselectiva de Mukaiyama aldol con aziridina-2-carboxaldehído quiral y su abertura de anillo de aziridina regio y estereoselectiva por hidroxinucléfilo interno para la síntesis total de (-)- epiallo-somuscarina (17)
- Síntesis de (S)-4-hidroxi-4-((R)-1-((R)-1-feniletil)aziridina-2-il)butan-2-ona (12)
- Transfiera (R)-1-((R)-1-feniletil)aziridina-2-carbaldehído (10) (140 mg, 0,8 mM, 1,0 equiv) y una barra de agitación magnética en un matraz de fondo redondo de dos cuellos de 25 mL secado al horno bajo atmósfera de N2 .
- Añadir CH3CN seco (4,0 ml) al matraz de reacción con una jeringa hermética.
- Luego enfríe la mezcla de reacción a -20 ° C con un baño de hielo y acetona y revuelva la mezcla de reacción durante 5 min.
- Añadir ZnCl2 anhidro (108 mg, 0,8 mM, 1,0 equiv) a la mezcla de reacción a -20 °C y dejar remover durante 5 min.
- A continuación, agregue trimetil(prop-1-en-2-iloxi)silano (11) (104 mg, 0,8 mM, 1,0 equiv) disuelto en CH3CN seco (3,0 ml) a la mezcla de reacción a -20 °C de forma superficial y deje que la mezcla de reacción se revuelva durante 1 h a la misma temperatura.
- Monitoree el progreso de la reacción utilizando cromatografía de capa delgada utilizando hexano 8: 2 v / v: acetato de etilo (EtOAc), Rf = 0.2) como eluyente.
- Apague la mezcla de reacción con NaHCO3 saturado (4 ml).
- Extraiga la capa orgánica con EtOAc (2 x 15 mL) dos veces utilizando un embudo de separación.
- Secar la capa orgánica combinada con 3,0 g de Na2SO4 anhidro y concentrarla en vacío (<15 mbar) utilizando un evaporador rotativo.
- Purificar el producto crudo con cromatografía de columna de fase normal sobre gel de sílice (malla 70-230) eluyendo con 80% etOAc/hexano, (Rf = 0,20) para permitir 58 mg de (S)-4-hidroxi-4-((R)-1-((R)-1-feniletil)aziridina-2-il)butan-2-ona) (12) (85% de rendimiento).
- Síntesis de (R)-N-(((2R,3 S,5 R)-3-((tert-butyldimethylsilyl)oxy)-5-methyltetra hydrofuran-2-yl)methyl)-1-phenylethanamine (15)
- Transfiera (S)-4-((tert-butyldimethylsilyl)oxy)-4-((R)-1-((R)-1-phenylethyl) aziridin-2-yl)butan-2-one (13) (400 mg, 1.15 mM, 1.0 equiv) y una barra de agitación magnética en un matraz de fondo redondo de dos cuellos seco de 25 mL secado al horno bajo atmósfera de N2 .
- Añadir THF anhidro (50 ml) al matraz de reacción con una jeringa hermética.
- Enfríe la mezcla de reacción a -78 °C con un baño seco de hielo y acetona y deje remover durante 5 min.
- Luego agregue litio tri-sec-butilborohidruro (L-selectride) (solución de 1 M en THF) (2.3 mL, 2.0 equiv) gota a gota en la mezcla de reacción a -78 ° C y deje remover durante otros 25 min.
- Caliente la mezcla de reacción a RT y deje remover durante 8 h.
- Monitorizar el progreso de la reacción por TLC utilizando EtOAc/hexano al 30% (Rf = 0,40) como eluyente.
- Después del consumo completo del compuesto 13, apague la mezcla de reacción con 0,1 M de NaOH (5 ml).
- Extraiga la capa orgánica con EtOAc (3 x 15 ml), luego lave con salmuera (15 ml).
- Secar la capa orgánica sobre 3,0 g de Na2SO4 anhidro y concentrar en vacío (<15 mbar) utilizando un evaporador rotativo.
- Purificar el producto crudo con cromatografía de columna de fase normal sobre gel de sílice (malla 70-230, eluyente, 8:2 v/v, hexano:acetato de etilo (EtOAc), Rf = 0,2) para proporcionar un compuesto puro (15) (382 mg, 95% de rendimiento).
- Síntesis de yoduro (-)-epiallo-isomuscarina (17)
- Transfiera el compuesto 16 (20 mg, 0.15 mM, 1.0 equiv) y una barra de agitación magnética a un matraz de fondo redondo de 10 ml secado al horno bajo atmósfera N2 .
- Añadir 3 ml de matraz de reacción EtOAc con una jeringa hermética.
- A continuación, agregue yoduro de metilo (0,4 ml, 3,0 mM) a la mezcla de reacción en RT.
- Añadir 1,2,2,6,6-pentametilpiperidina (PMP) (0,05 mL, 0,3 mM, 2,0 equiv) a la mezcla de reacción a 0 °C.
- Luego, mantenga la mezcla de reacción en RT y deje remover durante 16 h adicionales.
- Evaporar el disolvente en vacío (<15 mbar) utilizando un evaporador rotativo para dar el producto crudo.
- Lavar tres veces añadiendo (3 x 5 ml) 10% de MeOH en EtOAc a la mezcla de reacción cruda.
- Luego lave la mezcla cruda con n-pentano (5 ml) y concéntrese al vacío (<15 mbar) utilizando un evaporador rotativo para lograr yoduro puro (-)-epiallo-isomuscarina (17) (32 mg, 68%).
4. Caracterización de todos los productos
- Caracterizar todos los nuevos compuestos medianteespectroscopia de RMN de 1 H, 13C y espectrometría de masas de alta resolución (HRMS)7,8,11.
Resultados
Aquí, informamos la síntesis de aziridina-2-carboxilatos enantiopure. La mezcla diastereomérica de (R)-(1R,2 S,5 R)-2-isopropil-5-metilciclohexil1-((R)-1-feniletil)aziridina-2-carboxilato (2) y (S)-(1R,2S,5R)-2-isopropil-5-metilciclohexil1-((R)-1-feniletil)aziridina-2-carboxilato (3) (4,1 g, 90%) se prepararon en rendimiento cuantitativo a partir de 2,3 -dibromopropano (-)-éster de mentoloilo y (1R)-feniletilamin...
Discusión
Las aziridinas como heterociclos de tres miembros que contienen nitrógeno tienen un enorme potencial para que los marciales o intermedios sintéticos de inicio preparen moléculas orgánicas ricas en nitrógeno. Según el grupo que contiene nitrógeno en el anillo, se clasifican como aziridinas "activadas" y "no activadas" cuya reactividad química y selectividad son diferentes. Sin embargo, hay métodos muy limitados disponibles para preparar esta valiosa aziridina en una forma ópticamente activa.
Divulgaciones
Los autores declaran que no hubo conflicto de intereses en este estudio.
Agradecimientos
Esta investigación fue apoyada por la Fundación Nacional de Investigación de Corea (NRF-2020R1A2C1007102 y 2021R1A5A6002803) con el Centro de Nuevas Direcciones en Síntesis Orgánica y una Subvención HUFS 2022.
Materiales
| Name | Company | Catalog Number | Comments |
| (2R)-1-[(1R)-1-Phenylethyl]-2-aziridinecarboxylic acid (-)-menthol ester, 98% | Sigma-Aldrich | 57054-0 | |
| (2S)-1-[(1R)-1-Phenylethyl]-2-aziridinecarboxylic acid (-)-menthol ester | Sigma-Aldrich | 57051-6 | |
| 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride | TCI | 424331-25 g | CAS No: 25952-53-8 |
| 1,4-Dioxane | SAMCHUN | D0654-1 kg | CAS No: 123-91-1 |
| 1-Hydroxybenzotriazole hydrate | Aldrich | 219-989-7-50 g | CAS No: 123333-53-9 |
| 2,6-Lutidine | Alfa Aesar | A10478-AP, 500 mL | CAS No: 108-48-5 |
| Acetonitrile | SAMCHUN | A0127-18 L | CAS No: 75-05-8 |
| Acetonitrile-d3 | Cambridge Isotope Laboratories, | 15G-744-25 g | CAS No: 2206-26-0 |
| Aluminum chloride hexahydrate | Aldrich | 231-208-1, 500 g | CAS No : 7784-13-6 |
| Bruker AVANCE III HD (400 MHz) spectrometer | Bruker | NA | |
| Chloroform-d | Cambridge Isotope Laboratories, | 100 g | CAS No: 865-49-6 |
| Dichloromethane | SAMCHUN | M0822-18 L | CAS No: 75-09-2 |
| Dimethyl sulfoxide-d6 | Cambridge Isotope Laboratories, | 25 g | CAS No: 2206-27-1 |
| Ethanol | EMSURE | 1009831000,1L | CAS No: 64-17-5 |
| Ethyl acetate | SAMCHUN | E0191-18 L | CAS No: 141-78-6 |
| High resolution mass spectra/MALDI-TOF/TOF Mass Spectrometry | AB SCIEX | 4800 Plus | High resolution mass spectra |
| JASCO P-2000 | JASCO | P-2000 | For optical rotation |
| Lithium aluminum hydride | TCI | L0203-100 g | CAS No: 16853-85-3 |
| L-Selectride, 1 M solution in THF | Acros | 176451000, 100 mL | CAS No: 38721-52-7 |
| Methanol | SAMCHUN | M0585-18 L | CAS No: 67-56-1 |
| N-[(9H-Fluoren-9-ylmethoxy)carbonyl]-β-alanine | TCI | F08825G-5 g | CAS No: 35737-10-1 |
| N-Ethyldiisopropylamine | Aldrich | 230-392-0, 100 mL | CAS No: 7087-68-5 |
| n-Hexane | SAMCHUN | H0114-18 L | CAS No: 110-54-3 |
| Ninhydrin | Alfa Aesar | A10409-250 g | CAS No: 485-47-2 |
| p-Anisaldehyde | aldrich | A88107-5 g | CAS No: 123-11-5 |
| Phosphomolybdic acid hydrate | TCI | P1910-100 g | CAS No: 51429-74-4 |
| Sodium azide | D.S.P | 703301-500 g | CAS No: 26628-22-8 |
| Sodium Hydride 60% dispersion in mineral oil | Sigma-Aldrich | 452912-100 G | CAS No: 7646-69-7 |
| Sodium hydroxide | DUKSAN | A31226-1 kg | CAS No: 1310-73-2 |
| Sodium sulfate | SAMCHUN | S1011-1 kg | CAS No: 7757-82-6 |
| Thin Layer Chromatography (TLC) | Merck | 100390 | |
| Tert-Butyldimethylsilyl trifluoromethanesulfonate, 98% | Aldrich | 274-102-0, 25 g | CAS NO: 69739-34-0 |
| Tetrahydrofuran | SAMCHUN | T0148-18 L | CAS No: 109-99-9 |
| Triethylethylamine | DAEJUNG | 8556-4400-1 L | CAS No: 121-44-8 |
| UV light | Korea Ace Sci | TN-4C | 254 nm |
| Zinc chloride, anhydrous, 98+% | Alfa Aesar | A16281-22100 g | CAS No : 7646-85-7 |
Referencias
- Pitzer, K. S. Strain energies of cyclic hydrocarbons. Science. 101 (2635), 672 (1945).
- Dudev, T., Lim, C. Ring strain energies from ab initio calculations. Journal of the American Chemical Society. 120 (18), 4450-4458 (1998).
- D'hooghe, M., Ha, H. -. J. . Synthesis of 4- to 7-Membered Heterocycles by Ring Expansion. , (2016).
- Ranjith, J., Ha, H. -. J. Synthetic applications of aziridinium ions. Molecules. 26 (6), 1744 (2021).
- Sweeney, J. B. Aziridines: epoxides' ugly cousins. Chemical Society Reviews. 31 (5), 247-258 (2002).
- Stankovic, S., et al. Regioselectivity in the ring opening of non-activated aziridines. Chemical Society Reviews. 41 (2), 643-665 (2012).
- Lee, W. K., Ha, H. -. J. Highlights of the chemistry of enantiomerically pure aziridine-2-carboxylates. Aldrichimica Acta. 36 (2), 57-63 (2003).
- Tranchant, M. J., Dalla, V., Jabin, I., Decroix, B. Reaction of vinyl triflates of α-keto esters with primary amines: efficient synthesis of aziridine carboxylates. Tetrahedron. 58 (42), 8425-8432 (2002).
- Ha, H. -. J., Jung, J. -. H., Lee, W. K. Application of regio- and stereoselective functional group transformation of chiral aziridine-2-carboxylate. Asian Journal of Organic Chemistry. 3 (10), 1020-1035 (2014).
- Kim, Y., et al. Preparation of 2,3-diaminopropionate from ring opening of aziridine-2-carboxylate. Tetrahedron Letters. 46 (25), 4407-4409 (2005).
- Srivastava, N., Macha, L., Ha, H. -. J. Total synthesis and stereochemical revision of biemamides B and D. Organic Letters. 21 (22), 8992-8996 (2019).
- Zhang, F., et al. Biemamides A-E, inhibitors of the TGF-β pathway that block the epithelial to mesenchymal transition. Organic Letters. 20 (18), 5529-5532 (2018).
- Srivastava, N., Ha, H. -. J. Highly efficient and stereoselective Mukaiyama Aldol reaction with chiral aziridine-2-carboxaldehyde and its synthetic applications. Asian Journal of Organic Chemistry. 11 (1), 2021005671 (2021).
- Kempter, I., et al. Synthesis and structural characterization of the isomuscarines. Tetrahedron. 70 (10), 1918-1927 (2014).
- Pirrrung, M. C., DeAmicis, C. V. Total synthesis of the muscarines. Tetrahedron Letters. 29 (2), 159-162 (1988).
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoExplorar más artículos
This article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados