Для просмотра этого контента требуется подписка на Jove Войдите в систему или начните бесплатную пробную версию.
Method Article
Получение энантиопурных неактивированных азиридинов и синтез биемамида B, D и эпиалло-изомускарина
В этой статье
Резюме
В этом исследовании мы получаем как энантиомеры азиридина-2-карбоксилата, которые используются в асимметричном синтезе алкалоидов, включая биемамид B и D, так и (-)-эпиалло-изомускарин.
Аннотация
Азотсодержащие гетероцикловые азиридины синтетически очень ценны для получения азациклических и ациклических молекул. Однако очень трудно и трудоемко производить азиридины в оптически чистых формах в больших масштабах для применения асимметричного синтеза аза-соединений. К счастью, мы успешно достигли как энантиомеров (2R)-, так и (2S)-азиридин-2-карбоксилатов с электрон-донорной α-метилбензиловой группой на кольцевом азоте в качестве неактивированных азиридинов. Эти исходные азиридины имеют две различные функциональные группы - высокореактивное трехчленное кольцо и универсальный карбоксилат. Они применимы при кольцевом раскрытии или кольцевом превращении с азиридином и в функциональном групповом превращении в другие из карбоксилата. Оба этих энантиомера использовали при получении биологически важных аминоациклических и/или аза-гетероциклических соединений асимметричным образом. В частности, в настоящем отчете описывается первый целесообразный асимметричный синтез обоих энантиомеров 5, 6-дигидроурациловых морских натуральных продуктов биемамида B и D в качестве потенциальных ингибиторов TGF-β. Этот синтез состоял из регио- и стереоселективной реакции открытия кольца азиридина-2-карбоксилата и последующего образования 4-аминотетерагидропиримидина-2,4-диона. Еще один пример в этом протоколе касался высокостимаселективной реакции Мукайяма азиридина-2-карбоксилата и силиленолового эфира после внутримолекулярного открытия кольца азиридина для обеспечения легкого и легкого доступа к (-)-эпиалло-изомускарину.
Введение
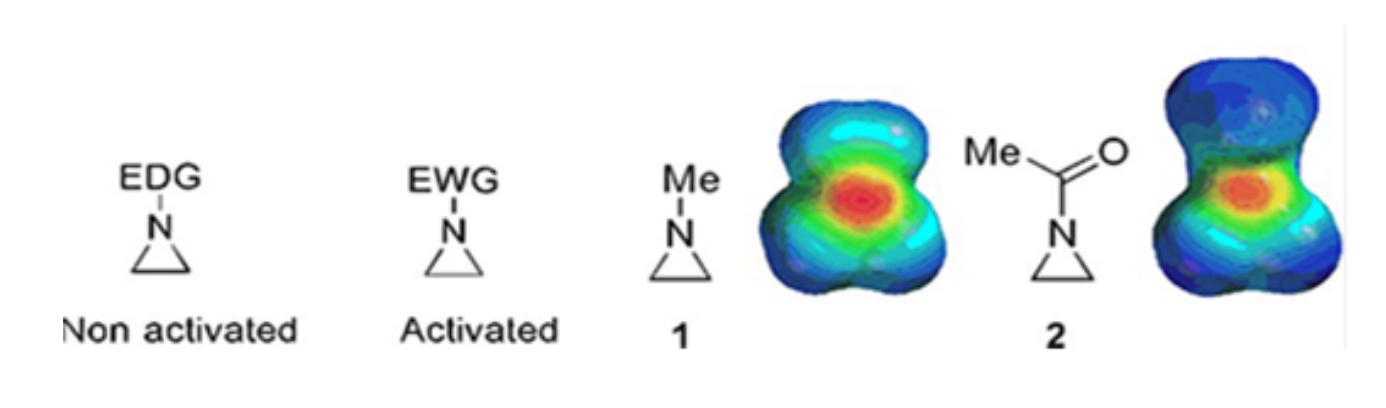
Небольшие кольца, состоящие из циклопропанов, оксиранов и азиридинов, содержатся в различных соединениях, таких как натуральные продукты и препараты 1,2. Они в основном используются в качестве исходных материалов, использующих их кольцевую деформацию. Среди трехкольцевых соединений азиридин изучен менее широко из-за его нестабильности и неконтролируемой реакционной способности3. Как показано на картах электростатических потенциалов (рисунок 1), группа, присоединенная к азиридиновому кольцу-азоту, будь то донорство электронов или притяжение электронов, делает основу азота различной. Это различие обеспечивает разительный контраст с реактивностью и селективностью соответствующих азиридинов.

Рисунок 1: Химические структуры «активированных» и «неактивированных» азиридинов и карты электростатических потенциалов их репрезентативных примеров N-метилазиридина и N-ацетилазиридина4. Эта цифра была изменена с разрешения Ranjith et al.4. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
{kind=link}
Когда кольцевой азот имеет электрон-выводящую группу, такую как сульфонат, фосфонат и карбамат, мы называем его «активированным» азиридином. Это легко реагирует с нуклеофилами, чтобы компенсировать его нестабильность с ограниченным объемом региохимии. Эти активированные азиридины получают различными каталитическими методами и используют в качестве исходного материала. Большая часть недавней химии азиридина имела дело с этими активированными азиридинами. Однако активированные азиридины страдают от определенных ограничений, обусловленных их нестабильностью и ограниченным объемом реакции на открытие кольца. С другой стороны, азиридины, несущие электрон-донорные заместители, такие как алкильные или замещенные алкильные группы, в кольцевом азоте, называемом «неактивированным»4, относительно стабильны в большинстве случаев и могут оставаться на стенде в течение длительного времени без значительного разложения. Нуклеофильные реакции открытия кольца неактивированного азиридина происходят путем образования ионов азиридиния. Большинство реакций открытия кольца азиридина и превращений колец протекают высокорегиохимическим образом. Однако очень немногие литературные отчеты обсуждают получение оптически чистых неактивированных азиридинов с заместителями в позициях С2 или С3 5,6.
В данной работе показано успешное получение α-метилбензилгрупп-содержащих хирал азиридин-2-карбоксилат производных, в частности (-)-ментолил(1R)-фенилэтилазиридин-2-карбоксилатов в качестве его диастереомерной смеси, из реакции 2,3-дибромпропионата и (1R)-фенилэтиламина. Из этой диастереомерной смеси энантиопура (1R)-фенилэтил-(2R)- и (2S)-азиридин-2-карбоксилаты в качестве их (-)-ментолиловых эфиров были получены в оптически чистых формах путем селективной рекристаллизации из MeOH и n-пентана на многосоткилометровых шкалах (рисунок 1)7. Эти (-)-ментолиловые эфиры могут быть легко преобразованы в их этиловые или метиловые эфиры путем переэтерификации в присутствии карбоната магния или калия7. Эти соединения также могут быть легко получены в лабораторных масштабах из реакций алкил-2,3-дибромпропионатов или винилэрифлата α-кетоэстера с хиральным 2-фенилэтиламином с последующим разделением диастереомерной смеси с помощью простой флэш-колоночной хроматографии8.
Получив энантиопуру хиральный азиридин-2-карбоксилат, можно синтезировать различные циклические и ациклические азотсодержащие биологически важные молекулы-мишени на основе функциональных групповых превращений карбоксилата и высокорегио- и стереоселективных реакций открытия азиридинового кольца 6,9,10. Первый целесообразный асимметричный синтез был применен для обоих энантиомеров 5, 6-дигидроурациловых морских натуральных продуктов биемамида В и D в качестве потенциальных ингибиторов TGF-β11,12. Во-вторых, диастереозелективный синтез β-(азиридин-2-ил)-β-гидроксикетонов был достигнут реакцией альдола Мукайяма оптически чистого 1-(1-фенилэтил)-азиридина-2-карбоксальдегида и различных еноловых силанов в присутствииZnCl2, в высоком выходе (>82%) с почти идеальной стереоселективностью (98:2 др) через хелатно-контролируемое переходное состояние. Они использовались для асимметричного синтеза эпиалло-изомускариновых алкалоидов 13,14,15.
протокол
1. Синтез диастереомерной смеси производного хирального азиридина (-)-ментолового эфира (1)
- Добавьте 2,3-дибромпропан (-)-ментоловый эфир 1a (5,0 г, 13,58 мМ, 1,0 экв) и магнитный перемешивающий стержень в высушенную в духовке 250 мл двухгорловую колбу с круглым дном в атмосфере азота (N2).
- Добавьте безводный ацетонитрил (60 мл) в реакционную колбу с помощью герметичного шприца.
- Затем охладите реакционную смесь при 0 °C с помощью ледяной ванны и перемешайте реакционную смесь в течение 5 мин.
- Добавить карбонат калия (5,6 г, 40,74 мМ, 3,0 экв) в реакционную смесь при той же температуре и дать помешивать в течение 30 мин.
- Добавить (2R)-фенилэтиламина (2,0 мл, 16,29 нМ, 1,2 эквив) по каплям при комнатной температуре (RT) и дать реакционной смеси перемешиваться в течение 12 ч.
- Мониторинг хода реакции с помощью тонкослойной хроматографии с использованием 9:1 v/v гексан:этилацетат (EtOAc; Rf = 0,4) в качестве элюента.
- После завершения реакции процедите смесь по фильтровальной бумаге (размер пор 70 мм).
- Затем добавьте воду (30 мл) в органический фильтрат и экстрагируйте органический слой Et2O (2 х 50 мл) два раза, используя разделительную воронку.
- Сушат комбинированные органические экстракты более 7,5 г безводного Na2SO4 и концентрируют в вакууме (<15 мбар) с помощью ротационного испарителя.
ПРИМЕЧАНИЕ: В настоящее время получена сырая смесь диастереомерного хирального азиридина, содержащая оба изомера (R)-(1R,2 S,5 R)-2-изопропил-5-метилциклогексил1-((R)-1-фенилэтил)азиридин-2-карбоксилат и (S)-(1R,2 S,5 R)-2-изопропил-5-метилциклогексил1-((R)-1-фенилэтил)азиридин-2-карбоксилат (4,1 г, 90%). - Выделение хирального азиридина (R)-(1R,2S,5R)-2-изопропил-5-метилциклогексил-1-(R)-1-фенилэтил)азиридина-2-карбоксилата (2) методом селективной кристаллизации
- Добавить 8,7 г сырой смеси производного хирального азиридина (-)-ментолового эфира 1 и растворить в 70 мл метанола в высушенную в духовке колбу с одним горловиной круглого дна объемом 250 мл.
- Теперь разогрейте реакционную смесь до 70 °C с помощью ванны с горячей водой, затем охладите реакционную смесь при -10 °C до образования твердого кристалла.
- Фильтруют твердое соединение над фильтровальной бумагой (размер пор 70 мм) для получения 2,2 г (R)-(1R,2S,5R)-2-изопропил-5-метилциклогексил 1-(R)-1-фенилэтил)эфира азиридина-2-карбоксилата(2).
- Снова концентрируют раствор фильтрата в вакууме (<15 мбар) с помощью роторного испарителя, растворяют 50 мл этанола в оставшейся реакционной смеси и перекристаллизуют при -10 °C с получением 1,2 г (R)-(1R,2S,5R)-2-изопропил-5-метилциклогексил-1-(R)-1-фенилэтил)азиридина-2-карбоксилата (2).
- В это время используют другой спиртовой этанол так же, как и метанол.
- После рекристаллизации снова процеживают фильтровальную бумагу (размер пор 70 мм), концентрируют оставшиеся сырые 5,3 г раствора фильтрата полностью в вакууме (<15 мбар) с помощью ротационного испарителя и добавляют 50 мл пентанового углеводородного растворителя.
- Оставшийся реакционный раствор держать при -15 °C.
ПРИМЕЧАНИЕ: В настоящее время получено твердое соединение почти 1,9 г (S)-(1R,2S,5R)-2-изопропил-5-метилциклогексил1-((R)-1-фенилэтил)азиридин-2-карбоксилатный эфир (3). - После получения кристаллов снова концентрируют раствор в вакууме (<15 мбар) с помощью ротационного испарителя и растворяют его в 30 мл пентанового углеводородного растворителя.
- Повторно перекристаллизуют при -15 °C с получением 0,8 г (S)-(1R,2S,5R)-2-изопропил-5-метилциклогексил1-((R)-1-фенилэтил)азиридина-2-карбоксилатного эфира (2').
- Получение (R)-этил1-((R)-1-фенилэтил)азиридина-2-карбоксилата (3)
- Добавьте (R)-(1R,2 S,5 R)-2-изопропил-5-метилциклогексил1-((R)-1-фенилэтил)азиридин-2-карбоксилат (2) (0,167 г, 0,57 мМ) и магнитный перемешиватель в высушенную в печи 25 мл двухшейную колбу с круглым дном в атмосфере азота (N2).
- Добавьте 1,8 мл этанола в реакционную колбу с помощью герметичного шприца и перемешайте его при РТ.
- Затем добавляют карбонат калия (0,40 г, 20,28 ммоль, 4,0 эквав) и перемешивают его при РТ в течение 2 дней.
- Контролируют ход реакции с помощью тонкослойной хроматографии (элюент, 8:2 об/об, гексан:этилацетат (EtOAc), Rf =0,6).
- После завершения реакции фильтруют смесь на фильтровальной бумаге (размер пор 70 мм), затем добавляют воду (5 мл) в органический фильтрат и экстрагируют органический слой с CH2Cl2 (2 x 15 мл) два раза с помощью разделительной воронки.
- Сушат комбинированные органические экстракты более 3,0 г безводного Na2SO4 и концентрируют в вакууме (<15 мбар) с помощью ротационного испарителя.
- Очищают сырой продукт с помощью обычной фазовой колоночной хроматографии на силикагеле (70-230 меш), чтобы получить чистый продукт (R)-этил-1-(R)-1-фенилэтил)азиридин-2-карбоксилат (3) (950 мг, 88%). Rf (30% EtOAc/гексан = 0,50).
2. Регио и стереоселективное кольцо азиридина-открытие азидным нуклеофилом для общего синтеза биемамида В и биемамида D
- Синтез (R)-этил2-азидо-3-(((R)-1-фенилэтил)амино)пропаноата (5)
- Перенос (500 мг, 2,20 мМ, 1,0 экв) хирального (S)-этил-1-((R)-1-фенилэтил)азиридина-2-карбоксилата (4) и магнитного перемешивателя в высушенную в духовке 50 мл двухшейную колбу круглого дна в открытой атмосфере.
- Добавьте 50% водного этанола (15 мл) в реакционную смесь.
- Охладите реакционную смесь при 0 °C и добавьте концентрированную серную кислоту (36 Н) по каплям для поддержания почти рН 4,0 и перемешайте в течение 5 мин.
- Добавьте азид натрия (370 мг, 5,70 мМ, 2,5 эквив) при 0 °C и дайте реакционной смеси перемешиваться в течение 10 мин при той же температуре, а затем нагреваться при РТ.
- Затем добавляют AlCl3∙6H2O (55 мг, 0,22 мМ, 0,1 эквив) в качестве катализатора на том же RT и дают перемешивать еще 3 ч.
- Контролируют ход реакции с помощью тонкослойной хроматографии (элюент, 6:4 об/об, гексан:этилацетат (EtOAc), Rf =0,2).
- Гасить реакционную смесь двумя порциями по 20 мл, насыщенными NaHCO3.
- Затем процедите сырую смесь на целиитовой прокладке с этанолом (2 х 10 мл).
- Концентрируйте реакционную смесь в вакууме (<15 мбар) с помощью роторного испарителя.
- Экстрагируйте органический слой с CH2Cl2 (2 x 50 мл) два раза, используя разделительную воронку.
- Затем высушите комбинированным органическим слоем более 5,0 г безводного Na2SO4 в течение 5 мин.
- Сконцентрируйте сырой органический слой в вакууме (<15 мбар) с помощью роторного испарителя для получения сырого азидного продукта.
- Очищают сырой продукт с помощью нормальной фазовой колоночной хроматографии на силикагеле (70-230 меш) путем элюирования 40% EtOAc/гексаном (Rf = 0,20), чтобы получить 490 мг (выход 90%) (R)-этил-2-азидо-3-(((R)-1-фенилэтил)амино)пропаноата (5) в виде вязкой жидкости.
- Синтез (9Н-фтор-9-ил)метил(3-((((R)-3-метил-2,4-диоксо-1-((R)-1 фенилэтил)гексагидропиримидин-5-ил)амино)-3-оксопропил)карбамата (7)
- Перенесите 150 мг хирального (R)-5-амино-3-метил-1-((R)-1-фенилэтил)дигидропиримидина-2,4(1H,3H)-диона (6) (150 мг, 0,60 мМ, 1,0 экв) и магнитного перемешивателя в высушенную в духовке двухшейную колбу с круглым дном объемом 25 мл под атмосферойN2 .
- Добавьте сухой CH2Cl2 (15,0 мл) в реакционную колбу с помощью герметичного шприца.
- Затем охладите реакционную смесь при 0 °C с помощью ледяной ванны и перемешайте реакционную смесь в течение 5 мин.
- Добавьте Fmoc-бета-аланин (377 мг, 1,20 мМ, 2,0 экв) и DIPEA (0,67 мл, 3,64 мМ, 6,0 экв) при 0 °C и перемешайте в течение 5 мин.
- Добавьте EDCI (347 мг, 1,82 мМ, 3,0 эквив) и HOBt (165 мг, 1,21 мМ, 2,0 экв) в реакционную смесь при 0 °C и дайте перемешивать в течение 10 мин при той же температуре.
- Держите реакционную смесь на RT и дайте помешиваться еще 8 ч.
- Контролируют ход реакции с помощью тонкослойной хроматографии с использованием (2:8 v/v, гексан:этилацетат (EtOAc), Rf = 0,4) в качестве элюента.
- Гасить реакционную смесь водой (10 мл).
- Промыть комбинированный органический слой рассолом (15 мл), затем дважды экстрагировать органический слой с CH2Cl2 (2 x 20 мл) с помощью разделительной воронки.
- Затем высушите комбинированный органический слой более 5,0 г безводного Na2SO4 в течение 5 мин и сконцентрируйте его в вакууме (<15 мбар) с помощью ротационного испарителя.
- Очистите сырой продукт с помощью нормальной фазовой колоночной хроматографии на силикагеле (70-230 меш) путем элюирования 80% EtOAc/гексана (Rf = 0,40), чтобы получить 295 мг (7) (90% выхода).
3. Стереоселективная альдольная реакция Мукайяма с хиральным азиридином-2-карбоксальдегидом и его регио и стереоселективное азиридиновое кольцо- открытие внутренним гидроксинуклеофилом для полного синтеза (-)- эпиалло-сомускарина (17)
- Синтез (S)-4-гидрокси-4-((R)-1-((R)-1-фенилэтил)азиридин-2-ил)бутана-2-он (12)
- Перенесите (R)-1-((R)-1-фенилэтил)азиридин-2-карбальдегид (10) (140 мг, 0,8 мМ, 1,0 экв)) и магнитный перемешину в высушенную в духовке 25 мл двухшейную колбу круглого дна подN2 атмосферы.
- Добавьте сухой CH3CN (4,0 мл) в реакционную колбу с помощью герметичного шприца.
- Затем охладите реакционную смесь при -20 °C с помощью ацетоновой ванны со льдом и перемешайте реакционную смесь в течение 5 мин.
- ДобавьтеZnCl2 безводный (108 мг, 0,8 мМ, 1,0 экв) в реакционную смесь при -20 °C и дайте помешивать в течение 5 мин.
- Затем добавляют триметил(проп-1-ен-2-илокси)силан (11) (104 мг, 0,8 мМ, 1,0 экв), растворенный в сухом CH3CN (3,0 мл), в реакционную смесь при -20 °C по каплям и дают реакционной смеси перемешиваться в течение 1 ч при той же температуре.
- Контролируют ход реакции с помощью тонкослойной хроматографии с использованием 8:2 v/v гексан:этилацетат (EtOAc), Rf = 0,2) в качестве элюента.
- Гасить реакционную смесь насыщенным NaHCO3 (4 мл).
- Извлеките органический слой с помощью EtOAc (2 x 15 мл) два раза, используя разделительную воронку.
- Высушите комбинированный органический слой 3,0 г безводного Na2SO4 и сконцентрируйте его в вакууме (<15 мбар) с помощью ротационного испарителя.
- Очищают сырой продукт с помощью нормальной фазовой колоночной хроматографии на силикагеле (70-230 меш) путем элюирования 80% EtOAc/гексана, (Rf = 0,20) для получения 58 мг (S)-4-гидрокси-4-((R)-1-((R)-1-фенилэтил)азиридина-2-ил)бутана-2-она) (12) (85% выхода).
- Синтез (R)-N-(((2R,3 S,5 R)-3-((трет-бутилдиметилсилил)окси)-5-метилтетра гидрофуран-2-ил)метил)-1-фенилетанамин (15)
- Перенос (S)-4-((трет-бутилдиметилсилил)окси)-4-((R)-1-((R)-1-фенилэтил)азиридин-2-ил)бутан-2-он(13)(400 мг, 1,15 мМ, 1,0 эквив) и магнитного перемешивателя в высушенную в печи двухшейную колбу круглого дна объемом 25 мл подN2 атмосферы.
- Добавьте безводный ТГФ (50 мл) в реакционную колбу с помощью герметичного шприца.
- Охладите реакционную смесь при -78 °C с помощью сухих льдо-ацетоновых ванн и дайте помешиваться в течение 5 мин.
- Затем добавляют три-сек-бутилборогидрид лития (L-селектрид) (1 М раствор в ТГФ) (2,3 мл, 2,0 экв) по каплям в реакционную смесь при -78 °С и дают перемешивать еще 25 мин.
- Разогреть реакционную смесь до RT и дать помешивать в течение 8 ч.
- Контролируют ход реакции с помощью TLC с использованием 30% EtOAc/гексана (Rf = 0,40) в качестве элюента.
- После полного расхода соединения 13 закаляют реакционную смесь 0,1 М NaOH (5 мл).
- Экстрагировать органический слой EtOAc (3 х 15 мл), затем промыть рассолом (15 мл).
- Высушите органический слой более 3,0 г безводного Na2SO4 и сконцентрируйте в вакууме (<15 мбар) с помощью роторного испарителя.
- Очистите сырой продукт с помощью нормальной фазовой колонковой хроматографии на силикагеле (70-230 меш, элюент, 8:2 v/v, гексан:этилацетат (EtOAc), Rf = 0,2), чтобы получить чистое соединение (15) (382 мг, выход 95%).
- Синтез (-)-эпиалло-изомускарина йодида (17)
- Перенесите соединение 16 (20 мг, 0,15 мМ, 1,0 экв) и магнитный перемешивание в высушенную в духовке колбу круглого дна объемом 10 мл под атмосферойN2 .
- Добавьте 3 мл реакционной колбы EtOAc с помощью герметичного шприца.
- Затем добавляют метилйодид (0,4 мл, 3,0 мМ) в реакционную смесь на РТ.
- Добавить 1,2,2,6,6-пентаметилпиперидин (ПМФ) (0,05 мл, 0,3 мМ, 2,0 экв) в реакционную смесь при 0°С.
- Затем держите реакционную смесь при RT и дайте помешивать еще 16 ч.
- Выпаривают растворитель в вакууме (<15 мбар) с помощью роторного испарителя для получения сырого продукта.
- Промыть три раза, добавив (3 х 5 мл) 10% MeOH в EtOAc к сырой реакционной смеси.
- Затем промыть сырую смесь н-пентаном (5 мл) и сконцентрировать в вакууме (<15 мбар) с помощью ротационного испарителя для получения чистого (-)-эпиалло-изомускарина йодида (17) (32 мг, 68%).
4. Характеристика всех продуктов
- Характеризуют все новые соединения с помощью 1H, ЯМР-спектроскопии 13C и масс-спектрометрии высокого разрешения (HRMS)7,8,11.
Результаты
Здесь мы сообщаем о синтезе энантиопуре азиридина-2-карбоксилатов. Диастереометрическая смесь (R)-(1R,2 S,5 R)-2-изопропил-5-метилциклогексил1-((R)-1-фенилэтил)азиридин-2-карбоксилат (2) и (S)-(1R,2S,5R)-2-изопропил-5-метилциклогексил1-((R)-1-фенилэтил)азиридин-2-ка?...
Обсуждение
Азиридины как азотсодержащие трехчленные гетероциклы обладают огромным потенциалом для синтетических стартовых боевых или промежуточных продуктов для получения богатых азотом органических молекул. Исходя из группы, несущей на кольцевом кольце азот, они классифицируются как «актив?...
Раскрытие информации
Авторы заявляют, что в данном исследовании не было конфликта интересов.
Благодарности
Это исследование было поддержано Национальным исследовательским фондом Кореи (NRF-2020R1A2C1007102 и 2021R1A5A6002803) с Центром новых направлений в органическом синтезе и грантом HUFS 2022.
Материалы
| Name | Company | Catalog Number | Comments |
| (2R)-1-[(1R)-1-Phenylethyl]-2-aziridinecarboxylic acid (-)-menthol ester, 98% | Sigma-Aldrich | 57054-0 | |
| (2S)-1-[(1R)-1-Phenylethyl]-2-aziridinecarboxylic acid (-)-menthol ester | Sigma-Aldrich | 57051-6 | |
| 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride | TCI | 424331-25 g | CAS No: 25952-53-8 |
| 1,4-Dioxane | SAMCHUN | D0654-1 kg | CAS No: 123-91-1 |
| 1-Hydroxybenzotriazole hydrate | Aldrich | 219-989-7-50 g | CAS No: 123333-53-9 |
| 2,6-Lutidine | Alfa Aesar | A10478-AP, 500 mL | CAS No: 108-48-5 |
| Acetonitrile | SAMCHUN | A0127-18 L | CAS No: 75-05-8 |
| Acetonitrile-d3 | Cambridge Isotope Laboratories, | 15G-744-25 g | CAS No: 2206-26-0 |
| Aluminum chloride hexahydrate | Aldrich | 231-208-1, 500 g | CAS No : 7784-13-6 |
| Bruker AVANCE III HD (400 MHz) spectrometer | Bruker | NA | |
| Chloroform-d | Cambridge Isotope Laboratories, | 100 g | CAS No: 865-49-6 |
| Dichloromethane | SAMCHUN | M0822-18 L | CAS No: 75-09-2 |
| Dimethyl sulfoxide-d6 | Cambridge Isotope Laboratories, | 25 g | CAS No: 2206-27-1 |
| Ethanol | EMSURE | 1009831000,1L | CAS No: 64-17-5 |
| Ethyl acetate | SAMCHUN | E0191-18 L | CAS No: 141-78-6 |
| High resolution mass spectra/MALDI-TOF/TOF Mass Spectrometry | AB SCIEX | 4800 Plus | High resolution mass spectra |
| JASCO P-2000 | JASCO | P-2000 | For optical rotation |
| Lithium aluminum hydride | TCI | L0203-100 g | CAS No: 16853-85-3 |
| L-Selectride, 1 M solution in THF | Acros | 176451000, 100 mL | CAS No: 38721-52-7 |
| Methanol | SAMCHUN | M0585-18 L | CAS No: 67-56-1 |
| N-[(9H-Fluoren-9-ylmethoxy)carbonyl]-β-alanine | TCI | F08825G-5 g | CAS No: 35737-10-1 |
| N-Ethyldiisopropylamine | Aldrich | 230-392-0, 100 mL | CAS No: 7087-68-5 |
| n-Hexane | SAMCHUN | H0114-18 L | CAS No: 110-54-3 |
| Ninhydrin | Alfa Aesar | A10409-250 g | CAS No: 485-47-2 |
| p-Anisaldehyde | aldrich | A88107-5 g | CAS No: 123-11-5 |
| Phosphomolybdic acid hydrate | TCI | P1910-100 g | CAS No: 51429-74-4 |
| Sodium azide | D.S.P | 703301-500 g | CAS No: 26628-22-8 |
| Sodium Hydride 60% dispersion in mineral oil | Sigma-Aldrich | 452912-100 G | CAS No: 7646-69-7 |
| Sodium hydroxide | DUKSAN | A31226-1 kg | CAS No: 1310-73-2 |
| Sodium sulfate | SAMCHUN | S1011-1 kg | CAS No: 7757-82-6 |
| Thin Layer Chromatography (TLC) | Merck | 100390 | |
| Tert-Butyldimethylsilyl trifluoromethanesulfonate, 98% | Aldrich | 274-102-0, 25 g | CAS NO: 69739-34-0 |
| Tetrahydrofuran | SAMCHUN | T0148-18 L | CAS No: 109-99-9 |
| Triethylethylamine | DAEJUNG | 8556-4400-1 L | CAS No: 121-44-8 |
| UV light | Korea Ace Sci | TN-4C | 254 nm |
| Zinc chloride, anhydrous, 98+% | Alfa Aesar | A16281-22100 g | CAS No : 7646-85-7 |
Ссылки
- Pitzer, K. S. Strain energies of cyclic hydrocarbons. Science. 101 (2635), 672 (1945).
- Dudev, T., Lim, C. Ring strain energies from ab initio calculations. Journal of the American Chemical Society. 120 (18), 4450-4458 (1998).
- D'hooghe, M., Ha, H. -. J. . Synthesis of 4- to 7-Membered Heterocycles by Ring Expansion. , (2016).
- Ranjith, J., Ha, H. -. J. Synthetic applications of aziridinium ions. Molecules. 26 (6), 1744 (2021).
- Sweeney, J. B. Aziridines: epoxides' ugly cousins. Chemical Society Reviews. 31 (5), 247-258 (2002).
- Stankovic, S., et al. Regioselectivity in the ring opening of non-activated aziridines. Chemical Society Reviews. 41 (2), 643-665 (2012).
- Lee, W. K., Ha, H. -. J. Highlights of the chemistry of enantiomerically pure aziridine-2-carboxylates. Aldrichimica Acta. 36 (2), 57-63 (2003).
- Tranchant, M. J., Dalla, V., Jabin, I., Decroix, B. Reaction of vinyl triflates of α-keto esters with primary amines: efficient synthesis of aziridine carboxylates. Tetrahedron. 58 (42), 8425-8432 (2002).
- Ha, H. -. J., Jung, J. -. H., Lee, W. K. Application of regio- and stereoselective functional group transformation of chiral aziridine-2-carboxylate. Asian Journal of Organic Chemistry. 3 (10), 1020-1035 (2014).
- Kim, Y., et al. Preparation of 2,3-diaminopropionate from ring opening of aziridine-2-carboxylate. Tetrahedron Letters. 46 (25), 4407-4409 (2005).
- Srivastava, N., Macha, L., Ha, H. -. J. Total synthesis and stereochemical revision of biemamides B and D. Organic Letters. 21 (22), 8992-8996 (2019).
- Zhang, F., et al. Biemamides A-E, inhibitors of the TGF-β pathway that block the epithelial to mesenchymal transition. Organic Letters. 20 (18), 5529-5532 (2018).
- Srivastava, N., Ha, H. -. J. Highly efficient and stereoselective Mukaiyama Aldol reaction with chiral aziridine-2-carboxaldehyde and its synthetic applications. Asian Journal of Organic Chemistry. 11 (1), 2021005671 (2021).
- Kempter, I., et al. Synthesis and structural characterization of the isomuscarines. Tetrahedron. 70 (10), 1918-1927 (2014).
- Pirrrung, M. C., DeAmicis, C. V. Total synthesis of the muscarines. Tetrahedron Letters. 29 (2), 159-162 (1988).
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеСмотреть дополнительные статьи
This article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены