
È necessario avere un abbonamento a JoVE per visualizzare questo. Accedi o inizia la tua prova gratuita.
Method Article
Preparazione di aziridine non attivate enantiopure e sintesi di biemamide B, D ed epiallo-isomuscarina
In questo articolo
Riepilogo
In questo studio, prepariamo sia enantiomeri di aziridina-2-carbossilato, che vengono utilizzati nella sintesi asimmetrica di alcaloidi, tra cui biemamide B e D, e (-)-epiallo-isomuscarina.
Abstract
Le aziridine eterocicliche contenenti azoto sono sinteticamente molto preziose per la preparazione di molecole azacicliche e acicliche. Tuttavia, è molto difficile e laborioso produrre aziridine in forme otticamente pure su larga scala per applicare la sintesi asimmetrica dei composti aza. Fortunatamente, abbiamo raggiunto con successo entrambi gli enantiomeri (2R) - e (2S) -aziridina-2-carbossilati con il gruppo α-metilbenzile che dona elettroni all'azoto dell'anello come aziridine non attivate. Queste aziridine di partenza hanno due gruppi funzionali distinti: anello a tre membri altamente reattivo e carbossilato versatile. Sono applicabili nell'apertura dell'anello o nella trasformazione dell'anello con aziridina e nella trasformazione del gruppo funzionale in altri dal carbossilato. Entrambi questi enantiomeri sono stati utilizzati nella preparazione di composti amminoaciclici e/o aza-eterociclici biologicamente importanti in modo asimmetrico. In particolare, questo rapporto descrive la prima sintesi asimmetrica espediente di entrambi gli enantiomeri di 5, prodotti naturali marini di tipo 6-diidrouracile biemamide B e D come potenziali inibitori del TGF-β. Questa sintesi consisteva nella reazione di apertura dell'anello regio- e stereoselettiva dell'aziridina-2-carbossilato e nella successiva formazione di 4-aminoteteraidropirimidina-2,4-dione. Un altro esempio in questo protocollo ha riguardato una reazione di Mukaiyama altamente stereoselettiva di aziridina-2-carbossilato e silil enol etere, seguendo l'apertura dell'anello aziridina intramolecolare per fornire un facile e facile accesso alla (-)-epiallo-isomuscarina.
Introduzione
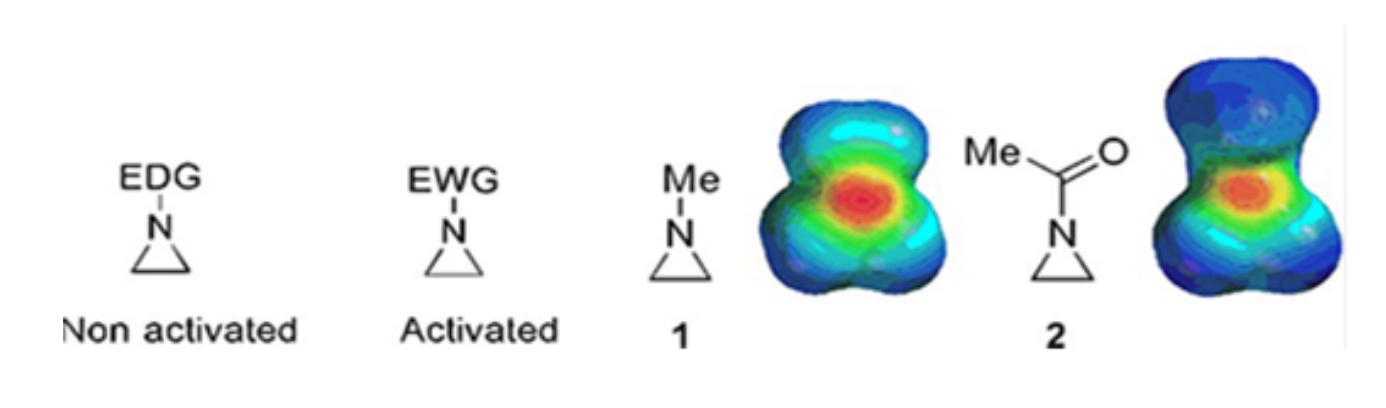
Piccoli anelli costituiti da ciclopropani, ossirani e aziridine si trovano in vari composti come prodotti naturali e farmaci 1,2. Sono utilizzati principalmente come materiali di partenza sfruttando la loro tensione ad anello. Tra i composti a tre anelli, l'aziridina è stata studiata meno ampiamente a causa della sua instabilità e reattività incontrollabile3. Come mostrato nelle mappe del potenziale elettrostatico (Figura 1), un gruppo attaccato all'anello aziridina-azoto, sia che doni elettroni o che attiri elettroni, rende diversa la basicità dell'azoto. Questa differenza fornisce un contrasto sorprendente con la reattività e la selettività delle aziridine corrispondenti.

Figura 1: Strutture chimiche di aziridine "attivate" e "non attivate" e mappe del potenziale elettrostatico dei loro esempi rappresentativi N-metilaziridina e N-acetilaziridina4. Questa cifra è stata modificata con il permesso di Ranjith et al.4. Fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
Quando l'azoto ad anello ha un gruppo di ritiro di elettroni, come solfonato, fosfonato e carbammato, lo chiamiamo aziridina "attivata". Questo è prontamente reattivo con i nucleofili per compensare la sua instabilità con un ambito limitato di regiochimica. Queste aziridine attivate vengono preparate attraverso vari metodi catalitici e utilizzate come materiale di partenza. Gran parte della recente chimica dell'aziridina si è occupata di queste aziridine attivate. Tuttavia, le aziridine attivate subiscono alcune restrizioni derivanti dalla loro instabilità e dalla limitata portata di reazione dell'apertura dell'anello. D'altra parte, le aziridine che portano sostituenti che donano elettroni, come i gruppi alchilici o alchilici sostituiti, all'anello azoto chiamato "non attivato"4, sono relativamente stabili nella maggior parte delle circostanze e possono essere lasciate sul banco per lungo tempo senza una decomposizione significativa. Le reazioni nucleofile di apertura dell'anello dell'aziridina non attivata avvengono attraverso la formazione di ioni aziridinio. La maggior parte delle reazioni di apertura dell'anello aziridina e delle trasformazioni dell'anello procedono in modo altamente regiochimico. Tuttavia, pochissimi rapporti di letteratura discutono la preparazione di aziridine non attivate otticamente pure con sostituenti nelle posizioni C2 o C3 5,6.
Questo documento mostra la preparazione di successo di derivati del gruppo α-metilbenzile contenente aziridina-2-carbossilato, in particolare (-)-mentoloil (1R)-feniletilaziridina-2-carbossilati come miscela diastereomerica, dalla reazione del 2,3-dibromopropionato e (1R)-feniletilammina. Da questa miscela diastereomerica, enantiopuro (1R)-feniletil-(2R)- e (2S)-aziridina-2-carbossilati come loro esteri (-)-mentololica sono stati ottenuti in forme otticamente pure mediante ricristallizzazione selettiva da MeOH e n-pentano su scale multi-cento chili (Figura 1)7. Questi esteri (-)-mentolilici possono essere facilmente convertiti nei loro esteri etilici o metilici mediante transesterificazione in presenza di magnesio o carbonato di potassio7. Questi composti possono anche essere preparati facilmente su scala di laboratorio dalle reazioni degli alchili 2,3-dibromopropionati o del vinile triflato di α-chetoestere con 2-feniletilammina chirale seguita dalla separazione della miscela diastereomerica utilizzando la semplice cromatografia a colonna flash8.
Una volta che abbiamo enantiopura chirale aziridina-2-carbossilato, possiamo sintetizzare varie molecole bersaglio cicliche e acicliche contenenti azoto biologicamente importanti basate su trasformazioni di gruppi funzionali di carbossilato e reazioni di apertura dell'anello aziridina altamente regio- e stereoselettivo 6,9,10. La prima sintesi asimmetrica espediente è stata applicata sia per gli enantiomeri di 5, 6-diidrouracile-tipo prodotti naturali marini biemamide B e D come potenziali inibitori del TGF-β11,12. In secondo luogo, la sintesi diastereoselettiva di β-(aziridin-2-il)-β-idrossi chetoni è stata ottenuta dalla reazione aldolica di Mukaiyama di 1-(1-feniletil)-aziridina-2-carbossildeide otticamente pura e vari silani di enolo in presenza di ZnCl2, in alta resa (>82%) con stereoselettività quasi perfetta (98:2 dr) attraverso uno stato di transizione controllato dalla chelazione. Questi sono stati utilizzati per la sintesi asimmetrica degli alcaloidi epiallo-isomuscarinici 13,14,15.
Access restricted. Please log in or start a trial to view this content.
Protocollo
1. Sintesi della miscela diastereomerica del derivato dell'estere chirale aziridina (-)-mentololica (1)
- Aggiungere 2,3-dibromopropano (-)-mentolo estere 1a (5,0 g, 13,58 mM, 1,0 equiv) e una barra di agitazione magnetica in un matraccio a fondo tondo a due colli da 250 mL essiccato al forno in atmosfera di azoto (N2).
- Aggiungere acetonitrile anidro (60 ml) al matraccio di reazione utilizzando una siringa ermetica.
- Quindi raffreddare la miscela di reazione a 0 °C usando un bagno di ghiaccio e mescolare la miscela di reazione per 5 minuti.
- Aggiungere carbonato di potassio (5,6 g, 40,74 mM, 3,0 equiv) nella miscela di reazione alla stessa temperatura e lasciare mescolare per 30 minuti.
- Aggiungere (2R)-feniletilammina (2,0 ml, 16,29 nM, 1,2 equiv) in modo goccioline a temperatura ambiente (RT) e lasciare che la miscela di reazione si mescoli per 12 ore.
- Monitorare l'andamento della reazione utilizzando la cromatografia su strato sottile utilizzando acetato di esano:etile 9:1 v/v (EtOAc; Rf = 0,4) come eluente.
- Al termine della reazione, filtrare la miscela su carta da filtro (dimensione dei pori 70 mm).
- Quindi, aggiungere acqua (30 ml) nel filtrato organico ed estrarre lo strato organico con Et2O (2 x 50 ml) due volte utilizzando un imbuto separatore.
- Asciugare gli estratti organici combinati oltre 7,5 g di Na2SO4 anidro e concentrare in vacuo (<15 mbar) utilizzando un evaporatore rotativo.
NOTA: Ora si ottiene una miscela grezza di aziridina chirale diastereomerica contenente entrambi gli isomeri di (R)-(1R,2 S,5 R)-2-isopropil-5-metilcicloesil1-((R)-1-feniletil)aziridina-2-carbossilato e (S)-(1R,2 S,5 R)-2-isopropil-5-metilcicloesil1-((R)-1-feniletil)aziridina-2-carbossilato (4,1 g, 90%). - Isolamento dell'aziridina chirale (R)-(1R,2S,5R)-2-isopropil-5-metilcicloesil 1-((R)-1- feniletil)aziridina-2-carbossilato (2) mediante un metodo di cristallizzazione selettiva
- Aggiungere 8,7 g della miscela grezza di derivato chirale dell'estere di mentolo 1 e sciogliere in 70 ml di metanolo in un matraccio a fondo tondo da 250 mL essiccato al forno.
- Ora riscaldare la miscela di reazione fino a 70 °C utilizzando un bagno di acqua calda, quindi raffreddare la miscela di reazione a -10 °C fino a formare cristalli solidi.
- Filtrare il composto solido su una carta da filtro (dimensione dei pori 70 mm) per ottenere 2,2 g di estere (R)-(1R,2S,5R)-2-isopropil-5-metilcicloesil 1-((R)-1-feniletil)aziridina-2-carbossilato (2).
- Concentrare nuovamente la soluzione filtrata in vacuo (<15 mbar) utilizzando un evaporatore rotativo, sciogliere 50 mL di etanolo nella miscela di reazione rimanente e ricristallizzare a -10 °C per ottenere 1,2 g di (R)-(1R,2S,5R)-2-isopropil-5-metilcicloesil 1-((R)-1-feniletil)aziridina-2-carbossilato (2).
- In questo momento, utilizzare l'altro etanolo alcolico allo stesso modo del metanolo.
- Dopo la ricristallizzazione, filtrare nuovamente su una carta da filtro (dimensione dei pori 70 mm), concentrare completamente in vacuo i restanti 5,3 g di soluzione filtrata grezza (<15 mbar) utilizzando un evaporatore rotante e aggiungere 50 ml di solvente idrocarburico pentanico.
- Mantenere la soluzione di reazione rimanente a -15 °C.
NOTA: Ora si ottiene un composto solido di quasi 1,9 g di (S)-(1R,2S,5R)-2-isopropil-5-metilcicloesil1-((R)-1-feniletil)estere aziridina-2-carbossilato (3). - Dopo aver ottenuto i cristalli, concentrare nuovamente la soluzione in vacuo (<15 mbar) utilizzando un evaporatore rotante e scioglierla in 30 ml di solvente idrocarburico pentanico.
- Ricristallizzare nuovamente a -15 °C per ottenere 0,8 g di estere (S)-(1R,2S,5R)-2-isopropil-5-metilcicloesil1-((R)-1-feniletil)aziridina-2-carbossilato (2').
- Ottenimento di (R)-etile 1-((R)-1-feniletil)aziridina-2-carbossilato (3)
- Aggiungere (R)-(1R,2 S,5 R)-2-isopropil-5-metilcicloesil1-((R)-1-feniletil)aziridina-2-carbossilato (2) (0,167 g, 0,57 mM) e una barra magnetica in un matraccio a fondo tondo a due colli da 25 mL essiccato al forno in atmosfera di azoto (N2).
- Aggiungere 1,8 ml di etanolo al matraccio di reazione usando una siringa ermetica e mescolare a RT.
- Quindi aggiungere carbonato di potassio (0,40 g, 20,28 mmol, 4,0 equiv) e mescolare a RT per 2 giorni.
- Monitorare l'avanzamento della reazione utilizzando la cromatografia su strato sottile (eluente, 8:2 v/v, esano:acetato di etile (EtOAc), Rf = 0,6).
- Al termine della reazione, filtrare la miscela su carta da filtro (dimensione dei pori 70 mm), quindi aggiungere acqua (5 ml) nel filtrato organico ed estrarre lo strato organico con CH2Cl2 (2 x 15 ml) due volte utilizzando un imbuto separatore.
- Asciugare gli estratti organici combinati oltre 3,0 g di Na2SO4 anidro e concentrare in vacuo (<15 mbar) utilizzando un evaporatore rotativo.
- Purificare il prodotto grezzo mediante cromatografia a colonna di fase normale su gel di silice (70-230 maglie) per ottenere un prodotto puro (R)-etil 1-((R)-1-feniletil)aziridina-2-carbossilato (3) (950 mg, 88%). Rf (30% EtOAc/esano = 0,50).
2. Apertura dell'anello di aziridina regio e stereoselettiva da parte di azide nucleofilo per la sintesi totale di biemamide B e biemamide D
- Sintesi di (R)-etile 2-azido-3-((((R)-1-feniletil)ammino)propanoato (5)
- Trasferimento (500 mg, 2,20 mM, 1,0 equiv) di chirale (S)-etil 1-((R)-1-feniletil)aziridina-2-carbossilato (4) e di una barra magnetica in un matraccio a fondo tondo a due colli da 50 mL essiccato al forno in atmosfera aperta.
- Aggiungere il 50% di etanolo acquoso (15 ml) alla miscela di reazione.
- Raffreddare la miscela di reazione a 0 °C e aggiungere acido solforico concentrato (36 N) a goccia per mantenere quasi il pH 4,0 e mescolare per 5 minuti.
- Aggiungere sodio azide (370 mg, 5,70 mM, 2,5 equiv) a 0 °C e lasciare che la miscela di reazione si mescoli per 10 minuti alla stessa temperatura e poi si riscaldi a RT.
- Quindi, aggiungere AlCl3∙6H2O (55 mg, 0,22 mM, 0,1 equiv) come catalizzatore allo stesso RT e lasciare mescolare per ulteriori 3 ore.
- Monitorare l'avanzamento della reazione utilizzando la cromatografia a strato sottile (eluente, 6:4 v/v, esano:acetato di etile (EtOAc), Rf = 0,2).
- Spegnere la miscela di reazione con due porzioni di NaHCO3 saturo da 20 ml.
- Quindi filtrare la miscela grezza su un tampone di celite con etanolo (2 x 10 ml).
- Concentrare la miscela di reazione in vacuo (<15 mbar) utilizzando un evaporatore rotante.
- Estrarre lo strato organico con CH2Cl2 (2 x 50 mL) due volte utilizzando un imbuto separatore.
- Quindi asciugare lo strato organico combinato su 5,0 g di Na2SO4 anidro per 5 minuti.
- Concentrare lo strato organico grezzo in vacuo (<15 mbar) utilizzando un evaporatore rotante per ottenere un prodotto azide grezzo.
- Purificare il prodotto grezzo con cromatografia su colonna di fase normale su gel di silice (70-230 mesh) eluendo con il 40% di EtOAc/esano (Rf = 0,20) per ottenere 490 mg (resa al 90%) di (R)-etil 2-azido-3-((R)-1-feniletil)ammino)propanoato (5) come liquido viscoso.
- Sintesi di (9H-Fluoren-9-il)metil(3-(((R)-3-metil-2,4-dioxo-1-((R)-1 feniletil)esaidro pirimidin-5-il)ammino)-3-ossopropil)carbammato (7)
- Trasferire 150 mg di chirale (R)-5-ammino-3-metil-1-((R)-1-feniletil) diidropirimidina-2,4(1H,3H)-dione (6) (150 mg, 0,60 mM, 1,0 equiv) e una barra magnetica in un matraccio a fondo tondo a due colli da 25 mL essiccato al forno in atmosfera N2 .
- Aggiungere CH2Cl2 (15,0 mL) secco al matraccio di reazione utilizzando una siringa ermetica.
- Quindi raffreddare la miscela di reazione a 0 °C usando un bagno di ghiaccio e mescolare la miscela di reazione per 5 minuti.
- Aggiungere Fmoc-beta-alanina (377 mg, 1,20 mM, 2,0 equiv) e DIPEA (0,67 mL, 3,64 mM, 6,0 equiv) a 0 °C e mescolare per 5 min.
- Aggiungere EDCI (347 mg, 1,82 mM, 3,0 equiv) e HOBt (165 mg, 1,21 mM, 2,0 equiv) alla miscela di reazione a 0 °C e lasciare mescolare per 10 minuti alla stessa temperatura.
- Mantenere la miscela di reazione a RT e lasciare mescolare per altre 8 ore.
- Monitorare l'avanzamento della reazione utilizzando la cromatografia a strato sottile utilizzando (2:8 v/v, esano:acetato di etile (EtOAc), Rf = 0,4) come eluente.
- Spegnere la miscela di reazione con acqua (10 ml).
- Lavare lo strato organico combinato con salamoia (15 mL), quindi estrarre lo strato organico con CH2Cl2 (2 x 20 mL) due volte utilizzando un imbuto separatore.
- Quindi asciugare lo strato organico combinato su 5,0 g di Na2SO4 anidro per 5 minuti e concentrarlo in vacuo (<15 mbar) utilizzando un evaporatore rotante.
- Purificare il prodotto grezzo con cromatografia a colonna di fase normale su gel di silice (70-230 mesh) eluendo con l'80% di EtOAc/esano (Rf = 0,40) per ottenere 295 mg di (7) (resa del 90%).
3. Reazione aldolica stereoselettiva di Mukaiyama con aziridina-2-carbossaldeide chirale e sua apertura ad anello aziridina regio e stereoselettiva da parte di idrossi nucleofilo interno per la sintesi totale di (-)- epiallo-somuscarina (17)
- Sintesi di (S)-4-idrossi-4-((R)-1-((R)-1-feniletil)aziridin-2-il)butan-2-one (12)
- Trasferire (R)-1-((R)-1-feniletil)aziridina-2-carbaldeide (10) (140 mg, 0,8 mM, 1,0 equiv) e una barra magnetica in un matraccio a fondo tondo a due colli da 25 mL essiccato al forno in atmosfera N2 .
- Aggiungere CH3CN secco (4,0 ml) al matraccio di reazione utilizzando una siringa ermetica.
- Quindi raffreddare la miscela di reazione a -20 °C usando un bagno di ghiaccio-acetone e mescolare la miscela di reazione per 5 minuti.
- Aggiungere ZnCl2 anidro (108 mg, 0,8 mM, 1,0 equiv) alla miscela di reazione a -20 °C e lasciare mescolare per 5 minuti.
- Aggiungere quindi trimetil(prop-1-en-2-ilossi)silano (11) (104 mg, 0,8 mM, 1,0 equiv) disciolto in CH3CN secco (3,0 ml) alla miscela di reazione a -20 °C in modo goccioso e lasciare che la miscela di reazione si mescoli per 1 ora alla stessa temperatura.
- Monitorare l'andamento della reazione utilizzando la cromatografia su strato sottile utilizzando 8:2 v/v esano:acetato di etile (EtOAc), Rf = 0,2) come eluente.
- Spegnere la miscela di reazione con NaHCO3 saturo (4 ml).
- Estrarre lo strato organico con EtOAc (2 x 15 mL) due volte utilizzando un imbuto separatore.
- Asciugare lo strato organico combinato con 3,0 g di Na2SO4 anidro e concentrarlo in vacuo (<15 mbar) utilizzando un evaporatore rotante.
- Purificare il prodotto grezzo con cromatografia a colonna di fase normale su gel di silice (70-230 mesh) eluendo con l'80% di EtOAc/esano, (Rf = 0,20) per ottenere 58 mg di (S)-4-idrossi-4-((R)-1-(R)-1-feniletil)aziridin-2-il)butan-2-one) (12) (resa dell'85%).
- Sintesi di (R)-N-(((2R,3 S,5 R)-3-((terz-butildimetilsilil)ossi)-5-metiltetra idrofurran-2-il)metil)-1-feniletanamina (15)
- Trasferire (S)-4-(((terz-butildimetilsilil)ossi)-4-((R)-1-((R)-1-feniletil) aziridin-2-il)butan-2-one (13) (400 mg, 1,15 mM, 1,0 equiv) e una barra magnetica in un matraccio a fondo tondo a due colli da 25 mL essiccato al forno in atmosfera N2 .
- Aggiungere THF anidro (50 mL) al pallone di reazione utilizzando una siringa ermetica.
- Raffreddare la miscela di reazione a -78 °C utilizzando un bagno secco di ghiaccio-acetone e lasciare mescolare per 5 minuti.
- Quindi aggiungere il litio tri-sec-butilboroidruro (L-selectride) (1 M soluzione in THF) (2,3 ml, 2,0 equiv) a goccia nella miscela di reazione a -78 °C e lasciare mescolare per altri 25 minuti.
- Riscaldare la miscela di reazione a RT e lasciare mescolare per 8 ore.
- Monitorare l'andamento della reazione da parte di TLC utilizzando il 30% di EtOAc/esano (Rf = 0,40) come eluente.
- Dopo il consumo completo del composto 13, spegnere la miscela di reazione con 0,1 M di NaOH (5 mL).
- Estrarre lo strato organico con EtOAc (3 x 15 mL), quindi lavare con salamoia (15 mL).
- Asciugare lo strato organico su 3,0 g di Na2SO4 anidro e concentrare in vacuo (<15 mbar) utilizzando un evaporatore rotativo.
- Purificare il prodotto grezzo con cromatografia a colonna di fase normale su gel di silice (70-230 mesh, eluente, 8:2 v/v, esano:acetato di etile (EtOAc), Rf = 0,2) per ottenere un composto puro (15) (382 mg, resa del 95%).
- Sintesi di ioduro di (-)-epiallo-isomuscariina (17)
- Trasferire il composto 16 (20 mg, 0,15 mM, 1,0 equiv) e una barra magnetica in un matraccio a fondo tondo da 10 mL essiccato al forno in atmosfera N2 .
- Aggiungere 3 mL di pallone di reazione EtOAc utilizzando una siringa ermetica.
- Quindi aggiungere ioduro di metile (0,4 mL, 3,0 mM) alla miscela di reazione a RT.
- Aggiungere 1,2,2,6,6-pentametilpiperidina (PMP) (0,05 mL, 0,3 mM, 2,0 equiv) alla miscela di reazione a 0 °C.
- Quindi, mantenere la miscela di reazione a RT e lasciare mescolare per ulteriori 16 ore.
- Evaporare il solvente in vacuo (<15 mbar) utilizzando un evaporatore rotante per dare il prodotto grezzo.
- Lavare tre volte aggiungendo (3 x 5 mL) il 10% di MeOH in EtOAc alla miscela di reazione grezza.
- Quindi lavare la miscela grezza con n-pentano (5 ml) e concentrare sotto vuoto (<15 mbar) utilizzando un evaporatore rotante per ottenere ioduro puro (-)-epiallo-isomuscarina (17) (32 mg, 68%).
4. Caratterizzazione di tutti i prodotti
- Caratterizzare tutti i nuovi composti con laspettroscopia NMR 1 H, 13C e la spettrometria di massa ad alta risoluzione (HRMS)7,8,11.
Access restricted. Please log in or start a trial to view this content.
Risultati
Qui, riportiamo la sintesi di enantiopure aziridina-2-carbossilati. La miscela diastereomerica di (R)-(1R,2 S,5 R)-2-isopropil-5-metilcicloesil1-((R)-1-feniletil)aziridina-2-carbossilato (2) e (S)-(1R,2S,5R)-2-isopropil-5-metilcicloesil1-((R)-1-feniletil)aziridina-2-carbossilato (3) (4,1 g, 90%) è stata preparata in resa quantitativa da estere 2,3 -dibromopropano (-)-mentololica estere e (1R)-feniletilammin...
Access restricted. Please log in or start a trial to view this content.
Discussione
Le aziridine come eterocicli a tre membri contenenti azoto hanno un enorme potenziale per le marziali di partenza sintetiche o intermedi per preparare molecole organiche ricche di azoto. Sulla base del gruppo che porta all'anello azoto, sono classificati come aziridine "attivate" e "non attivate" la cui reattività chimica e selettività sono diverse. Tuttavia, sono disponibili metodi molto limitati per preparare questa preziosa aziridina in una forma otticamente attiva.
Il protocollo in ques...
Access restricted. Please log in or start a trial to view this content.
Divulgazioni
Gli autori dichiarano che non c'era conflitto di interessi in questo studio.
Riconoscimenti
Questa ricerca è stata sostenuta dalla National Research Foundation of Korea (NRF-2020R1A2C1007102 e 2021R1A5A6002803) con il Center for New Directions in Organic Synthesis e una sovvenzione HUFS 2022.
Access restricted. Please log in or start a trial to view this content.
Materiali
| Name | Company | Catalog Number | Comments |
| (2R)-1-[(1R)-1-Phenylethyl]-2-aziridinecarboxylic acid (-)-menthol ester, 98% | Sigma-Aldrich | 57054-0 | |
| (2S)-1-[(1R)-1-Phenylethyl]-2-aziridinecarboxylic acid (-)-menthol ester | Sigma-Aldrich | 57051-6 | |
| 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride | TCI | 424331-25 g | CAS No: 25952-53-8 |
| 1,4-Dioxane | SAMCHUN | D0654-1 kg | CAS No: 123-91-1 |
| 1-Hydroxybenzotriazole hydrate | Aldrich | 219-989-7-50 g | CAS No: 123333-53-9 |
| 2,6-Lutidine | Alfa Aesar | A10478-AP, 500 mL | CAS No: 108-48-5 |
| Acetonitrile | SAMCHUN | A0127-18 L | CAS No: 75-05-8 |
| Acetonitrile-d3 | Cambridge Isotope Laboratories, | 15G-744-25 g | CAS No: 2206-26-0 |
| Aluminum chloride hexahydrate | Aldrich | 231-208-1, 500 g | CAS No : 7784-13-6 |
| Bruker AVANCE III HD (400 MHz) spectrometer | Bruker | NA | |
| Chloroform-d | Cambridge Isotope Laboratories, | 100 g | CAS No: 865-49-6 |
| Dichloromethane | SAMCHUN | M0822-18 L | CAS No: 75-09-2 |
| Dimethyl sulfoxide-d6 | Cambridge Isotope Laboratories, | 25 g | CAS No: 2206-27-1 |
| Ethanol | EMSURE | 1009831000,1L | CAS No: 64-17-5 |
| Ethyl acetate | SAMCHUN | E0191-18 L | CAS No: 141-78-6 |
| High resolution mass spectra/MALDI-TOF/TOF Mass Spectrometry | AB SCIEX | 4800 Plus | High resolution mass spectra |
| JASCO P-2000 | JASCO | P-2000 | For optical rotation |
| Lithium aluminum hydride | TCI | L0203-100 g | CAS No: 16853-85-3 |
| L-Selectride, 1 M solution in THF | Acros | 176451000, 100 mL | CAS No: 38721-52-7 |
| Methanol | SAMCHUN | M0585-18 L | CAS No: 67-56-1 |
| N-[(9H-Fluoren-9-ylmethoxy)carbonyl]-β-alanine | TCI | F08825G-5 g | CAS No: 35737-10-1 |
| N-Ethyldiisopropylamine | Aldrich | 230-392-0, 100 mL | CAS No: 7087-68-5 |
| n-Hexane | SAMCHUN | H0114-18 L | CAS No: 110-54-3 |
| Ninhydrin | Alfa Aesar | A10409-250 g | CAS No: 485-47-2 |
| p-Anisaldehyde | aldrich | A88107-5 g | CAS No: 123-11-5 |
| Phosphomolybdic acid hydrate | TCI | P1910-100 g | CAS No: 51429-74-4 |
| Sodium azide | D.S.P | 703301-500 g | CAS No: 26628-22-8 |
| Sodium Hydride 60% dispersion in mineral oil | Sigma-Aldrich | 452912-100 G | CAS No: 7646-69-7 |
| Sodium hydroxide | DUKSAN | A31226-1 kg | CAS No: 1310-73-2 |
| Sodium sulfate | SAMCHUN | S1011-1 kg | CAS No: 7757-82-6 |
| Thin Layer Chromatography (TLC) | Merck | 100390 | |
| Tert-Butyldimethylsilyl trifluoromethanesulfonate, 98% | Aldrich | 274-102-0, 25 g | CAS NO: 69739-34-0 |
| Tetrahydrofuran | SAMCHUN | T0148-18 L | CAS No: 109-99-9 |
| Triethylethylamine | DAEJUNG | 8556-4400-1 L | CAS No: 121-44-8 |
| UV light | Korea Ace Sci | TN-4C | 254 nm |
| Zinc chloride, anhydrous, 98+% | Alfa Aesar | A16281-22100 g | CAS No : 7646-85-7 |
Riferimenti
- Pitzer, K. S. Strain energies of cyclic hydrocarbons. Science. 101 (2635), 672(1945).
- Dudev, T., Lim, C. Ring strain energies from ab initio calculations. Journal of the American Chemical Society. 120 (18), 4450-4458 (1998).
- D'hooghe, M., Ha, H. -J. Synthesis of 4- to 7-Membered Heterocycles by Ring Expansion. , Springer. Berlin/Heidelberg, Germany. (2016).
- Ranjith, J., Ha, H. -J. Synthetic applications of aziridinium ions. Molecules. 26 (6), 1744(2021).
- Sweeney, J. B. Aziridines: epoxides' ugly cousins. Chemical Society Reviews. 31 (5), 247-258 (2002).
- Stankovic, S., et al. Regioselectivity in the ring opening of non-activated aziridines. Chemical Society Reviews. 41 (2), 643-665 (2012).
- Lee, W. K., Ha, H. -J. Highlights of the chemistry of enantiomerically pure aziridine-2-carboxylates. Aldrichimica Acta. 36 (2), 57-63 (2003).
- Tranchant, M. J., Dalla, V., Jabin, I., Decroix, B. Reaction of vinyl triflates of α-keto esters with primary amines: efficient synthesis of aziridine carboxylates. Tetrahedron. 58 (42), 8425-8432 (2002).
- Ha, H. -J., Jung, J. -H., Lee, W. K. Application of regio- and stereoselective functional group transformation of chiral aziridine-2-carboxylate. Asian Journal of Organic Chemistry. 3 (10), 1020-1035 (2014).
- Kim, Y., et al. Preparation of 2,3-diaminopropionate from ring opening of aziridine-2-carboxylate. Tetrahedron Letters. 46 (25), 4407-4409 (2005).
- Srivastava, N., Macha, L., Ha, H. -J. Total synthesis and stereochemical revision of biemamides B and D. Organic Letters. 21 (22), 8992-8996 (2019).
- Zhang, F., et al. Biemamides A-E, inhibitors of the TGF-β pathway that block the epithelial to mesenchymal transition. Organic Letters. 20 (18), 5529-5532 (2018).
- Srivastava, N., Ha, H. -J. Highly efficient and stereoselective Mukaiyama Aldol reaction with chiral aziridine-2-carboxaldehyde and its synthetic applications. Asian Journal of Organic Chemistry. 11 (1), 2021005671(2021).
- Kempter, I., et al. Synthesis and structural characterization of the isomuscarines. Tetrahedron. 70 (10), 1918-1927 (2014).
- Pirrrung, M. C., DeAmicis, C. V. Total synthesis of the muscarines. Tetrahedron Letters. 29 (2), 159-162 (1988).
Access restricted. Please log in or start a trial to view this content.
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneEsplora altri articoli
This article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati