
需要订阅 JoVE 才能查看此. 登录或开始免费试用。
Method Article
对映草非活化氮丙啶的制备及比咪咪唑B、D和 表皮异毒羯碱的合成
摘要
在这项研究中,我们制备了两种对映体-2-羧酸盐,用于生物碱的不对称合成,包括比咪胺B和D,以及(-)-表皮异毒羯碱。
摘要
含氮杂环氮丙啶在合成上对于制备氮杂环和非环分子非常有价值。然而,大规模地制造光学纯形式的氮丙啶以应用氮杂化合物的不对称合成是非常困难和费力的。幸运的是,我们成功地实现了对映异构体(2R)-和(2S)-氮丙啶-2-羧酸盐,环氮上具有α-甲基苄基的电子供体作为非活化的氮丙啶。这些起始氮丙啶具有两个不同的官能团 - 高反应性的三元环和多功能羧酸盐。它们适用于阿齐啶的开环或环转化,以及羧酸盐对其他物的官能团转化。这两种对映异构体都以不对称的方式用于制备生物学上重要的氨基无环和/或氮杂环化合物。具体而言,本报告描述了5,6-二氢尿嘧啶型海洋天然产物比咪酰胺B和D的对映异构体的首次权宜之计不对称合成作为潜在的TGF-β抑制剂。该合成包括氮丙啶-2-羧酸酯的管状和立体选择性开环反应以及随后形成4-氨基四氢嘧啶-2,4-二酮。该协议中的另一个例子涉及氮丙啶-2-羧酸酯和硅基烯醇醚的高度立体选择性Mukaiyama反应,在分子内齐啶环开口之后,提供容易和容易地获得(-)-表皮异毒羯碱。
引言
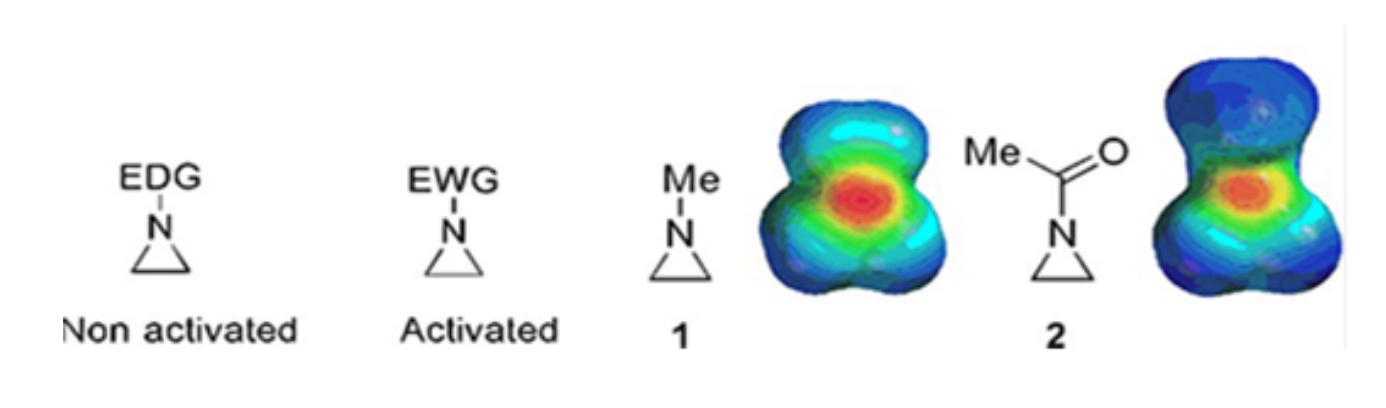
由环丙烷,环氧乙烷和氮丙啶组成的小环存在于各种化合物中,例如天然产物和药物1,2。它们主要用作利用其环应变的起始材料。在三环化合物中,由于丙啶的不稳定性和不可控的反应性,对阿齐啶的研究较少3.如静电势图(图1)所示,连接到氮丙啶环氮的基团,无论是电子供电子还是电子吸引,都使氮的碱度不同。这种差异与相应氮丙啶的反应性和选择性形成了鲜明的对比。

图1:"活化"和"非活化"氮丙啶的化学结构和它们代表性实例N-甲基氮丙啶和N-乙酰氮丙啶4的静电电位图。 这个数字是在Ranjith等人的许可下修改的。 请点击此处查看此图的大图。
{kind=link}
当环氮具有吸电子基团时,例如磺酸盐,膦酸盐和氨基甲酸酯,我们称之为"活化"的氮丙啶。这很容易与亲核试剂反应,以补偿其不稳定性,而区域化学范围有限。这些活化的氮丙啶是通过各种催化方法制备的,并用作原料。最近的许多阿齐啶化学都涉及这些活化的阿齐啶。然而,活化的氮丙啶由于其不稳定性和环开口的反应范围有限而受到某些限制。另一方面,在称为"非活化"4的环氮中,含有供电子取代基的氮丙啶,如烷基或取代的烷基,在大多数情况下相对稳定,可以长时间留在工作台上而不会有明显的分解。非活化叠氮啶的亲核开环反应 通过 叠氮啶离子的形成发生。阿齐啶开环和环转化的大多数反应以高度区域化学的方式进行。然而,很少有文献报道讨论制备光学纯的非活化氮丙啶,其取代基位于C2或C3位5,6。
本文展示了由2,3-二溴丙酸和(1 R)-苯乙胺反应制备成功制备含α甲基苄基的手性氮丙啶-2-羧酸酯衍生物,特别是(-)-薄荷醇(1R)-苯乙啶-2-羧酸酯作为其对映异构体的混合物。从这种非对映异构体混合物中,通过从MeOH和正戊烷在数百公斤尺度上的选择性重结晶,以光学纯形式获得对映体(1R)-苯乙基-(2R)-和(2S)-氮丙啶-2-羧酸酯(图1)7。这些(-)-薄荷醇酯可以在镁或碳酸钾7存在下通过酯交换容易地转化为它们的乙基或甲基酯。这些化合物也可以在实验室规模上由烷基2,3-二溴丙酸酯或α-酮酯的乙烯基三氟甲磺酸酯与手性2-苯基乙胺的反应轻松制备,然后使用简单的闪速柱色谱8分离非对映异构体混合物。
一旦我们有了对映卤手性氮丙啶-2-羧酸酯,我们就可以基于羧酸盐的官能团转化和高区域选择性的氮丙啶环开口反应6,9,10合成各种环状和非环状含氮生物重要的靶分子。首次采用权宜之计不对称合成5, 6-二氢尿嘧啶型海洋天然产物比咪酰胺B和D的对映异构体作为潜在的TGF-β抑制剂11,12.其次,在ZnCl 2存在下,通过螯合控制过渡态,通过光学纯的1-(1-苯基乙基)-氮丙啶-2-甲醛与各种烯醇硅烷反应,在高收率(>82%)和几乎完美的立体选择性(98:2 dr)下,实现了对β-(叠氮嘧啶-2-基 β)-羟基酮的对映选择性合成。这些用于表皮异毒羯碱生物碱13,14,15的不对称合成。
Access restricted. Please log in or start a trial to view this content.
研究方案
1.手性氮丙啶(-)-薄荷醇酯衍生物的非对映异构体混合物的合成(1)
- 在氮气(N 2)气氛下,将2,3-二溴丙烷(-)-薄荷醇酯1a(5.0克,13.58毫升,1.0当量)和磁力搅拌棒加入烘干的250毫升双颈圆底烧瓶中。
- 使用气密注射器将无水乙腈(60mL)加入反应瓶中。
- 然后使用冰浴将反应混合物在0°C下冷却,并将反应混合物搅拌5分钟。
- 在相同温度下将碳酸钾(5.6g,40.74mM,3.0等值)加入反应混合物中,并使其搅拌30分钟。
- 在室温(RT)下滴加(2 R)-苯乙胺(2.0毫升,16.29nM,1.2等价物),并使反应混合物搅拌12小时。
- 使用9:1 v / v己烷:乙酸乙酯(EtOAc;Rf = 0.4) 作为淋洗液。
- 反应完成后,在滤纸上过滤混合物(孔径70毫米)。
- 然后,将水(30 mL)加入有机滤液中,并使用分离漏斗用Et2O(2×50 mL)提取有机层两次。
- 干燥超过7.5克无水Na2SO4 的组合有机提取物,并使用旋转蒸发器 真空 浓缩(<15毫巴)。
注意:现在得到含有(R,2 S,5 R)-2-异丙基-5-甲基环己基1-(R)-5-甲基环己基1-(R)-1-苯基乙基)氮杂胺-2-羧酸酯和(S)-(1R,2 S,5R)-2-异丙基-5-甲基环己基1-(R)-1-苯基乙基)氮丙啶-2-羧酸酯(4.1克,90%)的两种异构体的对映异构体唾液氮丙啶的粗混合物。 - 通过选择性结晶法分离手性氮丙啶(R)-(1R,2S,5R)-2-异丙基-5-甲基环己基 1-(R)-1-苯乙基)氮丙啶-2-羧酸酯(2)
- 加入8.7g手性氮丙啶(-)-薄荷醇酯 1 衍生物的粗混合物,并溶于70mL甲醇中,进入烘干的250mL单颈圆底烧瓶中。
- 现在使用热水浴将反应混合物加热至70°C,然后将反应混合物冷却至-10°C直至形成固体晶体。
- 在滤纸(孔径70毫米)上过滤固体化合物,得到2.2克(R)-(1R,2S,5R)-2-异丙基-5-甲基环己基1-((R)-1-苯乙基)氮丙啶-2-羧酸酯(2)酯。
- 使用旋转蒸发 器真空( <15毫巴)再次浓缩滤液溶液,将50mL乙醇溶解在剩余的反应混合物中并在-10°C下重结晶,得到1.2g(R)-(1R,2S,5R)-2-异丙基-5-甲基环己基1-(R)-1-苯乙基)氮丙啶-2-羧酸酯(2)。
- 此时,以与甲醇相同的方式使用其他醇乙醇。
- 重结晶后,再次过滤滤纸(孔径70毫米),使用旋转蒸发器将剩余的粗5.3克滤液溶液完全 真空 (<15 mbar)浓缩,并加入50毫升戊烷烃溶剂。
- 将剩余的反应溶液保持在-15°C。
注:现在得到近1.9克(S)-(1R,2S,5R)-2-异丙基-5-甲基环己基1-(R)-1-苯基乙基)氮丙啶-2-羧酸酯(3)的固体化合物。 - 获得晶体后,使用旋转蒸发器再次 真空 (<15 mbar)浓缩溶液,并将其溶解在30 mL戊烷烃溶剂中。
- 在-15°C下再次重结晶,得到0.8克(S)-(1R,2S,5R)-2-异丙基-5-甲基环己基1-(R)-1-苯基乙基)氮丙啶-2-羧酸酯(2')。
- 获得 1-(R)-1-苯乙基)氮丙啶-2-羧酸乙酯 (3)
- 在氮气(N 2)气氛下,将(R)-(1R,2 S,5R)-2-异丙基-5-甲基环己基1-(R)-1-苯基乙基)氮丙啶-2-羧酸酯(2)(0.167克,0.57毫升)和磁力搅拌棒加入烘干的25毫升双颈圆底烧瓶中。
- 使用密闭注射器向反应瓶中加入1.8 mL乙醇,并在室温下搅拌。
- 然后加入碳酸钾(0.40克,20.28毫摩尔,4.0当量)并在室温下搅拌2天。
- 使用薄层色谱法监测反应过程(淋洗液,8:2 v / v,己烷:乙酸乙酯(EtOAc),Rf = 0.6)。
- 反应完成后,在滤纸上过滤混合物(孔径70mm),然后将水(5mL)加入有机滤液中,并使用分离漏斗用CH2Cl2 (2×15 mL)两次提取有机层。
- 干燥超过3.0克无水Na2SO4 的组合有机提取物,并使用旋转蒸发器 真空 浓缩(<15毫巴)。
- 在硅胶(70-230目)上用正相柱层析纯化粗产物,得到纯产物(R)-乙基1-(R)-1-苯乙基)氮丙啶-2-羧酸酯(3)(950mg,88%)。Rf (30% 环氧乙烷/己烷 = 0.50)。
2. 通过叠氮化物亲核试剂进行区域和立体选择性氮丙啶环开口,用于双乙酰胺B和双乙酰胺D的总合成
- 2-叠氮基-3-(R)-1-苯基乙基氨基丙酸乙酯的合成
- 将手性(S)-乙基1-((R)-1-苯乙基-2-羧酸(4)和磁性搅拌棒(4)转移到烤箱干燥的50mL双颈圆底烧瓶中(500mg,2.20mM,1.0当量)。
- 向反应混合物中加入50%乙醇水溶液(15mL)。
- 将反应混合物在0°C下冷却,滴加浓硫酸(36N)以保持接近pH 4.0,搅拌5分钟。
- 在0°C下加入叠氮化钠(370mg,5.70mM,2.5等价物),并让反应混合物在相同温度下搅拌10分钟,然后在室温下加热。
- 然后,在相同的室温下加入AlCl3∙6H2O(55mg,0.22mM,0.1等价物)作为催化剂,并允许再搅拌3小时。
- 使用薄层色谱法监测反应过程(淋洗液,6:4 v / v,己烷:乙酸乙酯(EtOAc),Rf = 0.2)。
- 用两份20毫升饱和的NaHCO3淬灭反应混合物。
- 然后用乙醇(2 x 10 mL)在硅藻土垫上过滤粗混合物。
- 使用旋转蒸发器 真空 (<15 mbar)浓缩反应混合物。
- 使用分离漏斗用CH2Cl2 (2 x 50 mL)提取有机层两次。
- 然后将组合的有机层干燥在5.0克无水Na2SO4 上5分钟。
- 使用旋转蒸发器 真空 (<15 mbar)浓缩粗有机层,以获得粗叠氮化物产品。
- 用硅胶(70-230目)用正相柱层析纯化粗产物,用40%的EtOAc/己烷(Rf = 0.20)洗脱,得到490mg(90%产率)的(R)-乙基2-叠氮基-3-((R)-1-苯乙基)氨基)丙酸酯(5)作为粘稠液体。
- 3-(R)-3-甲基-2,4-二氧代-1-苯基乙基六氢嘧啶-5-氨基)-3-氧代丙基氨基甲酸酯的合成 (7)
- 将150mg手性(R)-5-氨基-3-甲基-1-(R)-1-苯乙基)二氢嘧啶-2,4(1H,3H)-二酮(6)(150mg,0.60mM,1.0等价物)和磁性搅拌棒转移到N2 气氛下烘干的25mL双颈圆底烧瓶中。
- 使用密闭注射器将干燥的CH2 Cl 2(15.0 mL)加入反应瓶中。
- 然后使用冰浴将反应混合物在0°C下冷却,并将反应混合物搅拌5分钟。
- 在0°C下加入Fmoc-β-丙氨酸(377毫克,1.20毫升,2.0当量)和DIPEA(0.67毫升,3.64毫升,6.0等价物),搅拌5分钟。
- 在0°C下向反应混合物中加入EDCI(347毫克,1.82毫米,3.0当量)和HOBt(165毫克,1.21毫米,2.0当量),并在相同温度下搅拌10分钟。
- 将反应混合物保持在室温下,再搅拌8小时。
- 使用薄层色谱法(2:8 v / v,己烷:乙酸乙酯(EtOAc),Rf = 0.4)作为淋洗液来监测反应过程。
- 用水(10毫升)淬灭反应混合物。
- 用盐水(15 mL)洗涤组合的有机层,然后使用分离漏斗用CH2Cl2 (2 x 20 mL)提取有机层两次。
- 然后将组合的有机层干燥超过5.0g无水Na2SO4 5分钟,并使用旋转蒸发器将其 真空 (<15 mbar)浓缩。
- 用硅胶(70-230目)用正相柱层析纯化粗产物,用80%EtOAc /己烷(Rf = 0.40)洗脱,得到295mg的(7)(90%收率)。
3. 立体选择性木海山醛与手性氮丙啶-2-甲醛的反应及其通过内部羟基亲核试剂打开的雷焦和立体选择性氮丙啶环开口,用于(-)- 表皮-索羯碱的总合成 (17)
- 4-羟基-4-1-苯基乙基-2-丁烷的合成 (12)
- 在N2气氛下,将(R)-1-(R)-1-苯乙基)氮丙啶-2-甲醛(10)(140毫克,0.8毫升,1.0当量)和磁力搅拌棒转移到烤箱干燥的25毫升双颈圆底烧瓶中。
- 使用气密注射器将干燥的CH3CN(4.0mL)加入反应瓶中。
- 然后使用冰丙酮浴将反应混合物冷却至-20°C,并将反应混合物搅拌5分钟。
- 在-20 °C下向反应混合物中加入无水ZnCl 2(108mg,0.8mM,1.0等价物),并搅拌5分钟。
- 然后滴加三甲基(丙-1-烯-2-基氧基)硅烷(11)(104mg,0.8mM,1.0等值)溶解在-20°C下的反应混合物中,使反应混合物在相同温度下搅拌1小时。
- 使用薄层色谱法监测反应过程,使用8:2 v / v己烷:乙酸乙酯(EtOAc),Rf = 0.2)作为淋洗液。
- 用饱和的 NaHCO3 (4 mL) 淬灭反应混合物。
- 使用分离漏斗用EtOAc(2 x 15 mL)提取有机层两次。
- 用3.0克无水Na2SO4 干燥组合的有机层,并使用旋转蒸发器将其 真空 (<15毫巴)浓缩。
- 用硅胶(70-230目)用正相柱层析纯化粗产物,用80%的EtOAc/己烷洗脱(Rf = 0.20),得到58mg(S)-4-羟基-4-((R)-1-(R)-1-苯乙基)叠氮丁-2-基)丁烷-2-酮)(12)(85%产率)。
- 1-苯乙胺的合成
- 将(S)-4-(叔丁基二甲基硅氧基)-4-(R)-1-(R)-1-苯乙基)叠氮基丁-2-基)丁烷-2-酮(400毫克,1.15毫米,1.0当量)和磁性搅拌棒在N2 气氛下烘干的25毫升双颈圆底烧瓶中。
- 使用气密注射器将无水THF(50mL)加入反应瓶中。
- 使用干冰丙酮浴将反应混合物在-78°C下冷却,并搅拌5分钟。
- 然后加入三仲丁基硼氢化锂(L-选择剂)(THF中的1M溶液)(2.3mL,2.0等价物)滴入-78°C的反应混合物中,并再次搅拌25分钟。
- 将反应混合物加热至室温,搅拌8小时。
- 使用30%环氧乙烷/己烷(Rf = 0.40)作为淋洗液,通过TLC监测反应的进度。
- 在完全消耗化合物13后,用0.1M的NaOH(5mL)淬灭反应混合物。
- 用环氧乙烷(3 x 15毫升)提取有机层,然后用盐水(15毫升)洗涤。
- 干燥3.0克无水Na2SO4 的有机层,并使用旋转蒸发器 真空 浓缩(<15毫巴)。
- 在硅胶上用正相柱层析纯化粗产物(70-230目,淋洗液,8∶2 v/v,己烷:乙酸乙酯(EtOAc),Rf = 0.2),得到纯化合物(15)(382mg,95%收率)。
- (-)-表皮异毒蕈碱碘化物的合成 (17)
- 将化合物 16 (20mg,0.15mM,1.0等价物)和磁力搅拌棒转移到在N2 气氛下烘干的10mL圆底烧瓶中。
- 使用密闭注射器加入3 mL EtOAc反应瓶。
- 然后在室温下向反应混合物中加入碘甲烷(0.4 mL,3.0mM)。
- 在0°C下向反应混合物中加入1,2,2,6,6-五甲基哌啶(PMP)(0.05毫升,0.3立方米,2.0当量)。
- 然后,将反应混合物保持在室温下,再搅拌16小时。
- 使用旋转蒸发器 真空 蒸发溶剂(<15 mbar),得到粗产物。
- 通过在EtOAc中加入(3 x 5 mL)10%MeOH到粗反应混合物中洗涤三次。
- 然后用 正戊烷(5毫升)洗涤粗混合物,并使用旋转蒸发器在真空(<15毫巴)下浓缩,以获得纯(-)-表皮异毒蕈碱碘化物(17)(32毫克,68%)。
4. 所有产品的表征
- 通过 1 H、13C NMR 光谱和高分辨率质谱 (HRMS)7,8,11 表征所有新化合物。
Access restricted. Please log in or start a trial to view this content.
结果
在这里,我们报道了对映腥基吡啶-2-羧酸盐的合成。(R)-(1R,2S,5R)-2-异丙基-5-甲基环己基1-(R)-1-苯乙基)氮丙啶-2-羧酸酯(2)和(S)-(1R,2S,5R)-2-异丙基-5-甲基环己基1-(R)-1-苯基乙基)氮丙啶-2-羧酸酯(4.1 g, 90%)的异质异构体混合物,由2,3-二溴丙烷(-)-薄荷醇酯和(1R)-苯乙?...
Access restricted. Please log in or start a trial to view this content.
讨论
阿齐啶作为含氮的三元杂环具有巨大的潜力,可用于合成起始马具或中间体,以制备富含氮的有机分子。根据环氮的基团,它们被分类为"活化"和"非活化"氮丙啶,其化学反应性和选择性不同。然而,非常有限的方法可以以光学活性形式制备这种有价值的氮丙啶。
本文的方案描述了一种大规模制备对映腭非活化氮丙啶的方法。首先,将(2 R)和对映体纯薄荷醇酯(2R)?...
Access restricted. Please log in or start a trial to view this content.
披露声明
作者声明,本研究不存在利益冲突。
致谢
该研究得到了韩国国家研究基金会(NRF-2020R1A2C1007102和2021R1A5A6002803)的支持,有机合成新方向中心和HUFS拨款2022。
Access restricted. Please log in or start a trial to view this content.
材料
| Name | Company | Catalog Number | Comments |
| (2R)-1-[(1R)-1-Phenylethyl]-2-aziridinecarboxylic acid (-)-menthol ester, 98% | Sigma-Aldrich | 57054-0 | |
| (2S)-1-[(1R)-1-Phenylethyl]-2-aziridinecarboxylic acid (-)-menthol ester | Sigma-Aldrich | 57051-6 | |
| 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride | TCI | 424331-25 g | CAS No: 25952-53-8 |
| 1,4-Dioxane | SAMCHUN | D0654-1 kg | CAS No: 123-91-1 |
| 1-Hydroxybenzotriazole hydrate | Aldrich | 219-989-7-50 g | CAS No: 123333-53-9 |
| 2,6-Lutidine | Alfa Aesar | A10478-AP, 500 mL | CAS No: 108-48-5 |
| Acetonitrile | SAMCHUN | A0127-18 L | CAS No: 75-05-8 |
| Acetonitrile-d3 | Cambridge Isotope Laboratories, | 15G-744-25 g | CAS No: 2206-26-0 |
| Aluminum chloride hexahydrate | Aldrich | 231-208-1, 500 g | CAS No : 7784-13-6 |
| Bruker AVANCE III HD (400 MHz) spectrometer | Bruker | NA | |
| Chloroform-d | Cambridge Isotope Laboratories, | 100 g | CAS No: 865-49-6 |
| Dichloromethane | SAMCHUN | M0822-18 L | CAS No: 75-09-2 |
| Dimethyl sulfoxide-d6 | Cambridge Isotope Laboratories, | 25 g | CAS No: 2206-27-1 |
| Ethanol | EMSURE | 1009831000,1L | CAS No: 64-17-5 |
| Ethyl acetate | SAMCHUN | E0191-18 L | CAS No: 141-78-6 |
| High resolution mass spectra/MALDI-TOF/TOF Mass Spectrometry | AB SCIEX | 4800 Plus | High resolution mass spectra |
| JASCO P-2000 | JASCO | P-2000 | For optical rotation |
| Lithium aluminum hydride | TCI | L0203-100 g | CAS No: 16853-85-3 |
| L-Selectride, 1 M solution in THF | Acros | 176451000, 100 mL | CAS No: 38721-52-7 |
| Methanol | SAMCHUN | M0585-18 L | CAS No: 67-56-1 |
| N-[(9H-Fluoren-9-ylmethoxy)carbonyl]-β-alanine | TCI | F08825G-5 g | CAS No: 35737-10-1 |
| N-Ethyldiisopropylamine | Aldrich | 230-392-0, 100 mL | CAS No: 7087-68-5 |
| n-Hexane | SAMCHUN | H0114-18 L | CAS No: 110-54-3 |
| Ninhydrin | Alfa Aesar | A10409-250 g | CAS No: 485-47-2 |
| p-Anisaldehyde | aldrich | A88107-5 g | CAS No: 123-11-5 |
| Phosphomolybdic acid hydrate | TCI | P1910-100 g | CAS No: 51429-74-4 |
| Sodium azide | D.S.P | 703301-500 g | CAS No: 26628-22-8 |
| Sodium Hydride 60% dispersion in mineral oil | Sigma-Aldrich | 452912-100 G | CAS No: 7646-69-7 |
| Sodium hydroxide | DUKSAN | A31226-1 kg | CAS No: 1310-73-2 |
| Sodium sulfate | SAMCHUN | S1011-1 kg | CAS No: 7757-82-6 |
| Thin Layer Chromatography (TLC) | Merck | 100390 | |
| Tert-Butyldimethylsilyl trifluoromethanesulfonate, 98% | Aldrich | 274-102-0, 25 g | CAS NO: 69739-34-0 |
| Tetrahydrofuran | SAMCHUN | T0148-18 L | CAS No: 109-99-9 |
| Triethylethylamine | DAEJUNG | 8556-4400-1 L | CAS No: 121-44-8 |
| UV light | Korea Ace Sci | TN-4C | 254 nm |
| Zinc chloride, anhydrous, 98+% | Alfa Aesar | A16281-22100 g | CAS No : 7646-85-7 |
参考文献
- Pitzer, K. S. Strain energies of cyclic hydrocarbons. Science. 101 (2635), 672(1945).
- Dudev, T., Lim, C. Ring strain energies from ab initio calculations. Journal of the American Chemical Society. 120 (18), 4450-4458 (1998).
- D'hooghe, M., Ha, H. -J. Synthesis of 4- to 7-Membered Heterocycles by Ring Expansion. , Springer. Berlin/Heidelberg, Germany. (2016).
- Ranjith, J., Ha, H. -J. Synthetic applications of aziridinium ions. Molecules. 26 (6), 1744(2021).
- Sweeney, J. B. Aziridines: epoxides' ugly cousins. Chemical Society Reviews. 31 (5), 247-258 (2002).
- Stankovic, S., et al. Regioselectivity in the ring opening of non-activated aziridines. Chemical Society Reviews. 41 (2), 643-665 (2012).
- Lee, W. K., Ha, H. -J. Highlights of the chemistry of enantiomerically pure aziridine-2-carboxylates. Aldrichimica Acta. 36 (2), 57-63 (2003).
- Tranchant, M. J., Dalla, V., Jabin, I., Decroix, B. Reaction of vinyl triflates of α-keto esters with primary amines: efficient synthesis of aziridine carboxylates. Tetrahedron. 58 (42), 8425-8432 (2002).
- Ha, H. -J., Jung, J. -H., Lee, W. K. Application of regio- and stereoselective functional group transformation of chiral aziridine-2-carboxylate. Asian Journal of Organic Chemistry. 3 (10), 1020-1035 (2014).
- Kim, Y., et al. Preparation of 2,3-diaminopropionate from ring opening of aziridine-2-carboxylate. Tetrahedron Letters. 46 (25), 4407-4409 (2005).
- Srivastava, N., Macha, L., Ha, H. -J. Total synthesis and stereochemical revision of biemamides B and D. Organic Letters. 21 (22), 8992-8996 (2019).
- Zhang, F., et al. Biemamides A-E, inhibitors of the TGF-β pathway that block the epithelial to mesenchymal transition. Organic Letters. 20 (18), 5529-5532 (2018).
- Srivastava, N., Ha, H. -J. Highly efficient and stereoselective Mukaiyama Aldol reaction with chiral aziridine-2-carboxaldehyde and its synthetic applications. Asian Journal of Organic Chemistry. 11 (1), 2021005671(2021).
- Kempter, I., et al. Synthesis and structural characterization of the isomuscarines. Tetrahedron. 70 (10), 1918-1927 (2014).
- Pirrrung, M. C., DeAmicis, C. V. Total synthesis of the muscarines. Tetrahedron Letters. 29 (2), 159-162 (1988).
Access restricted. Please log in or start a trial to view this content.
转载和许可
请求许可使用此 JoVE 文章的文本或图形
请求许可探索更多文章
This article has been published
Video Coming Soon
版权所属 © 2025 MyJoVE 公司版权所有,本公司不涉及任何医疗业务和医疗服务。