
Aby wyświetlić tę treść, wymagana jest subskrypcja JoVE. Zaloguj się lub rozpocznij bezpłatny okres próbny.
Method Article
Preparation of Enantiopure Non-Activated Aziridines and Synthesis of Biemamide B, D, and epiallo-Isomuscarine
W tym Artykule
Podsumowanie
In this study, we prepare both enantiomers of aziridine-2-carboxylate, which are used in the asymmetric synthesis of alkaloids, including biemamide B and D, and (-)-epiallo-isomuscarine.
Streszczenie
Nitrogen-containing heterocycle aziridines are synthetically very valuable for the preparation of azacyclic and acyclic molecules. However, it is very difficult and laborious to make aziridines in optically pure forms on a large scale to apply asymmetric synthesis of aza compounds. Fortunately, we successfully achieved both enantiomers (2R)- and (2S)-aziridine-2-carboxylates with the electron-donating α-methylbenzyl group at the ring nitrogen as non-activated aziridines. These starting aziridines have two distinct functional groups-highly reactive three-membered ring and versatile carboxylate. They are applicable in ring-opening or ring-transformation with aziridine and in functional group transformation to others from carboxylate. Both of these enantiomers were utilized in the preparation of biologically important amino acyclic and/or aza-heterocyclic compounds in an asymmetric manner. Specifically, this report describes the first expedient asymmetric synthesis of both enantiomers of 5, 6-dihydrouracil-type marine natural products biemamide B and D as potential TGF-β inhibitors. This synthesis consisted of regio- and the stereoselective ring-opening reaction of aziridine-2-carboxylate and subsequent formation of 4-aminoteterahydropyrimidine-2,4-dione. One more example in this protocol dealt a highly stereoselective Mukaiyama reaction of aziridine-2-carboxylate and silyl enol ether, following intramolecular aziridine ring-opening to provide easy and facile access to (-)-epiallo-isomuscarine.
Wprowadzenie
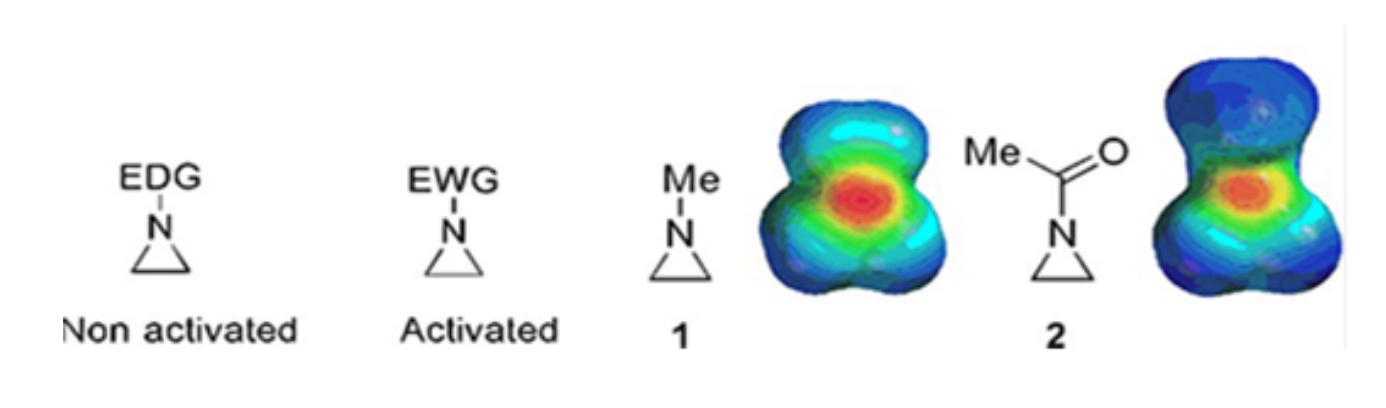
Small rings consisting of cyclopropanes, oxiranes, and aziridines are found in various compounds such as natural products and drugs1,2. They are primarily used as starting materials exploiting their ring strain. Among the three-ring compounds, aziridine has been studied less extensively due to its instability and uncontrollable reactivity3. As shown in the electrostatic potential maps (Figure 1), a group attached to the aziridine ring-nitrogen, whether electron-donating or electron-attracting, makes the basicity of nitrogen different. This difference provides a striking contrast to the reactivity and selectivity of the corresponding aziridines.

Figure 1: Chemical structures of "activated" and "non-activated" aziridines and electrostatic potential maps of their representative examples N-methylaziridine, and N-acetylaziridine4. This figure has been modified with permission from Ranjith et al.4. Please click here to view a larger version of this figure.
{kind=link}
When the ring nitrogen has an electron-withdrawing group, such as sulfonate, phosphonate, and carbamate, we call it "activated" aziridine. This is readily reactive with nucleophiles to compensate for its instability with a limited scope of regiochemistry. These activated aziridines are prepared through various catalytic methods and used as a starting material. Much of recent aziridine chemistry has dealt with these activated aziridines. However, activated aziridines suffer certain restrictions resulting from their instability and limited reaction scope of the ring opening. On the other hand, aziridines bearing electron-donating substituents, like alkyl or substituted alkyl groups, at the ring nitrogen called "non-activated"4, are relatively stable under most circumstances and can be left on the bench for a long time without significant decomposition. The nucleophilic ring-opening reactions of non-activated aziridine occur via the formation of aziridinium ions. Most reactions of aziridine ring-opening and ring transformations proceed in a highly regiochemical manner. However, very few literature reports discuss the preparation of optically pure non-activated aziridines with substituents at the C2 or C3 positions5,6.
This paper shows the successful preparation of α-methylbenzyl group-containing chiral aziridine-2-carboxylate derivatives, specifically (-)-mentholyl (1R)-phenylethylaziridine-2-carboxylates as its diastereomeric mixture, from the reaction of 2,3-dibromopropionate and (1R)-phenylethylamine. From this diastereomeric mixture, enantiopure (1R)-phenylethyl-(2R)- and (2S)-aziridine-2-carboxylates as their (-)-mentholyl esters were obtained in optically pure forms by selective recrystallization from MeOH and n-pentane on multi-hundred-kilo scales (Figure 1)7. These (-)-mentholyl esters can be easily converted into their ethyl or methyl esters by transesterification in the presence of magnesium or potassium carbonate7. These compounds can also be prepared easily on a laboratory scale from the reactions of alkyl 2,3-dibromopropionates or the vinyl triflate of α-ketoester with chiral 2-phenylethylamine followed by separation of the diastereomeric mixture using simple flash column chromatography8.
Once we have enantiopure chiral aziridine-2-carboxylate, we can synthesize various cyclic and acyclic nitrogen-containing biologically important target molecules based on functional group transformations of carboxylate and highly regio- and stereoselective aziridine-ring opening reactions6,9,10. The first expedient asymmetric synthesis was applied for both enantiomers of 5, 6-dihydrouracil-type marine natural products biemamide B and D as potential TGF-β inhibitors11,12. Secondly, the diastereoselective synthesis of β-(aziridin-2-yl)-β-hydroxy ketones was achieved by Mukaiyama aldol reaction of optically pure 1-(1-phenylethyl)-aziridine-2-carboxaldehyde and various enol silanes in the presence of ZnCl2, in high yield (>82%) with almost perfect stereoselectivity (98:2 dr) via a chelation-controlled transition state. These were used for the asymmetric synthesis of epiallo-isomuscarine alkaloids13,14,15.
Access restricted. Please log in or start a trial to view this content.
Protokół
1. Synthesis of the diastereomeric mixture of chiral aziridine (-)-mentholyl ester derivative (1)
- Add 2,3-dibromopropane (-)-menthol ester 1a (5.0 g, 13.58 mM, 1.0 equiv) and a magnetic stirring bar into an oven-dried 250 mL two-neck round-bottom flask under nitrogen (N2) atmosphere.
- Add anhydrous acetonitrile (60 mL) to the reaction flask using an airtight syringe.
- Then cool the reaction mixture at 0 °C using an ice bath and stir the reaction mixture for 5 min.
- Add potassium carbonate (5.6 g, 40.74 mM, 3.0 equiv) into the reaction mixture at the same temperature and allow to stir for 30 min.
- Add (2R)-phenylethylamine (2.0 mL, 16.29 nM, 1.2 equiv) in a dropwise manner at room temperature (RT) and allow the reaction mixture to stir for 12 h.
- Monitor the progress of reaction using thin layer chromatography by using 9:1 v/v hexane:ethyl acetate (EtOAc; Rf = 0.4) as an eluent.
- After the reaction is complete, filter the mixture over filter paper (pore size 70 mm).
- Then, add water (30 mL) into the organic filtrate and extract the organic layer with Et2O (2 x 50 mL) two times using a separating funnel.
- Dry the combined organic extracts over 7.5 g of anhydrous Na2SO4 and concentrate in vacuo (<15 mbar) using a rotary evaporator.
NOTE: Now a crude mixture of diastereomeric chiral aziridine containing both isomers of (R)-(1R,2S,5R)-2-isopropyl-5-methylcyclohexyl1-((R)-1-phenylethyl)aziridine-2-carboxylate and (S)-(1R,2S,5R)-2-isopropyl-5-methylcyclohexyl1-((R)-1-phenylethyl)aziridine-2-carboxylate (4.1 g, 90%) is obtained. - Isolation of chiral aziridine (R)-(1R,2S,5R)-2-isopropyl-5-methylcyclohexyl 1-((R)-1- phenylethyl)aziridine-2-carboxylate (2) by a selective crystallization method
- Add 8.7 g of the crude mixture of chiral aziridine (-)-Menthol ester 1 derivative and dissolve in 70 mL of methanol to an oven-dried 250 mL single-neck round-bottom flask.
- Now warm the reaction mixture up to 70 °C by using a hot water bath, then cool the reaction mixture at -10 °C until solid crystal forms.
- Filter the solid compound over a filter paper (pore size 70 mm) to obtain 2.2 g of (R)-(1R,2S,5R)-2-isopropyl-5-methylcyclohexyl 1-((R)-1-phenylethyl)aziridine-2-carboxylate (2) ester.
- Concentrate the filtrate solution again in vacuo (<15 mbar) using a rotary evaporator, dissolve 50 mL of ethanol in the remaining reaction mixture and recrystallize at -10 °C to obtain 1.2 g of (R)-(1R,2S,5R)-2-isopropyl-5-methylcyclohexyl 1-((R)-1-phenylethyl)aziridine-2-carboxylate (2).
- At this time, use the other alcohol ethanol in the same way as methanol.
- After recrystallization, again filter over a filter paper (pore size 70 mm), concentrate the remaining crude 5.3 g of filtrate solution completely in vacuo (<15 mbar) using a rotary evaporator, and add 50 mL of a pentane hydrocarbon solvent.
- Keep the remaining reaction solution at -15 °C.
NOTE: Now a solid compound of nearly 1.9 g of (S)-(1R,2S,5R)-2-isopropyl-5-methylcyclohexyl1-((R)-1-phenylethyl)aziridine-2-carboxylate ester (3) is obtained. - After obtaining crystals, concentrate the solution again in vacuo (<15 mbar) using a rotary evaporator and dissolve it in 30 mL of pentane hydrocarbon solvent.
- Recrystallize again at -15 °C to obtain 0.8 g of (S)-(1R,2S,5R)-2-isopropyl-5-methylcyclohexyl1-((R)-1-phenylethyl)aziridine-2-carboxylate ester (2').
- Obtaining (R)-ethyl 1-((R)-1-phenylethyl)aziridine-2-carboxylate (3)
- Add (R)-(1R,2S,5R)-2-isopropyl-5-methylcyclohexyl1-((R)-1-phenylethyl)aziridine-2-carboxylate (2) (0.167 g, 0.57 mM) and a magnetic stir-bar into an oven-dried 25 mL two-neck round-bottom flask under nitrogen (N2) atmosphere.
- Add 1.8 mL of ethanol to the reaction flask using an airtight syringe and stir it at RT.
- Then add potassium carbonate (0.40 g, 20.28 mmol, 4.0 equiv) and stir it at RT for 2 days.
- Monitor the progress of reaction using thin layer chromatography (eluent, 8:2 v/v, hexane:ethyl acetate (EtOAc), Rf = 0.6).
- After the reaction is completed, filter the mixture over filter paper (pore size 70 mm), then add water (5 mL) into the organic filtrate and extract the organic layer with CH2Cl2 (2 x 15 mL) two times by using a separating funnel.
- Dry the combined organic extracts over 3.0 g of anhydrous Na2SO4 and concentrate in vacuo (<15 mbar) using a rotary evaporator.
- Purify the crude product by normal phase column chromatography on silica gel (70-230 mesh) to afford a pure product (R)-ethyl 1-((R)-1-phenylethyl)aziridine-2-carboxylate (3) (950 mg, 88%). Rf (30% EtOAc/hexane = 0.50).
2. Regio and stereoselective aziridine ring-opening by azide nucleophile for the total synthesis of biemamide B and biemamide D
- Synthesis of (R)-ethyl 2-azido-3-(((R)-1-phenylethyl)amino)propanoate (5)
- Transfer (500 mg, 2.20 mM, 1.0 equiv) of chiral (S)-ethyl 1-((R)-1-phenylethyl)aziridine-2-carboxylate (4) and a magnetic stir-bar into an oven-dried 50 mL two-neck round-bottom flask under open atmosphere.
- Add 50% aqueous ethanol (15 mL) to the reaction mixture.
- Cool the reaction mixture at 0 °C and add concentrated sulfuric acid (36 N) dropwise to maintain nearly pH 4.0, and stir for 5 min.
- Add sodium azide (370 mg, 5.70 mM, 2.5 equiv) at 0 °C and allow the reaction mixture to stir for 10 min at the same temperature and then warm at RT.
- Then, add AlCl3∙6H2O (55 mg, 0.22 mM, 0.1 equiv) as a catalyst at the same RT and allow to stir for an additional 3 h.
- Monitor the progress of reaction using thin layer chromatography (eluent, 6:4 v/v, hexane:ethyl acetate (EtOAc), Rf = 0.2).
- Quench the reaction mixture with two portions of 20 mL saturated NaHCO3.
- Then filter the crude mixture over a celite pad with ethanol (2 x 10 mL).
- Concentrate the reaction mixture in vacuo (<15 mbar) using a rotary evaporator.
- Extract the organic layer with CH2Cl2 (2 x 50 mL) two times using a separating funnel.
- Then dry the combined organic layer over 5.0 g of anhydrous Na2SO4 for 5 min.
- Concentrate the crude organic layer in vacuo (<15 mbar) using a rotary evaporator to afford a crude azide product.
- Purify the crude product with normal phase column chromatography on silica gel (70-230 mesh) by eluting with 40% EtOAc/hexane (Rf = 0.20) to afford 490 mg (90% yield) of (R)-ethyl 2-azido-3-(((R)-1-phenylethyl)amino)propanoate (5) as a viscous liquid.
- Synthesis of (9H-Fluoren-9-yl)methyl(3-(((R)-3-methyl-2,4-dioxo-1-((R)-1 phenylethyl)hexahydro pyrimidin-5-yl)amino)-3-oxopropyl)carbamate (7)
- Transfer 150 mg of chiral (R)-5-amino-3-methyl-1-((R)-1-phenylethyl) dihydropyrimidine-2,4(1H,3H)-dione (6) (150 mg, 0.60 mM, 1.0 equiv) and a magnetic stir-bar into an oven-dried 25 mL two-neck round-bottom flask under N2 atmosphere.
- Add dry CH2Cl2 (15.0 mL) to the reaction flask using an airtight syringe.
- Then cool the reaction mixture at 0 °C using an ice bath and stir the reaction mixture for 5 min.
- Add Fmoc-beta-alanine (377 mg, 1.20 mM, 2.0 equiv) and DIPEA (0.67 mL, 3.64 mM, 6.0 equiv) at 0 °C and stir it for 5 min.
- Add EDCI (347 mg, 1.82 mM, 3.0 equiv) and HOBt (165 mg, 1.21 mM, 2.0 equiv) to the reaction mixture at 0 °C and allow to stir for 10 min at the same temperature.
- Keep the reaction mixture at RT and allow to stir for an additional 8 h.
- Monitor the progress of reaction using thin layer chromatography using (2:8 v/v, hexane:ethyl acetate (EtOAc), Rf = 0.4) as an eluent.
- Quench the reaction mixture with water (10 mL).
- Wash the combined organic layer with brine (15 mL), then extract the organic layer with CH2Cl2 (2 x 20 mL) two times using a separating funnel.
- Then dry the combined organic layer over 5.0 g of anhydrous Na2SO4 for 5 min and concentrate it in vacuo (<15 mbar) using a rotary evaporator.
- Purify the crude product with normal phase column chromatography on silica gel (70-230 mesh) by eluting with 80% EtOAc/hexane (Rf = 0.40) to afford 295 mg of (7) (90% yield).
3. Stereoselective Mukaiyama aldol reaction with chiral aziridine-2-carboxaldehyde and Its regio and stereoselective aziridine ring-opening by internal hydroxy nucleophile for the total synthesis of (-)- epiallo-somuscarine (17)
- Synthesis of (S)-4-hydroxy-4-((R)-1-((R)-1-phenylethyl)aziridin-2-yl)butan-2-one (12)
- Transfer (R)-1-((R)-1-phenylethyl)aziridine-2-carbaldehyde (10) (140 mg, 0.8 mM, 1.0 equiv) and a magnetic stir-bar into an oven-dried 25 mL two-neck round-bottom flask under N2 atmosphere.
- Add dry CH3CN (4.0 mL) to the reaction flask using an airtight syringe.
- Then cool the reaction mixture at -20 °C using an ice-acetone bath and stir the reaction mixture for 5 min.
- Add ZnCl2 anhydrous (108 mg, 0.8 mM, 1.0 equiv) to the reaction mixture at -20 °C and allow to stir for 5 min.
- Then add trimethyl(prop-1-en-2-yloxy)silane (11) (104 mg, 0.8 mM, 1.0 equiv) dissolved in dry CH3CN (3.0 mL) to the reaction mixture at -20 °C in a dropwise manner and allow the reaction mixture to stir for 1 h at the same temperature.
- Monitor the progress of reaction using thin layer chromatography using 8:2 v/v hexane:ethyl acetate (EtOAc), Rf = 0.2) as an eluent.
- Quench the reaction mixture with saturated NaHCO3 (4 mL).
- Extract the organic layer with EtOAc (2 x 15 mL) two times using a separating funnel.
- Dry the combined organic layer with 3.0 g of anhydrous Na2SO4 and concentrate it in vacuo (<15 mbar) using a rotary evaporator.
- Purify the crude product with normal phase column chromatography on silica gel (70-230 mesh) by eluting with 80% EtOAc/hexane, (Rf = 0.20) to afford 58 mg of (S)-4-hydroxy-4-((R)-1-((R)-1-phenylethyl)aziridin-2-yl)butan-2-one) (12) (85% yield).
- Synthesis of (R)-N-(((2R,3S,5R)-3-((tert-butyldimethylsilyl)oxy)-5-methyltetra hydrofuran-2-yl)methyl)-1-phenylethanamine (15)
- Transfer (S)-4-((tert-butyldimethylsilyl)oxy)-4-((R)-1-((R)-1-phenylethyl) aziridin-2-yl)butan-2-one (13) (400 mg, 1.15 mM, 1.0 equiv) and a magnetic stir-bar into an oven-dried 25 mL two-neck round-bottom flask under N2 atmosphere.
- Add anhydrous THF (50 mL) to the reaction flask using an airtight syringe.
- Cool the reaction mixture at -78 °C using a dry ice-acetone bath and allow to stir for 5 min.
- Then add lithium tri-sec-butylborohydride (L-selectride) (1 M solution in THF) (2.3 mL, 2.0 equiv) dropwise into the reaction mixture at -78 °C and allow to stir for another 25 min.
- Warm the reaction mixture to RT and allow to stir for 8 h.
- Monitor the progress of the reaction by TLC using 30% EtOAc/hexane (Rf = 0.40) as an eluent.
- After the complete consumption of compound 13, quench the reaction mixture with 0.1 M of NaOH (5 mL).
- Extract the organic layer with EtOAc (3 x 15 mL), then wash with brine (15 mL).
- Dry the organic layer over 3.0 g of anhydrous Na2SO4 and concentrate in vacuo (<15 mbar) using a rotary evaporator.
- Purify the crude product with normal phase column chromatography on silica gel (70-230 mesh, eluent, 8:2 v/v, hexane:ethyl acetate (EtOAc), Rf = 0.2) to afford a pure compound (15) (382 mg, 95% yield).
- Synthesis of (-)-epiallo-isomuscarine iodide (17)
- Transfer compound 16 (20 mg, 0.15 mM, 1.0 equiv) and a magnetic stir bar into an oven-dried 10 mL round-bottom flask under N2 atmosphere.
- Add 3 mL of EtOAc reaction flask using an airtight syringe.
- Then add Methyl iodide (0.4 mL, 3.0 mM) to the reaction mixture at RT.
- Add 1,2,2,6,6-pentamethylpiperidine (PMP) (0.05 mL, 0.3 mM, 2.0 equiv) to the reaction mixture at 0 °C.
- Then, keep the reaction mixture at RT and allow to stir for an additional 16 h.
- Evaporate the solvent in vacuo (<15 mbar) using a rotary evaporator to give the crude product.
- Wash three times by adding (3 x 5 mL) 10% MeOH in EtOAc to the crude reaction mixture.
- Then wash the crude mixture with n-Pentane (5 mL) and concentrate under vacuum (<15 mbar) using a rotary evaporator to achieve pure (-)-epiallo-isomuscarine iodide (17) (32 mg, 68%).
4. Characterization of all products
- Characterize all the new compounds by 1H, 13C NMR spectroscopy, and high-resolution mass spectrometry (HRMS)7,8,11.
Access restricted. Please log in or start a trial to view this content.
Wyniki
Here, we report the synthesis of enantiopure aziridine-2-carboxylates. The diastereomeric mixture of (R)-(1R,2S,5R)-2-isopropyl-5-methylcyclohexyl1-((R)-1-phenylethyl)aziridine-2-carboxylate (2) and (S)-(1R,2S,5R)-2-isopropyl-5-methylcyclohexyl1-((R)-1-phenylethyl)aziridine-2-carboxylate (3) (4.1 g, 90%) were prepared in quantitative yield from 2,3 -dibromopropane (-)-mentholyl ester and (1R
Access restricted. Please log in or start a trial to view this content.
Dyskusje
Aziridines as nitrogen-containing three-membered heterocycles have enormous potential for synthetic starting martials or intermediates to prepare nitrogen-rich organic molecules. Based on the group bearing at the ring nitrogen, they are classified as "activated" and "non-activated" aziridines whose chemical reactivity and selectivity are different. However, very limited methods are available to prepare this valuable aziridine in an optically active form.
The protocol in this paper describes a...
Access restricted. Please log in or start a trial to view this content.
Ujawnienia
The authors declare that there was no conflict of interest in this study.
Podziękowania
This research was supported by the National Research Foundation of Korea (NRF-2020R1A2C1007102 and 2021R1A5A6002803) with the Center for New Directions in Organic Synthesis and an HUFS Grant 2022.
Access restricted. Please log in or start a trial to view this content.
Materiały
| Name | Company | Catalog Number | Comments |
| (2R)-1-[(1R)-1-Phenylethyl]-2-aziridinecarboxylic acid (-)-menthol ester, 98% | Sigma-Aldrich | 57054-0 | |
| (2S)-1-[(1R)-1-Phenylethyl]-2-aziridinecarboxylic acid (-)-menthol ester | Sigma-Aldrich | 57051-6 | |
| 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride | TCI | 424331-25 g | CAS No: 25952-53-8 |
| 1,4-Dioxane | SAMCHUN | D0654-1 kg | CAS No: 123-91-1 |
| 1-Hydroxybenzotriazole hydrate | Aldrich | 219-989-7-50 g | CAS No: 123333-53-9 |
| 2,6-Lutidine | Alfa Aesar | A10478-AP, 500 mL | CAS No: 108-48-5 |
| Acetonitrile | SAMCHUN | A0127-18 L | CAS No: 75-05-8 |
| Acetonitrile-d3 | Cambridge Isotope Laboratories, | 15G-744-25 g | CAS No: 2206-26-0 |
| Aluminum chloride hexahydrate | Aldrich | 231-208-1, 500 g | CAS No : 7784-13-6 |
| Bruker AVANCE III HD (400 MHz) spectrometer | Bruker | NA | |
| Chloroform-d | Cambridge Isotope Laboratories, | 100 g | CAS No: 865-49-6 |
| Dichloromethane | SAMCHUN | M0822-18 L | CAS No: 75-09-2 |
| Dimethyl sulfoxide-d6 | Cambridge Isotope Laboratories, | 25 g | CAS No: 2206-27-1 |
| Ethanol | EMSURE | 1009831000,1L | CAS No: 64-17-5 |
| Ethyl acetate | SAMCHUN | E0191-18 L | CAS No: 141-78-6 |
| High resolution mass spectra/MALDI-TOF/TOF Mass Spectrometry | AB SCIEX | 4800 Plus | High resolution mass spectra |
| JASCO P-2000 | JASCO | P-2000 | For optical rotation |
| Lithium aluminum hydride | TCI | L0203-100 g | CAS No: 16853-85-3 |
| L-Selectride, 1 M solution in THF | Acros | 176451000, 100 mL | CAS No: 38721-52-7 |
| Methanol | SAMCHUN | M0585-18 L | CAS No: 67-56-1 |
| N-[(9H-Fluoren-9-ylmethoxy)carbonyl]-β-alanine | TCI | F08825G-5 g | CAS No: 35737-10-1 |
| N-Ethyldiisopropylamine | Aldrich | 230-392-0, 100 mL | CAS No: 7087-68-5 |
| n-Hexane | SAMCHUN | H0114-18 L | CAS No: 110-54-3 |
| Ninhydrin | Alfa Aesar | A10409-250 g | CAS No: 485-47-2 |
| p-Anisaldehyde | aldrich | A88107-5 g | CAS No: 123-11-5 |
| Phosphomolybdic acid hydrate | TCI | P1910-100 g | CAS No: 51429-74-4 |
| Sodium azide | D.S.P | 703301-500 g | CAS No: 26628-22-8 |
| Sodium Hydride 60% dispersion in mineral oil | Sigma-Aldrich | 452912-100 G | CAS No: 7646-69-7 |
| Sodium hydroxide | DUKSAN | A31226-1 kg | CAS No: 1310-73-2 |
| Sodium sulfate | SAMCHUN | S1011-1 kg | CAS No: 7757-82-6 |
| Thin Layer Chromatography (TLC) | Merck | 100390 | |
| Tert-Butyldimethylsilyl trifluoromethanesulfonate, 98% | Aldrich | 274-102-0, 25 g | CAS NO: 69739-34-0 |
| Tetrahydrofuran | SAMCHUN | T0148-18 L | CAS No: 109-99-9 |
| Triethylethylamine | DAEJUNG | 8556-4400-1 L | CAS No: 121-44-8 |
| UV light | Korea Ace Sci | TN-4C | 254 nm |
| Zinc chloride, anhydrous, 98+% | Alfa Aesar | A16281-22100 g | CAS No : 7646-85-7 |
Odniesienia
- Pitzer, K. S. Strain energies of cyclic hydrocarbons. Science. 101 (2635), 672(1945).
- Dudev, T., Lim, C. Ring strain energies from ab initio calculations. Journal of the American Chemical Society. 120 (18), 4450-4458 (1998).
- D'hooghe, M., Ha, H. -J. Synthesis of 4- to 7-Membered Heterocycles by Ring Expansion. , Springer. Berlin/Heidelberg, Germany. (2016).
- Ranjith, J., Ha, H. -J. Synthetic applications of aziridinium ions. Molecules. 26 (6), 1744(2021).
- Sweeney, J. B. Aziridines: epoxides' ugly cousins. Chemical Society Reviews. 31 (5), 247-258 (2002).
- Stankovic, S., et al. Regioselectivity in the ring opening of non-activated aziridines. Chemical Society Reviews. 41 (2), 643-665 (2012).
- Lee, W. K., Ha, H. -J. Highlights of the chemistry of enantiomerically pure aziridine-2-carboxylates. Aldrichimica Acta. 36 (2), 57-63 (2003).
- Tranchant, M. J., Dalla, V., Jabin, I., Decroix, B. Reaction of vinyl triflates of α-keto esters with primary amines: efficient synthesis of aziridine carboxylates. Tetrahedron. 58 (42), 8425-8432 (2002).
- Ha, H. -J., Jung, J. -H., Lee, W. K. Application of regio- and stereoselective functional group transformation of chiral aziridine-2-carboxylate. Asian Journal of Organic Chemistry. 3 (10), 1020-1035 (2014).
- Kim, Y., et al. Preparation of 2,3-diaminopropionate from ring opening of aziridine-2-carboxylate. Tetrahedron Letters. 46 (25), 4407-4409 (2005).
- Srivastava, N., Macha, L., Ha, H. -J. Total synthesis and stereochemical revision of biemamides B and D. Organic Letters. 21 (22), 8992-8996 (2019).
- Zhang, F., et al. Biemamides A-E, inhibitors of the TGF-β pathway that block the epithelial to mesenchymal transition. Organic Letters. 20 (18), 5529-5532 (2018).
- Srivastava, N., Ha, H. -J. Highly efficient and stereoselective Mukaiyama Aldol reaction with chiral aziridine-2-carboxaldehyde and its synthetic applications. Asian Journal of Organic Chemistry. 11 (1), 2021005671(2021).
- Kempter, I., et al. Synthesis and structural characterization of the isomuscarines. Tetrahedron. 70 (10), 1918-1927 (2014).
- Pirrrung, M. C., DeAmicis, C. V. Total synthesis of the muscarines. Tetrahedron Letters. 29 (2), 159-162 (1988).
Access restricted. Please log in or start a trial to view this content.
Przedruki i uprawnienia
Zapytaj o uprawnienia na użycie tekstu lub obrazów z tego artykułu JoVE
Zapytaj o uprawnieniaPrzeglądaj więcej artyków
This article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Wszelkie prawa zastrzeżone