
Se requiere una suscripción a JoVE para ver este contenido. Inicie sesión o comience su prueba gratuita.
Method Article
Plataforma analítica multimodal en un chip de resonancia de plasmón de superficie multiplexada para el análisis de subconjuntos de vesículas extracelulares
En este artículo
Resumen
Este artículo propone una nueva generación de plataformas analíticas multiparamétricas con mayor rendimiento para la caracterización de subconjuntos de vesículas extracelulares. El método se basa en una combinación de métodos de biodetección multiplexada con análisis metrológicos y morfomecánicos por microscopía de fuerza atómica, junto con espectroscopia Raman, para calificar objetivos vesiculares atrapados en un biochip de microarray.
Resumen
Las vesículas extracelulares (EV) son pequeñas vesículas derivadas de membrana producidas por todas las células que van desde 50 a varios cientos de nanómetros de diámetro y se utilizan como medio de comunicación intercelular. Están emergiendo como herramientas diagnósticas y terapéuticas prometedoras para una variedad de enfermedades. Hay dos procesos principales de biogénesis utilizados por las células para producir EV con diferencias en tamaño, composición y contenido. Debido a su alta complejidad en tamaño, composición y origen celular, su caracterización requiere una combinación de técnicas analíticas. Este proyecto implica el desarrollo de una nueva generación de plataformas analíticas multiparamétricas con mayor rendimiento para la caracterización de subpoblaciones de vehículos eléctricos. Para lograr este objetivo, el trabajo parte de la plataforma nanobioanalítica (NBA) establecida por el grupo, que permite una investigación original de EVs basada en una combinación de métodos de biodetección multiplexados con análisis metrológicos y morfomecánicos por microscopía de fuerza atómica (AFM) de objetivos vesiculares atrapados en un biochip de microarray. El objetivo era completar esta investigación EV con un análisis fenotípico y molecular mediante espectroscopia Raman. Estos desarrollos permiten proponer una solución analítica multimodal y fácil de usar para la discriminación de subconjuntos EV en fluidos biológicos con potencial clínico.
Introducción
El creciente interés en la investigación de EV en el diagnóstico y en la terapéutica 1,2,3,4,5, combinado con los desafíos que enfrenta este campo, ha resultado en el desarrollo e implementación de una gran variedad de enfoques y técnicas para cuantificar o caracterizar estas vesículas. Los métodos más utilizados para la identificación de EV son la inmunotransferencia específica de proteínas y la proteómica para confirmar el origen de las EV, la microscopía electrónica de transmisión (TEM) para confirmar su estructura y el análisis de seguimiento de nanopartículas (NTA) para cuantificar su número y distribución de tamaño en un volumen de muestra.
Sin embargo, ninguna de estas técnicas por sí sola proporciona toda la información requerida para caracterizar los subconjuntos de EV. La heterogeneidad inherente de los EV debido a la diversidad en sus propiedades bioquímicas y físicas impide los análisis globales que son confiables y reproducibles, especialmente para los EV contenidos en una mezcla (muestra cruda). Por lo tanto, los métodos de detección y caracterización son necesarios para los VE, tanto individualmente como en general para complementar otros métodos que son más rápidos pero no selectivos6.
La imagen de alta resolución por TEM (o cryoTEM) o AFM permite la determinación de la morfología y metrología de EVs con una resolución nanométrica 7,8,9,10,11,12. Sin embargo, la principal limitación del uso de la microscopía electrónica para objetos biológicos, como los EV, es la necesidad de un vacío para llevar a cabo el estudio que requiere la fijación y deshidratación de la muestra. Tal preparación hace que sea difícil de traducir de las estructuras observadas a la morfología EV en solución. Para evitar esta deshidratación de la muestra, la técnica de crioTEM es la más adecuada para la caracterización de EV13. Es ampliamente utilizado para determinar la ultraestructura de los vehículos eléctricos. El inmunomarcaje de vesículas mediante nanopartículas de oro biofuncionalizadas también permite identificar subpoblaciones específicas de EV y distinguirlas de otras partículas presentes en una muestra biológica compleja. Sin embargo, debido al bajo número de EV analizadas por microscopía electrónica, a menudo es difícil realizar una caracterización que sea representativa de una muestra compleja y heterogénea.
Para revelar esta heterogeneidad de tamaño, la Sociedad Internacional de Vesículas Extracelulares (ISEV) sugiere analizar un número suficiente de imágenes de campo amplio, acompañadas de imágenes más pequeñas, para revelar EV individuales con alta resolución14. AFM es una alternativa a los enfoques ópticos y las técnicas de difracción electrónica para el estudio de EVs. Esta técnica utiliza una punta afilada sostenida por un voladizo flexible que escanea la muestra depositada en un soporte, línea por línea, y ajusta la distancia entre la punta y los elementos presentes a través de un bucle de retroalimentación. Esto permite caracterizar la topografía de la muestra y recolectar información morfomecánica15,16,17,18. Los EVs pueden ser escaneados por AFM ya sea después de ser depositados sobre un sustrato atómicamente plano o después de haber sido capturados sobre un sustrato específico funcionalizado por anticuerpos, péptidos o aptámeros para caracterizar las diversas subpoblaciones18,19. Debido a su capacidad para cuantificar y sondear simultáneamente la estructura, la biomecánica y el contenido biomolecular membranoso de EV dentro de muestras biológicas complejas sin necesidad de pretratamiento, etiquetado o deshidratación, AFM ahora se usa cada vez más para caracterizar EV de manera fina y multiparamétrica en condiciones fisiológicas de temperatura y medio.
Este artículo propone una metodología que utiliza un biochip de oro central capaz de ser (bio)funcionalizado químicamente en un formato multiplexado. Este sustrato es la piedra angular de una potente plataforma analítica que combina la biodetección de subconjuntos de EV por resonancia de plasmones de superficie, y una vez que los EVs son adsorbidos/injertados o inmunocapturados en el chip, AFM permite la caracterización metrológica y morfomecánica de los EVs. Junto con la firma Raman de los subconjuntos EV capturados en el chip, esta plataforma analítica permite la calificación de los EV presentes en muestras biológicas de una manera libre de etiquetas y sin necesidad de pasos preanalíticos. Este documento muestra que la combinación de técnicas poderosas, asistidas por una metodología altamente rigurosa en la preparación de sustratos y adquisición de datos, hace que el análisis EV sea profundo, definitivo y robusto.
El principio del enfoque propuesto es preparar un sustrato de oro, adsorber / injertar o capturar los subtipos de EV, y escanearlos por AFM para estimar el tamaño y la morfología de cada subconjunto de EV. Además, esos EV adsorbidos se analizan mediante espectroscopia Raman. Este sustrato puede, de hecho, presentar tres tipos de interfaces de complejidad creciente: desnudas, químicamente funcionalizadas o microarrays de ligandos. Antes de describir los diferentes pasos del protocolo, se remite a los lectores a la presentación esquemática del enfoque de la plataforma nanobioanalítica (NBA) en la Figura 1, que combina imágenes de resonancia de plasmones de superficie (SPRi), AFM y espectroscopia.

Figura 1: La plataforma NanoBioAnalítica. El enfoque combina (A) imágenes de resonancia de plasmones de superficie, (B) microscopía de fuerza atómica y espectroscopía infrarroja / Raman (nano), todas involucradas en el mismo sustrato: un chip de oro multiplexado. Abreviaturas: NBA = plataforma NanoBioAnalítica; SPRi = resonancia de plasmón de superficie; AFM = microscopía de fuerza atómica; EV = vesícula extracelular. Haga clic aquí para ver una versión más grande de esta figura.
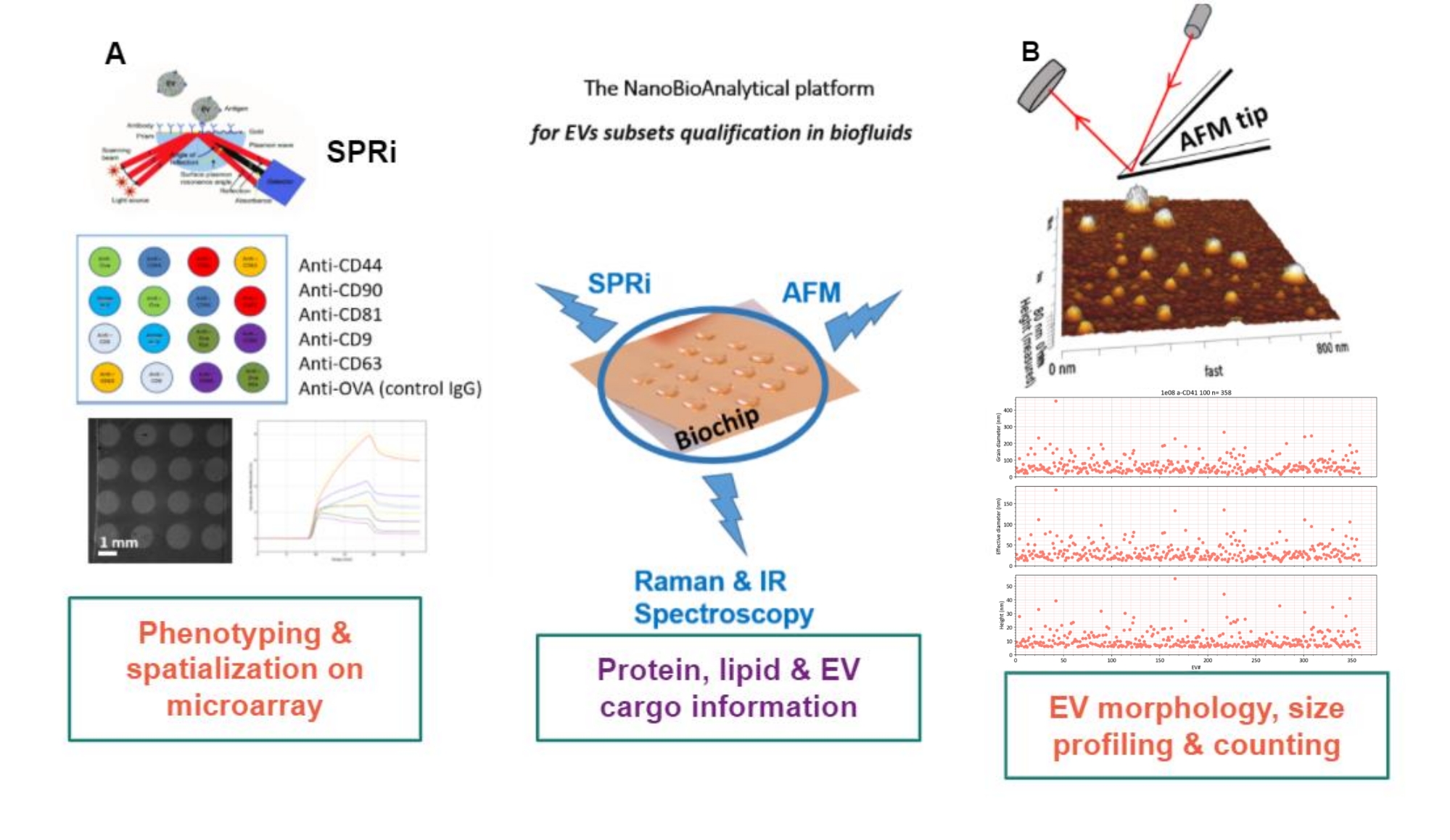{kind=link}
El biochip de oro central constituye el corazón de la plataforma, ya que todas las técnicas de caracterización sin etiquetas se llevan a cabo en este biochip. De acuerdo con las necesidades de la caracterización de EV (ya sea EV globales / totales o subconjuntos de EV) y las limitaciones / demandas de los métodos utilizados, se han desarrollado tres tipos de superficies de biochip de oro: "desnudas", químicamente funcionalizadas "C11 / C16" o ligando-biofuncionalizadas, llamadas superficie de oro "ligando".
El biochip desnudo, llamado "desnudo", permite la simple adsorción de vehículos eléctricos en oro. Es posible seleccionar el tampón utilizado y realizar esta adsorción de forma pasiva (pasos de incubación y luego enjuague) o monitorizarlo bajo flujo (en SPRi). Además, esta adsorción pasiva se puede realizar en todo el chip (como una macromatriz) o localizarse en micromatrices utilizando un observador de micropipetas. El "procedimiento bajo flujo" permite a los investigadores seguir la cinética y el nivel de adsorción EV. Este enfoque en el sustrato de oro desnudo se adopta cuando la interfaz de la capa química puede interferir con el método analítico (por ejemplo, para la espectroscopia Raman).
El biochip químicamente funcionalizado, llamado "C11/C16", se utiliza para crear una "alfombra" densa y robusta de EVs unidos covalentemente en la superficie de oro formando enlaces amida primarios con los tiolatos cuando el objetivo es tener una visión global de la muestra EV. De hecho, en este caso, el oro es funcionalizado por una mezcla de tiolato de mercapto-1-undecanol (11-MUOH: "C11") y ácido mercapto-1-hexadecanoico (16-MHA: "C16"), y una fracción de los tiolatos se activan químicamente para establecer la unión covalente con los objetivos. Una vez más, esta estrategia se puede realizar de forma pasiva (pasos de incubación y luego enjuague, ya sea en "macroarray" o en múltiples microarrays utilizando un observador de micropipetas) o bajo caudales (en SPRi) para seguir la cinética y el nivel de injerto EV en la superficie de oro.
El biochip ligando-biofuncionalizado, llamado "ligandos", se activa químicamente para injertar covalentemente diferentes ligandos (por ejemplo, anticuerpos, receptores) para capturar selectivamente (con afinidad) diferentes subconjuntos de EV que coexisten en la muestra biológica.
Access restricted. Please log in or start a trial to view this content.
Protocolo
1. Preparación del sustrato de oro
NOTA: Se producen tres tipos de superficies en fichas de oro: 1) superficie desnuda, 2) funcionalizada químicamente y 3) biofuncionalizada (ligandos injertados en la capa C11C16). Serán llamados "desnudos", "C11C16" y "ligandos", respectivamente, a partir de este momento.
- Preparación del sustrato de oro:
NOTA: Para este protocolo, los biochips de oro se fabricaron internamente en la sala limpia. Los biochips caseros estaban compuestos por portaobjetos de vidrio (SF11) con un recubrimiento de cromo (2 nm Cr) y oro (48 nm Au). La longitud del biochip era de 28 mm, el ancho era de 12,5 mm y el grosor era de 0,5 mm20.- Utilice la pulverización catódica de magnetrónDC 15 para recubrir los portaobjetos mediante deposición física de vapor (PVD).
- Funcionalización química:
- Funcionalizar las virutas desnudas incubándolas durante la noche en una mezcla de 90%/10% por mol de mercapto-1-undecanol (11-MUOH: C11) y ácido mercapto-1-hexadecanoico (16-MHA: C16) a 1 mM en etanol absoluto, bajo agitación y a temperatura ambiente.
NOTA: Este paso formará una monocapa estable y autoensamblada (SAM), que es útil en el injerto de los ligandos. - Limpie el biochip (lave suavemente) con etanol absoluto y agua ultrapura, séquelo bajo nitrógeno y guárdelo en condiciones de sala limpia.
- Activación del biochip químicamente funcionalizado:
NOTA: A partir de este paso, los experimentos deben realizarse en un laboratorio analítico.- Limpie el biochip con agua ultrapura lavándolo suavemente y luego seque el chip bajo un flujo de aire suave. Para activar los grupos carboxílicos C16, incubar el biochip en una mezcla de 200 mM de etilo (dimetilaminopropil) carbodiimida/N-hidroxisuccinida (EDC) y 50 mmol/L de N-hidroxisuccinimida (Sulfo-NHS) durante al menos 30 minutos en la oscuridad a temperatura ambiente. Luego enjuague con agua antes de los experimentos de injerto.
- Funcionalizar las virutas desnudas incubándolas durante la noche en una mezcla de 90%/10% por mol de mercapto-1-undecanol (11-MUOH: C11) y ácido mercapto-1-hexadecanoico (16-MHA: C16) a 1 mM en etanol absoluto, bajo agitación y a temperatura ambiente.
- Injerto de los ligandos en el biochip funcionalizado:
NOTA: La inmovilización de los ligandos (o EV para algunos experimentos) en el chip puede realizarse pasivamente fuera del instrumento SPRi (incubación de una gota en el chip activado) o dinámicamente bajo flujo en el instrumento SPRi. Esto constituye los chips modificados con EV o ligando. La Figura 2 presenta el biochip de oro, el observador de micropipetas y el biochip después de manchar con 16 gotas de ligando de 300 nL cada una.- Para el injerto de ligandos, use moléculas como anticuerpos (por ejemplo, inmunoglobulinas-antiCD41 [específicas para EV derivadas de plaquetas sanguíneas nativas, llamadas N-PEV], antiCD61, antiCD62P, antiCD9 y antiOVA [anticuerpo de control contra la ovoalbúmina]) y Annexin V. Dilúyalos a 200 μg / ml en una solución ácida (de pH 4.5 a 6 dependiendo del pH óptimo para la actividad o función del ligando).
NOTA: El pH óptimo para el injerto de anticuerpos se determinó previamente mediante experimentos de preconcentración realizados en un instrumento SPR para la determinación del pH óptimo para el injerto de ligando (ver Figura 3). Como las condiciones de injerto cambian con los clones de anticuerpos que se utilizan, se recomienda determinar estas condiciones antes de proceder con los experimentos SPRi. - Para el procedimiento de injerto, agregue 300 nL de EV / solución de ligando utilizando el observador.
NOTA: Un pedazo de papel sumergido en agua debe mantenerse en los pozos izquierdo y derecho para evitar la evaporación de las gotas. Este paso es importante para mantener los EVs/ligandos en sus condiciones óptimas de estabilidad y funcionalidad. - Después de la incubación, mantenga el biochip bajo un baño sónico (frecuencia: 37 kHz, potencia: 30%) durante una incubación de 30 minutos. Lave el biochip desde la parte superior con agua ultrapura y colóquelo suavemente en un prisma con el mismo índice de refractividad (IR) que el biochip. Mientras ajusta el biochip en la parte superior del prisma, agregue una gota (~ 2.3 μL) de aceite con el mismo RI que el prisma para crear una capa delgada y uniforme entre el biochip y el prisma.
NOTA: Este paso garantiza un medio continuo del mismo RI en la ruta óptica. Es importante evitar la incorporación de burbujas en la capa de aceite en este paso, ya que esto cambiará las propiedades ópticas en la trayectoria y dificultará el análisis posterior.
- Para el injerto de ligandos, use moléculas como anticuerpos (por ejemplo, inmunoglobulinas-antiCD41 [específicas para EV derivadas de plaquetas sanguíneas nativas, llamadas N-PEV], antiCD61, antiCD62P, antiCD9 y antiOVA [anticuerpo de control contra la ovoalbúmina]) y Annexin V. Dilúyalos a 200 μg / ml en una solución ácida (de pH 4.5 a 6 dependiendo del pH óptimo para la actividad o función del ligando).

Figura 2: Biochip y observador manual. Biochip de oro (izquierda), observador de micropipetas (centro) y el biochip después de manchar con gotitas de ligando de 300 nL cada una (derecha). Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 3: Pruebas de preconcentración para determinar el pH óptimo para el injerto de ligando. El sensorgrama presenta el nivel de interacción en función del tiempo para un ligando inyectado aleatoriamente (a diferentes valores de pH) a la misma concentración durante 2 min en la superficie. OG es el detergente, que permite recuperar la línea de base entre cada inyección. Aquí, el sensorgrama indica que el pH 6 permitió el mayor injerto de ligando, con una señal SPRi de 1091 RU. Abreviaturas: OG = glucósido octilo; RU = unidad de respuesta. Haga clic aquí para ver una versión más grande de esta figura.
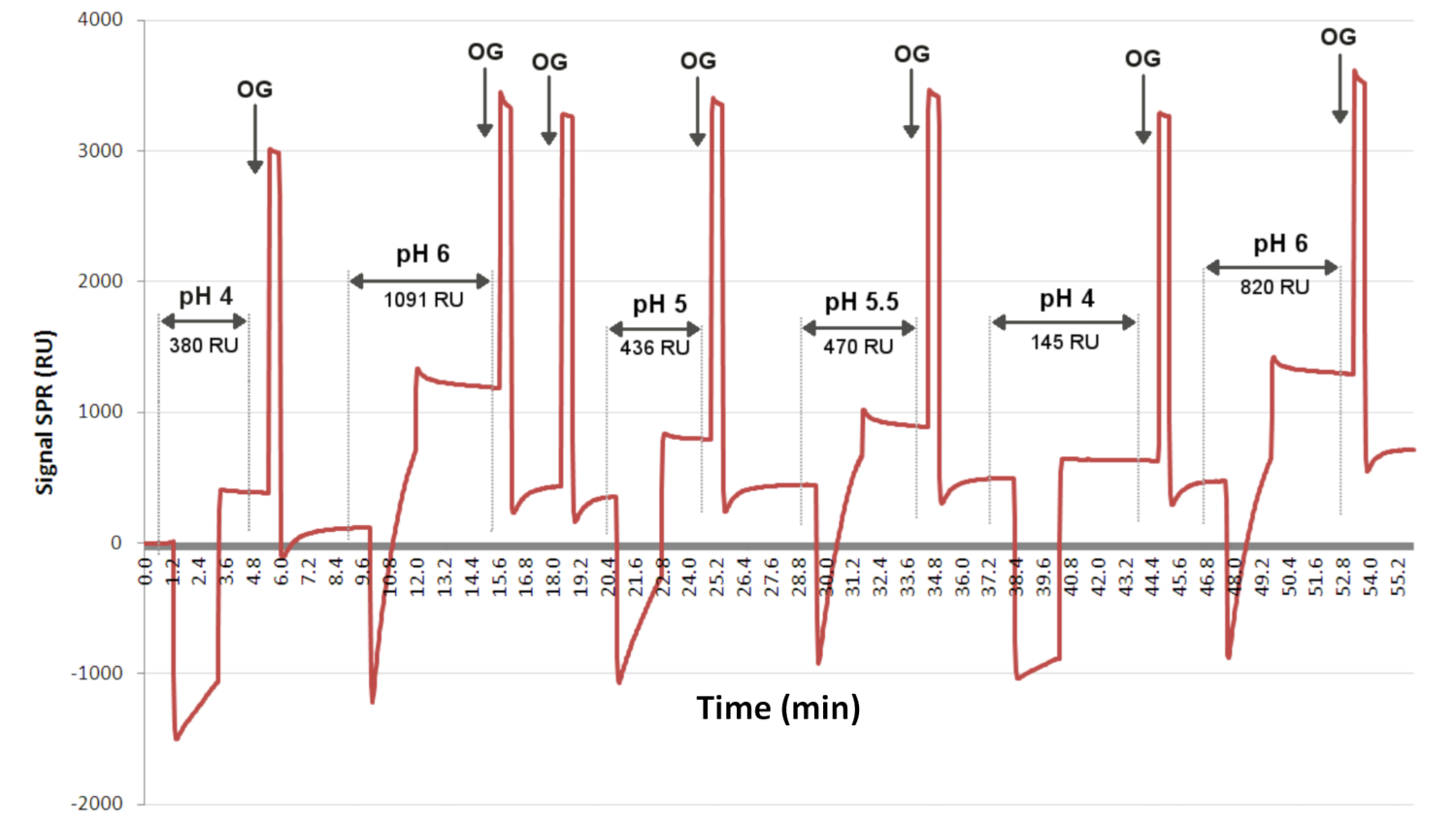{kind=link}
2. Resonancia de plasmones superficiales
- Monte el biochip en el sistema SPRi. Mantenga el caudal del tampón PBS (búfer de funcionamiento) a 50 μL/min.
NOTA: Si hay burbujas, aumente el caudal hasta 500-1,000 μL / min, e inyecte 40 mM de glucósido de octilo (OG) con frecuencia para eliminarlos lo antes posible. - Acondicionamiento del biochip de oro en el instrumento SPRi: Elección de los ángulos de trabajo
- Haga clic en el menú desplegable en el lado izquierdo del software y haga clic en el directorio de trabajo para definir la carpeta donde se guardarán los datos experimentales. Después, haga clic en Plasmon | Adquisición de imágenes.
- Busque la imagen (como se muestra en la Figura 4A) donde hay diferentes puntos visibles y haga clic para seleccionar esta imagen. Para definir la región de interés (ROI), como se representa en la Figura 4, haga clic en detección automática o definición manual dentro de los puntos (Figura 4B) o en el chip para referirse a una ubicación específica incluso sin puntos (Figura 4C).
- Cuando se utilicen biochips multiplexados, escriba el nombre de las familias de ligandos y luego haga clic en los puntos correspondientes.
NOTA: Una familia de ligandos corresponde a varios puntos funcionalizados con el mismo ligando. Por lo general, el chip presenta al menos duplicados e incluso a menudo triplicados de cada ligando. - Haga clic en la definición de la especie final y espere a que se le dirija a la ventana que contiene las curvas de plasmones obtenidas.
- Elija un ángulo de trabajo. Arrastre la línea negra con el cursor al ángulo de trabajo óptimo y haga clic en Mover espejo al ángulo de trabajo.
NOTA: La curva de plasmones consiste en el valor de la reflectividad (%) frente al ángulo, y el software da otra curva con el valor de la pendiente (%) frente al ángulo. Para seleccionar un buen ángulo de trabajo, elija el ángulo con el valor más alto de la pendiente.- En el caso de un paso pasivante (debido a la albúmina) realizado dentro del aparato, seleccione un ángulo de trabajo para obtener la sensibilidad óptima hacia la superficie, estableciendo así un control de calidad de la reactividad de la superficie.
NOTA: Este paso de pasivación es importante cuando el chip está preparado para la biodetección de afinidad/captura para reducir las interacciones no específicas entre la muestra y la superficie del biochip.
- En el caso de un paso pasivante (debido a la albúmina) realizado dentro del aparato, seleccione un ángulo de trabajo para obtener la sensibilidad óptima hacia la superficie, estableciendo así un control de calidad de la reactividad de la superficie.
- Haga clic en Kinetics para iniciar el monitoreo cinético en tiempo real. Una vez que el software solicite al usuario que defina el control negativo, elija ningún control negativo en este punto (ya que esto permitirá la observación de la cinética en los puntos de control negativo ).
- Inyectar albúmina sérica de rata (RSA, 200 μg/ml, preparada en tampón acetato, pH 4,5) a 50 μL/min durante 4 min para pasivar la superficie alrededor de las manchas y posiblemente para llenar espacios vacíos dentro de las manchas del ligando.
NOTA: El RSA se inyecta para cubrir el biochip, que no está unido a ningún ligando. - Inyectar etanolamina (1 M) a 20 μL/ min durante 10 min para desactivar los grupos carboxílicos aún presentes y reactivos en la superficie.
- Lave el biochip inyectando 40 mM OG a 50 μL/ min durante 4 min.
NOTA: Después de la etapa de pasivación, el ángulo de trabajo se ajusta (como una nueva determinación de referencia antes de la inyección de la muestra) para que tenga la sensibilidad más alta en los puntos de interés.
- Inyectar albúmina sérica de rata (RSA, 200 μg/ml, preparada en tampón acetato, pH 4,5) a 50 μL/min durante 4 min para pasivar la superficie alrededor de las manchas y posiblemente para llenar espacios vacíos dentro de las manchas del ligando.
- Inyección de muestra:
- Redefina las curvas de plasmones después de la pasivación, y elija el ángulo de trabajo de acuerdo con el ligando esta vez.
- En la monitorización cinética, reducir el caudal a 20 μL/min y esperar a que la línea de base sea estable.
- Inyecte la muestra a la concentración de su elección (de 1 x 10 8 EVs/mL a 1 x 10 10 EVs/mL dependiendo de la afinidad entre los EVs y los ligandos injertados), y haga clic en inyección manual o inyección automática. En el caso de la inyección manual, después de inyectar 200 μL de la muestra, haga clic en detener la inyección. Como la duración de la inyección es generalmente de 10 minutos, comience a seguir la cinética de la interacción y mida la variación de reflectividad calculando la diferencia en la reflectividad entre el inicio y el final de la inyección observada durante el monitoreo cinético (Figura 5).
NOTA: Las diferentes muestras que se inyectaron se describen en la sección de resultados representativos.
- Después de la inyección de la muestra, siga cualquiera de estos dos enfoques para finalizar el experimento SPRi:
- En el enfoque no fijo / en líquido, saque el biochip del aparato SPRi, agregue una gota de líquido sobre él y proceda a una caracterización adicional de AFM de la superficie en líquido.
- En el enfoque fijo , inyectar glutaraldehído (0,5%) diluido en agua a 20 μL/ min durante 10 min para fijar las moléculas capturadas en el biochip. Inyecte agua para enjuagar la superficie, saque el biochip, lávelo muy suavemente con agua destilada y séquelo al aire para analizarlo más a fondo bajo AFM.

Figura 4: Imagen CCD SPRi del biochip. (A,B) Biochip multiplexado después de la pasivación de la albúmina. (A) Un chip sin defecto; (B) algunos defectos que aparecieron en el chip: fusión de manchas (i), injerto débil (ii) o polvo o "contaminantes" (iii). Los ROIs, en color en las manchas (un color por familia de ligandos), fueron elegidos para evitar esos "contaminantes". Cuando las manchas se fusionaron, se observaron e ignoraron o se nombraron como "mezcla de ligandos 1 y 2". (C) Chip de oro desnudo sin microarrays para el experimento que examina la adsorción de EV en oro. La flecha azul indica la dirección del flujo. Este chip no presentaba manchas, y los ROI fueron elegidos para registrar la señal de reflectividad de la línea 1 (L1, círculos rojos) a la línea 4 (L4, círculos morados) durante la inyección de la muestra. Barra de escala = 1 mm para las tres imágenes. Abreviaturas: SPRi = imágenes de resonancia de plasmones de superficie; CCD = dispositivo de carga acoplada; ROIs = regiones de interés; EVs = vesículas extracelulares. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}

Figura 5: Experimentos SPRi de inyección de EV en un biochip. (A) Experimento de captura en un biochip multiplexado que muestra las señales de reflectividad de diferentes ligandos. Aquí, la relación señal-ruido para los diferentes ligandos fue muy buena (y especialmente en los puntos antiCD41) ya que la respuesta del control negativo fue insignificante. (B) Experimento de adsorción de EVs en un biochip desnudo. Sensorgrama que presenta el acondicionamiento del chip con dos descargas de tampón y limpieza OG (1), con la inyección de la muestra EV (2), y la señal de reflectividad después de la interacción EV (3). En este biochip, no hubo control negativo, pero la señal de reflectividad (su cinética, su estabilidad después de la inyección) fue alta, lo que significa que esos EV pudieron adsorber y permanecer estables en el chip de oro. Abreviaturas: EV = vesícula extracelular; OG = glucósido octilo. Haga clic aquí para ver una versión más grande de esta figura.
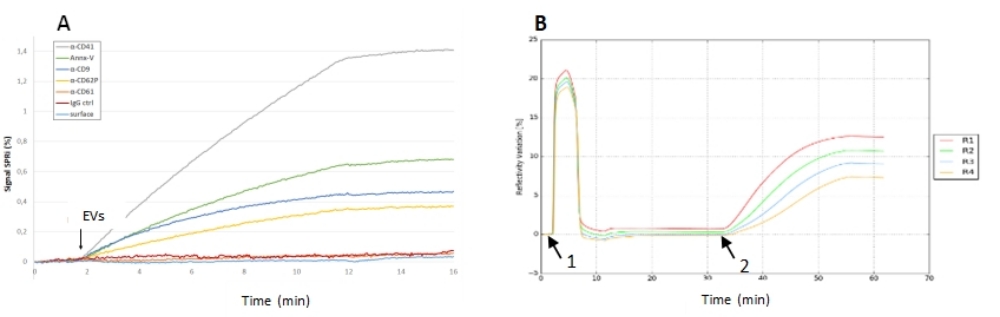{kind=link}
3. Microscopía de fuerza atómica
- Utilice el modo de contacto para escanear el biochip en el aire y el modo de imagen cuantitativa para escanear el biochip en condiciones líquidas.
- Alinee el biochip en la parte superior de la máscara en el portaobjetos de vidrio (desarrollado en el laboratorio) para identificar la posición de los micropuntos respectivos en el biochip (Figura 6A).
NOTA: La máscara contiene las marcas de las manchas, que corresponden a las posiciones de las manchas de los ligandos según el observador utilizado. Esta máscara consiste en un portaobjetos de vidrio sobre el que dos cuñas perpendiculares permiten la colocación del chip. Además, el portaobjetos de vidrio está marcado con 16 puntos de fieltro correspondientes a la localización puntual en el chip, lo que permite, por transparencia, localizar las manchas y escanear el área deseada. - Posicionamiento de la punta:
- Utilice una cámara CCD en la parte superior del AFM para localizar el voladizo en el lugar correcto que debe escanearse. Para seguir este protocolo, utilice voladizos triangulares de 200 μm de longitud, 28 μm de ancho y una constante de resorte de 0,08 N/m.
- Alinee el láser en la parte superior del voladizo en una posición que proporcione una respuesta óptima en el mecanismo de control de retroalimentación.
- Escaneo:
- Una vez activado y en contacto con la superficie del biochip, inicie la adquisición de AFM en modo de contacto o en modo de imagen cuantitativa de tres a cinco áreas grandes (típicamente 10 × 10 μm²) a áreas pequeñas (1 x 1 μm²).
NOTA: En la Figura 6D se muestra una representación de las diferentes áreas que se pueden escanear. Asegúrese de que la caracterización de AFM sea representativa de todo el punto mm² y que se visualicen suficientes EV a una buena resolución para un análisis robusto (un mínimo de 300 EV contados y analizados para cada condición), y realice las mediciones de metrología y morfología.
- Una vez activado y en contacto con la superficie del biochip, inicie la adquisición de AFM en modo de contacto o en modo de imagen cuantitativa de tres a cinco áreas grandes (típicamente 10 × 10 μm²) a áreas pequeñas (1 x 1 μm²).
- Tratamiento de imagen AFM:
- Trate las imágenes AFM con el software de procesamiento de datos JPK seleccionando primero el canal de altura.
- Elija un ajuste polinómico que se restará de cada línea para obtener líneas de escaneo enderezadas.
- Seleccione el umbral de altura en los granos de oro para eliminar la rugosidad de la superficie. En el software al que se hace referencia (consulte la Tabla de materiales), dentro del módulo de extracción de granos, marque los granos utilizando un valor umbral de altura de 8,5 nm. Una vez filtrados los granos, aparece el número de granos .
NOTA: Por lo general, el sustrato de oro en bruto (RMS de ~ 3 nm) y la presencia de las capas químicas y de ligando requieren que el umbral se establezca en 8.5 nm. - Para extraer varias propiedades de los granos marcados, como altura, volumen y diámetro, abra la imagen en el software al que se hace referencia y seleccione Tratamiento de datos | Granos | Marcar por umbral.
- Elija granos de filtro en función de las propiedades. Cuando aparezca una nueva ventana, elija los siguientes parámetros: Valor = máximo z-max, Superficie = superficie proyectada A0. Luego, elija los criterios A Y B.
- Distribución abierta de granos; En la ventana que aparece, elija Valor (máximo), Volumen (base: cero) y Límite (longitud). Observe la tabla (en formato .txt) que aparece, que presenta tres columnas con los valores de altura, volumen y diámetro para todos los granos detectados en el umbral establecido por imagen.
- A partir de la altura, h, y diámetro, D, calcule el radio de curvatura, Rc, de cada EV usando la ecuación (1)8, y luego calcule el volumen, V, usando la ecuación (2):
 (1)
(1) (2)
(2) - A partir de V, calcule el diámetro efectivo, d eff, de cada EV (el diámetro de una esfera con el mismo volumen) usando la ecuación (3):
 (3)
(3) - Trazar gráficos que muestren los tamaños (altura medida, diámetro medido y diámetro efectivo calculado) de los EV, con cada partícula contada representada por un punto.
NOTA: Por lo tanto, al final de la caracterización, la plataforma NBA permite la correlación de la señal de biodetección y luego, el fenotipado, con los números y tamaños de los subconjuntos EV.

Figura 6: Caracterización de biochips por AFM. Después del experimento SPRi, el chip se fijó y secó o se mantuvo en líquido para la caracterización de AFM. (A) El portaobjetos de vidrio mecanizado (con dos cuñas de posicionamiento perpendiculares, indicadas con una "w" en la imagen) que presenta una máscara que encaja con la localización de los 16 microarrays de biochips. Por exposición a la luz y transparencia, una vez instalado para la caracterización de AFM, el portaobjetos de vidrio permite colocar la punta AFM en el lugar deseado para caracterizarlo. (B) El biochip instalado en el portaobjetos de la "máscara" y debajo de una gota de tampón para escanear en condiciones líquidas. (C) Imagen SPRi de los 16 microarrays. (D) Un microarray fotografiado por microscopía óptica después de la inmunocaptura de nanopartículas de calibración biofuncionalizadas de 920 nm de diámetro. Los cuadrados blancos indican el muestreo de las diferentes áreas escaneadas por AFM en cada punto de interés para hacer robusta la caracterización de AFM. Barras de escala = (C) 1 mm, (D) 500 μm. Abreviaturas: AFM = microscopía de fuerza atómica; SPRi = resonancia de plasmón de superficie. Haga clic aquí para ver una versión más grande de esta figura.
{kind=link}
4. Espectroscopia Raman
NOTA: Para la espectroscopia Raman, reemplace el portaobjetos de vidrio utilizado como sustrato con un portaobjetos de CaF2, que tiene una firma Raman insignificante.
- Condiciones ópticas para la adquisición:
- Establezca las siguientes condiciones para el microscopio de imagen Raman: objetivo del microscopio: 50x; longitud de onda del láser: 532 nm; potencia del láser: 10 mW; tiempo de exposición: 500 ms; número de acumulaciones: 140; rango espectral: de 450 cm−1 a 3.200 cm−1.
NOTA: El uso de demasiada potencia y/o un tiempo de adquisición demasiado largo puede provocar daños en la muestra, evidenciados por espectros inestables a lo largo del tiempo. Comience con una cantidad baja de energía y aumente esto si la señal es demasiado débil. Se pueden usar longitudes de onda láser más altas (633 nm, 785 nm) para reducir la fluorescencia perjudicial para las mediciones Raman. Sin embargo, la intensidad disminuye con la cuarta potencia de la longitud de onda, y se debe considerar la sensibilidad espectral de la cámara.
- Establezca las siguientes condiciones para el microscopio de imagen Raman: objetivo del microscopio: 50x; longitud de onda del láser: 532 nm; potencia del láser: 10 mW; tiempo de exposición: 500 ms; número de acumulaciones: 140; rango espectral: de 450 cm−1 a 3.200 cm−1.
- Imágenes Raman:
- Primero, observe los espectros en vivo con un número reducido de acumulaciones (10) para encontrar el área con la mejor relación señal-ruido.
NOTA: Una señal fuerte en la región de alta frecuencia (2,800-3,000 cm−1) puede facilitar la detección de EV en la superficie con tiempos de exposición bajos, como se mostró anteriormente10. - Una vez seleccionado el ROI, elija la resolución espacial de acuerdo con el tiempo disponible para la adquisición.
NOTA: La resolución espacial está limitada por el límite de difracción (~500 nm). - Inicie la adquisición de mapeo Raman.
- Primero, observe los espectros en vivo con un número reducido de acumulaciones (10) para encontrar el área con la mejor relación señal-ruido.
- Preprocesamiento de datos:
- Usando un entorno de desarrollo integrado (IDE) de Python (por ejemplo, Spyder), abra el archivo que contiene los espectros.
- Reste la línea de base de los espectros para corregir la interferencia de la posible fluorescencia. Por ejemplo, utilice la función "arpls" del paquete "irfpy"23. Pruebe diferentes valores del parámetro "lam" para encontrar el que proporcione la mejor corrección de línea de base (generalmente entre 103 y 107).
- Normalizar los espectros, por ejemplo, dividiendo todas las intensidades de un espectro por su intensidad a 2.900 cm−1 o restando la media del espectro y luego dividiéndola por su desviación estándar (normalización "variable normal estándar").
NOTA: Este paso es necesario para comparar la composición molecular relativa de los EV.
Access restricted. Please log in or start a trial to view this content.
Resultados
Determinación de las condiciones óptimas de pH para el injerto de ligando
Los diferentes ligandos utilizados para preparar los biochips se prueban en función del pH y su disponibilidad para interactuar con la capa química de tiolato (Figura 3). Los ligandos se diluyen en tampón acetato a diferentes valores de pH y se inyectan en el biochip químicamente funcionalizado con una capa de C11C16. Las soluciones se inyectan aleatoriamente en la superficie, y se inyecta un ...
Access restricted. Please log in or start a trial to view this content.
Discusión
Los métodos recientes para la identificación de EV que son los más utilizados son el inmunoblotting específico de proteínas para confirmar el origen de los EV, TEM para confirmar su estructura y NTA para cuantificar su número y distribución de tamaño en un volumen de muestra3. Sin embargo, el gran interés en los vehículos eléctricos en la investigación (bio)médica y las limitaciones de las herramientas analíticas existentes han llevado a la comunidad científica a desarrollar nuevos ...
Access restricted. Please log in or start a trial to view this content.
Divulgaciones
Los autores no tienen conflictos de intereses que revelar.
Agradecimientos
Kelly Aubertin y Fabien Picot de la instalación central de IVETh (París) son reconocidos por los experimentos de imágenes Raman. Thierry Burnouf (Universidad Médica de Taipei, Taiwán) y Zuzana Krupova (de Helincourt, Francia) son reconocidos por proporcionar las muestras EV derivadas de muestras de plaquetas sanguíneas y leche bovina, respectivamente. El trabajo fue apoyado por la región Bourgogne Franche-Comté y la escuela de posgrado EUR EIPHI (proyecto NOVICE, 2021-2024). Parte de este trabajo se realizó utilizando la plataforma CLIPP y en las instalaciones de la sala limpia RENATECH en FEMTO-ENGINEERING, por lo que agradecemos a Rabah Zeggari.
Access restricted. Please log in or start a trial to view this content.
Materiales
| Name | Company | Catalog Number | Comments |
| CD41a antibody | Diaclone SAS (France) | 447528 | |
| CP920 | Microparticles GmbH, Germany | 448303 | |
| DXR3xi | Thermo Fisher Scientific | T1502 | |
| EDC | Sigma | A6272 | |
| Ethanolamine | Sigma | P5368-10PAK | |
| Evs derived from platelet concentrates | Collaboration : Pr T. Burnouf (TMU, Taipei) | S2889 | |
| Evs from bovine milk | Collaboration : Dr Z. Krupova (Excilone, Helincourt - France) | 3450 | |
| Glutaraldehyde | Sigma | 56845 | |
| Gwyddion | 853.223.020 | ||
| Magnetron sputtering | PLASSYS | SAB5300165 | |
| mercapto-1-hexadecanoic acid | Sigma | G5882 | |
| Mercapto-1-undecanol | Sigma | O8001 | |
| Mountains SPIP ones | Digital Surf | ||
| NanoWizard 3 Bioscience | Bruker-JPK | ||
| Octyl Glucoside (OG) | Sigma | ||
| Ovalbumine antibody | Sigma | ||
| Phosphate Buffer Saline (PBS) | Sigma | ||
| Rat Albumin Serum (RSA) | Sigma | ||
| Sodium acetate buffer | Sigma | ||
| SPR-Biacore 3000 | GE Healthcare/ Cytiva life sciences | ||
| SPRi Biochip | MIMENTO technology platform | The biochips were produced in-house in the clean room, Besancon | |
| SPRi Plex II | Horiba Scientific | ||
| Sulfo-NHS | Sigma |
Referencias
- Silva, A. K. A., et al. Development of extracellular vesicle-based medicinal products: A position paper of the group "Extracellular Vesicle translatiOn to clinicaL perspectiVEs - EVOLVE France". Advanced Drug Delivery Reviews. 179, 114001(2021).
- Xunian, Z., Kalluri, R. Biology and therapeutic potential of mesenchymal stem cell-derived exosomes. Cancer Science. 111 (9), 3100-3110 (2020).
- Hartjes, T. A., et al. Extracellular vesicle quantification and characterization: Common methods and emerging approaches. Bioengineering. 6 (1), 7(2019).
- Xing, Y., et al. Analysis of extracellular vesicles as emerging theranostic nanoplatforms. Coordination Chemistry Reviews. 424, 213506(2020).
- Wang, T., Xing, Y., Cheng, Z., Yu, F. Analysis of single extracellular vesicles for biomedical applications with especial emphasis on cancer investigations. Trends in Analytical Chemistry. 152, 116604(2022).
- Boireau, W., Elie-Caille, C. Extracellular vesicles: Definition, isolation and characterization. Medecine Sciences: M/S. 37 (12), 1092-1100 (2021).
- Brisson, A. R., et al. Extracellular vesicles from activated platelets: A semiquantitative cryo-electron microscopy and immuno-gold labeling study. Platelets. 28 (3), 263-271 (2017).
- Yuana, Y., et al. Atomic force microscopy: A novel approach to the detection of nanosized blood microparticles. Journal of Thrombosis and Haemostasis. 8 (2), 315-323 (2010).
- Sebaihi, N., de Boeck, B., Yuana, Y., Nieuwland, R., Pétry, J. Dimensional characterization of extracellular vesicles using atomic force microscopy. Measurement Science and Technology. 28 (3), 034006(2017).
- Beekman, P., et al. Immuno-capture of extracellular vesicles for individual multi-modal characterization using AFM, SEM and Raman spectroscopy. Lab on a Chip. 19 (15), 2526-2536 (2019).
- Malenica, M., et al. Perspectives of microscopy methods for morphology characterisation of extracellular vesicles from human biofluids. Biomedicines. 9 (6), 603(2021).
- Verweij, F. J., et al. The power of imaging to understand extracellular vesicle biology in vivo. Nature Methods. 18 (9), 1013-1026 (2021).
- Théry, C., et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. Journal of Extracellular Vesicles. 7 (1), 1535750(2018).
- Obeid, S., et al. NanoBioAnalytical characterization of extracellular vesicles in 75-nm nanofiltered human plasma for transfusion: A tool to improve transfusion safety. Nanomedicine: Nanotechnology, Biology, and Medicine. 20, 101977(2019).
- Obeid, S., et al. Development of a NanoBioAnalytical platform for «on-chip» qualification and quantification of platelet-derived microparticles. Biosensors and Bioelectronics. 93, 250-259 (2017).
- Ridolfi, A., et al. AFM-based high-throughput nanomechanical screening of single extracellular vesicles. Analytical Chemistry. 92 (15), 10274-10282 (2020).
- Vorselen, D., et al. The fluid membrane determines mechanics of erythrocyte extracellular vesicles and is softened in hereditary spherocytosis. Nature Communications. 9 (1), 4960(2018).
- Hardij, J., et al. Characterisation of tissue factor bearing extracellular vesicles with AFM: Comparison of air-tapping-mode AFM and liquid Peak Force AFM. Journal of Extracellular Vesicles. 2, 21045(2013).
- Jorgensen, M., et al. Extracellular Vesicle (EV) Array: Microarray capturing of exosomes and other extracellular vesicles for multiplexed phenotyping. Journal of Extracellular Vesicles. 2, 20920(2013).
- Remy-Martin, F., et al. Surface plasmon resonance imaging in arrays coupled with mass spectrometry (SUPRA-MS): Proof of concept of on-chip characterization of a potential breast cancer marker in human plasma. Analytical and Bioanalytical Chemistry. 404 (2), 423-432 (2012).
- Czamara, K., et al. Raman spectroscopy of lipids: A review. Journal of Raman Spectroscopy. 46 (1), 4-20 (2015).
- Penders, J., et al. Single particle automated Raman trapping analysis of breast cancer cell-derived extracellular vesicles as cancer biomarkers. ACS Nano. 15 (11), 18192-18205 (2021).
- Baek, S. J., Park, A., Ahn, Y. J., Choo, J. Baseline correction using asymmetrically reweighted penalized least squares smoothing. Analyst. 140 (1), 250-257 (2015).
- Daaboul, G. G., et al. Digital detection of exosomes by interferometric imaging. Scientific Reports. 6, 37246(2016).
- Ertsgaard, C. T., et al. Integrated nanogap platform for sub-volt dielectrophoretic trapping and real-time Raman imaging of biological nanoparticles. Nano Letters. 18 (9), 5946-5953 (2018).
- Maas, S. L., et al. Possibilities and limitations of current technologies for quantification of biological extracellular vesicles and synthetic mimics. Journal of Controlled Release. 200, 87-96 (2015).
Access restricted. Please log in or start a trial to view this content.
Reimpresiones y Permisos
Solicitar permiso para reutilizar el texto o las figuras de este JoVE artículos
Solicitar permisoExplorar más artículos
This article has been published
Video Coming Soon
ACERCA DE JoVE
Copyright © 2025 MyJoVE Corporation. Todos los derechos reservados