
Zum Anzeigen dieser Inhalte ist ein JoVE-Abonnement erforderlich. Melden Sie sich an oder starten Sie Ihre kostenlose Testversion.
Method Article
Multimodale Analyseplattform auf einem Multiplex-Oberflächenplasmonen-Resonanztomographie-Chip zur Analyse extrazellulärer Vesikel-Untergruppen
In diesem Artikel
Zusammenfassung
In diesem Artikel wird eine neue Generation multiparametrischer Analyseplattformen mit erhöhtem Durchsatz für die Charakterisierung von extrazellulären Vesikel-Untergruppen vorgeschlagen. Die Methode basiert auf einer Kombination von gemultiplexten Biosensormethoden mit metrologischen und morphomechanischen Analysen durch Rasterkraftmikroskopie, gekoppelt mit Raman-Spektroskopie, um vesikuläre Ziele zu qualifizieren, die auf einem Microarray-Biochip gefangen sind.
Zusammenfassung
Extrazelluläre Vesikel (EVs) sind membranabgeleitete, winzige Vesikel, die von allen Zellen mit einem Durchmesser von 50 bis mehreren hundert Nanometern produziert werden und als Mittel zur interzellulären Kommunikation verwendet werden. Sie entwickeln sich zu vielversprechenden diagnostischen und therapeutischen Instrumenten für eine Vielzahl von Krankheiten. Es gibt zwei Hauptbiogeneseprozesse, die von Zellen verwendet werden, um EVs mit Unterschieden in Größe, Zusammensetzung und Inhalt herzustellen. Aufgrund ihrer hohen Komplexität in Größe, Zusammensetzung und Zellherkunft erfordert ihre Charakterisierung eine Kombination von Analysetechniken. Dieses Projekt beinhaltet die Entwicklung einer neuen Generation von multiparametrischen Analyseplattformen mit erhöhtem Durchsatz für die Charakterisierung von Subpopulationen von Elektrofahrzeugen. Um dieses Ziel zu erreichen, geht die Arbeit von der von der Gruppe etablierten nanobioanalytischen Plattform (NBA) aus, die eine originelle Untersuchung von EVs auf der Grundlage einer Kombination von gemultiplexten Biosensormethoden mit metrologischen und morphomechanischen Analysen durch Rasterkraftmikroskopie (AFM) von vesikulären Targets ermöglicht, die auf einem Microarray-Biochip gefangen sind. Ziel war es, diese EV-Untersuchung mit einer phänotypischen und molekularen Analyse mittels Raman-Spektroskopie abzuschließen. Diese Entwicklungen ermöglichen den Vorschlag einer multimodalen und einfach zu bedienenden analytischen Lösung für die Unterscheidung von EV-Untergruppen in biologischen Flüssigkeiten mit klinischem Potenzial
Einleitung
Das wachsende Interesse an der EV-Forschung in der Diagnose und in der Therapie 1,2,3,4,5, kombiniert mit den Herausforderungen, mit denen dieses Feld konfrontiert ist, hat zur Entwicklung und Implementierung einer Vielzahl von Ansätzen und Techniken zur Quantifizierung oder Charakterisierung dieser Vesikel geführt. Die am weitesten verbreiteten Methoden zur Identifizierung von EVs sind proteinspezifisches Immunblotting und Proteomik, um die Herkunft von EVs zu bestätigen, Transmissionselektronenmikroskopie (TEM), um ihre Struktur zu bestätigen, und Nanopartikel-Tracking-Analyse (NTA), um ihre Anzahl und Größenverteilung in einem Probenvolumen zu quantifizieren.
Keine dieser Techniken allein liefert jedoch alle Informationen, die zur Charakterisierung von EV-Untergruppen erforderlich sind. Die inhärente Heterogenität von Elektrofahrzeugen aufgrund der Vielfalt ihrer biochemischen und physikalischen Eigenschaften erschwert globale Analysen, die zuverlässig und reproduzierbar sind, insbesondere für Elektrofahrzeuge, die in einem Gemisch (Rohprobe) enthalten sind. Detektions- und Charakterisierungsmethoden sind daher für Elektrofahrzeuge sowohl einzeln als auch allgemein erforderlich, um andere Methoden zu ergänzen, die schneller, aber nicht selektiv sind6.
Die hochauflösende Bildgebung mittels TEM (oder KryoTEM) oder AFM ermöglicht die Bestimmung der Morphologie und Metrologie von EVs mit einer nanometrischen Auflösungvon 7,8,9,10,11,12. Die Haupteinschränkung bei der Verwendung der Elektronenmikroskopie für biologische Objekte, wie z. B. Elektrofahrzeuge, ist jedoch die Notwendigkeit eines Vakuums zur Durchführung der Studie, die die Fixierung und Dehydratisierung der Probe erfordert. Eine solche Präparation macht es schwierig, von den beobachteten Strukturen auf die EV-Morphologie in Lösung zu übertragen. Um diese Dehydratisierung der Probe zu vermeiden, ist die Technik der KryoTEM für die EV-Charakterisierung am besten geeignet13. Es wird häufig zur Bestimmung der Ultrastruktur von Elektrofahrzeugen verwendet. Die Immunmarkierung von Vesikeln durch biofunktionalisierte Goldnanopartikel ermöglicht es auch, spezifische Subpopulationen von EVs zu identifizieren und sie von anderen Partikeln zu unterscheiden, die in einer komplexen biologischen Probe vorhanden sind. Aufgrund der geringen Anzahl von EVs, die durch Elektronenmikroskopie analysiert werden, ist es jedoch oft schwierig, eine Charakterisierung durchzuführen, die für eine komplexe und heterogene Probe repräsentativ ist.
Um diese Größenheterogenität aufzudecken, schlägt die International Society for Extracellular Vesicles (ISEV) vor, eine ausreichende Anzahl von Weitfeldbildern zu analysieren, begleitet von kleineren Bildern, um einzelne EVs mit hoher Auflösung zu enthüllen14. AFM ist eine Alternative zu optischen Ansätzen und elektronischen Beugungstechniken für die Untersuchung von Elektrofahrzeugen. Bei dieser Technik wird eine scharfe Spitze verwendet, die von einem flexiblen Ausleger gehalten wird, der die auf einem Träger abgelagerte Probe Zeile für Zeile abtastet und den Abstand zwischen der Spitze und den vorhandenen Elementen durch eine Rückkopplungsschleife anpasst. Dies ermöglicht es, die Topographie der Probe zu charakterisieren und morphomechanische Informationen zu sammeln15,16,17,18. Die EVs können mittels AFM gescannt werden, nachdem sie entweder auf einem atomar flachen Substrat abgeschieden wurden oder nachdem sie auf einem spezifischen Substrat eingefangen wurden, das durch Antikörper, Peptide oder Aptamere funktionalisiert wurde, um die verschiedenen Subpopulationen zu charakterisieren18,19. Aufgrund seiner Fähigkeit, die Struktur, die Biomechanik und den membranösen biomolekularen Gehalt von EVs in komplexen biologischen Proben zu quantifizieren und gleichzeitig zu untersuchen, ohne dass eine Vorbehandlung, Markierung oder Dehydratisierung erforderlich ist, wird AFM nun zunehmend zur feinen und multiparametrischen Charakterisierung von EVs unter physiologischen Bedingungen von Temperatur und Medium eingesetzt.
In diesem Artikel wird eine Methodik vorgeschlagen, bei der ein Kern-Gold-Biochip verwendet wird, der in einem Multiplex-Format (bio)chemisch funktionalisiert werden kann. Dieses Substrat ist der Eckpfeiler einer leistungsstarken analytischen Plattform, die die biologische Detektion von EV-Untergruppen durch Oberflächenplasmonenresonanz kombiniert, und sobald die EVs auf dem Chip adsorbiert/gepfropft oder immuneingefangen sind, ermöglicht AFM die metrologische und morphomechanische Charakterisierung der EVs. In Verbindung mit der Raman-Signatur der auf dem Chip erfassten EV-Untergruppen ermöglicht diese Analyseplattform die Qualifizierung der in biologischen Proben vorhandenen EVs markierungsfrei und ohne präanalytische Schritte. Dieses Papier zeigt, dass die Kombination aus leistungsstarken Techniken, unterstützt durch eine sehr strenge Methodik bei der Substratvorbereitung und Datenerfassung, die EV-Analyse tiefgreifend, definitiv und robust macht.
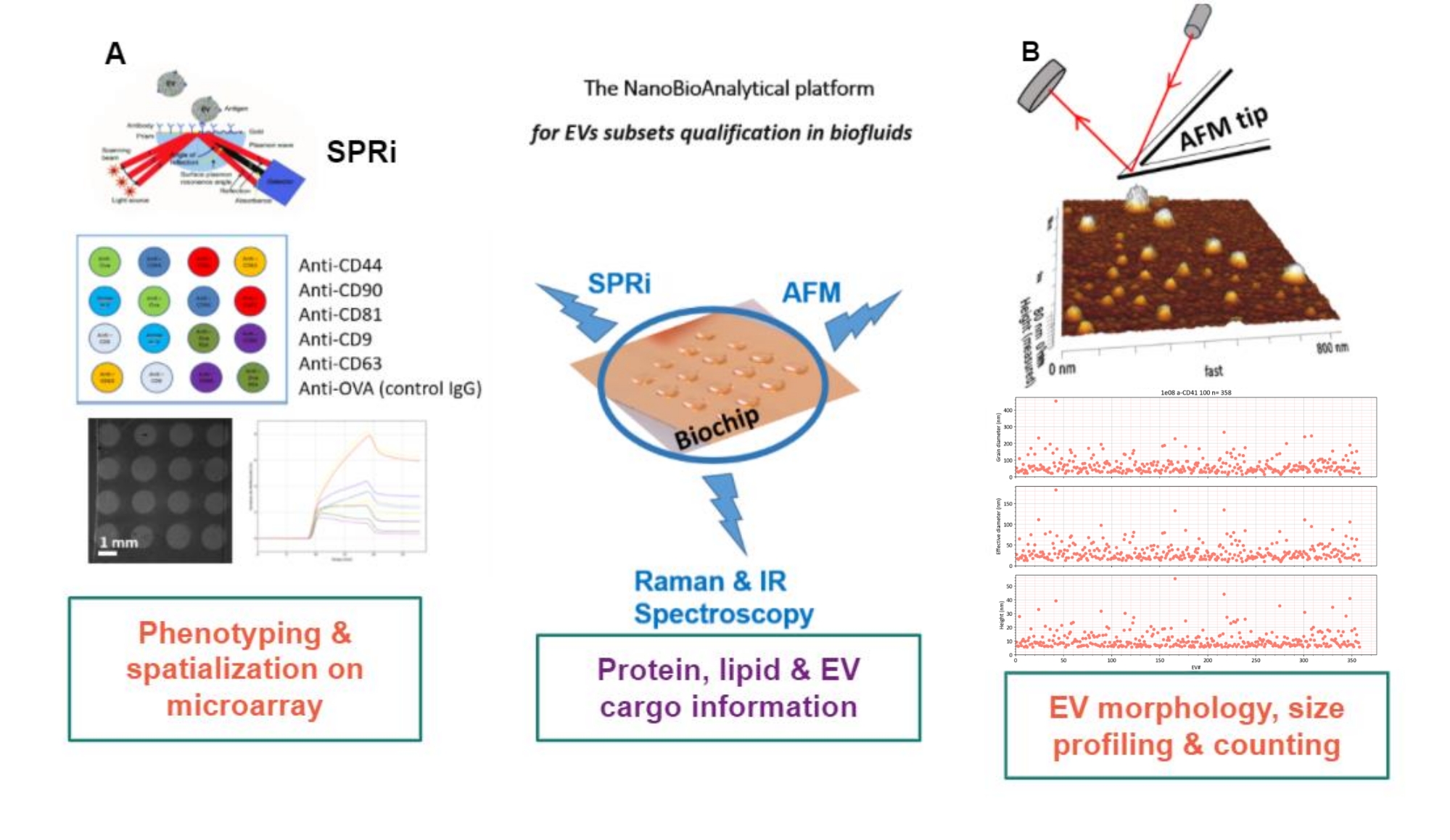
Das Prinzip des vorgeschlagenen Ansatzes besteht darin, ein Goldsubstrat herzustellen, die EV-Subtypen zu adsorbieren/pfropfen oder einzufangen und sie mit AFM zu scannen, um die Größe und Morphologie jeder EV-Untergruppe abzuschätzen. Zusätzlich werden diese adsorbierten EVs mittels Raman-Spektroskopie analysiert. Dieses Substrat kann in der Tat drei Arten von Grenzflächen mit wachsender Komplexität aufweisen: nackte, chemisch funktionalisierte oder Liganden-Microarrays. Bevor die verschiedenen Schritte des Protokolls beschrieben werden, wird auf die schematische Darstellung des Ansatzes der nanobioanalytischen Plattform (NBA) in Abbildung 1 verwiesen, der Oberflächenplasmonenresonanztomographie (SPRi), AFM und Spektroskopie kombiniert.

Abbildung 1: Die NanoBioAnalytical Plattform. Der Ansatz kombiniert (A) Oberflächenplasmonenresonanztomographie, (B) Rasterkraftmikroskopie und Infrarot/Raman-(Nano-)Spektroskopie, die alle auf demselben Substrat - einem Multiplex-Goldchip - eingesetzt werden. Abkürzungen: NBA = NanoBioAnalytical platform; SPRi = Oberflächenplasmonen-Resonanztomographie; AFM = Rasterkraftmikroskopie; EV = extrazelluläres Vesikel. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
Der Kern-Gold-Biochip bildet das Herzstück der Plattform, da alle markierungsfreien Charakterisierungstechniken auf diesem Biochip durchgeführt werden. Entsprechend den Anforderungen der EV-Charakterisierung (entweder globale/totale EVs oder EV-Untergruppen) und den Einschränkungen/Anforderungen der verwendeten Methoden wurden drei Arten von Gold-Biochip-Oberflächen entwickelt: entweder "nackte", chemisch funktionalisierte "C11/C16" oder ligandenbiofunktionalisierte, als "Liganden"-Goldoberfläche bezeichnete Goldoberfläche.
Der nackte Biochip, "nackt" genannt, ermöglicht die einfache Adsorption von Elektrofahrzeugen auf Gold. Es ist möglich, den verwendeten Puffer auszuwählen und diese Adsorption entweder passiv (Inkubations- und Spülschritte) oder unter Durchfluss (in SPRi) zu überwachen. Darüber hinaus kann diese passive Adsorption entweder auf dem gesamten Chip (als Makroarray) oder in Microarrays mit einem Mikropipetten-Spotter lokalisiert werden. Das "Under Flow-Verfahren" ermöglicht es den Ermittlern, die Kinetik und das Ausmaß der EV-Adsorption zu verfolgen. Dieser Ansatz auf dem nackten Goldsubstrat wird angewendet, wenn die chemische Schichtgrenzfläche die analytische Methode stören kann (z. B. für die Raman-Spektroskopie).
Der chemisch funktionalisierte Biochip mit der Bezeichnung "C11/C16" wird verwendet, um einen dichten und robusten "Teppich" aus EVs zu erzeugen, die kovalent an der Goldoberfläche gebunden sind, indem primäre Amidbindungen mit den Thiolaten gebildet werden, wenn das Ziel darin besteht, eine globale Sicht auf die EV-Probe zu erhalten. Tatsächlich wird das Gold in diesem Fall durch eine Thiolatmischung aus Mercapto-1-undecanol (11-MUOH: "C11") und Mercapto-1-hexadecansäure (16-MHA: "C16") funktionalisiert, und ein Teil der Thiolate wird chemisch aktiviert, um eine kovalente Bindung an die Ziele herzustellen. Auch diese Strategie kann entweder passiv (Inkubations- und anschließende Spülschritte, entweder in "Makroarrays" oder in mehreren Mikroarrays mit einem Mikropipetten-Spotter) oder unter Durchflussraten (in SPRi) realisiert werden, um die Kinetik und den Grad der EV-Pfropfung auf der Goldoberfläche zu verfolgen.
Der liganden-biofunktionalisierte Biochip, der als "Liganden" bezeichnet wird, wird chemisch aktiviert, um verschiedene Liganden (z. B. Antikörper, Rezeptoren) kovalent zu transplantieren, um selektiv (mit Affinität) verschiedene EV-Untergruppen zu erfassen, die in der biologischen Probe koexistieren.
Access restricted. Please log in or start a trial to view this content.
Protokoll
1. Vorbereitung des Goldsubstrats
HINWEIS: Drei Arten von Oberflächen werden auf Goldchips hergestellt: 1) nackte Oberfläche, 2) chemisch funktionalisiert und 3) biofunktionalisiert (Liganden, die auf die C11C16-Schicht gepfropft sind). Sie werden ab diesem Zeitpunkt als "nackt", "C11C16" bzw. "Liganden" bezeichnet.
- Vorbereitung des Goldsubstrats:
HINWEIS: Für dieses Protokoll wurden die Gold-Biochips im eigenen Haus im Reinraum hergestellt. Die selbstgebauten Biochips bestanden aus Objektträgern (SF11) mit einer Beschichtung aus Chrom (2 nm Cr) und Gold (48 nm Au). Die Länge des Biochips betrug 28 mm, die Breite 12,5 mm und die Dicke 0,5 mm20.- Verwenden Sie DC-Magnetron-Sputtern15 , um die Objektträger durch physikalische Gasphasenabscheidung (PVD) zu beschichten.
- Chemische Funktionalisierung:
- Die nackten Chips werden funktionalisiert, indem sie über Nacht in einer Mischung aus Mercapto-1-Undecanol (11-MUOH: C11) und Mercapto-1-Hexadecansäure (16-MHA: C16) bei 1 mM in absolutem Ethanol, unter Rühren und bei Raumtemperatur inkubiert werden.
HINWEIS: Dieser Schritt bildet eine stabile, selbstorganisierte Monoschicht (SAM), die beim Pfropfen der Liganden nützlich ist. - Reinigen Sie den Biochip (schonend waschen) mit absolutem Ethanol und Reinstwasser, trocknen Sie ihn unter Stickstoff und lagern Sie ihn unter Reinraumbedingungen.
- Aktivierung des chemisch funktionalisierten Biochips:
HINWEIS: Ab diesem Schritt müssen die Experimente in einem analytischen Labor durchgeführt werden.- Reinigen Sie den Biochip mit Reinstwasser, indem Sie ihn vorsichtig waschen, und trocknen Sie den Chip dann unter sanftem Luftstrom. Um die C16-Carboxylgruppen zu aktivieren, wird der Biochip in einer Mischung aus 200 mM Ethyl(dimethylaminopropyl)carbodiimid/N-Hydroxysuccinimid (EDC) und 50 mmol/L N-Hydroxysuccinimid (Sulfo-NHS) mindestens 30 min im Dunkeln bei Raumtemperatur inkubiert. Dann vor den Pfropfexperimenten mit Wasser abspülen.
- Die nackten Chips werden funktionalisiert, indem sie über Nacht in einer Mischung aus Mercapto-1-Undecanol (11-MUOH: C11) und Mercapto-1-Hexadecansäure (16-MHA: C16) bei 1 mM in absolutem Ethanol, unter Rühren und bei Raumtemperatur inkubiert werden.
- Pfropfen der Liganden auf den funktionalisierten Biochip:
HINWEIS: Die Immobilisierung der Liganden (oder EVs für einige Experimente) auf dem Chip kann entweder passiv außerhalb des SPRi-Instruments (Inkubation eines Tropfens auf dem aktivierten Chip) oder dynamisch in das SPRi-Instrument erfolgen. Dies stellt die EV- oder ligandenmodifizierten Chips dar. Abbildung 2 zeigt den Gold-Biochip, den Mikropipetten-Spotter und den Biochip nach der Spotting mit 16 Ligandentröpfchen von jeweils 300 nL.- Verwenden Sie für die Ligandentransplantation Moleküle wie Antikörper (z. B. Immunglobuline-AntiCD41 [spezifisch für EVs, die aus nativen Blutplättchen gewonnen werden, sogenannte N-PEVs], AntiCD61, AntiCD62P, AntiCD9 und AntiOVA [Kontrollantikörper gegen Ovalbumin]) und Annexin V. Verdünnen Sie sie mit 200 μg/ml in einer sauren Lösung (von pH 4,5 bis 6, abhängig vom optimalen pH-Wert für die Ligandenaktivität oder -funktion).
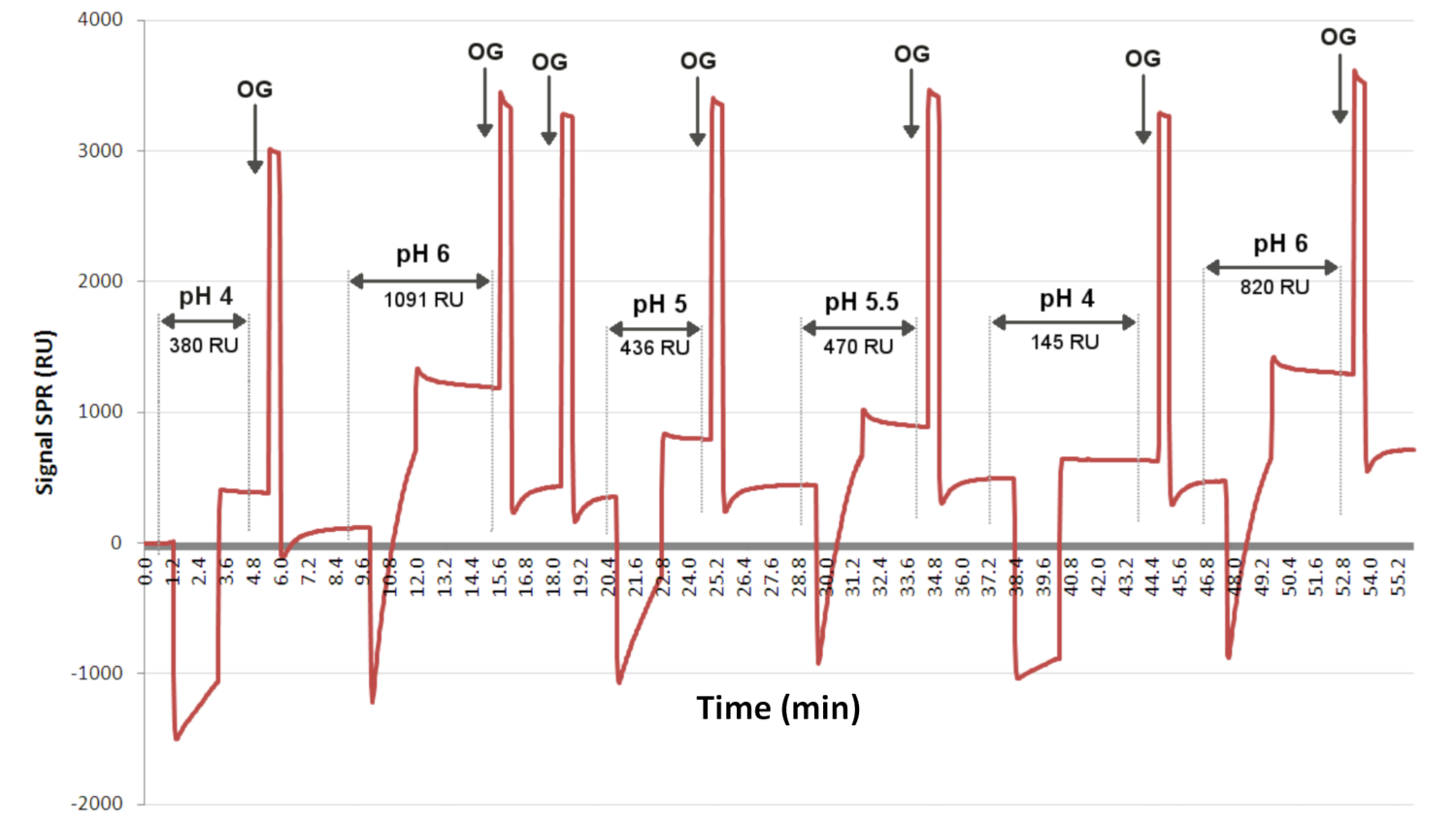
HINWEIS: Der optimale pH-Wert für die Transplantation von Antikörpern wurde zuvor durch Vorkonzentrationsexperimente bestimmt, die mit einem SPR-Gerät zur Bestimmung des optimalen pH-Werts für die Ligandentransplantation durchgeführt wurden (siehe Abbildung 3). Da sich die Pfropfbedingungen mit den verwendeten Antikörperklonen ändern, wird empfohlen, diese Bedingungen zu bestimmen, bevor mit den SPRi-Experimenten fortgefahren wird. - Für den Pfropfvorgang werden 300 nL EVs/Ligandenlösung mit dem Spotter hinzugefügt.
HINWEIS: Ein in Wasser getauchtes Stück Papier sollte sowohl in der linken als auch in der rechten Vertiefung aufbewahrt werden, um die Verdunstung von Tröpfchen zu vermeiden. Dieser Schritt ist wichtig, um die EVs/Liganden in ihren optimalen Bedingungen für Stabilität und Funktionalität zu erhalten. - Bewahren Sie den Biochip nach dem Spotting für eine 30-minütige Inkubationszeit unter einem Schallbad (Frequenz: 37 kHz, Leistung: 30 %) auf. Waschen Sie den Biochip von oben mit Reinstwasser und legen Sie ihn vorsichtig auf ein Prisma mit dem gleichen Brechungsindex (RI) wie der Biochip. Fügen Sie beim Einstellen des Biochips auf der Oberseite des Prismas ein Tröpfchen (~ 2,3 μl) Öl mit dem gleichen RI wie das Prisma hinzu, um eine gleichmäßige, dünne Schicht zwischen dem Biochip und dem Prisma zu erzeugen.
HINWEIS: Dieser Schritt stellt sicher, dass ein kontinuierliches Medium desselben RI im Strahlengang vorhanden ist. Es ist wichtig, bei diesem Schritt keine Blasen in die Ölschicht einzubauen, da dies die optischen Eigenschaften im Pfad verändert und die weitere Analyse behindert.
- Verwenden Sie für die Ligandentransplantation Moleküle wie Antikörper (z. B. Immunglobuline-AntiCD41 [spezifisch für EVs, die aus nativen Blutplättchen gewonnen werden, sogenannte N-PEVs], AntiCD61, AntiCD62P, AntiCD9 und AntiOVA [Kontrollantikörper gegen Ovalbumin]) und Annexin V. Verdünnen Sie sie mit 200 μg/ml in einer sauren Lösung (von pH 4,5 bis 6, abhängig vom optimalen pH-Wert für die Ligandenaktivität oder -funktion).

Abbildung 2: Biochip und manueller Spotter. Gold-Biochip (links), Mikropipetten-Spotter (Mitte) und der Biochip nach Spotting mit Ligandentröpfchen von jeweils 300 nL (rechts). Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

Abbildung 3: Vorkonzentrationstests zur Bestimmung des optimalen pH-Werts für die Ligandentransplantation. Das Sensorgramm stellt den Grad der Wechselwirkung als Funktion der Zeit für einen Liganden dar, der zufällig (bei unterschiedlichen pH-Werten) in der gleichen Konzentration über 2 Minuten auf die Oberfläche injiziert wurde. OG ist das Detergens, das es ermöglicht, die Grundlinie zwischen jeder Injektion wiederherzustellen. Hier zeigt das Sensorgramm an, dass pH 6 mit einem SPRi-Signal von 1091 RU die meisten Ligandentransplantationen ermöglichte. Abkürzungen: OG = Octylglucosid; RU = Antworteinheit. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
2. Oberflächenplasmonen-Resonanztomographie
- Montieren Sie den Biochip auf dem SPRi-System. Halten Sie die Durchflussrate des PBS-Puffers (Laufpuffer) bei 50 μl/min.
HINWEIS: Wenn Blasen vorhanden sind, erhöhen Sie die Flussrate auf 500-1.000 μl / min und injizieren Sie häufig 40 mM Octylglucosid (OG), um sie so schnell wie möglich zu entfernen. - Konditionierung des Gold-Biochips im SPRi-Instrument: Wahl der Arbeitswinkel
- Klicken Sie auf das Dropdown-Menü auf der linken Seite der Software und klicken Sie auf Arbeitsverzeichnis , um den Ordner zu definieren, in dem die experimentellen Daten gespeichert werden sollen. Klicken Sie anschließend auf Plasmon | Bildaufnahme.
- Suchen Sie das Bild (wie in Abbildung 4A gezeigt), auf dem verschiedene Punkte sichtbar sind, und klicken Sie, um dieses Bild auszuwählen. Um die Region of Interest (ROI) zu definieren, wie in Abbildung 4 dargestellt, klicken Sie entweder auf automatische Erkennung oder manuelle Definition innerhalb der Spots (Abbildung 4B) oder auf den Chip, um auch ohne Spots auf einen bestimmten Ort zu verweisen (Abbildung 4C).
- Wenn Multiplex-Biochips verwendet werden, schreiben Sie den Namen der Ligandenfamilien und klicken Sie dann auf die entsprechenden Stellen.
HINWEIS: Eine Ligandenfamilie entspricht mehreren Punkten, die mit demselben Liganden funktionalisiert sind. In der Regel weist der Chip mindestens Duplikate und sogar oft Verdreifachungen jedes Liganden auf. - Klicken Sie auf Speziesdefinition beenden und warten Sie, bis Sie zu dem Fenster mit den erhaltenen Plasmonenkurven weitergeleitet werden.
- Wählen Sie einen Arbeitswinkel. Ziehen Sie die schwarze Linie mit dem Cursor auf den optimalen Arbeitswinkel und klicken Sie auf Spiegel in Arbeitswinkel verschieben.
HINWEIS: Die Plasmonenkurve besteht aus dem Wert des Reflexionsvermögens (%) in Abhängigkeit vom Winkel, und die Software gibt eine weitere Kurve mit dem Wert der Steigung (%) in Abhängigkeit vom Winkel aus. Um einen guten Arbeitswinkel auszuwählen, wählen Sie den Winkel mit dem höchsten Wert der Neigung.- Im Falle eines Passivierungsschritts (wegen Albumin), der innerhalb der Vorrichtung durchgeführt wird, wählen Sie einen Arbeitswinkel , um die optimale Empfindlichkeit gegenüber der Oberfläche zu erhalten und so eine Qualitätskontrolle der Oberflächenreaktivität zu etablieren.
HINWEIS: Dieser Passivierungsschritt ist wichtig, wenn der Chip für die Affinitäts-/Capture-Biodetektion vorbereitet wird, um unspezifische Wechselwirkungen zwischen der Probe und der Biochip-Oberfläche zu reduzieren.
- Im Falle eines Passivierungsschritts (wegen Albumin), der innerhalb der Vorrichtung durchgeführt wird, wählen Sie einen Arbeitswinkel , um die optimale Empfindlichkeit gegenüber der Oberfläche zu erhalten und so eine Qualitätskontrolle der Oberflächenreaktivität zu etablieren.
- Klicken Sie auf Kinetik, um die kinetische Echtzeitüberwachung zu starten. Sobald die Software den Benutzer auffordert, die Negativkontrolle zu definieren, wählen Sie an dieser Stelle keine Negativkontrolle (da dies die Beobachtung der Kinetik an den Negativkontrollpunkten ermöglicht).
- Injizieren Sie Rattenserumalbumin (RSA, 200 μg/ml, hergestellt in Acetatpuffer, pH 4,5) bei 50 μl/min für 4 Minuten , um die Oberfläche um die Flecken herum zu passivieren und möglicherweise leere Räume innerhalb der Ligandenflecken zu füllen.
HINWEIS: Der RSA wird injiziert, um den Biochip zu bedecken, der nicht an Liganden gebunden ist. - Injizieren Sie Ethanolamin (1 M) mit 20 μl/min für 10 Minuten , um die noch vorhandenen und reaktiven Carboxylgruppen auf der Oberfläche zu deaktivieren.
- Waschen Sie den Biochip, indem Sie 40 mM OG bei 50 μl/ min für 4 min injizieren.
HINWEIS: Nach dem Passivierungsschritt wird der Arbeitswinkel (als neue Basislinienbestimmung vor der Probeninjektion) so eingestellt, dass er an den interessierenden Stellen die höchste Empfindlichkeit aufweist.
- Injizieren Sie Rattenserumalbumin (RSA, 200 μg/ml, hergestellt in Acetatpuffer, pH 4,5) bei 50 μl/min für 4 Minuten , um die Oberfläche um die Flecken herum zu passivieren und möglicherweise leere Räume innerhalb der Ligandenflecken zu füllen.
- Injektion der Probe:
- Definieren Sie die Plasmonenkurven nach der Passivierung neu und wählen Sie diesmal den Arbeitswinkel entsprechend dem Liganden.
- Reduzieren Sie bei der kinetischen Überwachung die Flussrate auf 20 μl/min und warten Sie, bis die Basislinie stabil ist.
- Injizieren Sie die Probe in einer Konzentration Ihrer Wahl (von 1 x 10 8 EVs/ml bis 1 x 10 10 EVs/ml, abhängig von der Affinität zwischen den EVs und den transplantierten Liganden) und klicken Sie entweder auf manuelle Injektion oder automatische Injektion. Bei manueller Injektion klicken Sie nach der Injektion von 200 μl der Probe auf Injektion stoppen. Da die Dauer der Injektion im Allgemeinen 10 Minuten beträgt, beginnen Sie, der Kinetik der Wechselwirkung zu folgen, und messen Sie die Reflexionsvariation, indem Sie die Differenz des Reflexionsvermögens zwischen dem Beginn und dem Ende der Injektion berechnen, die während der kinetischen Überwachung beobachtet wurde (Abbildung 5).
HINWEIS: Die verschiedenen Proben, die injiziert wurden, sind im Abschnitt "Repräsentative Ergebnisse" beschrieben.
- Befolgen Sie nach der Probeninjektion einen der beiden folgenden Ansätze, um das SPRi-Experiment abzuschließen:
- Nehmen Sie beim unfixierten/in-flüssigen Ansatz den Biochip aus der SPRi-Apparatur, fügen Sie einen Flüssigkeitstropfen hinzu und fahren Sie mit der weiteren AFM-Charakterisierung der Oberfläche in Flüssigkeit fort.
- Beim festen Ansatz wird Glutaraldehyd (0,5 %) 10 Minuten lang in Wasser mit 20 μl/min verdünnt, um die auf dem Biochip eingefangenen Moleküle zu fixieren. Injizieren Sie Wasser, um die Oberfläche zu spülen, nehmen Sie den Biochip heraus, waschen Sie ihn sehr vorsichtig mit destilliertem Wasser und trocknen Sie ihn an der Luft, um ihn unter AFM weiter zu analysieren.

Abbildung 4: SPRi-CCD-Aufnahme des Biochips. (A,B) Multiplex-Biochip nach Albuminpassivierung. (A) Ein Chip ohne Ausfall; (B) einige Defekte, die auf dem Chip aufgetreten sind: Verschmelzung von Flecken (i), schwache Pfropfung (ii) oder Staub oder "Verunreinigungen" (iii). Die ROIs, in Farbe in den Flecken (eine Farbe pro Ligandenfamilie), wurden gewählt, um diese "Verunreinigungen" zu vermeiden. Wenn Spots zusammengeführt wurden, wurden sie notiert und entweder ignoriert oder als "Mischung der Liganden 1 und 2" bezeichnet. (C) Nackter Goldchip ohne Microarrays für das Experiment zur Untersuchung der Adsorption von Elektrofahrzeugen auf Gold. Der blaue Pfeil zeigt die Fließrichtung an. Dieser Chip wies keine Flecken auf, und die ROIs wurden so gewählt, dass das Reflektivitätssignal von Zeile 1 (L1, rote Kreise) bis Zeile 4 (L4, violette Kreise) während der Probeninjektion registriert wurde. Maßstabsleiste = 1 mm für alle drei Bilder. Abkürzungen: SPRi = Oberflächenplasmonenresonanztomographie; CCD = ladungsgekoppeltes Gerät; ROIs = Regionen von Interesse; EVs = extrazelluläre Vesikel. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}

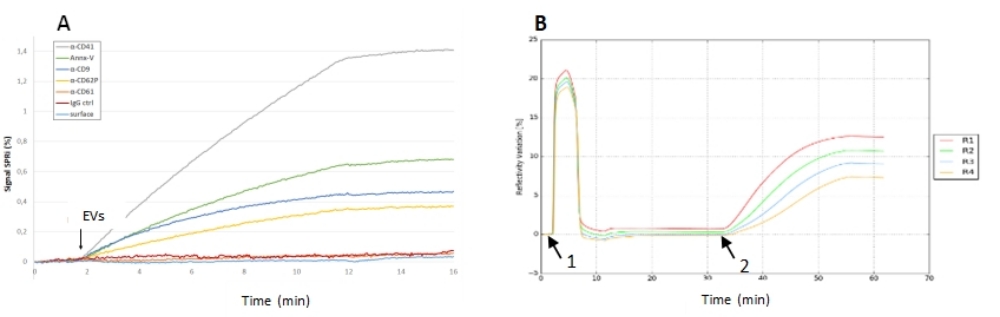
Abbildung 5: SPRi-Experimente der EV-Injektion auf einen Biochip. (A) Capture-Experiment auf einem Multiplex-Biochip, das die Reflektivitätssignale verschiedener Liganden zeigt. Hier war das Signal-Rausch-Verhältnis für die verschiedenen Liganden sehr gut (und insbesondere an den AntiCD41-Spots), da die Reaktion der Negativkontrolle vernachlässigbar war. (B) Adsorptionsexperiment von Elektrofahrzeugen auf einem nackten Biochip. Sensorgramm, das die Konditionierung des Chips mit zwei Pufferspülungen und OG-Reinigung (1), mit der EV-Probeninjektion (2) und dem Reflektivitätssignal nach EV-Wechselwirkung (3) darstellt. Auf diesem Biochip gab es keine Negativkontrolle, aber das Reflexionssignal (seine Kinetik, seine Stabilität nach der Injektion) war hoch, was bedeutet, dass diese EVs in der Lage waren, auf dem Goldchip zu adsorbieren und stabil zu bleiben. Abkürzungen: EV = extrazelluläres Vesikel; OG = Octylglucosid. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
3. Rasterkraftmikroskopie
- Verwenden Sie den Kontaktmodus , um den Biochip in Luft zu scannen, und den quantitativen Bildgebungsmodus , um den Biochip unter flüssigen Bedingungen zu scannen.
- Richten Sie den Biochip auf der Oberseite der Maske auf dem Glasobjektträger (im Labor entwickelt) aus, um die Position der jeweiligen Mikropunkte auf dem Biochip zu identifizieren (Abbildung 6A).
HINWEIS: Die Maske enthält die Markierungen der Spots, die den Positionen der Spots der Liganden entsprechend dem verwendeten Spotter entsprechen. Diese Maske besteht aus einem Glasobjektträger, auf dem zwei senkrechte Keile die Platzierung des Chips ermöglichen. Darüber hinaus ist der Objektträger mit 16 Filzpunkten markiert, die der Spotlokalisierung auf dem Chip entsprechen, was es ermöglicht, die Spots zu lokalisieren und den gewünschten Bereich zu scannen. - Positionierung der Spitze:
- Verwenden Sie eine CCD-Kamera auf dem AFM, um den Ausleger an der richtigen Stelle zu lokalisieren, die gescannt werden muss. Um diesem Protokoll zu folgen, verwenden Sie dreieckige Ausleger mit einer Länge von 200 μm, einer Breite von 28 μm und einer Federkonstante von 0,08 N/m.
- Richten Sie den Laser auf der Oberseite des Auslegers an einer Position aus, die eine optimale Reaktion im Rückkopplungssteuerungsmechanismus ergibt.
- Abtastung:
- Sobald Sie eingerastet sind und mit der Biochipoberfläche in Kontakt gekommen sind, starten Sie die AFM-Erfassung im Kontaktmodus oder im quantitativen Bildgebungsmodus von drei bis fünf großen Bereichen (typischerweise 10 × 10 μm²) bis hin zu kleinen Bereichen (1 x 1 μm²).
HINWEIS: Eine Darstellung der verschiedenen Bereiche, die gescannt werden können, ist in Abbildung 6D dargestellt. Stellen Sie sicher, dass die AFM-Charakterisierung für den gesamten mm²-Spot repräsentativ ist und dass genügend EVs mit einer guten Auflösung für eine robuste Analyse visualisiert werden (mindestens 300 EVs werden für jede Bedingung gezählt und analysiert), und führen Sie die Mess- und Morphologiemessungen durch.
- Sobald Sie eingerastet sind und mit der Biochipoberfläche in Kontakt gekommen sind, starten Sie die AFM-Erfassung im Kontaktmodus oder im quantitativen Bildgebungsmodus von drei bis fünf großen Bereichen (typischerweise 10 × 10 μm²) bis hin zu kleinen Bereichen (1 x 1 μm²).
- AFM-Bildbehandlung:
- Behandeln Sie die AFM-Bilder mit einer JPK-Datenverarbeitungssoftware, indem Sie zuerst den Höhenkanal auswählen.
- Wählen Sie eine Polynomanpassung , die von jeder Linie subtrahiert werden soll, um begradigte Scanlinien zu erhalten.
- Wählen Sie die Höhenschwelle für Goldkörner, um die Rauheit der Oberfläche zu beseitigen. Markieren Sie in der referenzierten Software (siehe Materialtabelle) innerhalb des Getreideextraktionsmoduls die Körner mit einem Höhenschwellenwert von 8,5 nm. Sobald die Körner gefiltert wurden, erscheint die Anzahl der Körner.
HINWEIS: Normalerweise erfordern das Rohgoldsubstrat (RMS von ~ 3 nm) und das Vorhandensein der chemischen und Ligandenschichten eine Einstellung des Schwellenwerts auf 8,5 nm. - Um verschiedene Eigenschaften der markierten Körner zu extrahieren, z. B. Höhe, Volumen und Durchmesser, öffnen Sie das Bild in der referenzierten Software und wählen Sie Datenbehandlung | Körner | Markieren Sie nach Schwellenwert.
- Wählen Sie Filterkörner in Abhängigkeit von Eigenschaften. Wenn ein neues Fenster erscheint, wählen Sie die folgenden Parameter: Wert = maximale Z-max, Fläche = projizierte Fläche A0. Wählen Sie dann die Kriterien A UND B.
- Offene Kornverteilung; Wählen Sie im angezeigten Fenster Wert (Maximum), Volume (Basis: Null) und Boundary (Länge) aus. Beachten Sie die angezeigte Tabelle (in .txt Format), die drei Spalten mit den Werten für Höhe, Volumen und Durchmesser für alle Körner enthält, die am eingestellten Schwellenwert pro Bild erkannt wurden.
- Berechnen Sie aus der Höhe, h und dem Durchmesser D den Krümmungsradius Rc jedes EV mit Gleichung (1)8 und berechnen Sie dann das Volumen V mit Gleichung (2):
 Bewertungen (1)
Bewertungen (1) Bewertungen (2)
Bewertungen (2) - Berechnen Sie aus V den effektiven Durchmesser d eff jedes EV (den Durchmesser einer Kugel mit gleichem Volumen) anhand von Gleichung (3):
 Bewertungen (3)
Bewertungen (3) - Zeichnen Sie Diagramme, die die Größen (gemessene Höhe, gemessener Durchmesser und berechneter effektiver Durchmesser) der EVs anzeigen, wobei jedes gezählte Partikel durch einen Punkt dargestellt wird.
HINWEIS: Somit ermöglicht die NBA-Plattform am Ende der Charakterisierung die Korrelation des Biodetektionssignals und der anschließenden Phänotypisierung mit der Anzahl und Größe der EV-Untergruppen.

Abbildung 6: Biochip-Charakterisierung mittels AFM. Nach dem SPRi-Experiment wurde der Chip entweder fixiert und getrocknet oder zur AFM-Charakterisierung in Flüssigkeit gehalten. (A) Der bearbeitete Objektträger (mit zwei senkrechten Positionierungskeilen, auf dem Bild mit einem "w" gekennzeichnet) zeigt eine Maske, die zur Lokalisierung der 16 Biochip-Microarrays passt. Durch Lichtbelichtung und Transparenz ermöglicht der Glasobjektträger, sobald er für die AFM-Charakterisierung installiert ist, die AFM-Spitze an der gewünschten Stelle zu platzieren, um sie zu charakterisieren. (B) Der Biochip, der auf dem Objektträger "Maske" und unter einem Tropfen Puffer installiert ist, um unter flüssigen Bedingungen zu scannen. (C) SPRi-Aufnahme der 16 Microarrays. (D) Ein Mikroarray, der durch optische Mikroskopie nach dem Immuneinfangen von biofunktionalisierten Kalibrierungsnanopartikeln mit einem Durchmesser von 920 nm abgebildet wurde. Die weißen Quadrate zeigen die Abtastung der verschiedenen Bereiche an, die von AFM an jedem interessierenden Punkt gescannt wurden, um die AFM-Charakterisierung robust zu machen. Maßstabsbalken = (C) 1 mm, (D) 500 μm. Abkürzungen: AFM = Rasterkraftmikroskopie; SPRi = Oberflächenplasmonen-Resonanztomographie. Bitte klicken Sie hier, um eine größere Version dieser Abbildung zu sehen.
{kind=link}
4. Raman-Spektroskopie
HINWEIS: Ersetzen Sie für die Raman-Spektroskopie den als Substrat verwendeten Objektträger durch einen Objektträger aus CaF2, der eine vernachlässigbare Raman-Signatur aufweist.
- Optische Bedingungen für die Erfassung:
- Stellen Sie die folgenden Bedingungen für das Raman-Bildgebungsmikroskop ein: Mikroskopobjektiv: 50x; Laserwellenlänge: 532 nm; Laserleistung: 10 mW; Belichtungszeit: 500 ms; Anzahl der Akkumulationen: 140; Spektralbereich: von 450 cm−1 bis 3.200 cm−1.
HINWEIS: Die Verwendung von zu viel Strom und/oder zu langer Erfassungszeit kann zu einer Beschädigung der Probe führen, die durch instabile Spektren im Laufe der Zeit belegt wird. Beginnen Sie mit einer geringen Energiemenge und erhöhen Sie diese, wenn das Signal zu schwach ist. Höhere Laserwellenlängen (633 nm, 785 nm) können verwendet werden, um die Fluoreszenz zu reduzieren, die für Raman-Messungen nachteilig ist. Die Intensität nimmt jedoch mit der vierten Potenz der Wellenlänge ab, und die spektrale Empfindlichkeit der Kamera sollte berücksichtigt werden.
- Stellen Sie die folgenden Bedingungen für das Raman-Bildgebungsmikroskop ein: Mikroskopobjektiv: 50x; Laserwellenlänge: 532 nm; Laserleistung: 10 mW; Belichtungszeit: 500 ms; Anzahl der Akkumulationen: 140; Spektralbereich: von 450 cm−1 bis 3.200 cm−1.
- Raman-Bildgebung:
- Beobachten Sie zunächst die Live-Spektren mit einer reduzierten Anzahl von Akkumulationen (10), um den Bereich mit dem besten Signal-Rausch-Verhältnis zu finden.
HINWEIS: Ein starkes Signal im Hochfrequenzbereich (2.800-3.000 cm−1) kann die Erkennung von Elektrofahrzeugen auf der Oberfläche mit geringen Belichtungszeiten erleichtern, wie zuvor gezeigt10. - Sobald der ROI ausgewählt ist, wählen Sie die räumliche Auflösung entsprechend der für die Erfassung verfügbaren Zeit.
HINWEIS: Die räumliche Auflösung ist durch die Beugungsgrenze (~500 nm) begrenzt. - Starten Sie die Raman-Mapping-Erfassung.
- Beobachten Sie zunächst die Live-Spektren mit einer reduzierten Anzahl von Akkumulationen (10), um den Bereich mit dem besten Signal-Rausch-Verhältnis zu finden.
- Vorverarbeitung der Daten:
- Öffnen Sie mit einer integrierten Entwicklungsumgebung (IDE) von Python (z. B. Spyder) die Datei mit den Spektren.
- Subtrahieren Sie die Basislinie der Spektren, um die Interferenz durch mögliche Fluoreszenz zu korrigieren. Verwenden Sie z.B. die Funktion "arpls" aus dem Paket "irfpy"23. Testen Sie verschiedene Werte des Parameters "lam", um denjenigen zu finden, der die beste Basislinienkorrektur liefert (normalerweise zwischen 10,3 und 10,7).
- Normalisieren Sie die Spektren beispielsweise, indem Sie alle Intensitäten eines Spektrums durch seine Intensität bei 2.900 cm−1 dividieren oder indem Sie den Mittelwert des Spektrums subtrahieren und dann durch seine Standardabweichung dividieren ("Standardnormalvariate"-Normalisierung).
HINWEIS: Dieser Schritt ist notwendig, um die relative molekulare Zusammensetzung von EVs zu vergleichen.
Access restricted. Please log in or start a trial to view this content.
Ergebnisse
Bestimmung der optimalen pH-Bedingungen für die Ligandentransplantation
Die verschiedenen Liganden, die zur Herstellung der Biochips verwendet werden, werden in Abhängigkeit vom pH-Wert und ihrer Verfügbarkeit für die Interaktion mit der chemischen Thiolatschicht getestet (Abbildung 3). Die Liganden werden in Acetatpuffer bei unterschiedlichen pH-Werten verdünnt und auf den mit einer C11C16-Schicht chemisch funktionalisierten Biochip injiziert. Die Lösungen werden n...
Access restricted. Please log in or start a trial to view this content.
Diskussion
Die neueren Methoden zur Identifizierung von EVs, die am weitesten verbreitet sind, sind proteinspezifisches Immunblotting, um die Herkunft von EVs zu bestätigen, TEM, um ihre Struktur zu bestätigen, und NTA, um ihre Anzahl und Größenverteilung in einem Probenvolumen3 zu quantifizieren. Nichtsdestotrotz haben das große Interesse an Elektrofahrzeugen in der (bio-)medizinischen Forschung und die Einschränkungen bestehender Analysewerkzeuge die wissenschaftliche Gemeinschaft dazu veranlasst, ne...
Access restricted. Please log in or start a trial to view this content.
Offenlegungen
Die Autoren haben keine Interessenkonflikte offenzulegen.
Danksagungen
Kelly Aubertin und Fabien Picot von der IVETh Core Facility (Paris) werden für die Raman-Bildgebungsexperimente ausgezeichnet. Thierry Burnouf (Taipei Medical University, Taiwan) und Zuzana Krupova (aus Helincourt, Frankreich) werden für die Bereitstellung der EV-Proben aus Blutplättchen- bzw. Rindermilchproben ausgezeichnet. Die Arbeit wurde von der Region Bourgogne Franche-Comté und der Graduiertenschule EUR EIPHI (NOVICE-Projekt, 2021-2024) unterstützt. Ein Teil dieser Arbeit wurde mit der CLIPP-Plattform und in RENATECH-Reinraumanlagen in FEMTO-ENGINEERING durchgeführt, wofür wir Rabah Zeggari danken.
Access restricted. Please log in or start a trial to view this content.
Materialien
| Name | Company | Catalog Number | Comments |
| CD41a antibody | Diaclone SAS (France) | 447528 | |
| CP920 | Microparticles GmbH, Germany | 448303 | |
| DXR3xi | Thermo Fisher Scientific | T1502 | |
| EDC | Sigma | A6272 | |
| Ethanolamine | Sigma | P5368-10PAK | |
| Evs derived from platelet concentrates | Collaboration : Pr T. Burnouf (TMU, Taipei) | S2889 | |
| Evs from bovine milk | Collaboration : Dr Z. Krupova (Excilone, Helincourt - France) | 3450 | |
| Glutaraldehyde | Sigma | 56845 | |
| Gwyddion | 853.223.020 | ||
| Magnetron sputtering | PLASSYS | SAB5300165 | |
| mercapto-1-hexadecanoic acid | Sigma | G5882 | |
| Mercapto-1-undecanol | Sigma | O8001 | |
| Mountains SPIP ones | Digital Surf | ||
| NanoWizard 3 Bioscience | Bruker-JPK | ||
| Octyl Glucoside (OG) | Sigma | ||
| Ovalbumine antibody | Sigma | ||
| Phosphate Buffer Saline (PBS) | Sigma | ||
| Rat Albumin Serum (RSA) | Sigma | ||
| Sodium acetate buffer | Sigma | ||
| SPR-Biacore 3000 | GE Healthcare/ Cytiva life sciences | ||
| SPRi Biochip | MIMENTO technology platform | The biochips were produced in-house in the clean room, Besancon | |
| SPRi Plex II | Horiba Scientific | ||
| Sulfo-NHS | Sigma |
Referenzen
- Silva, A. K. A., et al. Development of extracellular vesicle-based medicinal products: A position paper of the group "Extracellular Vesicle translatiOn to clinicaL perspectiVEs - EVOLVE France". Advanced Drug Delivery Reviews. 179, 114001(2021).
- Xunian, Z., Kalluri, R. Biology and therapeutic potential of mesenchymal stem cell-derived exosomes. Cancer Science. 111 (9), 3100-3110 (2020).
- Hartjes, T. A., et al. Extracellular vesicle quantification and characterization: Common methods and emerging approaches. Bioengineering. 6 (1), 7(2019).
- Xing, Y., et al. Analysis of extracellular vesicles as emerging theranostic nanoplatforms. Coordination Chemistry Reviews. 424, 213506(2020).
- Wang, T., Xing, Y., Cheng, Z., Yu, F. Analysis of single extracellular vesicles for biomedical applications with especial emphasis on cancer investigations. Trends in Analytical Chemistry. 152, 116604(2022).
- Boireau, W., Elie-Caille, C. Extracellular vesicles: Definition, isolation and characterization. Medecine Sciences: M/S. 37 (12), 1092-1100 (2021).
- Brisson, A. R., et al. Extracellular vesicles from activated platelets: A semiquantitative cryo-electron microscopy and immuno-gold labeling study. Platelets. 28 (3), 263-271 (2017).
- Yuana, Y., et al. Atomic force microscopy: A novel approach to the detection of nanosized blood microparticles. Journal of Thrombosis and Haemostasis. 8 (2), 315-323 (2010).
- Sebaihi, N., de Boeck, B., Yuana, Y., Nieuwland, R., Pétry, J. Dimensional characterization of extracellular vesicles using atomic force microscopy. Measurement Science and Technology. 28 (3), 034006(2017).
- Beekman, P., et al. Immuno-capture of extracellular vesicles for individual multi-modal characterization using AFM, SEM and Raman spectroscopy. Lab on a Chip. 19 (15), 2526-2536 (2019).
- Malenica, M., et al. Perspectives of microscopy methods for morphology characterisation of extracellular vesicles from human biofluids. Biomedicines. 9 (6), 603(2021).
- Verweij, F. J., et al. The power of imaging to understand extracellular vesicle biology in vivo. Nature Methods. 18 (9), 1013-1026 (2021).
- Théry, C., et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. Journal of Extracellular Vesicles. 7 (1), 1535750(2018).
- Obeid, S., et al. NanoBioAnalytical characterization of extracellular vesicles in 75-nm nanofiltered human plasma for transfusion: A tool to improve transfusion safety. Nanomedicine: Nanotechnology, Biology, and Medicine. 20, 101977(2019).
- Obeid, S., et al. Development of a NanoBioAnalytical platform for «on-chip» qualification and quantification of platelet-derived microparticles. Biosensors and Bioelectronics. 93, 250-259 (2017).
- Ridolfi, A., et al. AFM-based high-throughput nanomechanical screening of single extracellular vesicles. Analytical Chemistry. 92 (15), 10274-10282 (2020).
- Vorselen, D., et al. The fluid membrane determines mechanics of erythrocyte extracellular vesicles and is softened in hereditary spherocytosis. Nature Communications. 9 (1), 4960(2018).
- Hardij, J., et al. Characterisation of tissue factor bearing extracellular vesicles with AFM: Comparison of air-tapping-mode AFM and liquid Peak Force AFM. Journal of Extracellular Vesicles. 2, 21045(2013).
- Jorgensen, M., et al. Extracellular Vesicle (EV) Array: Microarray capturing of exosomes and other extracellular vesicles for multiplexed phenotyping. Journal of Extracellular Vesicles. 2, 20920(2013).
- Remy-Martin, F., et al. Surface plasmon resonance imaging in arrays coupled with mass spectrometry (SUPRA-MS): Proof of concept of on-chip characterization of a potential breast cancer marker in human plasma. Analytical and Bioanalytical Chemistry. 404 (2), 423-432 (2012).
- Czamara, K., et al. Raman spectroscopy of lipids: A review. Journal of Raman Spectroscopy. 46 (1), 4-20 (2015).
- Penders, J., et al. Single particle automated Raman trapping analysis of breast cancer cell-derived extracellular vesicles as cancer biomarkers. ACS Nano. 15 (11), 18192-18205 (2021).
- Baek, S. J., Park, A., Ahn, Y. J., Choo, J. Baseline correction using asymmetrically reweighted penalized least squares smoothing. Analyst. 140 (1), 250-257 (2015).
- Daaboul, G. G., et al. Digital detection of exosomes by interferometric imaging. Scientific Reports. 6, 37246(2016).
- Ertsgaard, C. T., et al. Integrated nanogap platform for sub-volt dielectrophoretic trapping and real-time Raman imaging of biological nanoparticles. Nano Letters. 18 (9), 5946-5953 (2018).
- Maas, S. L., et al. Possibilities and limitations of current technologies for quantification of biological extracellular vesicles and synthetic mimics. Journal of Controlled Release. 200, 87-96 (2015).
Access restricted. Please log in or start a trial to view this content.
Nachdrucke und Genehmigungen
Genehmigung beantragen, um den Text oder die Abbildungen dieses JoVE-Artikels zu verwenden
Genehmigung beantragenThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Alle Rechte vorbehalten