
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Plate-forme analytique multimodale sur une puce d’imagerie par résonance plasmonique de surface multiplexée pour l’analyse de sous-ensembles de vésicules extracellulaires
Dans cet article
Résumé
Cet article propose une nouvelle génération de plateformes analytiques multiparamétriques avec un débit accru pour la caractérisation des sous-ensembles de vésicules extracellulaires. La méthode est basée sur une combinaison de méthodes de biodétection multiplexées avec des analyses métrologiques et morphomécaniques par microscopie à force atomique, couplée à la spectroscopie Raman, pour qualifier des cibles vésiculaires piégées sur une biopuce de microréseau.
Résumé
Les vésicules extracellulaires (VE) sont de minuscules vésicules dérivées de la membrane produites par toutes les cellules qui vont de 50 à plusieurs centaines de nanomètres de diamètre et sont utilisées comme moyen de communication intercellulaire. Ils apparaissent comme des outils diagnostiques et thérapeutiques prometteurs pour diverses maladies. Il existe deux principaux processus de biogenèse utilisés par les cellules pour produire des VE avec des différences de taille, de composition et de contenu. En raison de leur grande complexité en taille, composition et origine cellulaire, leur caractérisation nécessite une combinaison de techniques analytiques. Ce projet comprend le développement d’une nouvelle génération de plateformes analytiques multiparamétriques avec un débit accru pour la caractérisation de sous-populations de VE. Pour atteindre cet objectif, les travaux partent de la plateforme nanobioanalytique (NBA) mise en place par le groupe, qui permet une étude originale des VE basée sur une combinaison de méthodes de biodétection multiplexées avec des analyses métrologiques et morphomécaniques par microscopie à force atomique (AFM) de cibles vésiculaires piégées sur une biopuce de microarray. L’objectif était de compléter cette étude EV par une analyse phénotypique et moléculaire par spectroscopie Raman. Ces développements permettent de proposer une solution analytique multimodale et facile à utiliser pour la discrimination des sous-ensembles de VE dans les fluides biologiques à potentiel clinique
Introduction
L’intérêt croissant pour la recherche sur les VE dans le diagnostic et la thérapeutique 1,2,3,4,5, combiné aux défis auxquels ce domaine est confronté, a entraîné le développement et la mise en œuvre d’une grande variété d’approches et de techniques pour quantifier ou caractériser ces vésicules. Les méthodes les plus largement utilisées pour l’identification des VE sont l’immunoblot et la protéomique spécifiques aux protéines pour confirmer l’origine des VE, la microscopie électronique à transmission (MET) pour confirmer leur structure et l’analyse de suivi des nanoparticules (NTA) pour quantifier leur nombre et leur distribution de taille dans un volume d’échantillon.
Cependant, aucune de ces techniques ne donne à elle seule toutes les informations nécessaires pour caractériser les sous-ensembles de véhicules électriques. L’hétérogénéité inhérente des VE due à la diversité de leurs propriétés biochimiques et physiques empêche les analyses globales fiables et reproductibles, en particulier pour les VE contenus dans un mélange (échantillon brut). Des méthodes de détection et de caractérisation sont donc nécessaires pour les véhicules électriques, à la fois individuellement et généralement pour compléter d’autres méthodes plus rapides mais non sélectives6.
L’imagerie haute résolution par TEM (ou cryoTEM) ou AFM permet de déterminer la morphologie et la métrologie des VE avec une résolution nanométrique 7,8,9,10,11,12. Cependant, la principale limitation de l’utilisation de la microscopie électronique pour les objets biologiques, tels que les véhicules électriques, est la nécessité d’un vide pour effectuer l’étude qui nécessite la fixation et la déshydratation de l’échantillon. Une telle préparation rend difficile la traduction des structures observées à la morphologie EV en solution. Pour éviter cette déshydratation de l’échantillon, la technique de cryoTEM est la plus adaptée à la caractérisation EV13. Il est largement utilisé pour déterminer l’ultrastructure des véhicules électriques. L’immunomarquage des vésicules par des nanoparticules d’or biofonctionnalisées permet également d’identifier des sous-populations spécifiques de VE et de les distinguer des autres particules présentes dans un échantillon biologique complexe. Cependant, en raison du faible nombre de VE analysés par microscopie électronique, il est souvent difficile d’effectuer une caractérisation représentative d’un échantillon complexe et hétérogène.
Pour révéler cette hétérogénéité de taille, l’International Society for Extracellular Vesicles (ISEV) suggère d’analyser un nombre suffisant d’images à grand champ, accompagnées d’images plus petites, pour révéler des VE individuels à haute résolution14. L’AFM est une alternative aux approches optiques et aux techniques de diffraction électronique pour l’étude des véhicules électriques. Cette technique utilise une pointe tranchante maintenue par un porte-à-faux flexible qui scanne l’échantillon déposé sur un support, ligne par ligne, et ajuste la distance entre la pointe et les éléments présents grâce à une boucle de rétroaction. Cela permet de caractériser la topographie de l’échantillon et de recueillir des informations morphomécaniques15,16,17,18. Les VE peuvent être scannés par AFM soit après avoir été déposés sur un substrat atomiquement plat, soit après avoir été capturés sur un substrat spécifique fonctionnalisé par des anticorps, des peptides ou des aptamères pour caractériser les différentes sous-populations18,19. En raison de sa capacité à quantifier et à sonder simultanément la structure, la biomécanique et le contenu biomoléculaire membraneux des VE dans des échantillons biologiques complexes sans avoir besoin de prétraitement, d’étiquetage ou de déshydratation, l’AFM est maintenant de plus en plus utilisé pour caractériser les VE de manière fine et multiparamétrique dans des conditions physiologiques de température et de milieu.
Cet article propose une méthodologie utilisant une biopuce d’or de base capable d’être (bio)chimiquement fonctionnalisée dans un format multiplexé. Ce substrat est la pierre angulaire d’une puissante plateforme analytique combinant la biodétection de sous-ensembles de VE par résonance plasmonique de surface, et une fois les VE adsorbés/greffés ou immunocapturés sur la puce, AFM permet la caractérisation métrologique et morphomécanique des VE. Couplée à la signature Raman des sous-ensembles EV capturés sur la puce, cette plateforme analytique permet la qualification des VE présents dans les échantillons biologiques de manière sans marquage et sans aucune étape préanalytique. Cet article montre que la combinaison de techniques puissantes, assistées par une méthodologie très rigoureuse dans la préparation du substrat et l’acquisition de données, rend l’analyse EV approfondie, définitive et robuste.
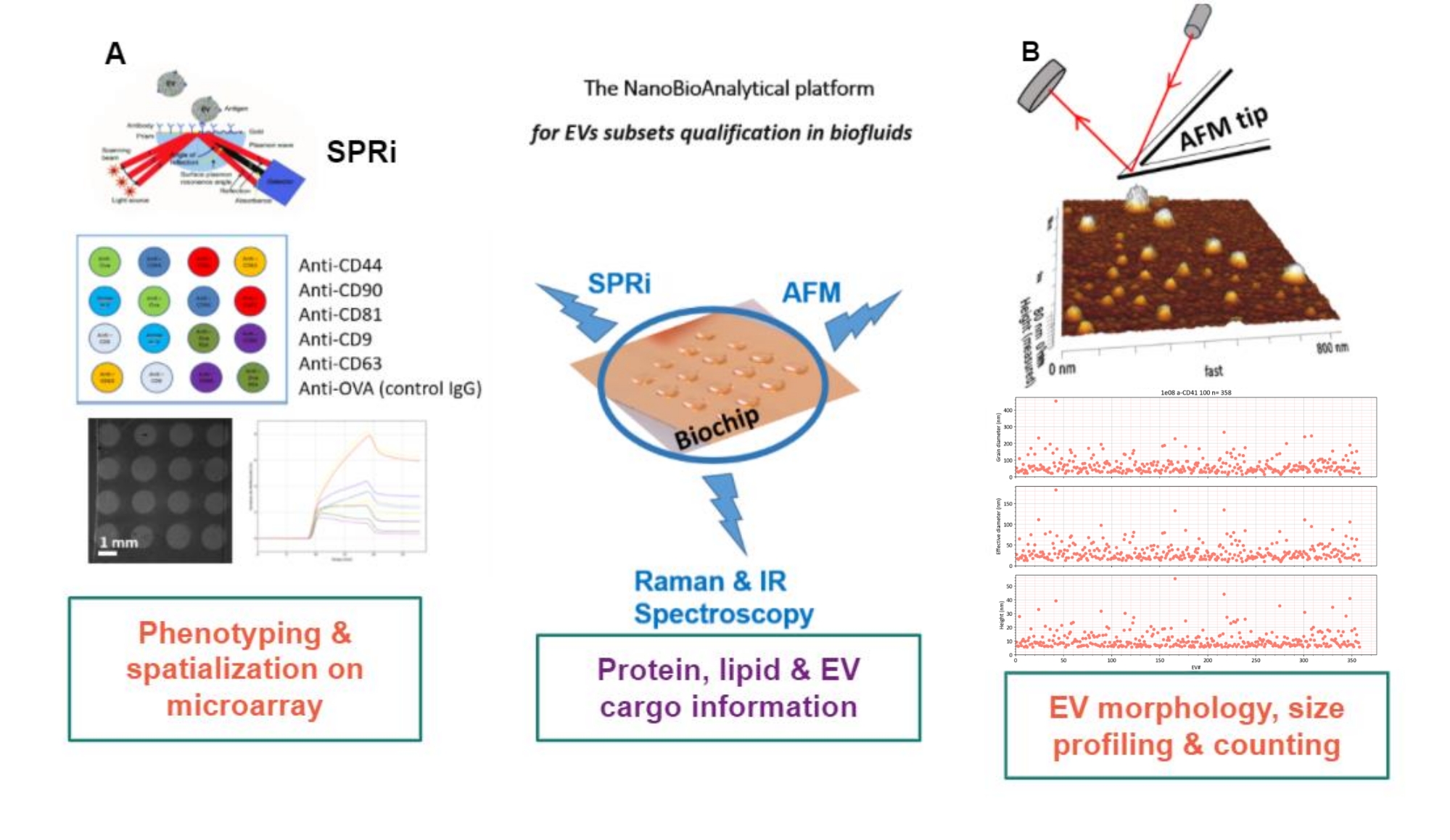
Le principe de l’approche proposée est de préparer un substrat aurifère, d’adsorber/greffer ou de capturer les sous-types de VE, et de les scanner par AFM pour estimer la taille et la morphologie de chaque sous-ensemble de VE. De plus, ces VE adsorbés sont analysés par spectroscopie Raman. Ce substrat peut, en effet, présenter trois types d’interfaces de plus en plus complexes : nues, fonctionnalisées chimiquement, ou puces à lignand. Avant de décrire les différentes étapes du protocole, les lecteurs sont invités à se reporter à la présentation schématique de l’approche de la plateforme nanobioanalytique (NBA) à la figure 1, combinant l’imagerie par résonance plasmonique de surface (SPRi), l’AFM et la spectroscopie.

Figure 1 : La plateforme NanoBioAnalytical L’approche combine (A) l’imagerie par résonance plasmonique de surface, (B) la microscopie à force atomique et la (nano)spectroscopie infrarouge / Raman, toutes engagées sur le même substrat - une puce d’or multiplexée. Abréviations : NBA = NanoBioAnalytical platform; SPRi =imagerie par résonance plasmonique de surface; AFM = microscopie à force atomique; EV = vésicule extracellulaire. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
La biopuce d’or de base constitue le cœur de la plateforme puisque toutes les techniques de caractérisation sans marquage sont réalisées sur cette biopuce. Selon les besoins de la caractérisation des VE (VE globaux/totaux ou sous-ensembles de VE) et les limites/exigences des méthodes utilisées, trois types de surfaces de biopuce d’or ont été développées : soit « nues », « C11/C16 » chimiquement fonctionnalisées ou biofonctionnalisées par ligand, appelées surface dorée « ligand ».
La biopuce nue, appelée « nue », permet l’adsorption simple de véhicules électriques sur l’or. Il est possible de sélectionner le tampon utilisé et de réaliser cette adsorption soit de manière passive (étapes d’incubation puis de rinçage), soit de la surveiller sous flux (en SPRi). De plus, cette adsorption passive peut être réalisée soit sur l’ensemble de la puce (sous forme de macroarray), soit localisée dans des microarrays à l’aide d’un observateur de micropipettes. La « procédure sous débit » permet aux enquêteurs de suivre la cinétique et le niveau d’adsorption de l’EV. Cette approche sur le substrat d’or nu est adoptée lorsque l’interface de la couche chimique peut interférer avec la méthode analytique (par exemple, pour la spectroscopie Raman).
La biopuce fonctionnalisée chimiquement, appelée « C11/C16 », est utilisée pour créer un « tapis » dense et robuste de véhicules électriques liés de manière covalente sur la surface de l’or en formant des liaisons amides primaires avec les thiolates lorsque l’objectif est d’avoir une vue globale de l’échantillon de VE. En effet, dans ce cas, l’or est fonctionnalisé par un mélange thiolate de mercapto-1-undécanol (11-MUOH : « C11 ») et d’acide mercapto-1-hexadécanoïque (16-MHA : « C16 »), et une fraction des thiolates est activée chimiquement pour établir une liaison covalente avec les cibles. Encore une fois, cette stratégie peut être réalisée soit passivement (étapes d’incubation puis de rinçage, soit dans un « macroarray » ou dans plusieurs microarrays à l’aide d’un observateur de micropipette) ou sous des débits (dans SPRi) pour suivre la cinétique et le niveau de greffage EV sur la surface de l’or.
La biopuce biofonctionnalisée par ligand, appelée « ligands », est activée chimiquement pour greffer par covalence différents ligands (p. ex. anticorps, récepteurs) afin de capturer sélectivement (avec affinité) différents sous-ensembles de VE qui coexistent dans l’échantillon biologique.
Access restricted. Please log in or start a trial to view this content.
Protocole
1. Préparation du substrat d’or
NOTE: Trois types de surfaces sont produites sur des copeaux d’or: 1) surface nue, 2) fonctionnalisée chimiquement et 3) biofonctionnalisée (ligands greffés sur couche C11C16). Ils seront appelés « nus », « C11C16 » et « ligands », respectivement, à partir de ce moment.
- Préparation du substrat d’or:
REMARQUE: Pour ce protocole, les biopuces d’or ont été fabriquées en interne dans la salle blanche. Les biopuces artisanales étaient composées de lames de verre (SF11) avec un revêtement de chrome (2 nm Cr) et d’or (48 nm Au). La longueur de la biopuce était de 28 mm, la largeur était de 12,5 mm et l’épaisseur était de 0,5 mm20.- Utilisez un magnétron à courant continupulvérisant 15 pour recouvrir les lames par dépôt physique en phase vapeur (PVD).
- Fonctionnalisation chimique :
- Fonctionnaliser les copeaux nus en les incubant pendant une nuit dans un mélange à 90%/10% par mole de mercapto-1-undécanol (11-MUOH: C11) et d’acide mercapto-1-hexadécanoïque (16-MHA: C16) à 1 mM dans de l’éthanol absolu, sous agitation et à température ambiante.
REMARQUE: Cette étape formera une monocouche stable et auto-assemblée (SAM), ce qui est utile dans la greffe des ligands. - Nettoyez la biopuce (lavez doucement) avec de l’éthanol absolu et de l’eau ultrapure, séchez-la sous azote et conservez-la dans des conditions de salle blanche.
- Activation de la biopuce fonctionnalisée chimiquement :
NOTE: À partir de cette étape, les expériences doivent être effectuées dans un laboratoire d’analyse.- Nettoyez la biopuce avec de l’eau ultrapure en la lavant doucement, puis séchez-la sous un flux d’air doux. Pour activer les groupes carboxyliques C16, incuber la biopuce dans un mélange de 200 mM d’éthyl(diméthylaminopropyl)carbodiimide/N-hydroxysuccinimide (EDC) et de 50 mmol/L de N-hydroxysuccinimide (Sulfo-NHS) pendant au moins 30 min dans l’obscurité à température ambiante. Rincez ensuite à l’eau avant les expériences de greffe.
- Fonctionnaliser les copeaux nus en les incubant pendant une nuit dans un mélange à 90%/10% par mole de mercapto-1-undécanol (11-MUOH: C11) et d’acide mercapto-1-hexadécanoïque (16-MHA: C16) à 1 mM dans de l’éthanol absolu, sous agitation et à température ambiante.
- Greffe des ligands sur la biopuce fonctionnalisée :
REMARQUE: L’immobilisation des ligands (ou EV pour certaines expériences) sur la puce peut être effectuée passivement en dehors de l’instrument SPRi (incubation d’une goutte sur la puce activée) ou dynamiquement sous l’écoulement dans l’instrument SPRi. Il s’agit des puces modifiées par EV ou ligand. La figure 2 présente la biopuce d’or, l’observateur de micropipette et la biopuce après repérage avec 16 gouttelettes de ligand de 300 nL chacune.- Pour la greffe de ligand, utilisez des molécules telles que des anticorps (p. ex. immunoglobulines-antiCD41 [spécifiques aux VE dérivés de plaquettes sanguines natives, appelées N-PEV], antiCD61, antiCD62P, antiCD9 et antiOVA [anticorps de contrôle contre l’ovalbumine]) et l’annexine V. Diluez-les à 200 μg/mL dans une solution acide (de pH 4,5 à 6 selon le pH optimal pour l’activité ou la fonction du ligand).
NOTE: Le pH optimal pour la greffe d’anticorps a été déterminé précédemment par des expériences de préconcentration effectuées sur un instrument SPR pour la détermination du pH optimal pour la greffe de ligand (voir Figure 3). Comme les conditions de greffe changent avec les clones d’anticorps utilisés, il est recommandé de déterminer ces conditions avant de procéder aux expériences SPRi. - Pour la procédure de greffe, ajoutez 300 nL de solution EVs/ligand en utilisant l’observateur.
REMARQUE: Un morceau de papier immergé dans l’eau doit être conservé dans les puits gauche et droit pour éviter l’évaporation des gouttelettes. Cette étape est importante pour maintenir les VE/ligands dans leurs conditions optimales de stabilité et de fonctionnalité. - Après le repérage, conserver la biopuce sous un bain sonique (fréquence : 37 kHz, puissance : 30 %) pendant une incubation de 30 minutes. Lavez la biopuce par le haut avec de l’eau ultrapure et placez-la délicatement sur un prisme ayant le même indice de réfraction (IR) que la biopuce. Tout en ajustant la biopuce au sommet du prisme, ajoutez une gouttelette (~ 2,3 μL) d’huile avec le même RI que le prisme pour créer une couche mince uniforme entre la biopuce et le prisme.
Remarque : Cette étape garantit un support continu du même RI dans le chemin optique. Il est important d’éviter d’incorporer des bulles dans la couche d’huile à cette étape, car cela modifierait les propriétés optiques du trajet et entraverait une analyse ultérieure.
- Pour la greffe de ligand, utilisez des molécules telles que des anticorps (p. ex. immunoglobulines-antiCD41 [spécifiques aux VE dérivés de plaquettes sanguines natives, appelées N-PEV], antiCD61, antiCD62P, antiCD9 et antiOVA [anticorps de contrôle contre l’ovalbumine]) et l’annexine V. Diluez-les à 200 μg/mL dans une solution acide (de pH 4,5 à 6 selon le pH optimal pour l’activité ou la fonction du ligand).

Figure 2 : Biopuce et observateur manuel. Biopuce d’or (à gauche), observateur de micropipette (au milieu) et la biopuce après repérage avec des gouttelettes de ligand de 300 nL chacune (à droite). Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

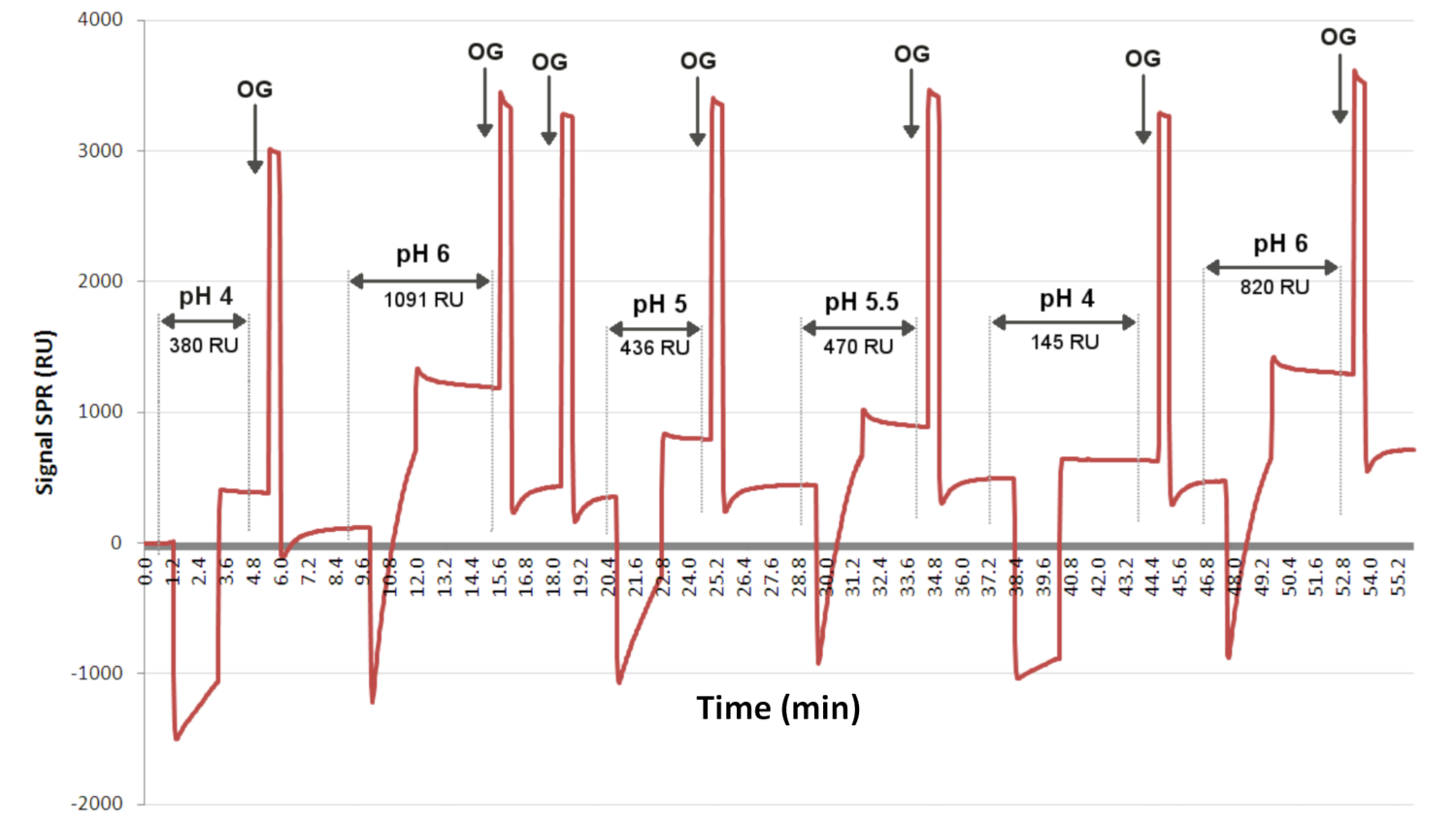
Figure 3 : Tests de préconcentration pour déterminer le pH optimal pour la greffe de ligand. Le sensorgramme présente le niveau d’interaction en fonction du temps pour un ligand injecté aléatoirement (à différentes valeurs de pH) à la même concentration sur 2 min en surface. OG est le détergent, ce qui permet de récupérer la ligne de base entre chaque injection. Ici, le sensorgramme indique que pH 6 a permis le plus de greffes de ligands, avec un signal SPRi de 1091 RU. Abréviations : OG = glucoside d’octyle; EF = unité d’intervention. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
2. Imagerie par résonance plasmonique de surface
- Montez la biopuce sur le système SPRi. Maintenir le débit du tampon PBS (tampon courant) à 50 μL/min.
REMARQUE: S’il y a des bulles, augmenter le débit jusqu’à 500-1 000 μL / min, et injecter 40 mM octyl glucoside (OG) fréquemment pour les enlever dès que possible. - Conditionnement de la biopuce d’or dans l’instrument SPRi : Choix des angles de travail
- Cliquez sur le menu déroulant sur le côté gauche du logiciel, puis cliquez sur le répertoire de travail pour définir le dossier dans lequel les données expérimentales doivent être enregistrées. Ensuite, cliquez sur Plasmon | Acquisition d’images.
- Recherchez l’image (comme illustré à la figure 4A) où différents points sont visibles, puis cliquez pour sélectionner cette image. Pour définir la région d’intérêt (ROI), telle que représentée à la figure 4, cliquez sur détection automatique ou définition manuelle à l’intérieur des spots (Figure 4B) ou sur la puce pour faire référence à un emplacement spécifique même sans spots (Figure 4C).
- Lorsque des biopuces multiplexées sont utilisées, écrivez le nom des familles de ligands, puis cliquez sur les taches correspondantes.
NOTE: Une famille de ligands correspond à plusieurs taches fonctionnalisées avec le même ligand. Habituellement, la puce présente au moins des doublons et même souvent des triplicates de chaque ligand. - Cliquez sur la définition de l’espèce de finition et attendez d’être dirigé vers la fenêtre contenant les courbes plasmoniques obtenues.
- Choisissez un angle de travail. Faites glisser la ligne noire avec le curseur vers l’angle de travail optimal, puis cliquez sur Déplacer le miroir à l’angle de travail.
REMARQUE: La courbe plasmonique se compose de la valeur de la réflectivité (%) par rapport à l’angle, et le logiciel donne une autre courbe avec la valeur de la pente (%) par rapport à l’angle. Pour sélectionner un bon angle de travail, choisissez l’angle avec la valeur la plus élevée de la pente.- Dans le cas d’une étape de passivation (à cause de l’albumine) effectuée à l’intérieur de l’appareil, sélectionner un angle de travail pour obtenir la sensibilité optimale vers la surface, établissant ainsi un contrôle de qualité de la réactivité de surface.
REMARQUE : Cette étape de passivation est importante lorsque la puce est préparée pour la biodétection d’affinité/capture afin de réduire les interactions non spécifiques entre l’échantillon et la surface de la biopuce.
- Dans le cas d’une étape de passivation (à cause de l’albumine) effectuée à l’intérieur de l’appareil, sélectionner un angle de travail pour obtenir la sensibilité optimale vers la surface, établissant ainsi un contrôle de qualité de la réactivité de surface.
- Cliquez sur Cinétique pour lancer la surveillance cinétique en temps réel. Une fois que le logiciel invite l’utilisateur à définir le contrôle négatif, choisissez aucun contrôle négatif à ce stade (car cela permettra l’observation de la cinétique sur les points de contrôle négatif ).
- Injecter de l’albumine sérique de rat (RSA, 200 μg/mL, préparée dans un tampon d’acétate, pH 4,5) à 50 μL/min pendant 4 min pour passiver la surface autour des taches et éventuellement pour remplir les espaces vides à l’intérieur des taches de ligand.
REMARQUE: Le RSA est injecté pour couvrir la biopuce, qui n’est liée à aucun ligand. - Injecter de l’éthanolamine (1 M) à 20 μL/ min pendant 10 min pour désactiver les groupes carboxyliques encore présents et réactifs en surface.
- Laver la biopuce en injectant 40 mM OG à 50 μL/ min pendant 4 min.
NOTE: Après l’étape de passivation, l’angle de travail est ajusté (comme une nouvelle détermination de base avant l’injection de l’échantillon) pour être à la sensibilité la plus élevée sur les points d’intérêt.
- Injecter de l’albumine sérique de rat (RSA, 200 μg/mL, préparée dans un tampon d’acétate, pH 4,5) à 50 μL/min pendant 4 min pour passiver la surface autour des taches et éventuellement pour remplir les espaces vides à l’intérieur des taches de ligand.
- Injection d’échantillon :
- Redéfinissez les courbes plasmoniques après passivation, et choisissez l’angle de travail en fonction du ligand cette fois.
- Dans la surveillance cinétique, réduire le débit à 20 μL/min et attendre que la ligne de base soit stable.
- Injectez l’échantillon à la concentration de votre choix (de 1 x 10 8 EV/mL à 1 x 10 10 EV/mL selon l’affinité entre les VE et les ligands greffés), puis cliquez sur injection manuelle ou injection automatique. Dans le cas d’une injection manuelle, après injection de 200 μL de l’échantillon, cliquez sur arrêter l’injection. Comme la durée de l’injection est généralement de 10 min, commencer en suivant la cinétique de l’interaction, et mesurer la variation de réflectivité en calculant la différence de réflectivité entre le début et la fin de l’injection observée lors du suivi cinétique (Figure 5).
REMARQUE : Les différents échantillons qui ont été injectés sont décrits dans la section des résultats représentatifs.
- Après l’injection de l’échantillon, suivez l’une de ces deux approches pour terminer l’expérience SPRi :
- Dans l’approche non fixée/ dans un liquide, retirer la biopuce de l’appareil SPRi, y ajouter une goutte liquide et procéder à une caractérisation AFM plus poussée de la surface dans un liquide.
- Dans l’approche fixe , injecter du glutaraldéhyde (0,5%) dilué dans de l’eau à 20 μL/ min pendant 10 min pour fixer les molécules capturées sur la biopuce. Injectez de l’eau pour rincer la surface, retirez la biopuce, lavez-la très doucement avec de l’eau distillée et séchez-la à l’air libre pour une analyse plus approfondie sous AFM.

Figure 4 : Image CCD SPRi de la biopuce. (A,B) Biopuce multiplexée après passivation de l’albumine. (A) Une puce sans défaut ; (B) quelques défauts apparus sur la puce : fusion de taches (i), greffe faible (ii), poussières ou « contaminants » (iii). Les ROIs, en couleur dans les taches (une couleur par famille de ligands), ont été choisis pour éviter ces « contaminants ». Lorsque les taches fusionnaient, elles étaient notées et ignorées ou nommées comme « mélange de ligands 1 et 2 ». (C) Des puces d’or nues sans microréseaux pour l’expérience examinant l’adsorption des VE sur l’or. La flèche bleue indique la direction de l’écoulement. Cette puce n’a pas présenté de taches, et les ROI ont été choisis pour enregistrer le signal de réflectivité de la ligne 1 (L1, cercles rouges) à la ligne 4 (L4, cercles violets) pendant l’injection de l’échantillon. Barre d’échelle = 1 mm pour les trois images. Abréviations : SPRi = imagerie par résonance plasmonique de surface; CCD = dispositif à couplage de charge; ROI = régions d’intérêt; EVs = vésicules extracellulaires. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}

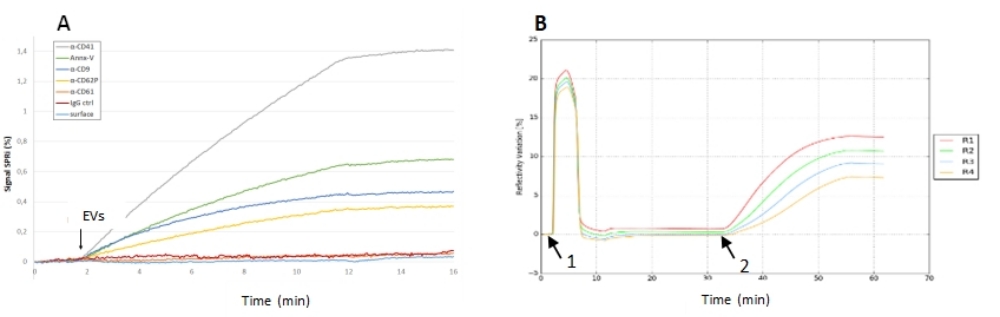
Figure 5 : Expériences SPRi d’injection de VE sur une biopuce. (A) Expérience de capture sur une biopuce multiplexée montrant les signaux de réflectivité de différents ligands. Ici, le rapport signal sur bruit pour les différents ligands était très bon (et surtout sur les spots antiCD41) puisque la réponse du témoin négatif était négligeable. (B) Expérience d’adsorption de VE sur une biopuce nue. Sensorgramme présentant le conditionnement de la puce avec deux buffers de tampon et un nettoyage OG (1), avec l’injection d’échantillon EV (2) et le signal de réflectivité après interaction EV (3). Sur cette biopuce, il n’y avait pas de contrôle négatif, mais le signal de réflectivité (sa cinétique, sa stabilité après injection) était élevé, ce qui signifie que ces VE étaient capables de s’adsorber et de rester stables sur la puce d’or. Abréviations : EV = vésicule extracellulaire; OG = octyl glucoside. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
3. Microscopie à force atomique
- Utilisez le mode contact pour scanner la biopuce dans l’air et le mode d’imagerie quantitative pour scanner la biopuce dans des conditions liquides.
- Alignez la biopuce sur le dessus du masque sur la lame de verre (développée en laboratoire) pour identifier la position des microspots respectifs sur la biopuce (Figure 6A).
REMARQUE: Le masque contient les marques des taches, qui correspondent aux positions des taches des ligands selon l’observateur utilisé. Ce masque consiste en une lame de verre sur laquelle deux coins perpendiculaires permettent le placement de la puce. De plus, la lame de verre est marquée de 16 points de feutre correspondant à la localisation du spot sur la puce, ce qui permet, par transparence, de localiser les spots et de scanner la zone souhaitée. - Positionnement de la pointe :
- Utilisez une caméra CCD au-dessus de l’AFM pour localiser le porte-à-faux au bon endroit qui doit être scanné. Pour suivre ce protocole, utilisez des porte-à-faux triangulaires de 200 μm de longueur, 28 μm de largeur et une constante de ressort de 0,08 N/m.
- Alignez le laser sur le dessus du porte-à-faux à une position qui donne une réponse optimale dans le mécanisme de contrôle de rétroaction.
- Balayage:
- Une fois engagé et en contact avec la surface de la biopuce, commencer l’acquisition AFM en mode contact ou en mode imagerie quantitative de trois à cinq grandes zones (généralement 10 × 10 μm²) à de petites zones (1 x 1 μm²).
REMARQUE: Une représentation des différentes zones qui peuvent être numérisées est montrée à la figure 6D. S’assurer que la caractérisation AFM est représentative de l’ensemble du point mm² et que suffisamment de VE sont visualisés à une bonne résolution pour une analyse robuste (un minimum de 300 EV comptés et analysés pour chaque condition), et effectuer les mesures métrologiques et morphologiques.
- Une fois engagé et en contact avec la surface de la biopuce, commencer l’acquisition AFM en mode contact ou en mode imagerie quantitative de trois à cinq grandes zones (généralement 10 × 10 μm²) à de petites zones (1 x 1 μm²).
- Traitement d’image AFM :
- Traitez les images AFM avec le logiciel de traitement de données JPK en sélectionnant d’abord le canal de hauteur.
- Choisissez un ajustement polynomial à soustraire de chaque ligne pour obtenir des lignes de balayage redressées.
- Sélectionnez le seuil de hauteur sur les grains d’or pour éliminer la rugosité de la surface. Dans le logiciel référencé (voir le tableau des matériaux), dans le module d’extraction des grains, marquez les grains en utilisant une valeur seuil de hauteur à 8,5 nm. Une fois les grains filtrés, le nombre de grains apparaît.
REMARQUE: Habituellement, le substrat d’or brut (RMS de ~3 nm) et la présence des couches chimiques et ligands nécessitent que le seuil soit fixé à 8,5 nm. - Pour extraire plusieurs propriétés des grains marqués, telles que la hauteur, le volume et le diamètre, ouvrez l’image dans le logiciel référencé et sélectionnez Traitement des données | Céréales | Marquer par seuil.
- Choisissez les grains filtrants dans la fonction des propriétés. Lorsqu’une nouvelle fenêtre apparaît, choisissez les paramètres suivants : Valeur = maximum z-max, Surface = surface projetée A0. Ensuite, choisissez les critères A ET B.
- Distribution ouverte des grains; Dans la fenêtre qui s’affiche, choisissez Valeur (maximum), Volume (base : zéro) et Limite (longueur). Observez le tableau (au format .txt) qui s’affiche, qui présente trois colonnes avec les valeurs de hauteur, de volume et de diamètre pour tous les grains détectés au seuil défini par image.
- À partir de la hauteur, h, et du diamètre, D, calculez le rayon de courbure, Rc, de chaque EV à l’aide de l’équation (1)8, puis calculez le volume, V, à l’aide de l’équation (2) :
 (1)
(1) (2)
(2) - À partir de V, calculer le diamètre effectif, d eff, de chaque EV (le diamètre d’une sphère de même volume) en utilisant l’équation (3):
 (3)
(3) - Graphiques de tracé montrant les tailles (hauteur mesurée, diamètre mesuré et diamètre effectif calculé) des VE, chaque particule comptée étant représentée par un point.
NOTE: Ainsi, à la fin de la caractérisation, la plateforme NBA permet la corrélation du signal de biodétection puis du phénotypage, avec les nombres et les tailles des sous-ensembles EV.

Figure 6 : Caractérisation des biopuces par AFM. Après l’expérience SPRi, la puce a été fixée et séchée ou maintenue dans un liquide pour la caractérisation AFM. (A) La lame de verre usiné (avec deux coins de positionnement perpendiculaires, indiqués par un « w » sur la photo) présentant un masque ajusté à la localisation des 16 puces à puces. Par exposition à la lumière et transparence, une fois installée pour la caractérisation AFM, la lame de verre permet de placer la pointe AFM à l’endroit souhaité pour la caractériser. (B) La biopuce installée sur la lame « masque » et sous une goutte de tampon pour scanner dans des conditions liquides. (C) Image SPRi des 16 puces. (D) Un microréseau imagé par microscopie optique après immunocapture de nanoparticules d’étalonnage biofonctionnalisées de 920 nm de diamètre. Les carrés blancs indiquent l’échantillonnage des différentes zones balayées par AFM dans chaque point d’intérêt pour rendre la caractérisation AFM robuste. Barres d’échelle = (C) 1 mm, (D) 500 μm. Abréviations : AFM = microscopie à force atomique; SPRi = imagerie par résonance plasmonique de surface. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
4. Spectroscopie Raman
REMARQUE: Pour la spectroscopie Raman, remplacez la lame de verre utilisée comme substrat par une lame de CaF2, qui a une signature Raman négligeable.
- Conditions optiques pour l’acquisition :
- Réglez les conditions suivantes pour le microscope imageur Raman : objectif du microscope : 50x ; longueur d’onde laser : 532 nm ; puissance laser : 10 mW ; temps d’exposition : 500 ms ; nombre d’accumulations : 140 ; gamme spectrale : de 450 cm−1 à 3 200 cm−1.
REMARQUE: L’utilisation d’une trop grande puissance et / ou d’un temps d’acquisition trop long peut endommager l’échantillon, comme en témoignent des spectres instables au fil du temps. Commencez avec une faible quantité d’énergie et augmentez-la si le signal est trop faible. Des longueurs d’onde laser plus élevées peuvent être utilisées (633 nm, 785 nm) pour réduire la fluorescence préjudiciable aux mesures Raman. Cependant, l’intensité diminue avec la quatrième puissance de la longueur d’onde, et la sensibilité spectrale de la caméra doit être prise en compte.
- Réglez les conditions suivantes pour le microscope imageur Raman : objectif du microscope : 50x ; longueur d’onde laser : 532 nm ; puissance laser : 10 mW ; temps d’exposition : 500 ms ; nombre d’accumulations : 140 ; gamme spectrale : de 450 cm−1 à 3 200 cm−1.
- Imagerie Raman:
- Tout d’abord, observez les spectres en direct avec un nombre réduit d’accumulations (10) pour trouver la zone avec le meilleur rapport signal sur bruit.
REMARQUE: Un signal fort dans la région des hautes fréquences (2 800-3 000 cm-1) peut faciliter la détection des véhicules électriques à la surface avec de faibles temps d’exposition, comme indiqué précédemment10. - Une fois le ROI sélectionné, choisissez la résolution spatiale en fonction du temps disponible pour l’acquisition.
REMARQUE: La résolution spatiale est limitée par la limite de diffraction (~500 nm). - Démarrez l’acquisition de mappage Raman.
- Tout d’abord, observez les spectres en direct avec un nombre réduit d’accumulations (10) pour trouver la zone avec le meilleur rapport signal sur bruit.
- Prétraitement des données :
- À l’aide d’un environnement de développement intégré (IDE) Python (par exemple, Spyder), ouvrez le fichier contenant les spectres.
- Soustrayez la ligne de base des spectres pour corriger l’interférence de la fluorescence possible. Par exemple, utilisez la fonction « arpls » du paquet « irfpy"23. Testez différentes valeurs du paramètre « lam » pour trouver celle qui donne la meilleure correction de base (généralement entre 103 et 107).
- Normaliser les spectres, par exemple, en divisant toutes les intensités d’un spectre par son intensité à 2 900 cm−1 ou en soustrayant la moyenne du spectre, puis en la divisant par son écart-type (normalisation de la variable normale standard).
REMARQUE: Cette étape est nécessaire pour comparer la composition moléculaire relative des VE.
Access restricted. Please log in or start a trial to view this content.
Résultats
Détermination des conditions optimales de pH pour la greffe de ligand
Les différents ligands utilisés pour préparer les biopuces sont testés en fonction du pH et de leur disponibilité à interagir avec la couche chimique thiolate (Figure 3). Les ligands sont dilués dans un tampon acétate à différentes valeurs de pH et injectés sur la biopuce fonctionnalisée chimiquement avec une couche C11C16. Les solutions sont injectées aléatoirement en surface, et un dét...
Access restricted. Please log in or start a trial to view this content.
Discussion
Les méthodes récentes d’identification des VE les plus largement utilisées sont l’immunoblot spécifique aux protéines pour confirmer l’origine des VE, la TEM pour confirmer leur structure et la NTA pour quantifier leur nombre et leur distribution granulométrique dans un échantillonde volume 3. Néanmoins, le grand intérêt pour les véhicules électriques dans la recherche (bio)médicale et les limites des outils analytiques existants ont incité la communauté scientifique à dével...
Access restricted. Please log in or start a trial to view this content.
Déclarations de divulgation
Les auteurs n’ont aucun conflit d’intérêts à divulguer.
Remerciements
Kelly Aubertin et Fabien Picot de l’IVETh (Paris) sont reconnus pour les expériences d’imagerie Raman. Thierry Burnouf (Taipei Medical University, Taïwan) et Zuzana Krupova (d’Helincourt, France) sont reconnus pour avoir fourni les échantillons EV dérivés d’échantillons de plaquettes sanguines et de lait bovin, respectivement. Les travaux ont été soutenus par la région Bourgogne Franche-Comté et l’école doctorale EUR EIPHI (projet NOVICE, 2021-2024). Une partie de ce travail a été réalisée à l’aide de la plateforme CLIPP et dans les salles blanches RENATECH de FEMTO-ENGINEERING, pour lesquelles nous remercions Rabah Zeggari.
Access restricted. Please log in or start a trial to view this content.
matériels
| Name | Company | Catalog Number | Comments |
| CD41a antibody | Diaclone SAS (France) | 447528 | |
| CP920 | Microparticles GmbH, Germany | 448303 | |
| DXR3xi | Thermo Fisher Scientific | T1502 | |
| EDC | Sigma | A6272 | |
| Ethanolamine | Sigma | P5368-10PAK | |
| Evs derived from platelet concentrates | Collaboration : Pr T. Burnouf (TMU, Taipei) | S2889 | |
| Evs from bovine milk | Collaboration : Dr Z. Krupova (Excilone, Helincourt - France) | 3450 | |
| Glutaraldehyde | Sigma | 56845 | |
| Gwyddion | 853.223.020 | ||
| Magnetron sputtering | PLASSYS | SAB5300165 | |
| mercapto-1-hexadecanoic acid | Sigma | G5882 | |
| Mercapto-1-undecanol | Sigma | O8001 | |
| Mountains SPIP ones | Digital Surf | ||
| NanoWizard 3 Bioscience | Bruker-JPK | ||
| Octyl Glucoside (OG) | Sigma | ||
| Ovalbumine antibody | Sigma | ||
| Phosphate Buffer Saline (PBS) | Sigma | ||
| Rat Albumin Serum (RSA) | Sigma | ||
| Sodium acetate buffer | Sigma | ||
| SPR-Biacore 3000 | GE Healthcare/ Cytiva life sciences | ||
| SPRi Biochip | MIMENTO technology platform | The biochips were produced in-house in the clean room, Besancon | |
| SPRi Plex II | Horiba Scientific | ||
| Sulfo-NHS | Sigma |
Références
- Silva, A. K. A., et al. Development of extracellular vesicle-based medicinal products: A position paper of the group "Extracellular Vesicle translatiOn to clinicaL perspectiVEs - EVOLVE France". Advanced Drug Delivery Reviews. 179, 114001(2021).
- Xunian, Z., Kalluri, R. Biology and therapeutic potential of mesenchymal stem cell-derived exosomes. Cancer Science. 111 (9), 3100-3110 (2020).
- Hartjes, T. A., et al. Extracellular vesicle quantification and characterization: Common methods and emerging approaches. Bioengineering. 6 (1), 7(2019).
- Xing, Y., et al. Analysis of extracellular vesicles as emerging theranostic nanoplatforms. Coordination Chemistry Reviews. 424, 213506(2020).
- Wang, T., Xing, Y., Cheng, Z., Yu, F. Analysis of single extracellular vesicles for biomedical applications with especial emphasis on cancer investigations. Trends in Analytical Chemistry. 152, 116604(2022).
- Boireau, W., Elie-Caille, C. Extracellular vesicles: Definition, isolation and characterization. Medecine Sciences: M/S. 37 (12), 1092-1100 (2021).
- Brisson, A. R., et al. Extracellular vesicles from activated platelets: A semiquantitative cryo-electron microscopy and immuno-gold labeling study. Platelets. 28 (3), 263-271 (2017).
- Yuana, Y., et al. Atomic force microscopy: A novel approach to the detection of nanosized blood microparticles. Journal of Thrombosis and Haemostasis. 8 (2), 315-323 (2010).
- Sebaihi, N., de Boeck, B., Yuana, Y., Nieuwland, R., Pétry, J. Dimensional characterization of extracellular vesicles using atomic force microscopy. Measurement Science and Technology. 28 (3), 034006(2017).
- Beekman, P., et al. Immuno-capture of extracellular vesicles for individual multi-modal characterization using AFM, SEM and Raman spectroscopy. Lab on a Chip. 19 (15), 2526-2536 (2019).
- Malenica, M., et al. Perspectives of microscopy methods for morphology characterisation of extracellular vesicles from human biofluids. Biomedicines. 9 (6), 603(2021).
- Verweij, F. J., et al. The power of imaging to understand extracellular vesicle biology in vivo. Nature Methods. 18 (9), 1013-1026 (2021).
- Théry, C., et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. Journal of Extracellular Vesicles. 7 (1), 1535750(2018).
- Obeid, S., et al. NanoBioAnalytical characterization of extracellular vesicles in 75-nm nanofiltered human plasma for transfusion: A tool to improve transfusion safety. Nanomedicine: Nanotechnology, Biology, and Medicine. 20, 101977(2019).
- Obeid, S., et al. Development of a NanoBioAnalytical platform for «on-chip» qualification and quantification of platelet-derived microparticles. Biosensors and Bioelectronics. 93, 250-259 (2017).
- Ridolfi, A., et al. AFM-based high-throughput nanomechanical screening of single extracellular vesicles. Analytical Chemistry. 92 (15), 10274-10282 (2020).
- Vorselen, D., et al. The fluid membrane determines mechanics of erythrocyte extracellular vesicles and is softened in hereditary spherocytosis. Nature Communications. 9 (1), 4960(2018).
- Hardij, J., et al. Characterisation of tissue factor bearing extracellular vesicles with AFM: Comparison of air-tapping-mode AFM and liquid Peak Force AFM. Journal of Extracellular Vesicles. 2, 21045(2013).
- Jorgensen, M., et al. Extracellular Vesicle (EV) Array: Microarray capturing of exosomes and other extracellular vesicles for multiplexed phenotyping. Journal of Extracellular Vesicles. 2, 20920(2013).
- Remy-Martin, F., et al. Surface plasmon resonance imaging in arrays coupled with mass spectrometry (SUPRA-MS): Proof of concept of on-chip characterization of a potential breast cancer marker in human plasma. Analytical and Bioanalytical Chemistry. 404 (2), 423-432 (2012).
- Czamara, K., et al. Raman spectroscopy of lipids: A review. Journal of Raman Spectroscopy. 46 (1), 4-20 (2015).
- Penders, J., et al. Single particle automated Raman trapping analysis of breast cancer cell-derived extracellular vesicles as cancer biomarkers. ACS Nano. 15 (11), 18192-18205 (2021).
- Baek, S. J., Park, A., Ahn, Y. J., Choo, J. Baseline correction using asymmetrically reweighted penalized least squares smoothing. Analyst. 140 (1), 250-257 (2015).
- Daaboul, G. G., et al. Digital detection of exosomes by interferometric imaging. Scientific Reports. 6, 37246(2016).
- Ertsgaard, C. T., et al. Integrated nanogap platform for sub-volt dielectrophoretic trapping and real-time Raman imaging of biological nanoparticles. Nano Letters. 18 (9), 5946-5953 (2018).
- Maas, S. L., et al. Possibilities and limitations of current technologies for quantification of biological extracellular vesicles and synthetic mimics. Journal of Controlled Release. 200, 87-96 (2015).
Access restricted. Please log in or start a trial to view this content.
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.