
Method Article
Intégration de Wet and Processes Banc sec Optimise ciblés Séquençage de nouvelle génération de faible qualité et à faible quantité biopsies de tumeurs
Dans cet article
Résumé
An integrated system for targeted next-generation sequencing of oncology specimens is described. This cross-platform system is optimized for low-quality and low-quantity tumor biopsies, accommodates low DNA inputs, includes well-characterized multi-variant controls, and features a novel variant caller that is informed by quantitative pre-analytical quality control measures.
Résumé
Toutes les procédures de séquençage de nouvelle génération (NGS) comprennent des analyses effectuées sur le banc de laboratoire ( «banc humide») et les données analyses effectuées en utilisant la bioinformatique pipelines ( «banc sec»). Les deux éléments sont essentiels pour produire des résultats précis et fiables, qui sont particulièrement critiques pour les laboratoires cliniques. technologies NGS ciblées ont de plus en plus trouvé grâce à des applications en oncologie pour aider les objectifs de la médecine de précision avance, mais les méthodes impliquent souvent déconnecté et variable workflows banc humides et secs et des ensembles de réactifs non coordonnées. Dans ce rapport, nous décrivons une méthode pour le séquençage contestant spécimens de cancer avec un panel de 21 gènes, par exemple, d'un système NGS complète et ciblée. Le système intègre la quantification fonctionnelle de l'ADN et de la qualification, l'enrichissement par PCR multiplexée à tube unique, et la purification de la bibliothèque et de la normalisation en utilisant analytiquement-vérifiés, des réactifs de source unique avec une suite de bioinformatique autonomes.En conséquence, la variante précise des appels de faible qualité et à faible quantité fixés au formol, enrobés de paraffine (FFPE) et l'aiguille fine (FNA) biopsies tumorales peuvent être atteints. Le procédé peut évaluer systématiquement les variantes associées au cancer provenant d'une entrée de 400 copies d'ADN amplifiables, et il est conçu de façon modulaire pour accueillir un nouveau contenu génétique. Deux types de contrôles analytiquement définis différentes fournissent l'assurance de la qualité et de l'aide sauvegarde la précision des appels avec des échantillons cliniquement pertinents. Une étape flexible "tag" de PCR intègre adaptateurs spécifiques à la plate-forme et les codes d'index pour permettre barcoding de l'échantillon et la compatibilité avec paillasse commun instruments NGS. Fait important, le protocole est simplifié et peut produire 24 bibliothèques de séquence prêt en une seule journée. Enfin, l'approche relie les processus de banc humide et sèche en incorporant les résultats de contrôle de la qualité de l'échantillon pré-analytique directement dans la variante d'appel des algorithmes pour améliorer la précision de la détection de mutations et de se différencier de faux négatifs et indetermappels onnées. Cette méthode NGS ciblée utilise les progrès dans les deux wetware et de logiciels pour atteindre haute profondeur, le séquençage et l'analyse multiplexée sensible des échantillons de cancer hétérogènes pour des applications de diagnostic.
Introduction
la médecine de précision repose sur l'individualisation des options diagnostiques et thérapeutiques pour les patients. La promesse de traitements adaptés est une conséquence directe d'une meilleure compréhension des voies de maladies qui peuvent éclairer la liaison des diagnostics moléculaires et des thérapies ciblées. Par exemple, l'utilisation des thérapies moléculaires ciblées est passé de 11% à 46% entre 2003 et 2013 1, et les médicaments anti-cancer tels que le vemurafenib et crizotinib sont approuvés par la FDA avec des tests de diagnostic compagnon. Avec sa capacité à récupérer avec précision des cibles de séquence faible abondance dans les ensembles d'échantillons hautement multiplexées, séquençage de nouvelle génération (NGS) a émergé comme une méthode de choix pour évaluer les aberrations génétiques associées au cancer et à l'identification des cibles moléculaires pour la médecine de précision.
Les biopsies de tumeurs solides les plus courants pour les tests moléculaires comprennent fixés au formol, enrobés de paraffine (FFPE) et aspiration à l'aiguille fine (FNA) SPECIMens. Ces échantillons sont lourdes de faible quantité et / ou de faible qualité des acides nucléiques qui défient des évaluations précises NGS 2-5. NGS commerciales actuelles méthodes pour l'analyse de ces échantillons sont basés sur un patchwork de différents réactifs, protocoles et outils informatiques qui représentent des cibles mobiles d'améliorations continues. Par exemple, les changements de chimies et / ou logiciels essai ont eu lieu tous les 1-2 mois pour NGS kits les plus couramment utilisés ciblés 6. Cette instabilité reflète un manque de cohérence dans la construction et la vérification d'un système unifié pour NGS types d'échantillons difficiles, en particulier pour le dépistage du cancer, et met un fardeau indu aux laboratoires d'élaborer des protocoles cohérents qui sont optimisés à partir d'échantillons en résultats. En effet, un récent sondage auprès des utilisateurs NGS a mis en évidence les difficultés de ces technologies "évolution rapide", ainsi que les exigences pour établir, le contenu médical à une action, une expertise en bio-informatique ancrée, un Solidified et procédure intégrée qui peut être rapidement mis en œuvre, et la rationalisation des flux de travail et des protocoles simplifiés qui facilitent sur le tas 7. Dans cet article, un système complet de NGS ciblée est décrite qui aborde ces lacunes.
La méthodologie présentée intègre toutes les étapes de la procédure de pré-analytique post-analytique à la fois des bancs humides et sèches pour améliorer la précision, la sensibilité et la fiabilité de la quantification de la cible et la détection pour NGS de loci des gènes du cancer cliniquement pertinentes. Cette approche commence par la quantification de l' ADN "fonctionnel" 4 pour évaluer la qualité de l' ADN, guide d' entrée dans l'étape d'enrichissement PCR, et se prémunir contre les appels faux positifs qui peuvent découler de l'interrogatoire de très faibles copies de modèles. Un multiplex unique tube PCR enrichit ensuite pour les gènes du cancer 46 loci à 21 en utilisant seulement 400 copies d'ADN amplifiables, suivie par l'incorporation de séquences spécifiques à la plateforme pour NGS using bureau commun instruments de séquençage. Les bibliothèques sont purifiés en utilisant une procédure de bille magnétique simple et quantifiées avec un roman, essai qPCR sans calibrage. Une suite de bioinformatique autonomes, informés par l'ADN QC résultats de l'échantillon pour améliorer les performances d'appel, fournit une analyse de séquence suivante NGS. Nous présentons des données en utilisant cette approche systémique NGS ciblée pour révéler des mutations de base de substitution, insertion / délétions (indels), et le nombre de copies variantes (CNV) dans des biopsies de faible qualité et à faible quantité tumorales tels que FFPE et des échantillons de cytoponction, et courir contrôles.
Protocole
Remarque: Ce protocole décrit le traitement simultané d'échantillons à l'aide d'un système de centrale nucléaire MiSeq mais peut être adapté pour l'instrument personnel du génome machine (PGM). Pour l'entrée d'ADN minimum recommandé de 400 amplifiables copies de modèles, le dosage est capable de produire au moins 3,000x couverture médiane pour chacun des 96 échantillons par NGS terme, et la profondeur de couverture équivalente pour 24 échantillons à l'aide du PGM sur une puce 318. Le procédé nécessite également l'utilisation d'un instrument de PCR en temps réel.
Quantification fonctionnelle 1. ADN et contrôle de la qualité (QC)
- réactifs Décongeler: 2x Master Mix, Primer Probe Mix, Inhibition Primer Probe Mix, 6-carboxy-X-rhodamine (ROX), Diluant, et les quatre normes de la courbe d'étalonnage de l'ADN génomique humain (ADN standard (50 ng / pl), l'ADN standard (10 ng / ul), de l' ADN standard (2 ng / ul) et de l' ADN standard (0,4 ng / pl)) (Tableau 1). Tourbillonnaire tous les réactifs pendant 10 secondes et centrifuger à la vitesse maximale pendant 10 secondes pour récupérer le contenu.Gardez le mélange maître 2x sur la glace.
- Préparer une quantité suffisante de mélange maître pour le nombre total d'échantillons à tester et comprennent 10% plus de volume pour éviter les pénuries dues à pipetage. Préparer le mélange maître dans un tube à centrifuger en utilisant les volumes suivants par échantillon: 5 pi 2x Master Mix, 0,5 ul Primer Probe Mix, 0,5 ul Inhibition Primer Probe Mix, 0,05 ul ROX et 2,95 Diluant ul. Vortex pendant 10 secondes et centrifuger à la vitesse maximale pendant 10 secondes pour récupérer le contenu.
- Ajouter 9 pi mélange maître dans les puits d'une plaque de 96 puits.
- Ajouter 1 pi des normes d'ADN en double exemplaire pour générer une courbe d'étalonnage. Mélanger par aspiration et vers le bas 5 fois.
- Vérifiez que l'échantillon d'acide nucléique est bien mélangé avant utilisation. Ajouter échantillon de 1 ul au mélange maître et mélanger par pipetage up-and-down 5 fois.
- Sceller la plaque, vortex pendant 10 s et centrifuger à la vitesse maximale pendant 10 s pour recueillir son contenu.
- Placer la plaque dans le système de PCR. Attribuer à la fois FAM (de quantification fonctionnelle) et VIC (Inhibition fonctionnelle) détecteurs pour chaque échantillon selon les instructions du fabricant. Effectuer des cycles de PCR de 10 min à 95 ° C et 40 cycles (15 s à 95 ° C, 1 min à 60 ° C).
- Analyser les données qPCR en générant une courbe de régression linéaire pour chacune des normes d'ADN en double en utilisant des protocoles de logiciels.
- Tracer le journal 10 du nombre de copies pour chaque standard d'ADN sur l'axe des x et la valeur de q FAM C correspondant à l'axe des ordonnées.
- Assurez-vous que les résultats de l'nucléique échantillon d'acide se situent dans la plage dynamique de la courbe d'étalonnage des normes ADN, puis calculer la concentration de l'ADN inconnu en «fonctionnelle» ou amplifiable nombre de copies par ml de sa position correspondante sur la courbe étalon de référence. la figure 1 montre des exemples de courbes d'étalonnage qui sont passés et ont échoué.
- Déterminer si une amplification a eu lieu dans chaque réaction en vérifiant la présence de l'amplicon cible non-humain dans le canal CIV.
Remarque: Comme témoin positif, la sonde Primer Mix d'inhibition contient des amorces spécifiques pour une cible exogène non-humain et le gabarit correspondant. Le Inhibition Primer Probe Mix est un composant du mélange maître qui est ajouté à chaque réaction, y compris le contrôle sans matrice (NTC). En l'absence d'un inhibiteur, le produit de PCR pour la cible non-humain doit toujours être détecté dans le canal CIV. Un «non détecté» Cq pour un échantillon dans le canal CIV indique la présence d'inhibiteurs de PCR qui peuvent bénéficier de nettoyage ultérieur de l'échantillon avant le traitement ultérieur.
2. Bibliothèque Préparation: Gene spécifique (GS) PCR
- Préparer une quantité suffisante de mélange maître pour le nombre total d'échantillons à tester et comprennent 10% plus de volume pour éviter les pénuries dues à pipetting. Préparer le mélange maître GS PCR dans un tube à centrifuger en utilisant les volumes suivants par échantillon: 5 pi 2x Amplification Master Mix (tableau 1) et 1 ul Pan Cancer Panel Primer (tableau 1). Mélanger par pipetage vers le haut et vers le bas, vortex pendant 10 secondes et centrifuger à la vitesse maximale pendant 10 s pour recueillir son contenu.
- Aliquoter le 6 pi GS PCR mélange maître dans les puits d'une plaque de 96 puits. Ajouter 4 ul de chaque échantillon d'acide nucléique dans des puits individuels. Pour d' autres puits, ajouter 4 pi du contrôle FFPE (tableau 1), 4 pl de la commande multi-Variant (tableau 1), et 4 ul d'eau sans nucléase pour une NTC procédurale. Pour chaque addition, mélanger par pipetage up-and-down 5 fois.
- Sceller la plaque, vortex pendant 10 s et centrifuger à la vitesse maximale pendant 10 s pour recueillir son contenu.
- Placer la plaque dans le thermocycleur pour des cycles de PCR suivants: 5 min à 95 ° C,2 cycles (15 s à 95 ° C, 4 min à 60 ° C), 23 cycles (15 s à 95 ° C, 4 min à 72 ° C), et une extension finale de 10 min à 72 ° C. Tenir à 4 ° C.
Remarque: Après l'achèvement de l'étape 2.4, la plaque sera appelée la plaque GS PCR.
3. Bibliothèque Préparation: Tag PCR
- Réactifs Décongeler: 2x Index Master Mix (tableau 1) et des codes Index (tableau 1). Vortex pendant 10 secondes et centrifuger à la vitesse maximale pendant 10 secondes pour récupérer le contenu.
Remarque: Les codes index sont prémélangés pour fournir un ensemble unique d'indices appariées (codes barres) pour chaque échantillon. - Dans une plaque de 96 puits, ajouter 7,5 pi de 2x Index Master Mix et 5,5 pi d'un code Index à un bien spécifié et mélanger par pipetage up-and-down 5 fois.
- Ouvrez soigneusement la plaque GS PCR, et ajouter 2 produit ul GS PCR à la nouvelle plaque avec le mélange maître. Mélanger par aspiration et vers le bas 5 fois. Pour chaque échantillon, enregistrer l'ID de l'échantillon et les codes correspondants appariées Index. Sceller la plaque, vortex pendant 10 s et centrifuger à la vitesse maximale pendant 10 s pour recueillir son contenu.
- Placer la plaque dans le thermocycleur et la PCR pendant 5 min à 95 ° C, 10 cycles (30 s à 95 ° C, 30 s à 55 ° C, 1 min à 72 ° C), et une extension finale de 10 min à 72 ° C. Tenir à 4 ° C.
Remarque: Après l'achèvement de l'étape 3.4, la plaque sera appelée la plaque PCR Tag.
4. Bibliothèque Purification et Sélection de la taille
- Enlever les billes magnétiques Bibliothèque pure Prep (tableau 1) et Elution Buffer (tableau 1) 2-8 ° C et laisser se stabiliser à température ambiante pendant 30 min. Ajouter 9,6 ml d' éthanol à 100% au tampon de lavage (tableau 1) récipient, bouchon et mélanger en inversant la bouteille plusieurs fois.
- Vortex les perles magnétiques pendant 10 secondes et ajouter 11 ul dans des puits séparés d'un 9plaque de 6 puits.
- Ouvrez la plaque PCR de Tag et ajouter 10 pl de produit PCR Tag aux billes et pipette mélange 5 fois. Laisser incuber le mélange pendant 4 min à température ambiante.
- Placer la plaque de 96 puits sur le support magnétique (tableau 1) pendant 4 min. Avec la plaque de 96 puits encore sur le stand, enlever et jeter le surnageant avec une pipette.
- Retirez la plaque de 96 puits à partir du support magnétique et ajouter 100 pi de tampon de lavage contenant de l'éthanol à chaque puits et mélanger par pipetage up-and-down 5 fois. Incuber pendant 2 min.
- Placer la plaque de 96 puits sur le support magnétique pendant 2 min, puis retirer et jeter le surnageant avec une pipette.
- Répétez l'étape 4.5, pour un total de 2 lavages à l'éthanol, retirer la solution de lavage autant possible après le second lavage.
- Avec la plaque de 96 puits sur le support magnétique, sécher les perles pendant 2 min à température ambiante, puis retirez la plaque du support.
- Resuspendre les billes en ajoutant 20 pi de tampon d'élution à chaque well et pipeter vers le haut et vers le bas 5 fois.
- Incuber pendant 2 minutes à température ambiante.
- Placer la plaque de 96 puits de retour sur le support magnétique pendant 4 min, et soigneusement enlever et transférer 18 pi du surnageant clair pour un nouveau puits.
Remarque: La procédure peut être arrêtée sans risque à cette étape et les échantillons stockés à -15 à -30 ° C. Pour redémarrer, dégeler les échantillons congelés sur de la glace avant de poursuivre.
5. Quantification Bibliothèque
- Thaw Library Quant (LQ) réactifs: 2x LQ Master Mix, Primer LQ / Probe Mix, LQ Standard, LQ Contrôle positif, LQ Diluant et LQ ROX (tableau 1). Vortex pendant 10 secondes et centrifuger à la vitesse maximale pendant 10 secondes pour récupérer le contenu.
- L'utilisation de produits de bibliothèque purifiés, effectuer une dilution en série de chaque échantillon individuel LQ Diluant.
- Ajouter 2 ul (bibliothèque de produit purifié) à 198 ul LQ Diluant et mélanger jusqu'à et vers le bas avec une pipette 10 fois.
- Ajouter 2 pi (dilution 1: 100) à 198 ul LQ Diluant et mélanger vers le haut et vers le bas avec une pipette 10 fois.
- Préparer une quantité suffisante de LQ master mix pour le nombre total d'échantillons à tester et comprennent 10% plus de volume pour éviter les pénuries dues à pipetage. Préparer le LQ Master Mix dans un tube à centrifuger en utilisant les volumes suivants par échantillon: 5 ul 2X LQ Master Mix, 2 pl LQ Primer / Probe Mix, 0,5 ul LQ standard et 0,5 ul LQ ROX. Mélanger par pipetage vers le haut et vers le bas, vortex pendant 10 secondes et centrifuger à la vitesse maximale pendant 10 s pour recueillir son contenu.
- Ajouter 8 ul maître LQ mélange à un puits d'une plaque à 96 puits optique.
- Dans les puits séparés, ajouter 2 pi de bibliothèque diluée, 2 pl LQ contrôle positif et 2 pl LQ Diluant (NTC) et mélanger par pipetage up-and-down 5 fois. Sceller la plaque avec un film adhésif optique, vortex pendant 10 secondes et centrifuger à 400 xg pendant 10 secondes pour recueillir le contenu.
- Attribuer à la fois FAM et VIC detectors pour chaque échantillon. Effectuer une amplification par PCR en utilisant des conditions de cycle de 5 min à 95 ° C, et 40 cycles (15 sec à 95 ° C, 1 min à 60 ° C).
- Déterminer la concentration de chaque échantillon (nm) en utilisant la méthode comparative C q. Calculez la différence de l'connue LQ standard (C q VIC) à la bibliothèque inconnue (C q FAM). La concentration de l'échantillon dilué (P) est calculée en utilisant l'équation suivante:
[Lib Conc] nM = 12,5 x 2 Δ Cq
Si on utilise un autre facteur de dilution de 10 000, calculer le rapport entre le facteur de dilution de la cible à 10 000, et multiplier ce facteur par le résultat de l'équation 5.7.
6. Bibliothèque Normalisation et Pooling Sample
- Déterminer la concentration médiane (nM) dans tous les échantillons (contenant chacun un indice pairwise uniques) être mis en commun.
- Déterminer le volum d'échantillon individuele (ul) à la piscine, en multipliant la concentration médiane dans tous les échantillons en 5, puis en divisant par la concentration individuelle (nM). Autour de la valeur résultante à l'entier le plus proche. volumes ronds avec des valeurs de <2 pi à 2 pi, et des volumes> 15 pi à 15 pi.
- Ajouter le volume normalisé (pi) pour chaque échantillon dans un tube à centrifuger unique pour créer le pool d'échantillons.
- Calculer la nouvelle concentration pour chaque échantillon en utilisant des valeurs entières arrondies et enregistrer les résultats.
- Pour déterminer la concentration de la piscine de l'échantillon, calculer la somme de toutes les concentrations individuelles et enregistrer la valeur résultante (nM).
- Diluer la piscine de l' échantillon à 1,25 nM en utilisant le séquençage Diluant (tableau 1).
Remarque: La procédure peut être arrêtée sans risque à cette étape et les échantillons stockés à -15 à -30 ° C. Pour redémarrer, dégeler les échantillons congelés sur de la glace avant de poursuivre.
7. séquençage
- Denature la piscine de l' échantillon en présence du v3 contrôle PhiX (tableau 1) en ajoutant les suivantes: 15 pi de 1,25 nM Sample Pool, 3 pi de 0,5 nM PhiX et 2 pi de 1 N NaOH. Vortex brièvement suivi par une brève centrifugation et incuber pendant 5 min à température ambiante.
- Placez la piscine de l'échantillon dénaturé sur la glace.
- Ajouter 8 ul de bibliothèque dénaturé à 992 ul de tampon HT1-Hyb prérefroidi dans un tube de microcentrifugation. Vortex brièvement pour mélanger, suivie d'une brève centrifugation pour recueillir le contenu. Gardez sur la glace.
- Ajouter 600 ul de la bibliothèque dénaturé et on dilue à la position # 17 de la cartouche de réactif.
- Thaw Lisez 1 Séquençage Amorces (tableau 1), Index Lisez Séquençage Amorces (tableau 1), et Lire 2 Séquençage Amorces (tableau 1). Dans les tubes à centrifuger, diluer séparément 4 pi séquençage Amorces avec 636 tampon HT1-Hyb ul. Remarque: tampon HT1-Hyb est fourni with , le kit de réactifs de séquençage (tableau 1).
- Mélanger par tourbillonnement pendant 10 secondes et centrifuger à vitesse maximale pendant 10 secondes pour recueillir le contenu. Ajouter 600 pl de dilution Lire 1 Amorces de séquençage à la position # 18 de la cartouche de réactif de séquençage, 600 pl de dilution index lues Amorces à la position # 19, et 600 ul de dilution Lire 2 Amorces de séquençage à la position # 20.
- Charger les réactifs sur l'instrument NGS (tableau 1) et la séquence selon les instructions du fabricant. Effectuer un jumelé fin 2 x 150 séquençage cyclique terme.
Analyse 8. Données
Remarque: Le logiciel de l'instrument NGS convertit les images en grappe aux appels de base et les scores de qualité, et démultiplexe indices appariées pour générer individuelle FASTQ gzip compressé (* .fastq.gz) fichiers pour chaque échantillon. Avant d'analyser les fichiers démultiplexés, le lecteur doit télécharger et installer le logiciel de bioinformatique associé (Tableau1). Le logiciel peut être installé sur un consommateur de qualité PC Windows et ne nécessite pas de matériel informatique spécialisé ou une connexion Internet pour effectuer l'analyse des données.
- Double-cliquez sur l'icône du bureau du logiciel.
- Connectez-vous au système en utilisant le nom d'utilisateur et mot de passe fourni dans le manuel du logiciel.
- Ouvrez le tableau de bord du projet, puis cliquez sur "Nouveau projet".
- Nommez le projet et fournir une description du projet en option. Sélectionnez le type de panneau NGS ciblé et NGS type d'instrument. Cliquez sur "Enregistrer et continuer".
- Téléchargez les fichiers FASTQ compressés pour la marche avant et arrière se lit. Ne téléchargez pas les FASTQs "non affectés", qui contiennent lit qui n'a pas réussi à démultiplexer. Cliquez sur "Enregistrer et continuer".
- Entrez le nombre de copies d'entrée fonctionnels utilisés pour préparer chaque bibliothèque tel que déterminé par la quantification de l'ADN fonctionnelle essai (voir l'étape 1 ci-dessus). Ajoutez manuellement des valeurs ou des valeurs de copier et coller à partir d'une propagationfeuille dans la table d'annotation. Cliquez sur "Enregistrer et continuer."
- Passez en revue les bibliothèques annotés téléchargées pour l'analyse et cliquez sur «Soumettre une analyse" pour lancer l'analyse.
- Surveiller les progrès de l'analyse affichée par le tableau de bord du projet.
Note: Une analyse complète indication d'état est présenté lorsque les résultats sont prêts pour examen. - Passez en revue les résultats analysés pour l'échantillon QC métriques, y compris la couverture totale par bibliothèque, le pourcentage de lectures filtres passant, la profondeur de la couverture de l'amplicon et de l'uniformité. Examiner la variante appelle à chaque bibliothèque séquencée avec dbSNP, COSMIC, 1000 génomes et autres sources d'annotation de niveau fonctionnel et de la population.
- Exporter les résultats bruts sous forme de tableaux sommaires de feuille de calcul, les fichiers * .bam et fichiers * .vcf pour le stockage à long terme ou de l'analyse en aval avec des outils informatiques complémentaires.
Résultats
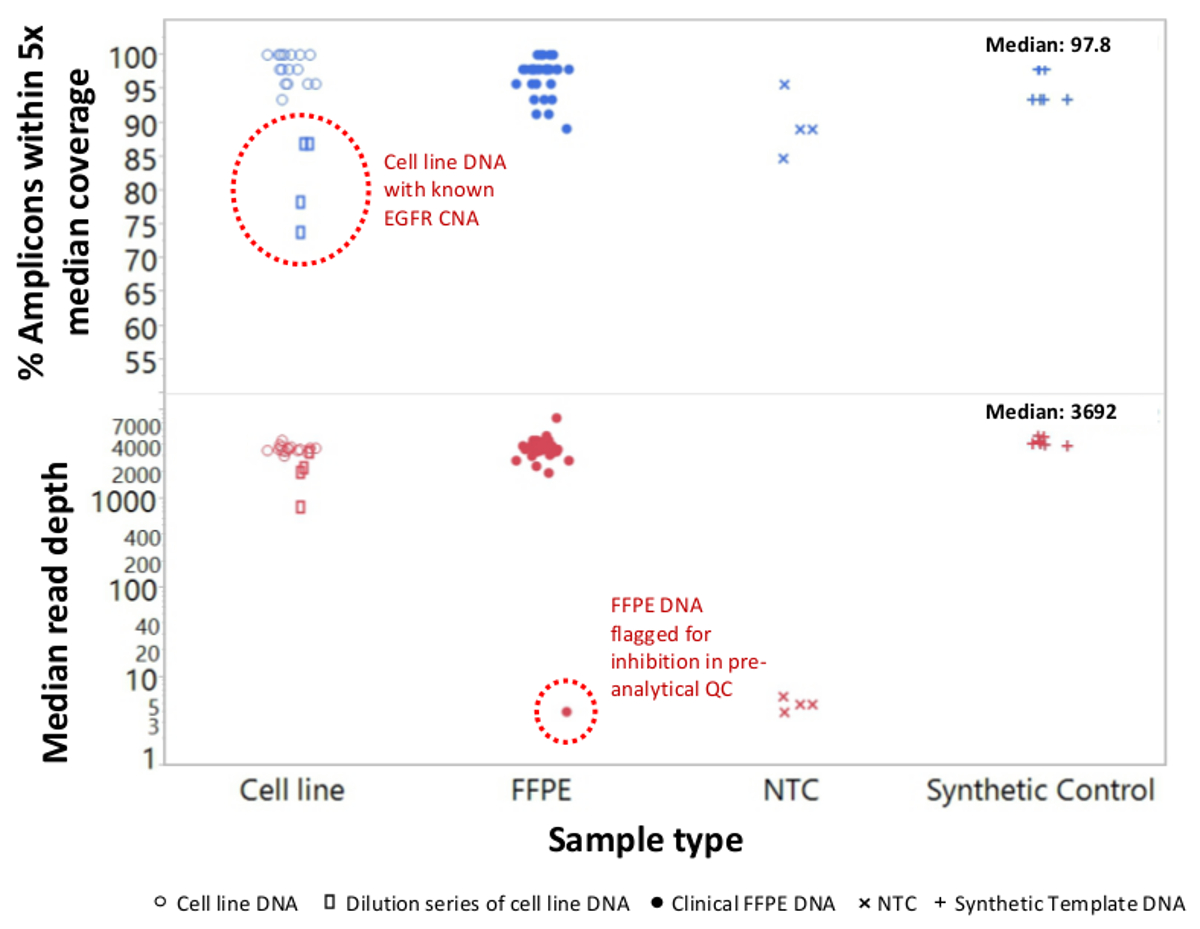
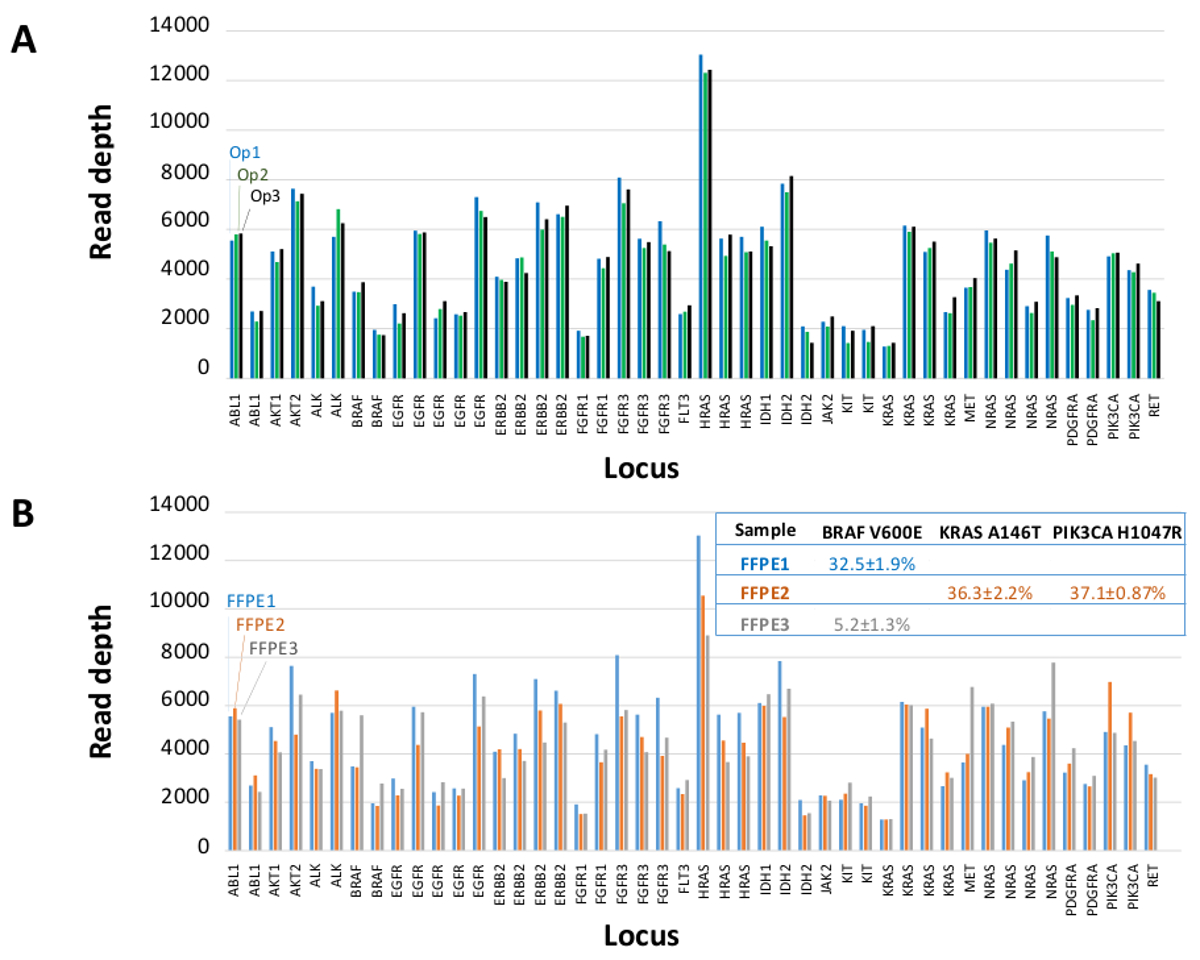
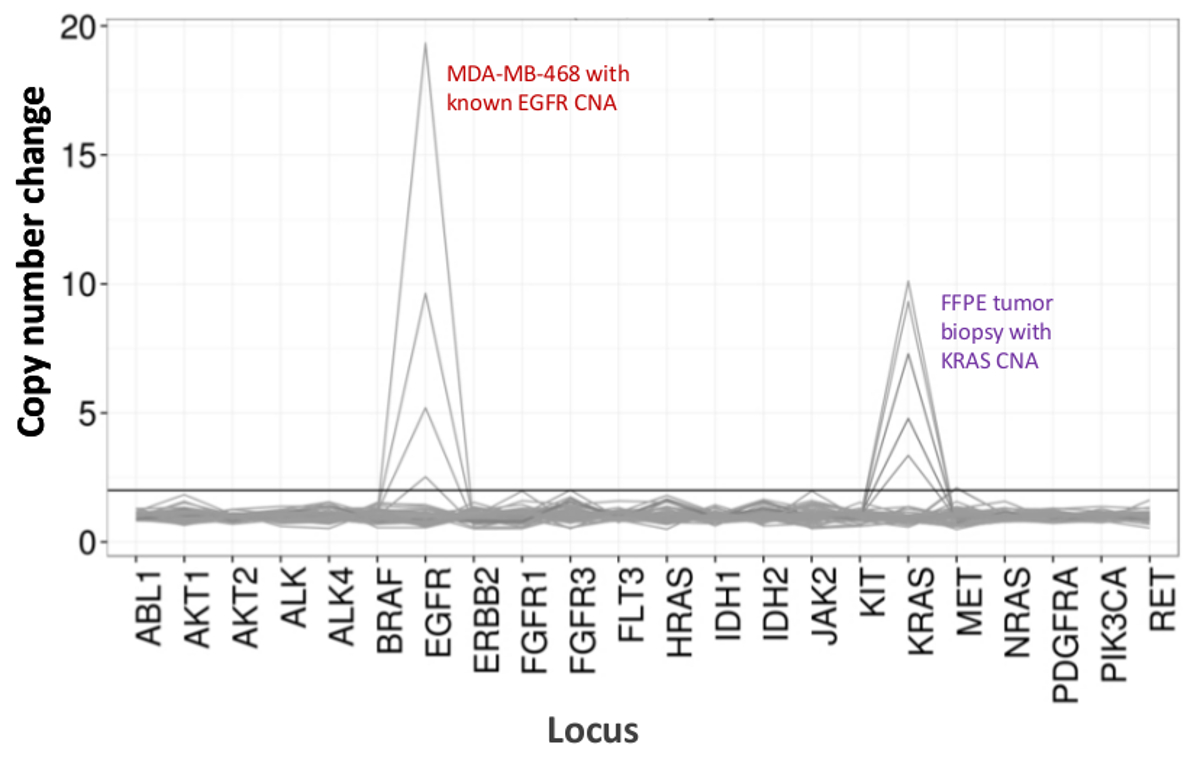
Un total de 90 échantillons (74 uniques) représentant des contrôles positifs et négatifs, des lignées cellulaires précédemment caractérisées, et des biopsies tumorales FFPE cliniques résiduels ont été évalués pour l'ADN amplifiable, entrée dans l'enrichissement PCR multiplex, marqués avec des adaptateurs de séquence, code-barres, et analysé en une seule paillasse NGS instrument run (Figure 2) qui a produit 19,1 M lit filtre passe. échantillon équimolaire mise en commun a abouti à un séquençage de grande profondeur (3,692x lectures) et une couverture uniforme (97,8% des amplicons couverts à moins de 5 fois de la profondeur de lecture médiane). Outliers composés sans matrice de contrôle, un ADN cellulaire en ligne avec un grand nombre de copies d' amplification et un ADN FFPE qui a été marqué pour inhibition de la PCR par le QC dosage pré-analytique (Figure 3). Uniformité de la couverture dans les 46 amplicons a été maintenu à l' aide de trois opérateurs différents (figure 4A), et pour différents échantillons d'ADN de FFPE de faible qualité ( Figure 4B). Une commande de l'ADN tumoral FFPE, formulé à partir d'un mélange d'échantillons cliniques résiduels pour atteindre 5% BRAF V600E (quantifié par gouttelettes PCR numérique), a été rapporté que la mutation BRAF cible à abondances de 3,9, 5,3 et 6,5% en trois opérateurs qui utilisent une entrée de 400 copies amplifiables (et donc seulement 20 copies mutantes) (figure 4B et "FFPE3" du tableau en encadré). En outre, un mélange de 12 modèles d'ADN synthétiques, chacun représentant un "pilote" base-substitution mutation connue, a révélé les mutations attendues dans la gamme prévue de 9-17% Fréquence allèle moyenne (tableau 2). Dilution de la lignée cellulaire et des échantillons FFPE ADN avec nombre de copies amplifications démontré dose-dépendance pour les variantes dans l' EGFR et KRAS, respectivement (figure 5). Fait important, l'entrée d'ADN FFPE pourrait être réduite à aussi peu que 50 copies amplifiables ou 1,2 ng d'ADN en vrac tout en préservant la détection de mutations connues sans faux positifsappels (figure 6). Entrées d'ADN ont été logés dans une plage de 100 fois jusqu'à au moins 50.000 copies amplifiables (tableau 3). Dans ce domaine et connexes expériences, variante appelle chez 22 FFPE et 20 spécimens cytoponction ont été signalés en accord avec des méthodes indépendantes avec une couverture de mutation partagée (tableau 4).
La sensibilité et la valeur prédictive positive pour le dosage a été déterminée à partir d'une analyse de 97 échantillons, y compris FFPE, AAF, frais congelé, et l'ADN cellulaire en ligne, et un total de 195 résultats de séquençage. Les résultats ont révélé 365 vrais appels positifs variantes, 4 appels faux négatifs et 1 appel faux positif pour une sensibilité de 98,9% (IC à 95%: 97,1 à 99,7%) et une valeur prédictive positive (VPP) de 99,7% (IC à 95% : 98,2 à 99,99%). Les analyses des indels ont été effectuées pour deux variantes de l'EGFR commun (de p.E746_A750delELREA et p.V769_D770insASV) dans 33 échantillons-pistes, ce qui démontre une sensibilité de 93,9% (IC à 95%: 78,4 à 98,9%) et un PPV de 100% (IC à 95%: 86,3 à 100%) avec des variantes détectées sur une plage de 2,4 à 84,8%.

Figure 1: Exemple de Quantification étalonnage ADN Curves que les critères d' acceptation et de Fail QC (A) une courbe standard qui passe.. (B) Une courbe standard défaillante. Dans ce cas, le problème a été causé par pipetage double de la norme de l' ADN d'entrée le plus bas. S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
{kind=link}

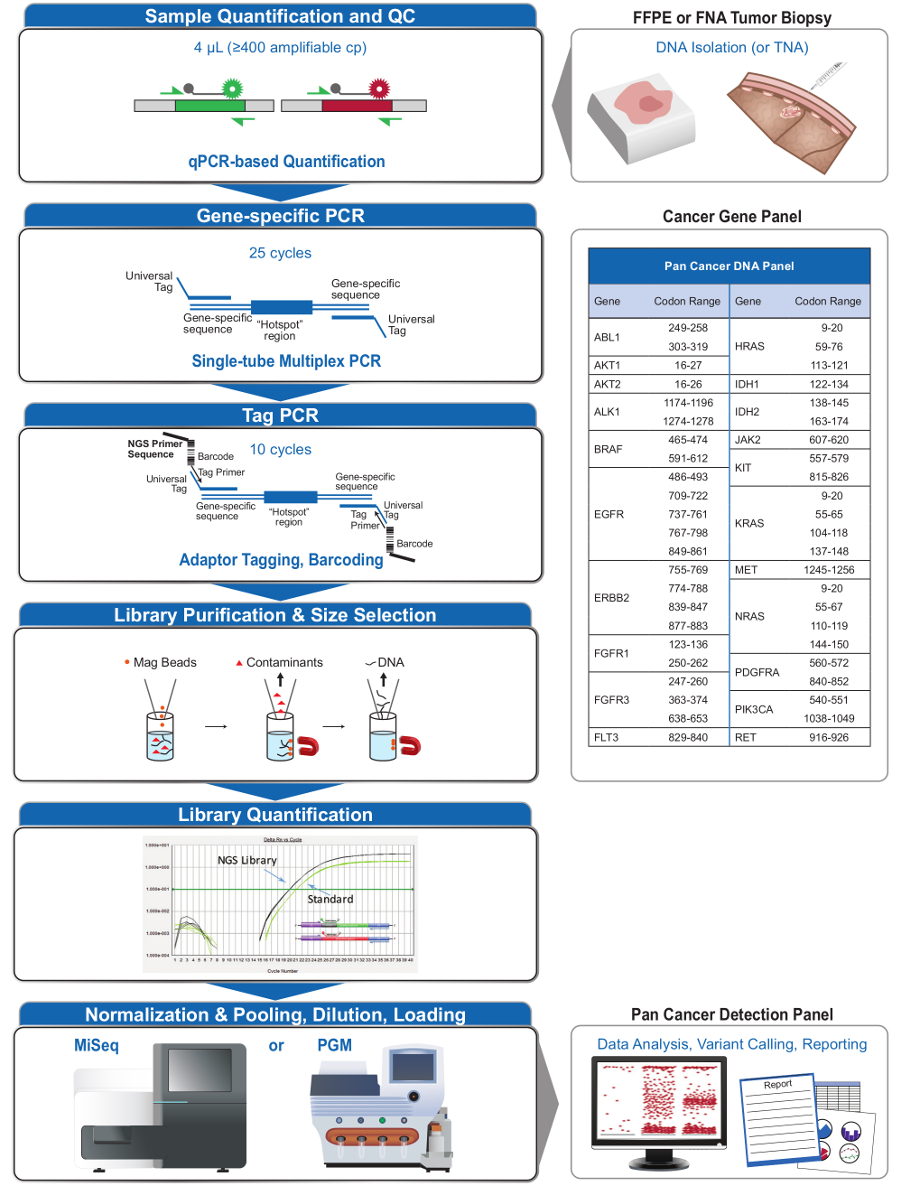
Figure 2: Vue d' ensemble d'un système de NGS complète ciblée pour Oncologie applications qui Intègre pré-analytiqueWorkflows al, Analytical, et post-analytiques. S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
{kind=link}

Figure 3: Lire la couverture et Uniformité pour NGS ciblée des gènes du cancer dans l' ADN de FFPE faible qualité par rapport à l' ADN et des contrôles Cell-line Intact Un total de 90 échantillons inclus résiduelle FFPE clinique (cercles pleins), la lignée cellulaire (cercles ouverts. ), et l'ADN synthétique de modèle (plus symboles) ont été traitées en utilisant un seul tube, multiplex PCR enrichissement 21 génique. Chaque bibliothèque d'amplicon a été marqué avec des séquences d'adaptateurs pour l'instrument, NGS code-barres avec un code distinct à double indice, purifié, quantifié et normalisé à une concentration de 2,5 nM. La bibliothèque d'ADN a été séquencé et analysé par le logiciel de bioinformatique de compagnie. exemples deviations inclus une série de dilution de l'ADN MDA-MB-468 lignée cellulaire portant un numéro d'amplification à grande copie de l'EGFR (en haut, rectangles ouverts au sein du cercle en pointillés) qui faussent la couverture uniformité et un échantillon de mélanome FFPE qui ont échoué à générer un nombre appréciable de lit en raison pour le report des inhibiteurs de la PCR à partir de l'extraction de l'ADN. L'échec de l'échantillon de mélanome (en bas, cercle en pointillés) a été prédit par le test ADN qPCR QC pré-analytique. NTC, sans matrice de contrôle (x symboles). S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
{kind=link}

Figure 4: amplicon par amplicon Lire la couverture, l' homogénéité et de la variante de détection dans l' ADN de la tumeur FFPE clinique résiduelle. (A) Lire la couverture pour tous les loci enrichi dans un échantillon représentatif de l' ADN FFPE mesuréeà travers trois opérateurs différents. Opérateur 1, Op1 (barres bleues); Opérateur 2, Op2 (barres vertes); Opérateur 3, Op3 (barres noires). (B) l' uniformité de la couverture et des variantes appels évaluées à l' aide de trois échantillons de tumeurs FFPE, y compris un mélange de contrôle (FFPE3, barres grises et texte) composés d'un connu 5% BRAF c.1799T> Une mutation. FFPE1 (barres bleues et texte), FFPE2 (barres oranges et texte). S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
{kind=link}

Figure 5:. Détection dose-dépendante de la Copy Number Variants in Cell-line et l' ADN de lignée cellulaire FFPE DNA MDA-MB-468 avec un nombre de copies d'amplification EGFR bien caractérisé a été dilué progressivement dans un fond d'une cellule de référence non muté ADN -ligne pour illustrer la diminution de la copie changement de numéro en tant quefonction de la dilution. Le pourcentage de chaque échantillon d'ADN de lignée cellulaire est représenté par une ligne distincte (0, 12,5, 25, 50 et 100%). Dilution d'un échantillon de tumeur FFPE ovarien avec une amplification du gène KRAS connu a révélé un profil similaire en utilisant la même série de titrage , mais avec répétitions d'ADN FFPE 100% pour les deux premières lignes. S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
{kind=link}

Figure 6: Détection précise Mutation et Quantification 50 copies amplifiable ADN FFPE, soit 1,2 ng d' ADN en vrac Le nombre de copies amplifiable d'un cancer du côlon FFPE ADN a été déterminée par le QC test à base de qPCR, et dilué de 400 à 25 exemplaires en tant que. entrée dans l'enrichissement PCR multiplex avant le séquençage. Le pipeline de bioinformatique correctement appelée à la fois de l'varian connuts jusqu'à 50 exemplaires, soit l'équivalent de ~ 10 modèles mutantes. S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
{kind=link}
| Nom du matériel / Matériel | Compagnie | Numéro de catalogue |
| 2x Quantidex Master Mix | Asuragen | 145345 |
| Quant Primer Probe Mix | Asuragen | 145336 |
| Inhibition Primer Probe Mix | Asuragen | 145344 |
| ROX | Asuragen | 145346 |
| Diluant | Asuragen | 145339 |
| ADN standard (50) | Asuragen | 145340 |
| ADN standard (10) | Asuragen | 145341 |
| ADN Standard (2) | Asuragen | 145342 |
| ADN standard (0,4) | Asuragen | 145343 |
| 2x Amplification Master Mix | Asuragen | 145348 |
| Panel Primer Cancer Pan | Asuragen | 145347 |
| Contrôle Pan Cancer FFPE | Asuragen | 145349 |
| Contrôle Pan Cancer Multi-Variant | Asuragen | 145350 |
| Perles Prep pures Bibliothèque | Asuragen | 145351 |
| Wash Buffer | Asuragen | 145352 |
| Elution Buffer | Asuragen | 145353 |
| 2x LQ Master Mix | Asuragen | 145358 |
| LQ diluant | Asuragen | 145354 |
| LQ positiveContrôle | Asuragen | 145355 |
| LQ standard | Asuragen | 145356 |
| LQ Primer Probe Mix / (de ILMN) | Asuragen | 145357 |
| LQ ROX | Asuragen | 145359 |
| Codes Index (ILMN) - Set A | Asuragen | 150004 |
| AIL001 - AIL048 (48) | ||
| Codes Index (ILMN) - Set B | Asuragen | 150005 |
| AIL049 - AIL096 (48) | ||
| 2x Index Master Mix | Asuragen | 145361 |
| Lire 1 Séquençage Amorces | Asuragen | 150001 |
| Index Lisez Séquençage Amorces | Asuragen | 150002 |
| Lire 2 Séquençage Amorces | Asuragen | 150003 |
| séquençage diluant | Asuragen | 145365 |
| Illumina MiSeq | Illumina | |
| MiSeq Réactif Kit v3 (600 cycles) | Illumina | MS-102-3003 |
| MiSeq Réactif Nano Kit v2 (300 cycles) | Illumina | MS-103-1001 |
| PhiX contrôle v3 | Illumina | FC-110-3001 |
| Magnétique stand-96 (ou un dispositif équivalent) | Ambion | AM10027 |
| Quantidex Software Reporter | Asuragen |
Tableau 1:. Réactifs et Kits Lors de la première utilisation du ROX, stocker le flacon à 2-8 ° C. Ne pas recongeler. Le logiciel peut être téléchargé à l'adresse www.asuragen.com.
| gène | variante COSMIC | Acide aminé COSMIC | % Une variante |
| NRAS | c.182A> G | p.Q61R | 13.3 |
| NRAS | c.35G> A | p.G12D | 15.2 |
| HRAS | c.182A> G | p.Q61R | 17,8 |
| HRAS | c.35G> A | p.G12D | 9.2 |
| KRAS | c.182A> G | p.Q61R | 13.5 |
| KRAS | c.35G> A | p.G12D | 19.1 |
| PIK3CA | c.1633G> A | p.E545K | 9.3 |
| PIK3CA | c.3140A> G | p.H1047R | 9.1 |
| TROUSSE | c.2447A> T | p.D816V | 14.6 |
| EGFR | c.2369C> T | p.T790M | 11.3 |
| EGFR | c.2573T> G | p.L858R | 14.9 |
| BRAF | c.1799T> A | p.V600E | 17.3 |
Tableau 2: Un contrôle synthétique Pooled est composé de 12 "Driver" Cancer Gene Variants qui sont quantifiées à 9-17% Abondance Un mélange de 12 modèles synthétiques double brin différents portant 12 mutations distinctes a été évaluée suivant le séquençage.. Toutes les variantes ont été correctement appelées sans faux positifs.
>% Variant| ID échantillon | Fonctionnel cps | Gène | variante COSMIC | Acide aminé COSMIC | Profondeur lecture médian | 5x% à l' intérieur de la médiane | |
| BCPAP | 400 | BRAF | c.1799T> A | p.V600E | 99,5 | 3289 | 96% |
| BCPAP | 10.000 | BRAF | c.1799T> A | p.V600E | 99,7 | 4040 | 98% |
| BCPAP | 25000 | BRAF | c.1799T> A | p.V600E | 99,4 | 3687 | 96% |
| BCPAP | 50.000 | BRAF | c.1799T> A | p.V600E | 99,7 | 4611 | 93% |
Tableau 3: Couverture et Variant Calling sont conservés sur une plage> 100 fois de l' ADN d' entrée. ADN amplifiable à partir d'une lignée de cellules BCPAP était entrée dans l'enrichissement PCR multiplex à 400 à 50.000 exemplaires et séquencés. Lire la profondeur, la couverture d'uniformité, la détection de mutation, et la précision de mutation ont été préservés à travers la gamme d'entrée.

Tableau 4: Variant Appels à 22 FFPE et 20 cytoponction biopsies de tumeurs d' accord avec les résultats de Independent Mutation Assays Un ensemble de 22 l' ADN tumoral FFPE avec le statut de mutation préalablement déterminée par orthogonales NGS tests ciblés était entrée à 400 à 2928 copies amplifiables dans l'enrichissement PCR. étape et séquence en utilisant le panneau Cancer Pan 21-gène. En outre, une cohorte de 20 échantillons d' ADN préalablement AAF , caractérisé en utilisant un liquide réseau de billes essai de mutation 8 a été amplifiée par PCR en utilisant 156 à 36 080 copies amplifiables entrée et séquencées. Tous les appels qui se chevauchent entre le panneau Pan Cancer NGS et le refereméthodes nce étaient d'accord. S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
{kind=link}
Discussion
Technologies NGS ont redéfini les attentes pour interroger les profils moléculaires de biopsies tumorales dans les milieux cliniques 9. Un certain nombre de panneaux NGS ciblés ont été mis au point des technologies de recherche, des tests de laboratoire mis au point, et des produits disponibles dans le commerce et des panneaux personnalisés pour évaluer plusieurs types d'échantillons cliniques 3,5,10-15. Les rapports de plusieurs études ont démontré la valeur de NGS comme outil clinique sensible et spécifique pour la détection des altérations génomiques 3,10-12,14. Pourtant , les études ont également démontré le risque d'artefacts qui peuvent causer des résultats faussement positifs provenant de biopsies de cancer difficiles telles que FFPE spécimens 2-5,12,14. En outre, des publications récentes ont mis en évidence des taux élevés d'échec pour NGS en utilisant l' oncologie spécimens 16 et de faux positifs appels avec des panneaux NGS commerciales ciblées qui sont aggravées par l'utilisation de l' ADN à faible entrée 12. Par conséquent, certains laboratories ont modifié ou ajouté des freins et contrepoids supplémentaires disponibles dans le commerce des technologies NGS ciblées pour améliorer les performances, souvent pour assurer l' exactitude ou de confirmer les résultats des analyses de la bioinformatique 5,10,11.
Le panneau 21-gène (figure 2) a été développé comme un contenu ciblé pour un système NGS global pour interroger, des mutations à une action sur la base des données probantes dans les types d'échantillons difficiles tels que FFPE et les biopsies tumorales cytoponction. Le flux de travail a un certain nombre d'avantages: 1) la cohérence en fournissant une couverture de amplicon uniforme (figures 3 et 4, tableau 3); 2) la facilité d'utilisation en fournissant des pré-formulé, des ensembles de réactifs optimisés, et de simplifier les exigences en matière de bio-informatique; 3) l'efficacité en rationalisant le flux de travail et en réduisant le nombre d'étapes de pipetage par rapport à d'autres méthodes de NGS commerciales; et 4) la précision en incorporant un test ADN QC pour évaluer l'amplifimesure le nombre de copies d'ADN pour assurer la diversité de modèles acceptables et éviter les fluctuations stochastiques dans la détection de la variante 4. L'intégration des données de QC pré-analytique avec l'analyse bioinformatique permet de faibles entrées de FFPE ADN. Ceci a été réalisé par la formation d'un algorithme d'arbre de décision en utilisant différentes copies fonctionnelles de l'entrée de l'ADN dans 400 échantillons FFPE avec des mesures indépendantes de la vérité, et en intégrant cet algorithme dans le logiciel bioinformatique. Par conséquent, l'entrée recommandée de 400 copies amplifiables, typiquement équivalente à ~ 5 à 20 ng d'ADN FFPE, se compare favorablement à d' autres méthodes, y compris l' enrichissement de 10-12 à base d' hybridation , où ~ 250 ng d'ADN FFPE est recommandé 17,18 . Bien que la technique est décrit pour une utilisation sur une plate-forme de MiSeq, il peut être modifié en utilisant des amorces de PCR Tag avec des adaptateurs spécifiques à l'instrument pour permettre une analyse de séquence sur d'autres plates-formes NGS.
Plusieurs étapes sont essentielles pour assurer le succèsde la procédure. CQ dosage pré-analytique détermine le nombre de copies de l'ADN et amplifiable rapports d'inhibition fonctionnelle. Cependant, si elle est inférieure à 400 copies d'ADN amplifiables sont utilisés dans l'étape d'enrichissement par PCR, il existe un risque accru d'un appel de faux négatifs à partir d' échantillons présentant des mutations de faible abondance (figure 6). En outre, il faut prendre soin lors de la purification bibliothèque pour éviter une sur-séchage des billes magnétiques pendant le lavage ou élution étapes. De plus, le succès de quantification de la bibliothèque est fortement dépendante de la dilution précise de la banque d'ADN. Pour le meilleur résultat, la différence de qPCR résultats pour la bibliothèque de l'échantillon (Cq FAM) par rapport à la LQ standard (Cq VIC) devrait être ≤3.3 Cq. Si la différence est supérieure à 3,3 Cq, re-dilution et le test de l'échantillon est recommandée. Bien qu'une excellente corrélation a été observée entre cette méthode concurrentielle qPCR et des kits commerciaux qui offrent une quantification absolue en utilisant une courbe standardUn décalage de l'entrée de bibliothèque dans l'étape d'amplification clonale par rapport à d'autres méthodes peuvent être nécessaires pour obtenir une densité d'ensemencement optimale.
Certains spécimens de cancer sont particulièrement difficiles à séquencer en raison des inhibiteurs qui persistent après l'isolement de l'ADN. Pour identifier ces échantillons avant la préparation bibliothèque, le test qPCR QC détecte également l'inhibition de l'amplification en incluant un modèle exogène qui sert à la fois un contrôle interne et une sentinelle pour l'inhibition fonctionnelle. Un exemple est présenté dans la figure 3 , dans lequel un échantillon d'ADN de mélanome n'a pas réussi à passer le QC inhibition métrique avant le séquençage et n'a pas réussi à générer une bibliothèque qui pourrait être séquencé. L'échec était probablement une conséquence de la contamination de la mélanine, un inhibiteur de PCR connu, reporté de l'étape d'isolement FFPE ADN. Les échantillons qui sont identifiés par le test QC pour être à risque d'échec de l'amplification peut être récupéré grâce à un supplément de nettoyage étape to éliminer les inhibiteurs potentiels.
Le panneau 21-gène ciblé se concentre sur les points chauds de gènes basés sur des données probantes et fournit un système complet avec des réactifs et des contrôles optimisés pour NGS et la bioinformatique logiciel qui est informé par pré-analytiques des résultats "fonctionnels" ADN quantification de l'ADN QC,. La méthode permet de détecter avec précision les mutations de base de substitution et indels à partir d'ADN faible apport d'intrants, et fournit un exemple d'un système NGS avec la possibilité d'élargir le contenu du panneau, pour détecter des variantes supplémentaires telles que CNV et être adapté pour le séquençage de l'ARN ciblé.
Déclarations de divulgation
JH, AH, RZ, BCH, et GJL sont employés et ont la propriété d'actions dans Asuragen, Inc. RZ, BCH et GJL sont co-inventeurs sur une demande de brevet pour l'amélioration de la variante d'appel en utilisant amplifiable informations du nombre de copies déterminé pour chaque échantillon.
Remerciements
Nous remercions le Dr Annette Schlageter pour examen du manuscrit. Ce travail a été soutenu en partie par la subvention CP120017 de la prévention du cancer et de l'Institut de recherche du Texas (PI: GJL).
matériels
| Name | Company | Catalog Number | Comments |
| 2x Quantidex Master Mix | Asuragen | 145345 | |
| Quant Primer Probe Mix | Asuragen | 145336 | |
| Inhibition Primer Probe Mix | Asuragen | 145344 | |
| ROX | Asuragen | 145346 | |
| Diluent | Asuragen | 145339 | |
| DNA Standard (50) | Asuragen | 145340 | |
| DNA Standard (10) | Asuragen | 145341 | |
| DNA Standard (2) | Asuragen | 145342 | |
| DNA Standard (0.4) | Asuragen | 145343 | |
| 2X Amplification Master Mix | Asuragen | 145348 | |
| Pan Cancer Primer Panel | Asuragen | 145347 | |
| Pan Cancer FFPE Control | Asuragen | 145349 | |
| Pan Cancer Multi-Variant Control | Asuragen | 145350 | |
| Library Pure Prep Beads | Asuragen | 145351 | |
| Wash Buffer | Asuragen | 145352 | |
| Elution Buffer | Asuragen | 145353 | |
| 2X LQ Master Mix | Asuragen | 145358 | |
| LQ Diluent | Asuragen | 145354 | |
| LQ Positive Control | Asuragen | 145355 | |
| LQ Standard | Asuragen | 145356 | |
| LQ Primer/Probe Mix (ILMN) | Asuragen | 145357 | |
| LQ ROX | Asuragen | 145359 | |
| Index Codes (ILMN) - Set A, AIL001 - AIL048 (48) | Asuragen | 150004 | |
| Index Codes (ILMN) - Set B, AIL049 - AIL096(48) | Asuragen | 150005 | |
| 2x Index Master Mix | Asuragen | 145361 | |
| Read 1 Sequencing Primers | Asuragen | 150001 | |
| Index Read Sequencing Primers | Asuragen | 150002 | |
| Read 2 Sequencing Primers | Asuragen | 150003 | |
| Sequencing Diluent | Asuragen | 145365 | |
| Illumina MiSeq | Illumina | ||
| MiSeq Reagent Kit v3 (600-cycle) | Illumina | MS-102-3003 | |
| MiSeq Reagent Nano Kit v2 (300-cycle) | Illumina | MS-103-1001 | |
| PhiX Control v3 | Illumina | FC-110-3001 | |
| Magnetic Stand-96 (Or equivalent device) | Ambion | AM10027 | |
| Quantidex Reporter Software | Asuragen |
Références
- Chen, G., Mosier, S., Gocke, C. D., Lin, M. T., Eshleman, J. R. Cytosine deamination is a major cause of baseline noise in next-generation sequencing. Mol Diagn Ther. 18 (5), 587-593 (2014).
- Choudhary, A., et al. Evaluation of an integrated clinical workflow for targeted next-generation sequencing of low-quality tumor DNA using a 51-gene enrichment panel. BMC Med Genomics. 7, 62 (2014).
- Sah, S., et al. Functional DNA quantification guides accurate next-generation sequencing mutation detection in formalin-fixed, paraffin-embedded tumor biopsies. Genome Med. 5 (8), 77 (2013).
- Zhang, L., et al. Profiling cancer gene mutations in clinical formalin-fixed, paraffin-embedded colorectal tumor specimens using targeted next-generation sequencing. Oncologist. 19 (4), 336-343 (2014).
- Latham, G. J. Next-generation sequencing of formalin-fixed, paraffin-embedded tumor biopsies: navigating the perils of old and new technology to advance cancer diagnosis. Expert Rev Mol Diagn. 13 (8), 769-772 (2013).
- Crawford, J. M., et al. The business of genomic testing: a survey of early adopters. Genet Med. 16 (12), 954-961 (2014).
- Smith, D. L., et al. A multiplex technology platform for the rapid analysis of clinically actionable genetic alterations and validation for BRAF p.V600E detection in 1549 cytologic and histologic specimens. Arch Pathol Lab Med. 138 (3), 371-378 (2014).
- Thomas, F., Desmedt, C., Aftimos, P., Awada, A. Impact of tumor sequencing on the use of anticancer drugs. Curr Opin Oncol. 26 (3), 347-356 (2014).
- Singh, R. R., et al. Clinical validation of a next-generation sequencing screen for mutational hotspots in 46 cancer-related genes. J Mol Diagn. 15 (5), 607-622 (2013).
- Beadling, C., et al. Combining highly multiplexed PCR with semiconductor-based sequencing for rapid cancer genotyping. J Mol Diagn. 15 (2), 171-176 (2013).
- McCall, C. M., et al. False positives in multiplex PCR-based next-generation sequencing have unique signatures. J Mol Diagn. 16 (5), 541-549 (2014).
- Schleifman, E. B., et al. Next generation MUT-MAP, a high-sensitivity high-throughput microfluidics chip-based mutation analysis panel. PLoS One. 9 (3), e90761 (2014).
- Wong, S. Q., et al. Sequence artefacts in a prospective series of formalin-fixed tumours tested for mutations in hotspot regions by massively parallel sequencing. BMC Med Genomics. 7, 23 (2014).
- Narayan, A., et al. Ultrasensitive measurement of hotspot mutations in tumor DNA in blood using error-suppressed multiplexed deep sequencing. Cancer Res. 72 (14), 3492-3498 (2012).
- Hagemann, I. S., et al. Clinical next-generation sequencing in patients with non-small cell lung cancer. Cancer. 121 (4), 631-639 (2015).
- Won, H. H., Scott, S. N., Brannon, A. R., Shah, R. H., Berger, M. F. Detecting somatic genetic alterations in tumor specimens by exon capture and massively parallel sequencing. J Vis Exp. (80), e50710 (2013).
- Simen, B. B., et al. Validation of a next-generation-sequencing cancer panel for use in the clinical laboratory. Arch Pathol Lab Med. 139 (4), 508-517 (2015).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.