
Method Article
Integrazione di Wet and Dry processi banco Ottimizza Targeted sequenziamento di prossima generazione di bassa qualità e di biopsie di tumori a bassa quantità
In questo articolo
Riepilogo
An integrated system for targeted next-generation sequencing of oncology specimens is described. This cross-platform system is optimized for low-quality and low-quantity tumor biopsies, accommodates low DNA inputs, includes well-characterized multi-variant controls, and features a novel variant caller that is informed by quantitative pre-analytical quality control measures.
Abstract
Tutte le procedure di sequenziamento di prossima generazione (NGS) comprendono esperimenti eseguiti presso il banco di laboratorio ( "bench bagnato") ei dati analisi condotte utilizzando bioinformatica condotte ( "banco secco"). Entrambi gli elementi sono essenziali per produrre risultati precisi e affidabili, che sono particolarmente critico per laboratori clinici. tecnologie NGS mirate hanno sempre trovato grazia in applicazioni di oncologia per aiutare gli obiettivi della medicina precisione anticipo, ma i metodi spesso comportano scollegato e variabili i flussi di lavoro banco umido e secco e set di reagenti non coordinate. In questa relazione, si descrive un metodo per il sequenziamento impugnare campioni di cancro con un pannello 21-gene come un esempio di un sistema NGS mirato completo. Il sistema integra quantificazione funzionale del DNA e la qualificazione, a tubo singolo multiplex arricchimento PCR, e la purificazione biblioteca e la normalizzazione utilizzando analiticamente-verificati, reagenti singolo-source con una suite bioinformatica standalone.Di conseguenza, la variante preciso chiamate da bassa qualità e basso-quantità fissati in formalina, incluso in paraffina (FFPE) e agoaspirato (FNA) biopsie di tumori possono essere raggiunti. Il metodo può ordinariamente valutare le varianti di cancro-associata da un ingresso di 400 copie di DNA amplificabile, e ha un design modulare per accogliere nuovi contenuti gene. Due diversi tipi di controlli analiticamente definiti forniscono una garanzia di qualità e la salvaguardia di accuratezza delle chiamate con i campioni clinicamente rilevanti. A "tag" step PCR flessibile incorpora adattatori specifici della piattaforma e dei codici di indice per consentire codici a barre campione e la compatibilità con i comuni da banco strumenti NGS. È importante sottolineare che il protocollo è snella e in grado di produrre 24 librerie sequenza-ready in un solo giorno. Infine, l'approccio collega processi banco umido e secco, integrando i risultati dei controlli pre-analitica qualità del campione direttamente nella variante chiamando algoritmi per migliorare la mutazione precisione del rilevamento e differenziare falsi negativi e indetermchiamate dere i. Questo metodo NGS mirata utilizza progressi sia in wetware e software per realizzare ad alta profondità, il sequenziamento e l'analisi multiplex sensibile dei campioni tumorali eterogenee per applicazioni diagnostiche.
Introduzione
medicina di precisione si basa sulla individuazione di opzioni diagnostiche e terapeutiche per i pazienti. La promessa di trattamenti su misura è una conseguenza diretta di una migliore comprensione di percorsi malattia che può informare il collegamento di diagnostica molecolare e terapie mirate. Ad esempio, l'uso di terapie molecolarmente mirati è aumentata dal 11% al 46% 2003-2013 1, e farmaci anti-cancro, come vemurafenib e crizotinib sono approvato dalla FDA con test diagnostici da compagnia. Con la sua capacità di recuperare con precisione gli obiettivi sequenza a bassa abbondanza attraverso set di campioni altamente multiplex, sequenziamento di prossima generazione (NGS) è emerso come un metodo di scelta per la valutazione aberrazioni genetiche associate con il cancro e identificare bersagli molecolari per la medicina di precisione.
Le biopsie di tumori solidi più comuni per i test molecolari sono fissati in formalina, incluso in paraffina (FFPE) e agoaspirato (FNA) SPECIMENS. Questi campioni sono carichi di bassa quantità e / o di bassa qualità acidi nucleici che sfidano accurata NGS a valutazioni 2-5. Gli attuali metodi di NGS commerciali per l'analisi di questi campioni si basano su un mosaico di diversi reagenti, protocolli e strumenti informatici che rappresentano bersagli in movimento di continui miglioramenti. Ad esempio, cambiamenti nella chimiche di analisi e / o software si sono verificati ogni 1-2 mesi per i più comunemente usati mirati NGS kit 6. Questa instabilità riflette una mancanza di coerenza nella costruzione e verifica di un sistema di NGS unificata per tipi di campioni difficili, in particolare per il test del cancro, e mette un onere eccessivo per i laboratori per lo sviluppo di protocolli coesivi che sono ottimizzati da campione a risultati. Infatti, una recente indagine di utenti NGS ha evidenziato le difficoltà di questi "rapida evoluzione" tecnologie, insieme con i requisiti per la stabilita, il contenuto medico-perseguibile, esperienza bioinformatica radicata, un solidifiED e procedura integrata che può essere implementata rapidamente, e razionalizzato i flussi di lavoro e protocolli semplificati che facilitano on-the-job training 7. In questo articolo, un sistema completo per mirato NGS è descritto che risponde a queste lacune.
La metodologia presentata integra tutte le fasi procedurali da pre-analitica di post-analitica a banchi sia umido e secco per migliorare l'accuratezza, la sensibilità e l'affidabilità di quantificazione di destinazione e di rilevamento per NGS di clinicamente rilevante loci gene del cancro. Questo approccio inizia con la quantificazione del DNA "funzionale" 4 per valutare la qualità del DNA, guidare in ingresso nella fase di arricchimento PCR, e la protezione contro le chiamate falsi positivi che possono derivare da l'interrogatorio di copie molto bassi di modello. Un multiplex monotubo PCR arricchisce poi per 46 loci in 21 geni del cancro usando solo 400 copie di DNA amplificabile, seguito da incorporazione di sequenze specifiche della piattaforma per NGS using desktop comuni strumenti di sequenziamento. Le biblioteche sono purificati mediante una semplice procedura tallone magnetico e quantificati con un romanzo, saggio qPCR senza calibrazione. Una suite bioinformatica standalone, informato dal campione risultati QC DNA per migliorare le prestazioni delle chiamate, fornisce analisi di sequenza seguente NGS. Vi presentiamo i dati che utilizzano questo approccio di sistema per il mirato NGS per rivelare mutazioni base-sostituzione, inserimento / delezioni (indels), e copiare il numero di varianti (CNV) nelle biopsie di bassa qualità e basso numero di tumori, come FFPE e campioni Fna e corsa controlli.
Protocollo
Nota: Questo protocollo descrive l'elaborazione simultanea di campioni utilizzando un sistema MiSeq NGS ma può essere adattato per lo strumento Personal Genome automatico (PGM). Per l'ingresso DNA minima raccomandata di 400 copie template amplificabile, il saggio è in grado di produrre almeno 3,000x copertura mediano per ciascuno dei 96 campioni per corsa NGS, e la profondità copertura equivalenti per 24 campioni utilizzando il PGM su un chip 318. Il metodo richiede anche l'uso di uno strumento di PCR in tempo reale.
1. DNA quantificazione funzionale e controllo qualità (QC)
- Reagenti disgelo: 2x Master Mix, Primer sonda Mix, Inibizione Primer sonda Mix, 6-carbossi-X-rodamina (ROX), diluente, e le quattro norme curva di calibrazione del DNA genomico umano (DNA standard (50 ng / ml), DNA standard (10 ng / ml), DNA standard (2 ng / ml), e il DNA standard (0,4 ng / ml)) (Tabella 1). Vortex tutti i reagenti per 10 secondi e centrifugare alla massima velocità per 10 secondi per raccogliere il contenuto.Tenere il master mix 2x su ghiaccio.
- Preparare una quantità sufficiente di master mix per il numero totale di campioni da testare e comprendono il 10% in più di volume per evitare carenze dovute alla dispensazione. Preparare la master mix in una provetta utilizzando i seguenti volumi per campione: 5 ml 2x Master Mix, 0,5 ml Primer miscela di sonde, 0,5 ml Inibizione Primer miscela di sonde, 0,05 ml ROX e 2,95 ml diluente. Vortex per 10 secondi e centrifugare a massima velocità per 10 secondi per raccogliere il contenuto.
- Aggiungere 9 ml master mix in pozzetti di una piastra da 96 pozzetti.
- Aggiungere 1 ml di norme di DNA in duplicato per generare una curva di calibrazione. Mescolare pipettando su e giù per 5 volte.
- Assicurarsi che il campione di acido nucleico è ben miscelato prima dell'uso. Aggiungere campione 1 ml al master mix e mescolare pipettando su e giù per 5 volte.
- Sigillare la piastra, vortex per 10 secondi e centrifugare alla massima velocità per 10 secondi per raccogliere il contenuto.
- Collocare la piastra nel sistema PCR. Assegnare sia FAM (quantificazione funzionale) e VIC (inibizione funzionale) rivelatori per ogni campione in base alle istruzioni del produttore. Eseguire cicli di PCR di 10 min a 95 ° C e 40 cicli (15 sec a 95 ° C, 1 min a 60 ° C).
- Analizzare i dati qPCR generando un grafico di regressione lineare per ciascuno degli standard DNA duplicati utilizzando protocolli software.
- Tracciare il registro 10 del numero di copie per ogni standard di DNA su l'asse x ed il corrispondente valore q FAM C on l'asse y.
- Verificare che i risultati dalla caduta campione di acido nucleico all'interno della gamma dinamica della curva di taratura standard di DNA, e quindi calcolare la concentrazione del DNA sconosciuto in numero di copie "funzionale" o amplifiable per microlitri dalla sua posizione corrispondente sulla curva standard di riferimento. La figura 1 mostra esempi di curve di calibrazione che passavano e non è riuscito.
- Determinare se l'amplificazione è verificato in ciascuna reazione controllando la presenza dell'amplicone destinazione non umano nel canale VIC.
Nota: Come controllo positivo, l'inibizione Primer sonda Mix contiene primer specifici per un target esogena non umano e il corrispondente modello. L'inibizione Primer miscela di sonde è un componente del master mix che viene aggiunto a ciascun reazione, compreso il controllo non-template (NTC). In assenza di un inibitore, il prodotto di PCR per il target non umano dovrebbe sempre essere rilevato nel canale VIC. Un "rilevata" C q per un campione nel canale VIC indica la presenza di inibitori della PCR che possono beneficiare successiva pulizia del campione prima dell'ulteriore lavorazione.
2. Biblioteca Preparazione: Gene-specifico (GS) PCR
- Preparare una quantità sufficiente di master mix per il numero totale di campioni da testare e comprendono il 10% in più di volume per evitare carenze dovute a pipetting. Preparare la master mix GS PCR in una provetta utilizzando i seguenti volumi per campione: 5 ml 2x Amplification Master Mix (Tabella 1) e 1 ml Pan Cancer Primer Panel (Tabella 1). Mescolare pipettando su e giù, vortex per 10 secondi e centrifugare alla massima velocità per 10 secondi per raccogliere il contenuto.
- Aliquota del 6 ml GS PCR master mix in pozzetti di una piastra da 96 pozzetti. Aggiungere 4 ml di ogni campione di acido nucleico in singoli pozzetti. Per altri pozzi, aggiungere 4 ml di controllo FFPE (Tabella 1), 4 ml di controllo Multi-Variante (Tabella 1), e 4 ml di acqua priva di nucleasi per un NTC procedurale. Per ogni aggiunta, mescolare pipettando su e giù per 5 volte.
- Sigillare la piastra, vortex per 10 secondi e centrifugare alla massima velocità per 10 secondi per raccogliere il contenuto.
- Posizionare la piastra nel termociclatore per i seguenti cicli di PCR: 5 min a 95 ° C,2 cicli (15 sec a 95 ° C, 4 min a 60 ° C), 23 cicli (15 sec a 95 ° C, 4 minuti a 72 ° C), e una estensione finale di 10 min a 72 ° C. Tenere a 4 ° C.
Nota: Dopo il completamento della fase 2.4, la piastra sarà indicata come piastra GS PCR.
3. Biblioteca Preparazione: Tag PCR
- Reagenti disgelo: 2x Indice Master Mix (tabella 1) e Codici dell'Indice (Tabella 1). Vortex per 10 secondi e centrifugare a massima velocità per 10 secondi per raccogliere il contenuto.
Nota: Codici dell'Indice sono premiscelati per fornire un unico insieme di indici a coppie (codici a barre) per ogni campione. - In una piastra da 96 pozzetti, aggiungere 7,5 ml di 2x Index Master Mix e 5,5 ml di un codice di indice per un determinato bene e mescolare pipettando su e giù per 5 volte.
- Aprire con cautela la piastra GS PCR, e aggiungere 2 ml di prodotto GS PCR per la nuova piastra con il master mix. Mescolare pipettando su e giù 5 volte. Per ogni campione, registrare l'ID campione e le corrispondenti Codici dell'Indice a due a due. Sigillare la piastra, vortex per 10 secondi e centrifugare alla massima velocità per 10 secondi per raccogliere il contenuto.
- Porre la piastra in termociclatore e PCR per 5 min a 95 ° C, 10 cicli (30 sec a 95 ° C, 30 sec a 55 ° C, 1 min a 72 ° C), ed una estensione finale di 10 min a 72 ° C. Tenere a 4 ° C.
Nota: Dopo il completamento della fase 3.4, la piastra viene denominato Tag piastra di PCR.
4. Biblioteca Purificazione e Selezione del formato
- Rimuovere le sfere magnetiche Biblioteca Pure Prep (Tabella 1) e Elution Buffer (Tabella 1) 2-8 ° C e lasciare equilibrare a temperatura ambiente per 30 min. Aggiungere 9,6 ml di etanolo al 100% per il tampone di lavaggio (tabella 1) del contenitore, tappo e miscelare capovolgendo il flacone diverse volte.
- Vortex le perline magnetiche per 10 secondi e aggiungere 11 ml in pozzetti separati di un 96-pozzetti.
- Aprire il Tag piastra di PCR e aggiungere 10 ml di Tag prodotto di PCR per il mix perline e pipetta per 5 volte. Incubare la miscela per 4 minuti a temperatura ambiente.
- Posizionare la piastra a 96 pozzetti sul supporto magnetico (Tabella 1) per 4 min. Con la piastra a 96 pozzetti ancora sul cavalletto, rimuovere ed eliminare il surnatante con una pipetta.
- Rimuovere la piastra a 96 pozzetti dal supporto magnetico e aggiungere 100 ml di tampone di lavaggio contenente etanolo in ciascun pozzetto e miscelare pipettando su e giù per 5 volte. Incubare per 2 min.
- Posizionare la piastra a 96 pozzetti sul supporto magnetico per 2 minuti, quindi rimuovere e scartare il surnatante con una pipetta.
- Ripetere passaggio 4.5, per un totale di 2 lavaggi etanolo, rimuovendo soluzione di lavaggio il più possibile dopo il secondo lavaggio.
- Con la piastra a 96 pozzetti sul supporto magnetico, asciugare le perline per 2 minuti a temperatura ambiente, quindi rimuovere la piastra dal supporto.
- Risospendere le sfere con l'aggiunta di 20 microlitri tampone di eluizione a ciascuna well e pipetta su e giù per 5 volte.
- Incubare per 2 minuti a temperatura ambiente.
- Posizionare la piastra a 96 pozzetti di nuovo sul supporto magnetico per 4 min, e con attenzione rimuovere e trasferire i 18 ml di surnatante chiaro per un nuovo pozzo.
Nota: La procedura può essere arrestato in modo sicuro a questo passaggio e campioni conservati a -15 e -30 ° C. Per riavviare, scongelare i campioni congelati in ghiaccio prima di procedere.
5. Quantificazione Biblioteca
- Thaw Biblioteca Quant (LQ) Reagenti: 2x LQ Master Mix, LQ Primer / Sonda Mix, LQ standard, LQ controllo positivo, LQ diluente, e LQ ROX (Tabella 1). Vortex per 10 secondi e centrifugare a massima velocità per 10 secondi per raccogliere il contenuto.
- Utilizzando prodotti biblioteca purificati, eseguire una diluizione seriale di ogni singolo campione LQ diluente.
- Aggiungere 2 ml (biblioteca prodotto purificato) a 198 ml LQ diluente e mescolare up-and-down con una pipetta 10 volte.
- Aggiungere 2 microlitri (diluizione 1: 100) per 198 ml LQ diluente e mescolare up-and-down con una pipetta 10 volte.
- Preparare una quantità sufficiente di LQ master mix per il numero totale di campioni da testare e comprendono il 10% in più di volume per evitare carenze dovute alla dispensazione. Preparare la LQ Master Mix in una provetta utilizzando i seguenti volumi per campione: 5 ml 2X LQ Master Mix, 2 microlitri LQ Primer / Sonda Mix, 0,5 ml LQ standard e 0,5 ml LQ ROX. Mescolare pipettando su e giù, vortex per 10 secondi e centrifugare alla massima velocità per 10 secondi per raccogliere il contenuto.
- Aggiungere 8 ml master mix LQ ad un pozzo di una piastra da 96 pozzetti ottica.
- In pozzetti separati, aggiungere 2 ml di libreria diluita, 2 ml LQ controllo positivo e 2 ml LQ diluente (NTC) e mescolare pipettando su e giù per 5 volte. Sigillare la piastra con pellicola adesiva ottica, vortex per 10 secondi e centrifugare a 400 xg per 10 secondi per raccogliere il contenuto.
- Assegnare sia FAM e VIC Detectors per ogni campione. Eseguire amplificazione usando condizioni di ciclo di 5 min a 95 ° C e 40 cicli (15 sec a 95 ° C, 1 min a 60 ° C).
- Determinare la concentrazione di ogni campione (nm) utilizzando il metodo C q comparativo. Calcolare la differenza della nota LQ standard (C q VIC) alla libreria sconosciuta (C q FAM). La concentrazione del campione diluito (PM) viene calcolata utilizzando la seguente equazione:
[Lib Conc] Nm = 12.5 x 2 Δ Cq
Se si usa un fattore di diluizione diversa 10.000, calcolare il rapporto tra il fattore di diluizione di destinazione a 10.000, e moltiplicare questo fattore dal risultato della equazione 5.7.
6. Biblioteca normalizzazione e la condivisione del campione
- Determinare la concentrazione mediana (nm) per tutti i campioni (ognuno dei quali contiene un indice a coppie univoco) per essere messe in comune.
- Determinare l'individuo volum campionee (ml) per piscina moltiplicando la concentrazione mediana in tutti i campioni da 5, quindi dividendo per la sua concentrazione individuale (nm). Intorno al valore risultante al numero intero più vicino. volumi rotondi con valori di <2 ml a 2 ml, e volumi> 15 microlitri a 15 ml.
- Aggiungere il volume normalizzato (microlitri) per ciascun campione da un unico tubo microcentrifuga per creare il pool di esempio.
- Calcolare la nuova concentrazione per ciascun campione utilizzando valori interi arrotondati e registrare i risultati.
- Per determinare la concentrazione del pool di esempio, calcolare la somma dei singoli concentrazioni e registrare il valore risultante (nM).
- Diluire la piscina del campione a 1,25 nm Sequencing diluente (Tabella 1).
Nota: La procedura può essere arrestato in modo sicuro a questo passaggio e campioni conservati a -15 e -30 ° C. Per riavviare, scongelare i campioni congelati in ghiaccio prima di procedere.
7. Sequencing
- Denature la piscina del campione in presenza di Phix controllo v3 (Tabella 1) aggiungendo le seguenti: 15 ml di 1,25 nM Pool Campione, 3 ml di 0,5 Nm Phix e 2 ml di 1 N NaOH. Vortex brevemente seguito da una breve centrifugazione e incubare per 5 minuti a temperatura ambiente.
- Posizionare la piscina del campione denaturato sul ghiaccio.
- Aggiungere 8 ml di biblioteca denaturato a 992 ml di pre-raffreddata tampone HT1-Ibrida in una provetta. Vortex brevemente per miscelare, seguito da una breve centrifugazione per raccogliere contenuti. Tenere in ghiaccio.
- Aggiungere 600 microlitri della biblioteca denaturato e diluita in posizione # 17 della cartuccia reagenti.
- Scongelare Leggi 1 Sequencing Primer (Tabella 1), indice letto Sequencing Primer (Tabella 1), e leggere 2 sequenziamento Primer (Tabella 1). In provette da microcentrifuga, separatamente diluire 4 primer di sequenziamento microlitri con 636 microlitri di buffer HT1-ibridazione. Nota: tampone HT1-Ibrida è fornito di spiritoh il kit di sequenziamento del reagente (Tabella 1).
- Mescolare vortexando per 10 secondi e centrifugare alla massima velocità per 10 secondi per raccogliere il contenuto. Aggiungere 600 ml di diluito Leggi 1 sequenziamento Primer per la posizione # 18 della cartuccia del sequenziamento del reagente, 600 ml di diluito indice letto Primer per la posizione # 19, e 600 ml di diluito Primer Leggi 2 sequenziamento alla posizione # 20.
- Caricare i reagenti sullo strumento NGS (Tabella 1) e sequenza secondo le istruzioni del produttore. Eseguire un abbinato-end 2 x 150 Ciclo di esecuzione di sequenziamento.
Analisi 8. Dati
Nota: il software dello strumento NGS converte le immagini a grappolo alle chiamate di base e punteggi di qualità, e demultiplexes indici a coppie per generare singoli FASTQ gzip-compresso (* .fastq.gz) file per ogni campione. Prima di analizzare i file demultiplexati, il lettore deve scaricare e installare il software di bioinformatica associato (Tabella1). Il software può essere installato su un tipo consumer PC Windows e non richiede hardware informatico specializzato o una connessione internet per eseguire l'analisi dei dati.
- Fare doppio clic sull'icona del desktop software.
- Effettua il login al sistema utilizzando il nome utente e la password forniti nel manuale del software.
- Aprire il dashboard del progetto, e fare clic su "Nuovo progetto".
- Nome del progetto e fornire una descrizione facoltativa del progetto. Selezionare il tipo di pannello di NGS mirato e NGS tipo di strumento. Fai clic su "Salva e continua".
- Caricare i file compressi FASTQ per la marcia avanti e retromarcia legge. Non caricare le FASTQs "non assegnati", che contengono legge che non è riuscito a demultiplex. Fai clic su "Salva e continua".
- Inserire il numero di copie di input funzionali utilizzati per preparare ogni libreria come determinato mediante il saggio quantificazione funzionale DNA (vedi passaggio 1). aggiungere manualmente i valori o valori di copia e incolla da uno spreadfoglio nella tabella di annotazione. Fai clic su "Salva e continua".
- Rivedere le librerie annotati caricati per l'analisi e fare clic su "Submit analisi" per avviare l'analisi.
- Monitorare l'avanzamento dell'analisi visualizzata attraverso il cruscotto progetto.
Nota: L'indicazione di stato completo di analisi è presentata quando i risultati sono pronti per la revisione. - Esaminare i risultati analizzati per il campione di controllo di qualità metriche, tra cui la copertura totale per biblioteca, la percentuale di legge filtri di passaggio, approfondimenti amplicone e uniformità. Rivedere la variante prevede per ogni libreria sequenziato con dbSNP, COSMIC, 1.000 genomi e altre fonti di annotazione funzionale e la popolazione di livello.
- Esportare i risultati grezzi come tabelle di sintesi dei fogli di calcolo, file * .bam e file * .vcf per la conservazione a lungo termine o l'analisi a valle informatici complementari strumenti.
Risultati
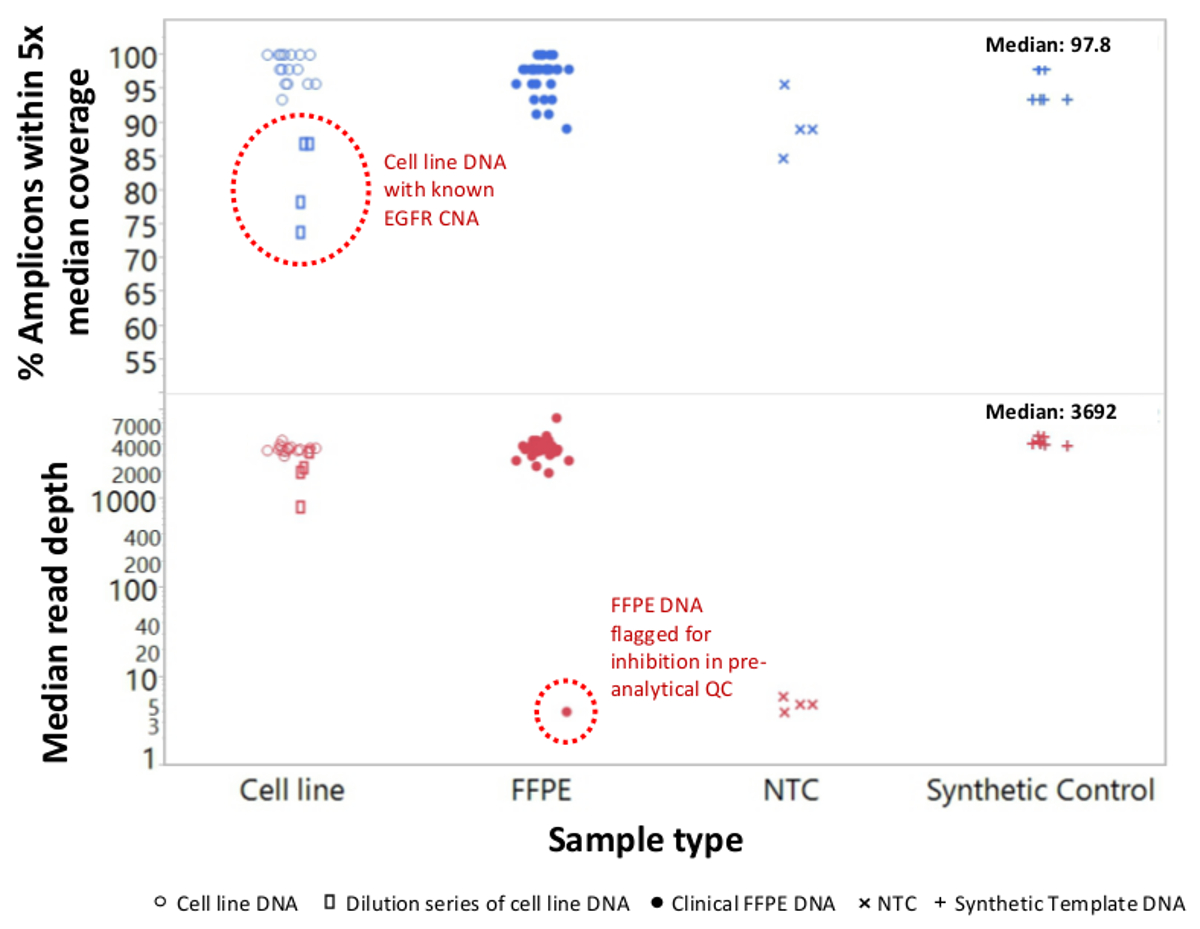
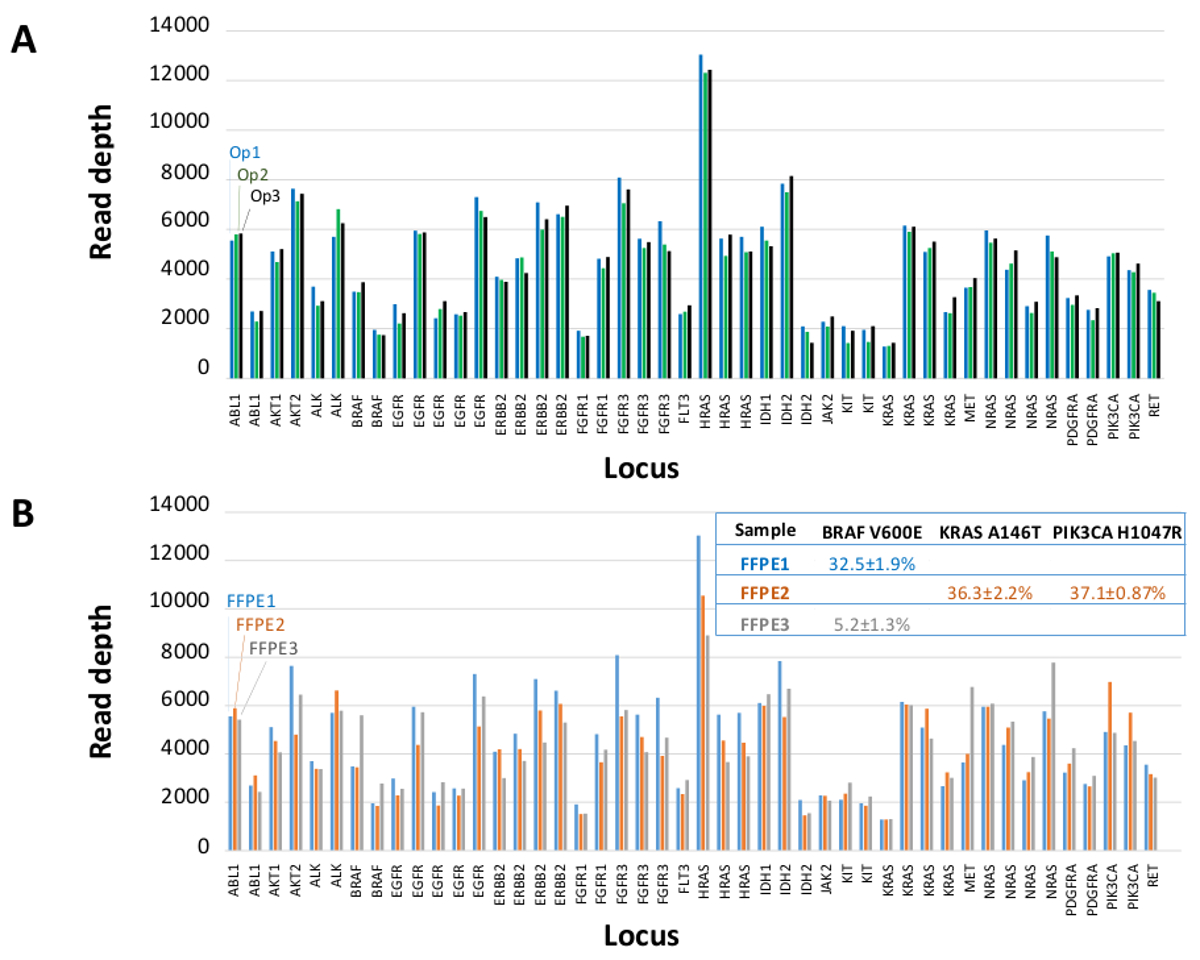
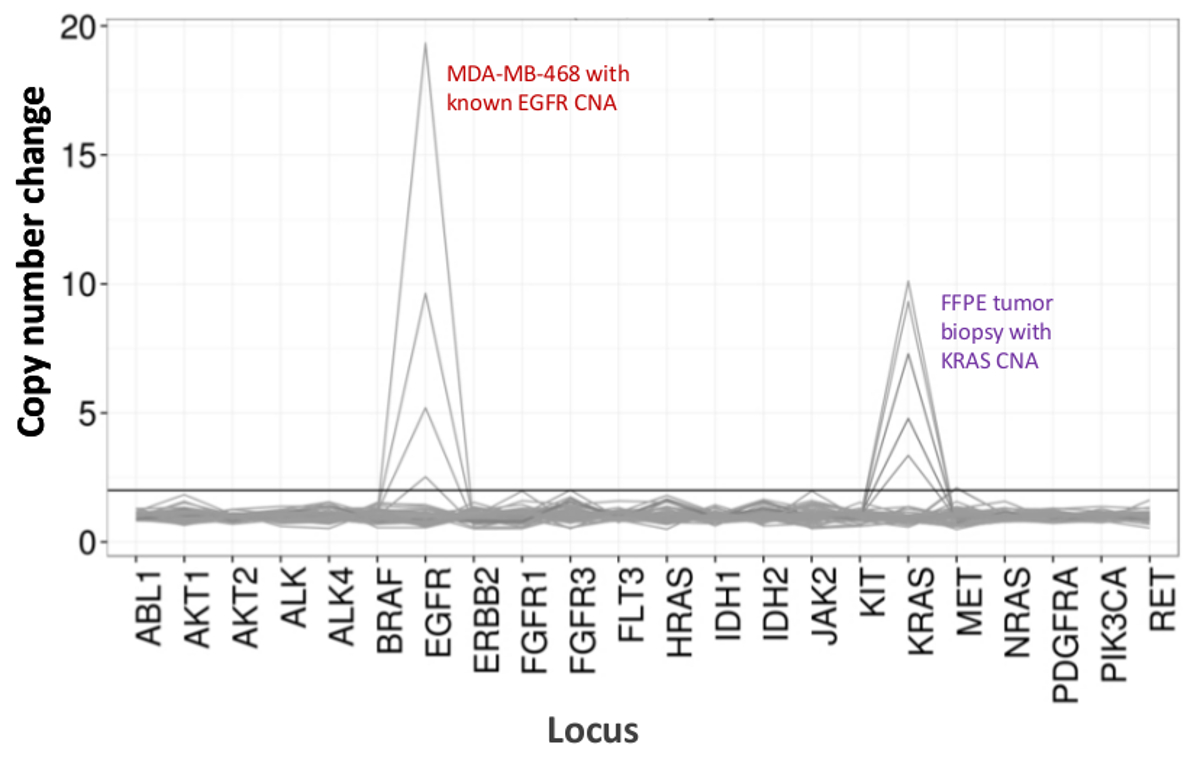
Un totale di 90 campioni (74 unici) che rappresentano controlli positivi e negativi, linee cellulari precedentemente caratterizzate e clinici biopsie tumorali FFPE residui sono stati valutati per il DNA amplificabile, ingresso in multiplex PCR arricchimento, etichettato con adattatori di sequenza, codice a barre, e analizzato in un unico da banco NGS run strumento (Figura 2) che ha prodotto il 19,1 M legge di passaggio del filtro. Equimolare pooling campione provocato alta sequenziamento profondità (3,692x letture) e una copertura uniforme (97,8% di ampliconi coperti all'interno di 5 volte della profondità di lettura mediana). Valori anomali composti non-template controlli, un DNA delle cellule-line con un gran numero di copie di amplificazione, e uno DNA FFPE che è stato fermato in posizione di inibizione della PCR con il test QC pre-analitica (Figura 3). La copertura uniformità dei 46 ampliconi è stata mantenuta utilizzando tre diversi operatori (figura 4a), e per diversi campioni FFPE DNA di bassa qualità ( Figura 4B). Un controllo del DNA del tumore FFPE, formulato da una miscela di campioni clinici residue per raggiungere il 5% V600E BRAF (quantificato da goccioline PCR digitale), è stato segnalato per avere il bersaglio BRAF mutazione a abbondanza di 3,9, 5,3 e 6,5% da tre operatori che utilizzano un ingresso di 400 copie amplificabile (e quindi solo 20 copie mutanti) (Figura 4B e "FFPE3" di tabella nel riquadro). Inoltre, una miscela di 12 modelli di DNA sintetico, ciascuna rappresentante un noto "driver" base-sostituzione mutazione, rivela le mutazioni attesi alla gamma prevista di 9 - 17% allele frequenza medio (Tabella 2). La diluizione di cellula-line e campioni FFPE DNA con numero di copia amplificazioni dimostrato dose-dipendenza per le varianti di EGFR e KRAS, rispettivamente (Figura 5). È importante sottolineare che, inseriti DNA FFPE potrebbe essere ridotto a come pochi come 50 copie amplificabile o 1,2 ng di DNA massa preservando la rilevazione di mutazioni note, senza falsi positivichiamate (Figura 6). Ingressi DNA sono stati sistemati in un range di 100 volte fino ad almeno 50.000 copie amplificabile (Tabella 3). In questo e relativi esperimenti, variante chiama 22 FFPE e 20 campioni FNA sono stati segnalati in accordo con metodi indipendenti con copertura mutazione comune (tabella 4).
La sensibilità e il valore predittivo positivo per il test è stato determinato da una analisi di 97 campioni, tra cui FFPE, FNA, fresco congelato, e di linee cellulari del DNA, e un totale di 195 risultati di sequenziamento. I risultati hanno rivelato 365 veri positivi chiamate variante, 4 chiamate falsi negativi, e 1 chiamata falso positivo per una sensibilità del 98,9% (95% CI: 97,1-99,7%) e un valore predittivo positivo (VPP) del 99,7% (95% CI : 98,2-99,99%). Le analisi di indels sono stati eseguiti per due varianti comune EGFR (p.E746_A750delELREA e p.V769_D770insASV) in 33 campioni-corre, dimostrando una sensibilità del 93,9% (95% CI: 78,4-98,9%) e un VPP del 100% (95% CI: 86,3-100%) con varianti rilevate in un range di 2,4-84,8%.

Figura 1: Esempio di curve DNA quantificazione di calibrazione che passano e Fail QC Criteria (A) una curva standard di passaggio.. (B) una curva standard fallendo. In questo caso, il guasto è stato causato da pipettaggio duplicato dei più bassi standard di DNA di ingresso. Si prega di cliccare qui per vedere una versione più grande di questa figura.
{kind=link}

Figura 2: Panoramica di un sistema globale mirato NGS per le applicazioni Oncology che integra pre-analiticaI flussi di lavoro al, analitica e post-analitica. Fai clic qui per vedere una versione più grande di questa figura.
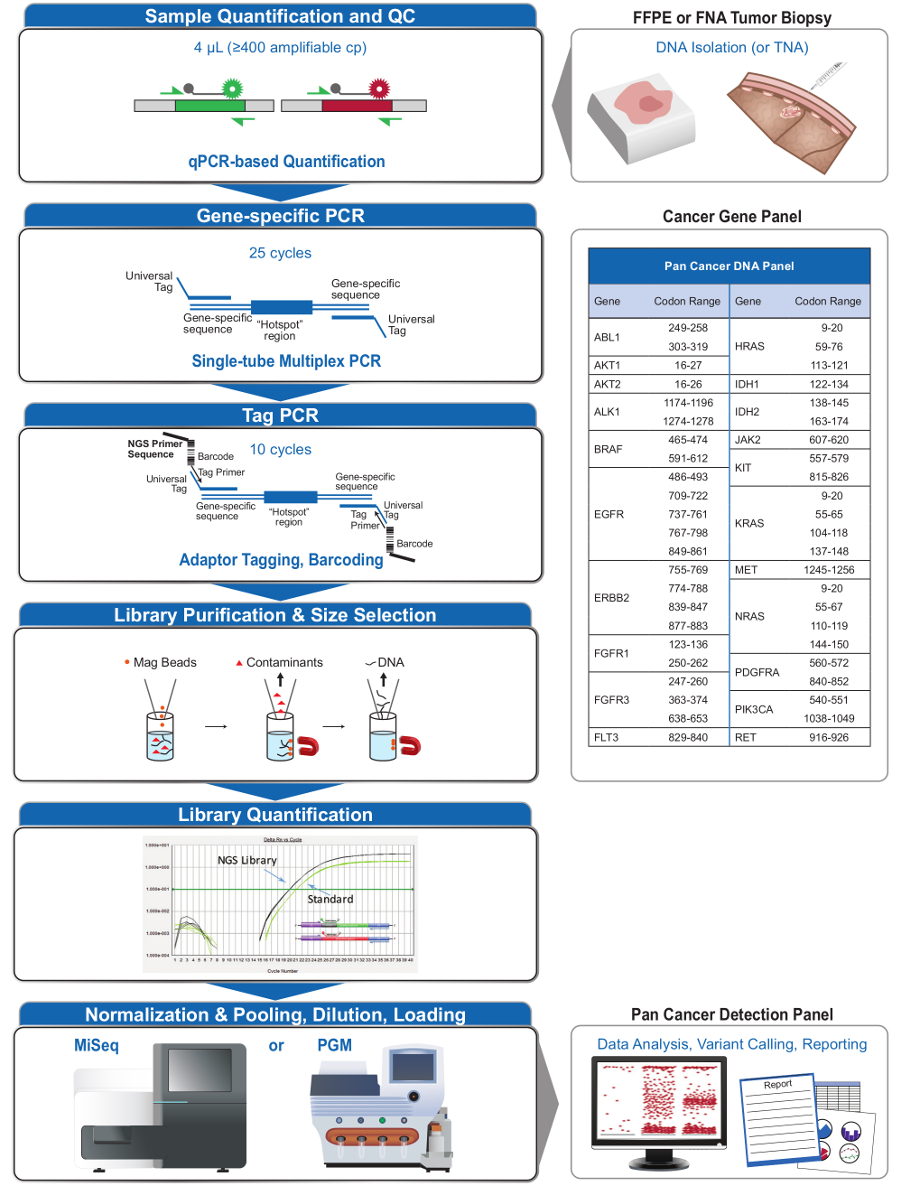{kind=link}

Figura 3: Leggi copertura e l'uniformità per NGS mirata di geni del cancro nel DNA FFPE bassa qualità rispetto a Intact cellulare-line DNA e controlla un totale di 90 campioni che comprendeva residua FFPE clinica (circoli chiusi), linea cellulare (circoli aperti. ), e il modello di DNA sintetico (più simboli) sono stati elaborati utilizzando monotubo, 21-gene multiplex PCR arricchimento. Ogni libreria amplicone è stato etichettato con sequenze adattatore per lo strumento NGS, di codici a barre con un codice distinta dual-index, purificato, quantificato e normalizzato ad una concentrazione di 2,5 nM. La biblioteca del DNA è stato sequenziato e analizzato dal software di bioinformatica compagno. devi campionezioni hanno incluso una serie di diluizioni di MDA-MB-468 cellule-line DNA recante un numero di gran copia di EGFR di amplificazione (in alto, rettangoli aperti all'interno cerchio tratteggiata) che hanno distorto la copertura uniformità e un campione del melanoma FFPE che non è riuscito a generare un numero apprezzabile di legge a causa a riporto di inibitori della PCR dalla estrazione del DNA. Il fallimento del campione del melanoma (in basso, cerchio tratteggiata) è stato previsto dal saggio qPCR DNA QC pre-analitica. NTC, il controllo non-template (simboli x). Si prega di cliccare qui per vedere una versione più grande di questa figura.
{kind=link}

Figura 4: Amplicon-by-amplicone Leggi copertura, uniformità e rilevamento Variante in residuo Clinica DNA tumorale FFPE. (A) Leggi la copertura in tutti i loci arricchito in un campione rappresentativo FFPE DNA misuratiin tre diversi operatori. Operatore 1, Op1 (blu bar); Operatore 2, Op2 (barre verdi); Operatore 3, Op3 (barre nere). Chiamate (B) Copertura di uniformità e variante valutati utilizzando tre campioni di tumore FFPE, tra cui una miscela di controllo (FFPE3, barre grigie e il testo), composto da una nota 5% BRAF c.1799T> Una mutazione. FFPE1 (barre blu e testo), FFPE2 (barre arancioni e testo). Clicca qui per vedere una versione più grande di questa figura.
{kind=link}

Figura 5:. Rilevamento dose-dipendente di Copy Number Varianti in Cell-line e il DNA delle cellule-line FFPE DNA MDA-MB-468 con un numero di EGFR copia di amplificazione ben caratterizzato è stato progressivamente diluito in uno sfondo di una cella di riferimento non mutato DNA-line per illustrare la diminuzione in copia cambiamento numero comefunzione della diluizione. La percentuale di ogni campione di DNA linea cellulare è mostrato con una linea distinta (0, 12,5, 25, 50 e 100%). La diluizione di un campione ovarica tumore FFPE con una amplificazione del KRAS nota rivelato un profilo simile utilizzando la stessa serie di titolazione, ma con repliche di 100% DNA FFPE per le due linee principali. Si prega di cliccare qui per vedere una versione più grande di questa figura.
{kind=link}

Figura 6: Preciso mutazioni individuazione e la quantificazione a 50 copie amplifiable FFPE DNA, ovvero il 1,2 ng DNA Bulk Il numero di copie amplifiable di un cancro al colon FFPE DNA è stata determinata mediante il test QC qPCR-based, e diluito da 400 a 25 copie come. input in multiplex PCR arricchimento prima di sequenziamento. Il gasdotto bioinformatica chiamato correttamente sia della Varian notats fino a 50 copie, o l'equivalente di ~ 10 modelli mutanti. Cliccate qui per vedere una versione più grande di questa figura.
{kind=link}
| Nome del materiale / Materiale | Società | Numero di catalogo |
| 2x Quantidex Master Mix | Asuragen | 145.345 |
| Quant Primer miscela di sonde | Asuragen | 145336 |
| Inibizione Primer miscela di sonde | Asuragen | 145.344 |
| ROX | Asuragen | 145346 |
| Diluente | Asuragen | 145339 |
| DNA standard (50) | Asuragen | 145.340 |
| DNA standard (10) | Asuragen | 145341 |
| DNA standard (2) | Asuragen | 145342 |
| DNA standard (0.4) | Asuragen | 145343 |
| 2x Amplification Master Mix | Asuragen | 145.348 |
| Pannello Pan Cancer Primer | Asuragen | 145347 |
| Controllo Pan Cancer FFPE | Asuragen | 145349 |
| Controllo pan Cancer Multi-Variante | Asuragen | 145350 |
| Perline Prep Pure Biblioteca | Asuragen | 145.351 |
| tampone di lavaggio | Asuragen | 145352 |
| Elution Buffer | Asuragen | 145353 |
| 2x LQ Master Mix | Asuragen | 145358 |
| LQ diluente | Asuragen | 145.354 |
| LQ positivoControllo | Asuragen | 145.355 |
| LQ standard | Asuragen | 145356 |
| LQ Primer / Sonda Mix (ILMN) | Asuragen | 145357 |
| LQ ROX | Asuragen | 145.359 |
| Codici dell'Indice (ILMN) - Set A | Asuragen | 150004 |
| AIL001 - AIL048 (48) | ||
| Codici dell'Indice (ILMN) - Set B | Asuragen | 150005 |
| AIL049 - AIL096 (48) | ||
| 2x Index Master Mix | Asuragen | 145361 |
| Leggi 1 sequenziamento Primer | Asuragen | 150001 |
| Indice Leggere sequenziamento Primer | Asuragen | 150002 |
| Leggi 2 sequenziamento Primer | Asuragen | 150003 |
| sequencing diluente | Asuragen | 145365 |
| Illumina MiSeq | Illumina | |
| MiSeq kit reagenti v3 (600 cicli) | Illumina | MS-102-3003 |
| MiSeq reagente Nano Kit v2 (300 cicli) | Illumina | MS-103-1001 |
| Phix controllo v3 | Illumina | FC-110-3001 |
| Magnetic Stand-96 (o un dispositivo equivalente) | Ambion | AM10027 |
| Reporter Software Quantidex | Asuragen |
Tabella 1:. Reagenti e kit Al primo utilizzo del ROX, conservare il flacone a 2-8 ° C. Non ricongelare. Il software può essere scaricato dal sito www.asuragen.com.
| solene | variante COSMIC | Aminoacido COSMIC | Variante% |
| NRAS | c.182A> G | p.Q61R | 13.3 |
| NRAS | c.35G> A | p.G12D | 15.2 |
| HRAS | c.182A> G | p.Q61R | 17.8 |
| HRAS | c.35G> A | p.G12D | 9.2 |
| KRAS | c.182A> G | p.Q61R | 13.5 |
| KRAS | c.35G> A | p.G12D | 19.1 |
| PIK3CA | c.1633G> A | p.E545K | 9.3 |
| PIK3CA | c.3140A> G | p.H1047R | 9.1 |
| KIT | c.2447A> T | p.D816V | 14.6 |
| EGFR | c.2369C> T | p.T790M | 11.3 |
| EGFR | c.2573T> G | p.L858R | 14.9 |
| BRAF | c.1799T> A | p.V600E | 17,3 |
Tabella 2: Un pool sintetico di controllo è composto da 12 "Driver" gene del cancro di varianti che sono quantificate al 9-17% Abbondanza Una miscela di 12 diversi modelli a doppio filamento sintetici cuscinetto 12 mutazioni distinte è stata valutata dopo il sequenziamento.. Tutte le varianti sono state correttamente chiamati senza falsi positivi.
| ID campione | Funzionale cps | Gene | variante COSMIC | Aminoacido COSMIC | Variante% | Mediana profondità di lettura | % Entro 5x della mediana |
| BCPAP | 400 | BRAF | c.1799T> A | p.V600E | 99.5 | 3289 | 96% |
| BCPAP | 10.000 | BRAF | c.1799T> A | p.V600E | 99,7 | 4040 | 98% |
| BCPAP | 25.000 | BRAF | c.1799T> A | p.V600E | 99,4 | 3687 | 96% |
| BCPAP | 50.000 | BRAF | c.1799T> A | p.V600E | 99,7 | 4611 | 93% |
Tabella 3: Copertura e Variante Calling sono conservati in un intervallo> 100 volte di ingresso del DNA. DNA amplificabile da una linea cellulare BCPAP era input in multiplex arricchimento PCR a 400 a 50.000 copie e sequenziato. Leggi la profondità, la copertura uniformità, l'identificazione della mutazione, e la precisione di mutazione sono stati conservati in tutta la gamma di ingresso.

Tabella 4: Variante di chiamate in 22 FFPE e 20 FNA biopsie di tumori D'accordo con i risultati di Independent mutazione saggi Un insieme di 22 FFPE DNA tumorale con lo status di mutazione precedentemente determinato da ortogonali saggi NGS mirata di ingresso a 400 a 2.928 copie amplificabile nel arricchimento PCR. sTEP e sequenziato utilizzando il 21-gene pannello Pan cancro. Inoltre, una coorte di 20 campioni di DNA FNA precedentemente caratterizzato utilizzando un test di mutazione serie tallone liquido 8 era PCR amplificato utilizzando da 156 a 36.080 copie di ingresso amplificabile e sequenziato. Tutte le chiamate sovrapposizione tra il pannello Pan Cancer NGS e la RIFERIMENTOmetodi SNO erano d'accordo. Si prega di cliccare qui per vedere una versione più grande di questa figura.
{kind=link}
Discussione
Tecnologie NGS hanno ridefinito le aspettative per interrogare i profili molecolari di biopsie di tumori in ambito clinico 9. Un certo numero di pannelli NGS mirati sono stati sviluppati come tecnologie di ricerca, test di laboratorio sviluppato, e prodotti disponibili in commercio e pannelli personalizzati per valutare diversi tipi di campioni clinici 3,5,10-15. Rapporti da diversi studi hanno dimostrato il valore di NGS come strumento clinico sensibile e specifico per la rilevazione di alterazioni genomico 3,10-12,14. Ma gli studi hanno anche dimostrato il rischio di artefatti che possono causare risultati falsi-positivi da biopsie tumorali impegnative come FFPE campioni 2-5,12,14. Inoltre, recenti pubblicazioni hanno evidenziato elevati tassi di fallimento per NGS utilizzando oncologia provini 16 e falsi positivi chiamate con i pannelli commerciali NGS mirati che sono aggravati con l'uso di DNA a basso input 12. Come risultato, alcuni laboraTories hanno o modificati o aggiunti pesi e contrappesi supplementari per commercialmente disponibili tecnologie NGS mirate per migliorare le prestazioni, spesso per garantire l'accuratezza o confermare i risultati dalla bioinformatica analisi 5,10,11.
Il pannello 21-gene (Figura 2) è stato sviluppato come contenuto mirato per un sistema completo NGS per interrogare evidence-based, mutazioni attuabili in tipi di campioni impegnative come FFPE e biopsie di tumori FNA. Il flusso di lavoro ha una serie di vantaggi: 1) la coerenza, fornendo una copertura uniforme amplicone (figure 3 e 4, Tabella 3); 2) la facilità d'uso, fornendo pre-formulata, set di reagenti ottimizzati, e semplificando i requisiti di bioinformatica; 3) l'efficienza semplificando il flusso di lavoro e riducendo il numero di fasi di pipettamento rispetto ad altri metodi di NGS commerciali; e 4) la precisione incorporando un test del DNA di controllo di qualità per valutare l'amplificatorein grado numero di copie di DNA per garantire modello diversità accettabili ed evitare fluttuazioni stocastiche a rilevazione alla variante 4. L'integrazione dei dati QC pre-analitica con l'analisi bioinformatica consente basse ingressi FFPE DNA. Ciò è stato ottenuto con la formazione di un algoritmo decisionale albero utilizzando diverse copie funzionali di ingresso del DNA in tutto 400 campioni FFPE con misure indipendenti di verità, e incorporando questo algoritmo nel software bioinformatico. Di conseguenza, l'ingresso raccomandata di 400 copie amplificabile, tipicamente pari a ~ 5-20 ng di DNA FFPE, confronta favorevolmente ad altri metodi 10-12, arricchimento ibridazione a base cui si raccomanda ~ 250 ng di DNA FFPE 17,18 . Anche se la tecnica è descritta per l'uso su una piattaforma MiSeq, può essere modificato utilizzando tag primer PCR con adattatori specifici dello strumento per consentire l'analisi di sequenza su altre piattaforme NGS.
Diversi passaggi sono fondamentali per garantire il successodella procedura. Il test QC pre-analitica determina il numero di copie amplifiable del DNA e le relazioni di inibizione funzionale. Tuttavia, se vengono utilizzati meno di 400 copie di DNA amplificabile nella fase di arricchimento PCR, vi è un aumento del rischio di una chiamata falsi negativi da campioni con mutazioni a bassa abbondanza (Figura 6). Inoltre, bisogna fare attenzione durante la purificazione libreria per impedire l'eccessivo essiccamento delle perline magnetiche durante le fasi di lavaggio o eluizione. Inoltre, biblioteca successo quantificazione è fortemente dipendente dalla diluizione accurata di DNA library. Per il miglior risultato, la differenza della qPCR risultati per la libreria di campioni (Cq FAM) rispetto al LQ standard (Cq VIC) dovrebbe essere ≤3.3 Cq. Se la differenza è maggiore di 3,3 Cq, ri-diluizione e test del campione è raccomandato. Anche se un eccellente correlazione è stata osservata tra questo metodo qPCR competitivo e kit commerciali che offrono la quantificazione assoluta utilizzando una curva standard, Un offset dell'ingresso libreria nel clonale fase di amplificazione rispetto ad altri metodi possono essere necessarie per raggiungere densità ottimale di semina.
Alcuni campioni di cancro sono particolarmente difficili da sequenziare a causa di inibitori che persistono dopo l'isolamento del DNA. Per identificare questi campioni prima della preparazione biblioteca, il saggio qPCR QC rileva anche l'inibizione di amplificazione includendo un modello esogeno che serve sia come controllo interno e una sentinella per l'inibizione funzionale. Un esempio è illustrato nella Figura 3, in cui un campione di DNA melanoma riuscito a passare l'inibizione QC metrica prima sequenza e poi riuscito a generare una libreria che potrebbe essere sequenziato. Il fallimento era probabilmente una conseguenza della contaminazione melanina, un inibitore della PCR nota, derivanti dal passaggio isolamento FFPE DNA. I campioni che sono identificati con il test di controllo di qualità per essere a rischio di insufficienza di amplificazione può essere recuperato attraverso un ulteriore passaggio t clean-upo eliminare potenziali inibitori.
Il pannello 21-genica mirata concentra su hotspot gene evidence-based e fornisce un sistema completo di reagenti ottimizzati e controlli per il DNA di controllo di qualità, NGS e la bioinformatica un software che è informato da risultati di quantificazione del DNA "funzionali" pre-analitiche. Il metodo rileva con precisione le mutazioni di base di sostituzione e indels dal DNA a basso input e fornisce un esempio di un sistema di NGS con la possibilità di espandere il contenuto del pannello, per rilevare le varianti aggiuntive come CNVs ed essere adattato per il sequenziamento di RNA mirato.
Divulgazioni
JH, AH, RZ, BCH, e GJL sono dipendenti e hanno possesso di azioni di Asuragen, Inc. RZ, BCH, e GJL sono co-inventori su una domanda di brevetto per il miglioramento della variante chiamata utilizzando amplifiable informazioni numero di copie determinato per ogni campione.
Riconoscimenti
Ringraziamo il Dr. Annette Schlageter per la revisione del manoscritto. Questo lavoro è stato sostenuto in parte da sovvenzioni CP120017 dalla prevenzione del cancro e Research Institute del Texas (PI: GJL).
Materiali
| Name | Company | Catalog Number | Comments |
| 2x Quantidex Master Mix | Asuragen | 145345 | |
| Quant Primer Probe Mix | Asuragen | 145336 | |
| Inhibition Primer Probe Mix | Asuragen | 145344 | |
| ROX | Asuragen | 145346 | |
| Diluent | Asuragen | 145339 | |
| DNA Standard (50) | Asuragen | 145340 | |
| DNA Standard (10) | Asuragen | 145341 | |
| DNA Standard (2) | Asuragen | 145342 | |
| DNA Standard (0.4) | Asuragen | 145343 | |
| 2X Amplification Master Mix | Asuragen | 145348 | |
| Pan Cancer Primer Panel | Asuragen | 145347 | |
| Pan Cancer FFPE Control | Asuragen | 145349 | |
| Pan Cancer Multi-Variant Control | Asuragen | 145350 | |
| Library Pure Prep Beads | Asuragen | 145351 | |
| Wash Buffer | Asuragen | 145352 | |
| Elution Buffer | Asuragen | 145353 | |
| 2X LQ Master Mix | Asuragen | 145358 | |
| LQ Diluent | Asuragen | 145354 | |
| LQ Positive Control | Asuragen | 145355 | |
| LQ Standard | Asuragen | 145356 | |
| LQ Primer/Probe Mix (ILMN) | Asuragen | 145357 | |
| LQ ROX | Asuragen | 145359 | |
| Index Codes (ILMN) - Set A, AIL001 - AIL048 (48) | Asuragen | 150004 | |
| Index Codes (ILMN) - Set B, AIL049 - AIL096(48) | Asuragen | 150005 | |
| 2x Index Master Mix | Asuragen | 145361 | |
| Read 1 Sequencing Primers | Asuragen | 150001 | |
| Index Read Sequencing Primers | Asuragen | 150002 | |
| Read 2 Sequencing Primers | Asuragen | 150003 | |
| Sequencing Diluent | Asuragen | 145365 | |
| Illumina MiSeq | Illumina | ||
| MiSeq Reagent Kit v3 (600-cycle) | Illumina | MS-102-3003 | |
| MiSeq Reagent Nano Kit v2 (300-cycle) | Illumina | MS-103-1001 | |
| PhiX Control v3 | Illumina | FC-110-3001 | |
| Magnetic Stand-96 (Or equivalent device) | Ambion | AM10027 | |
| Quantidex Reporter Software | Asuragen |
Riferimenti
- Chen, G., Mosier, S., Gocke, C. D., Lin, M. T., Eshleman, J. R. Cytosine deamination is a major cause of baseline noise in next-generation sequencing. Mol Diagn Ther. 18 (5), 587-593 (2014).
- Choudhary, A., et al. Evaluation of an integrated clinical workflow for targeted next-generation sequencing of low-quality tumor DNA using a 51-gene enrichment panel. BMC Med Genomics. 7, 62 (2014).
- Sah, S., et al. Functional DNA quantification guides accurate next-generation sequencing mutation detection in formalin-fixed, paraffin-embedded tumor biopsies. Genome Med. 5 (8), 77 (2013).
- Zhang, L., et al. Profiling cancer gene mutations in clinical formalin-fixed, paraffin-embedded colorectal tumor specimens using targeted next-generation sequencing. Oncologist. 19 (4), 336-343 (2014).
- Latham, G. J. Next-generation sequencing of formalin-fixed, paraffin-embedded tumor biopsies: navigating the perils of old and new technology to advance cancer diagnosis. Expert Rev Mol Diagn. 13 (8), 769-772 (2013).
- Crawford, J. M., et al. The business of genomic testing: a survey of early adopters. Genet Med. 16 (12), 954-961 (2014).
- Smith, D. L., et al. A multiplex technology platform for the rapid analysis of clinically actionable genetic alterations and validation for BRAF p.V600E detection in 1549 cytologic and histologic specimens. Arch Pathol Lab Med. 138 (3), 371-378 (2014).
- Thomas, F., Desmedt, C., Aftimos, P., Awada, A. Impact of tumor sequencing on the use of anticancer drugs. Curr Opin Oncol. 26 (3), 347-356 (2014).
- Singh, R. R., et al. Clinical validation of a next-generation sequencing screen for mutational hotspots in 46 cancer-related genes. J Mol Diagn. 15 (5), 607-622 (2013).
- Beadling, C., et al. Combining highly multiplexed PCR with semiconductor-based sequencing for rapid cancer genotyping. J Mol Diagn. 15 (2), 171-176 (2013).
- McCall, C. M., et al. False positives in multiplex PCR-based next-generation sequencing have unique signatures. J Mol Diagn. 16 (5), 541-549 (2014).
- Schleifman, E. B., et al. Next generation MUT-MAP, a high-sensitivity high-throughput microfluidics chip-based mutation analysis panel. PLoS One. 9 (3), e90761 (2014).
- Wong, S. Q., et al. Sequence artefacts in a prospective series of formalin-fixed tumours tested for mutations in hotspot regions by massively parallel sequencing. BMC Med Genomics. 7, 23 (2014).
- Narayan, A., et al. Ultrasensitive measurement of hotspot mutations in tumor DNA in blood using error-suppressed multiplexed deep sequencing. Cancer Res. 72 (14), 3492-3498 (2012).
- Hagemann, I. S., et al. Clinical next-generation sequencing in patients with non-small cell lung cancer. Cancer. 121 (4), 631-639 (2015).
- Won, H. H., Scott, S. N., Brannon, A. R., Shah, R. H., Berger, M. F. Detecting somatic genetic alterations in tumor specimens by exon capture and massively parallel sequencing. J Vis Exp. (80), e50710 (2013).
- Simen, B. B., et al. Validation of a next-generation-sequencing cancer panel for use in the clinical laboratory. Arch Pathol Lab Med. 139 (4), 508-517 (2015).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati