Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Fusion SNARE-médiée de Single protéoliposomes avec bicouches supportées Tethered dans une cellule microfluidique débit contrôlé par Polarized FRBR Microscopie
Dans cet article
Résumé
Ici, nous présentons un protocole pour détecter un seul, les événements de fusion SNARE médiation entre liposomes et bicouches pris en charge dans des canaux microfluidiques utilisant TIRFM polarisée, avec une sensibilité et une seule molécule ~ 15 résolution temporelle msec. Lipid et la libération de la cargaison soluble peuvent être détectés simultanément. la taille des liposomes, des lipides diffusivité et la fusion des pores propriétés sont mesurées.
Résumé
Dans le procédé omniprésent de fusion membranaire de l'ouverture d'un pore de fusion établit la première liaison entre les deux compartiments séparés auparavant. Pendant neurotransmetteur ou la libération d'hormones par exocytose, le pore de fusion peut et fermer à plusieurs reprises, la régulation cargaison cinétique de libération transitoirement ouverts. la dynamique de Pore déterminent également le mode de recyclage des vésicules; Résultats de réapposition de sceau irréversibles transitoire fusion, "kiss-and-run", alors que la dilatation conduit à pleine fusion. Pour mieux comprendre les facteurs qui régissent la dynamique des pores, nous avons développé un test pour contrôler la fusion membranaire en utilisant polarisée totale fluorescence à réflexion interne (FRBR) microscopie avec une sensibilité seule molécule et ~ 15 résolution temporelle msec dans un biochimiquement bien défini dans le système in vitro. Fusion de marquée par fluorescence de petites vésicules unilamellaires contenant des protéines v-v-SNARE (SUV) avec une double couche plane portant le t-SNARE, supporté sur un coussin de polymère mou (t-SBL, t-supporté bicouches) Est surveillée. Le test utilise des canaux d'écoulement microfluidiques qui assurent la consommation minimale de l'échantillon tout en fournissant une densité constante de VUS. Exploiter le signal amélioration rapide lors du transfert d'étiquettes lipidiques du SUV à la SBL lors de la fusion, la cinétique de transfert lipidique de colorant est surveillée. La sensibilité de la microscopie TIRF permet de suivre simples étiquettes lipidiques fluorescentes, à partir de laquelle des lipides diffusivité et la taille SUV peuvent être déduites pour chaque événement de fusion. Lipid fois colorant de libération peuvent être beaucoup plus longtemps que prévu pour le passage sans entrave à travers les pores ouverts en permanence. L'utilisation d'un modèle qui suppose un retard de lipide libération est due à des pores de scintillement, un pore «ouverture», la fraction du temps le pore reste ouvert pendant la fusion, peut être estimée. Un marqueur soluble peut être encapsulé dans les VUS pour la surveillance simultanée des lipides et de la libération de la cargaison soluble. Ces mesures indiquent des pores peuvent reboucher après avoir perdu une partie de la cargaison soluble.
Introduction
Fusion membranaire est un processus biologique universel requis pour le trafic intracellulaire de lipides et de protéines, la sécrétion, la fertilisation, le développement, et enveloppait entrée du virus dans des organismes hôtes 1-3. Pour la plupart des réactions de fusion intracellulaire , y compris la libération d'hormones et de neurotransmetteurs par exocytose, l'énergie nécessaire pour faire fondre les deux bicouches lipidiques est assurée par la formation d'un faisceau de quatre hélices entre les récepteurs de la protéine de fixation du facteur apparenté soluble dans le N-éthylmaléimide sensible à des protéines (SNARE), ancrée dans la vésicule (v-SNARE) et la membrane cible (t-SNARE) 4, respectivement. Synaptic vésiculaire exocytose est la réaction de la fusion la plus étroitement régulée et se produit au sein d' une milliseconde après l'arrivée d'une action 1,4,5 potentiel. Le pore de fusion, la connexion initiale entre les deux compartiments de fusion, peut scintiller ouvert et fermé à plusieurs reprises avant refermeture ou l' expansion irréversible 5-7. Les résultats antérieursen transitoire, "kiss & run" fusion, tandis que le second conduit à la pleine fusion. Les facteurs qui régissent l'équilibre entre ces deux modes de fusion et des mécanismes de régulation des pores scintillements ne sont pas bien comprises 5,8.
protéines SNARE sont nécessaires pour l'exocytose; fusion des vésicules synaptiques est abolie lors du clivage de SNAREs par des neurotoxines 9. Expériences de fusion en vrac à l' aide de petites vésicules unilamellaires (SUV) ont montré que SNAREs sont non seulement nécessaires, mais aussi suffisante pour entraîner la fusion de la membrane 10. Dans cet essai en vrac, SUVs reconstitués avec v-SNARE (v-SUV) ont été dopés avec des phospholipides fluorescents (N - (7-nitro-2-1,3-benzoxadiazol-4-yl) -phosphoethanolamine (NBD-PE) et ( N -. (lissamine rhodamine B sulfonyl) -phosphoethanolamine (LR-PE) et on mélange avec des vésicules non marquées contenant du t-SNARE (t-SUV) dans un premier temps la fluorescence du NBD-PE en v-SUV est désactivé par Förster transfert d'énergie par résonance (FRET ) pour LR-PE. En laboratoireELED v-VUS fusionnent avec sans étiquette t-SUV, la densité de surface de fluorophore dans la membrane maintenant combinée est réduite et l'augmentation résultante de NBD-PE fluorescence rapporte l'étendue des lipides de mélange 10. Comme le test en vrac est facile à configurer et à analyser, il a été largement utilisé pour étudier les mécanismes de SNARE médiée par fusion 10-14. Cependant, il présente plusieurs limites, telles que la faible sensibilité et une mauvaise résolution temporelle. Plus important encore, en tant que mesure d'ensemble, il est en moyenne des résultats sur tous les événements qui font une discrimination entre l'amarrage et la fusion, ainsi que la détection des intermédiaires difficiles de hemifusion.
Au cours de la dernière décennie , plusieurs groupes, dont le nôtre, ont mis au point de nouveaux tests pour surveiller les événements de fusion au niveau de la vésicule unique 15-27. Ha et ses collègues ont utilisé v-VUS attachés sur une surface et un suivi de leur fusion avec libres t-VUS 18,19. Le mélange de lipides a été contrôlée en utilisant le FRET entre une paire de fluorophores lié aux lipides emlits dans le v- et t-SUV, respectivement, en utilisant la fluorescence totale de réflexion interne (FRBR) microscopie 18. Plus tard, le laboratoire de Brunger utilisé une seule espèce de lipide-étiquette avec un marqueur de contenu pour la détection simultanée des teneurs en lipides et le mélange 20,28. Tant le lipide et les marqueurs de contenu ont été inclus à des concentrations élevées, auto-extinction; fusion avec VUS non marqués a donné lieu à fluorescence dequenching 20,28.
D' autres ont fusionné v-VUS à bicouches planes reconstituées avec t-SNAREs 15-17,21-27,29. La géométrie plane de la cible (t-SNARE contenant) bicouches imite mieux le processus de fusion physiologique de petites vésicules, à forte courbure d'une membrane plasmique à plat. Le groupe Steinem utilise des membranes à pores transmembranaires reconstituées avec du t-SNARE, suspendue sur un substrat de nitrure de silicium poreux et détecté la fusion avec v-SUV individuels en utilisant laser microscopie confocale à balayage 23. f Autresv-VUS d' occasion à bicouches planes reconstituées avec t-SNAREs, pris en charge sur un substrat de verre 15-17,21,22,24-27,29. Le grand avantage de l'utilisation de bicouches pris en charge (SBLS) est que la microscopie TIRF peut être utilisé pour détecter accueil et de fusion des événements avec un excellent rapport signal-bruit et sans interférence de v-VUS libres, bien que l'utilisation microfluidique fournit également la résolution d'événement unique à l'aide champ lointain norme microscopie épifluorescence 24.
Une préoccupation majeure est de savoir si et comment les interactions substrat-bicouche affectent pris en charge la qualité de bicouche et le processus de fusion. Les premiers travaux a fait usage de LTTS planes qui ont été directement pris en charge sur un verre ou de quartz substrat 15-17. Ces LTTS ont été faites par adsorption, à l'éclatement d'étalement et de la fusion des membranes du t-SUV sur le substrat. Il a été vite rendu compte, cependant, que l'omission d'un élément clé t-SNARE, SNAP25, de LTTS préparés de cette manière a donné lieu à v-SUV accueil et fusion cinétique indistinguishable de ceux obtenus en utilisant la complète t-SNAREs 17. Parce que SNAP25 est absolument nécessaire pour la fusion in vivo 30,31, la pertinence physiologique de ces premières tentatives a été mis en question. Le groupe de Tamm a surmonté ce défi en utilisant mieux contrôlé appuyé la formation de bicouche 21. Il a utilisé un dépôt de Langmuir-Blodgett , pour la première plaquette exempt de protéines du SBL, suivi par la fusion de cette monocouche avec des T-SUV 21. Cela a abouti à la fusion SNAP25-dépendante.
Afin d' éviter des artefacts potentiels associés à un bi - couche directement supporté sur un substrat de verre sans qu'il soit nécessaire d'utiliser des méthodes de Langmuir-Blodgett, Karatekin et al. , A présenté un poly hydraté (éthylène glycol) (PEG) amortit entre la double couche et le substrat 24. Cette modification a aussi entraîné SNAP25 dépendant fusion 24. Bicouches rembourrées sur une couche de polymère mou avaient été connus pour mieux préserver transmembranairela mobilité de la protéine et de la fonction 32, et ont été utilisées dans des études de fusion avec 33 les virus. En outre, les bicouches PEGylés semblent conserver une certaine capacité d'auto-guérison et sont très robustes 34,35. Tout d'abord, une fraction de disponibles dans le commerce, les chaînes de PEG lipidiques liées sont inclus dans la membrane t-SUV. Lorsque ces T-VUS éclatent et forment une bicouche plane sur un substrat de verre, une brosse PEG couvre les deux feuillets de la bicouche planaire. Comme la formation de bicouches plane est entraînée par adhérence des chaînes de PEG entourant t-SUV sur la surface du verre hydrophile, d'un liposome éclatement et la formation de deux couches planes sont relativement insensibles à la composition lipidique utilisée. Cependant, lorsque de grandes quantités de cholestérol sont inclus, ce qui augmente les propriétés de cohésion des VUS, les VUS ne peuvent pas éclater spontanément. Si tel est le cas, les ions divalents ou choc osmotique peuvent être utilisés pour aider à la formation de deux couches planes 25.
Comme il est mentionné ci-dessus, dans la présente apapproche une brosse PEG couvre les deux côtés de la plane, soutenue bicouche. La brosse en face du canal d'écoulement microfluidique aide à prévenir l'adhérence non spécifique de v-VUS entrants qui sont aussi généralement recouvertes d'une couche de PEG. Formation de complexes V- et t-SNARE commence à partir de l' extrémité N-terminales et procède à la membrane distale progressivement vers les domaines de la membrane proximale 36. Pour les v-VUS d'interagir avec le t-SBL, le v- et le besoin t-SNARE N-terminales pour faire saillie au-dessus des brosses PEG, ce qui semble être le cas dans les conditions de l'essai. La hauteur de brosse peut être adapté à l' étude des protéines autres que SNAREs en faisant varier la densité des lipides pégylés et la longueur de la chaîne de PEG 37,38. Un autre avantage des brosses PEG couvrant les surfaces proximales des bicouches de fusion est qu'ils imitent l'environnement encombré des membranes biologiques qui sont emballés avec 30.000-40.000 protéines membranaires intégrales par micron carré 39. Tout comme les chaînes de PEG dans cet essai, le repulsive couche de protéines portant sur des membranes biologiques doit être mis de côté pour permettre un contact entre les deux bicouches phospholipidiques pour la fusion se produise.
des canaux d'écoulement fluidiques sont utilisés dans ce test, car ils offrent des avantages uniques. Tout d'abord, le flux microfluidique permet un dépôt plus uniforme de t-VUS à se répandre et fusible pour former le t-SBL. Deuxièmement, le petit volume de canal (<1 pi) minimise la consommation d'échantillon. Troisièmement, les petits volumes requis permettent à l'ensemble de l'expérience effectuée en vertu du débit constant. Débit supprime faiblement, probablement non spécifique, adhéré v-VUS de la SBL 16. Il maintient également une densité constante de v-VUS au- dessus du t-SBL, ce qui simplifie l' analyse cinétique 17. Enfin, les vésicules accostés se distinguent facilement de celles qui sont libres portés par le flux 25. Quatrièmement, plusieurs canaux microfluidiques peuvent être utilisés sur le même lamelle, sonder chacun un état différent. Ceci permet la comparaison des conditions during le même essai expérimental. Une approche similaire a été utilisée par le groupe van Oijen pour étudier la fusion entre le virus de la grippe et LTTS rembourrées 33.
En microscopie TIRF, la décroissance exponentielle du champ évanescent (avec une constante de désintégration ~ 100 nm) confine l'excitation de fluorescence à ces molécules qui sont très près de l'interface verre-tampon. Cela réduit la contribution des molécules fluorescentes qui sont plus loin, augmente le rapport signal-bruit, et permet une sensibilité molécule unique avec des temps d'exposition de vues de 10-40 msec. Le champ évanescent conduit également à une augmentation du signal lors de la fusion: le transfert marqué des lipides du SUV dans la SBL, ils se trouvent, en moyenne, dans un champ d'excitation plus forte. Cette augmentation de la fluorescence est plus forte pour les grands liposomes.
Si la lumière polarisée est utilisée pour générer le champ évanescent, d'autres effets contribuent à des changements de fluorescence lors de la transfert des étiquettes du SUV dans la SBL. Certains colorants lipidiques ont un dipôle de transition orienté avec un angle moyen préféré par rapport à la double couche dans laquelle elles sont incorporées. Cela crée une différence dans la quantité de fluorescence émise par les fluorophores quand ils sont dans le SUV par rapport à la SBL, étant donné que le faisceau polarisé excitera les colorants dans les deux membranes différentes. Pour les premiers, le faisceau d'excitation va interagir avec dipôles de transition orientés autour du SUV sphérique, alors que pour ce dernier, les orientations dipolaires seront limitées par la géométrie SBL plat. Par exemple, quand on utilise de lumière polarisée incidente (polarisation perpendiculaire au plan d'incidence), l' excitation est plus efficace lorsque le colorant est dans la SBL que dans le SUV à un lipide colorant transition dipôle orienté parallèlement à la membrane 29,40 (comme celui de Dil ou DiD 41-43). Un SUV dopé avec un tel fluorophore apparaît dim quand il docks sur la SBL (Figure 7, représentat ive résultats). Comme un pore de fusion ouvre et relie les membranes de SUV et SBL, les sondes fluorescentes diffusent dans la SBL et deviennent plus susceptibles d'être excités par le champ évanescent 25,27,29 polarisée s. Par conséquent, le signal de fluorescence intégrée autour du site de fusion augmente fortement au cours du transfert de colorant depuis le SUV dans la (figure 7 et figure 3) SBL 27. Un autre facteur qui contribue à signaler des changements qui l'accompagnent fusion est dequenching de marqueurs fluorescents tels qu'ils sont dilués lorsqu'ils sont transférés dans le SBL. La contribution de dequenching est généralement mineure par rapport aux effets évanescents de décroissance du champ et de polarisation dans l'essai décrit ici, car seule une petite fraction (  ) Des lipides sont marqués.
) Des lipides sont marqués.
L'augmentation du signal lors de la fusion peut être exploitée pour déduire des propriétés de fusion des pores en comparant le temps,1 "src =" / files / ftp_upload / 54349 / 54349eq2.jpg "/>, requis pour un lipide pour échapper à travers un pore qui est librement perméable aux lipides à l'instant réel de libération,  . Si les deux échelles de temps sont comparables, on en conclut que le pore présente une faible résistance à l'écoulement des lipides. Toutefois, si le temps de libération réelle est beaucoup plus longue que le temps pour la libération de diffusion limitée, cela indiquerait un processus, tels que les pores vacillement, retardant la libération des lipides. Le temps de libération limitée par diffusion,
. Si les deux échelles de temps sont comparables, on en conclut que le pore présente une faible résistance à l'écoulement des lipides. Toutefois, si le temps de libération réelle est beaucoup plus longue que le temps pour la libération de diffusion limitée, cela indiquerait un processus, tels que les pores vacillement, retardant la libération des lipides. Le temps de libération limitée par diffusion,  , Dépend de la taille du liposome de fusion et de lipides diffusivité; son estimation exige que ces deux paramètres à quantifier. La sensibilité seule molécule de l'essai permet lipidique diffusivité à mesurer au moyen de plusieurs fluorophores unique de lipides après leur libération dans la SBL pour chaque événement de fusion 26. La taille de chaque vésicule fusionpeut être estimée 27 en combinant : (i) l'intensité d'un colorant unique de lipides, (ii) la modification de la fluorescence totale autour d' un site d'accueil après que tous les fluorophores sont transférés dans la SBL lors de la fusion, (iii) la densité de marquage connu de SUV les lipides, et (iv) la surface par lipide. Pour de nombreux événements de fusion, les temps des lipides de libération réels se sont révélés être beaucoup plus lent que prévu par la diffusion à libération contrôlée 27, comme cela a été indiqué précédemment en supposant que la taille de SUV uniforme 44. En supposant un retard de lipide libération est due à des pores de scintillement, un modèle quantitatif permet d' estimer « l' ouverture des pores", la fraction du temps le pore reste ouvert pendant la fusion 27.
, Dépend de la taille du liposome de fusion et de lipides diffusivité; son estimation exige que ces deux paramètres à quantifier. La sensibilité seule molécule de l'essai permet lipidique diffusivité à mesurer au moyen de plusieurs fluorophores unique de lipides après leur libération dans la SBL pour chaque événement de fusion 26. La taille de chaque vésicule fusionpeut être estimée 27 en combinant : (i) l'intensité d'un colorant unique de lipides, (ii) la modification de la fluorescence totale autour d' un site d'accueil après que tous les fluorophores sont transférés dans la SBL lors de la fusion, (iii) la densité de marquage connu de SUV les lipides, et (iv) la surface par lipide. Pour de nombreux événements de fusion, les temps des lipides de libération réels se sont révélés être beaucoup plus lent que prévu par la diffusion à libération contrôlée 27, comme cela a été indiqué précédemment en supposant que la taille de SUV uniforme 44. En supposant un retard de lipide libération est due à des pores de scintillement, un modèle quantitatif permet d' estimer « l' ouverture des pores", la fraction du temps le pore reste ouvert pendant la fusion 27.
À chaque fois que possible, il est important de tester les mécanismes de fusion en utilisant à la fois solubles dans les lipides et les contenus des étiquettes. Par exemple, le lipide libération pourrait être retardée par des procédés autres que le scintillement des pores, telles que la restriction de la diffusion des lipides par les protéines SNARE qui entourent til pore. Si tel était le cas, alors la libération de contenus précéderait libération des étiquettes lipidiques, à condition que le pore est assez grand pour permettre le passage de sondes solubles. Une faille plus fondamentale dans l'approche pourrait être dans l'hypothèse que le transfert des lipides marqués à la SBL se produit à travers une fusion étroite pores reliant la SBL à une vésicule qui a conservé en grande partie sa forme pré-fusion. Transfert de Lipid dans le SBL pourrait également résulter de la dilatation rapide du pore de fusion avec une concomitante, extrêmement rapide effondrement du SUV dans la membrane SBL, comme précédemment suggéré sur la base de données des lipides de libération seuls 29. Contrôler à la fois des lipides et le contenu de libérer en même temps, on a constaté que de nombreux pores refermés après libération de leurs marqueurs de lipides, mais a conservé une partie de leur cargaison soluble 27. Ceci indique qu'au moins certains des liposomes ne fusionnent pas dans la SBL après la fusion, et que le transfert des lipides de colorant dans le SBL se produit à travers un pore de fusion. En outre, lipid et le contenu de presse a eu lieu simultanément 27, ce qui rend peu probable que le retard de lipide libération était due à l' entrave de la diffusion des lipides par les protéines SNARE entourant le pore 45.
Un protocole de fusion SUV-SBL qui ne surveille pas le contenu soluble communiqué a été publié par Karatekin et Rothman 25. Ici, des développements plus récents sont inclus, la surveillance soit simultanée des teneurs en lipides et libèrent et l' estimation des propriétés SUV, lipides, et la fusion des pores 27. Le protocole commence avec des instructions pour la préparation des cellules microfluidiques, réalisés par collage d' un poly (diméthylsiloxane) , des rainures (PDMS) élastomère à blocs contenant une lamelle de verre 25. Ensuite, la préparation de v-SUV à la fois avec les lipides et le contenu des marqueurs est expliqué. Les articles 4 et 5 fournissent des instructions pour l' assemblage des cellules microfluidiques, formant les LTTS in situ et la vérification des défauts et la fluidité, l' introduction de vSUVS dans les cellules d'écoulement et la détection d'événements de fusion. Section 6 fournit des instructions pour l'analyse des données.
Protocole
1. Préparation d'un bloc PDMS pour former le canal microfluidique

La figure 1. La microfabrication de matrice de cellules d'écoulement et la préparation des blocs PDMS. (A) la conception d'une cellule d'écoulement à quatre canaux qui se fixe sur une lamelle de verre de 24 x 60 mm ( en bas). Six designs identiques sont disposés pour tenir sur une plaquette de silicium de 10 cm (en haut). (B) Découpez bloc d'environ 5-8 mm d' épaisseur PDMS sur un perforatrice. (C) L' insertion du tube dans le trou percé à l' aide d' une paire de pinces. S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
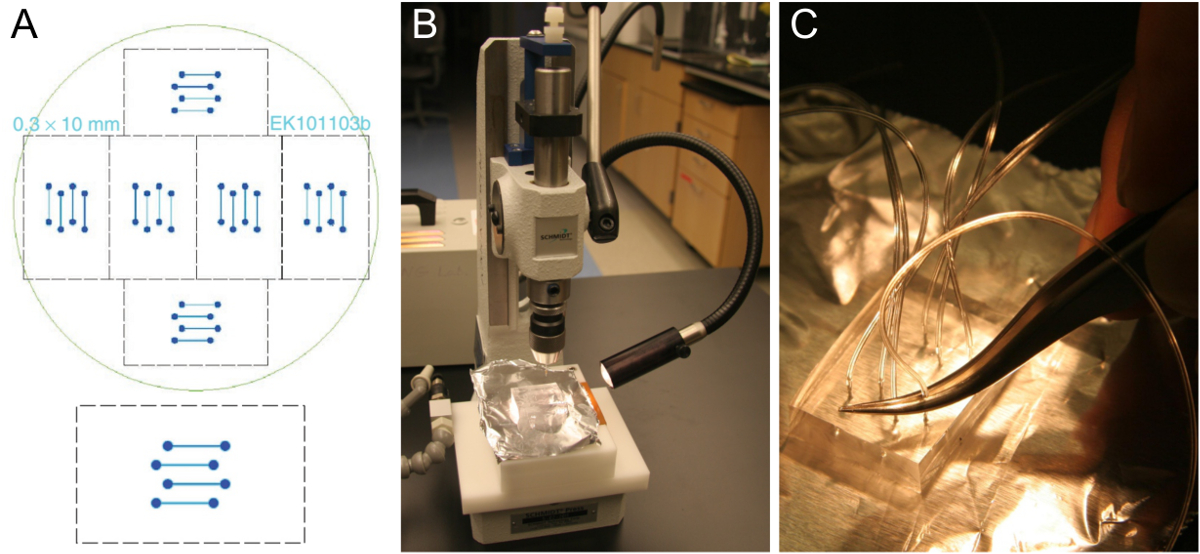{kind=link}
- Obtenir un modèle de cellule d'écoulement tel que celui de la figure 1A. canaux typiques sont 0,3-1 mm de large, 70-100 um de haut, et 1-2 cm de long. Fabriquez un modèle noustion des techniques de photolithographie standards dans une salle blanche de 25 ou de l' ordre si l' accès des salles blanches ne sont pas disponibles. En variante, la machine d'un modèle avec des dimensions plus grandes à partir d'un matériau approprié.
Remarque: le personnel Cleanroom peut former et guider les utilisateurs inexpérimentés (accès à une salle blanche est toujours limité aux utilisateurs avec une formation adéquate) dans la conception et la commande d'un masque, le nettoyage de la plaquette, et photolithographie. Une fois qu'un modèle est obtenu, il peut être utilisé en dehors de la salle blanche à plusieurs reprises, à condition qu'il soit conservé dans un plat fermé et on prend soin d'exclure la poussière. - Préparer un mélange de ~ 100 ml (PDMS silicone de base d'élastomère) et ~ 10 ml d'agent de reticulation (agent de durcissement) dans une coupelle en plastique jetable. Casser la pointe d'une pipette jetable pour gérer les PDMS visqueux plus facilement. Peser les PDMS dans la tasse en plastique comme pipetage est pas exact.
- Incorporer le mélange bien. Supprimer les nombreuses bulles d'air de dégazage dans un dessicateur sous vide (environ 20mn). Que le godet est placé sous vide, les bulles seront d'abord croître en taille, en augmentant le volume du mélange considérablement. Contrôler le vide qui est appliqué et assurez-vous la coupe est assez profonde pour éviter un déversement.
- Verser une grande goutte du mélange PDMS dégazé dans une boîte de Pétri en verre (150 mm x 20 mm) et appuyez sur la plaquette sur le PDMS avec le modèle vers le haut. Cela évite les bulles étant piégés sous la plaquette. bulles Trapped élargiront et peuvent basculer la plaquette lorsque le plat est placé dans le four. Maintenant, versez PDMS dégazés sur le modèle au-dessus de la plaquette jusqu'à ce qu'elle soit couverte par environ 5-8 mm de PMDS. Si une bulle d'air se produit et retirez délicatement avec une pointe de pipette.
- Cuire le PMDS dans un four à 60 ° C pendant 3 h. Assurez-vous que le plat est de niveau.
- Utilisez une nouvelle lame de scalpel pour découper un bloc de PDMS contenant les structures de canaux moulés. Le bloc découpé doit tenir sur une lamelle (voir étape 4.1.9).
- Peler le bloc découpé dans le plat et place sur un morceau de papier d'aluminium propre.
Remarque: Les blocs PMDS réticulés peuvent être conservés pendant quelques mois. Sur une plaquette unique, il existe 6 ensembles de canaux d'écoulement (figure 1A), de façon à 6 PDMS blocs peuvent être fabriqués en une seule pièce de moulage PDMS. Après avoir coupé tous les 6 des blocs de PDMS, pièces propres lâches de PDMS, mais sinon, ne pas retirer les PDMS restantes avant la coulée et la réticulation du prochain lot, ce qui nécessitera seulement environ la moitié des PDMS utilisés pour le premier lot. Un bon modèle peut durer quelques années. - Utilisez une perforatrice pour percer à travers le bloc de PDMS dans un mouvement rectiligne (figure 1B). Démarrer sur le côté des rainures de canal. Assurez-vous de retirer le morceau de PDMS poinçonné. Répétez cette opération pour tous les huit trous de la conception à quatre canaux.
- Placez le côté du canal de bloc PDMS vers le bas sur une nouvelle et sans rides morceau de papier d'aluminium. Rangez le bloc jusqu'à quelques mois dans une boîte sèche, idéalement dans un dessiccateur.
- Poussez le tube (0,25 mm ID, 0,76 mm OD) d' environ un tiers dans le trou percé à l' aide d' une paire de pinces (Figure 1C). Couper le tube incliné pour faciliter l'insertion. Laissez tubes assez long pour atteindre le réservoir SUV et la pompe de seringue, respectivement. Après avoir placé la puce assemblé sur le microscope, couper les tubes à nouveau si elles sont trop longues.
- Pour se connecter à la seringue de la pompe, couper un petit morceau de tube en silicone plus grande (0,51 mm ID, 2,1 mm de diamètre extérieur) et insérer le tube plus mince dans un côté. Ce bloc prêt-à-utilisation de PDMS peut être conservé pendant quelques mois.
2. Coverslip Nettoyage
- Nettoyer les lamelles , selon le protocole décrit dans l' Karatekin et Rothman 25 en utilisant un mélange oxydant fort de l' acide sulfurique (H 2 SO 4) et du peroxyde d'hydrogène (H 2 O 2). Effectuez cette procédure dans une salle blanche, et d'observer les précautions de sécurité adéquates. Sinon, utilisez un cleanroom nettoyé coverslip qui est disponible dans le commerce (voir la liste des matériaux).
3. Préparation de v-SUVS contenant à la fois Lipid et étiquettes de contenu

Figure 2. Schéma de préparation SUV. Les lipides sont mélangés dans un tube de verre (1) et le solvant est évaporé pour former un film lipidique en faisant tourner le tube dans un bain d'eau (2). Les traces restantes de solvant sont éliminées sous vide (3). Le film lipidique est hydraté dans le tampon de reconstitution contenant un détergent et la protéine pendant tourbillonnement (4). Si le contenu colorant est à encapsuler, il est inclus dans cette étape, ainsi que dans l'étape de dilution (5). La dilution de la concentration en détergent en dessous de sa concentration micellaire critique conduit à la formation des liposomes. Le détergent est dialysée du jour au lendemain (6). Pour T-VUS pour la formation SBL y compris NBD-PE (vert), les vésicules flottent dans un gradient de densité et recueillies àl'interface entre les deux couches (7a). Pour séparer v-SUV avec le marqueur contenu encapsulé de colorant libre l'échantillon est exécuté sur une colonne d'exclusion de taille et recueilli dans des fractions de 0,5 ml (7b). S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
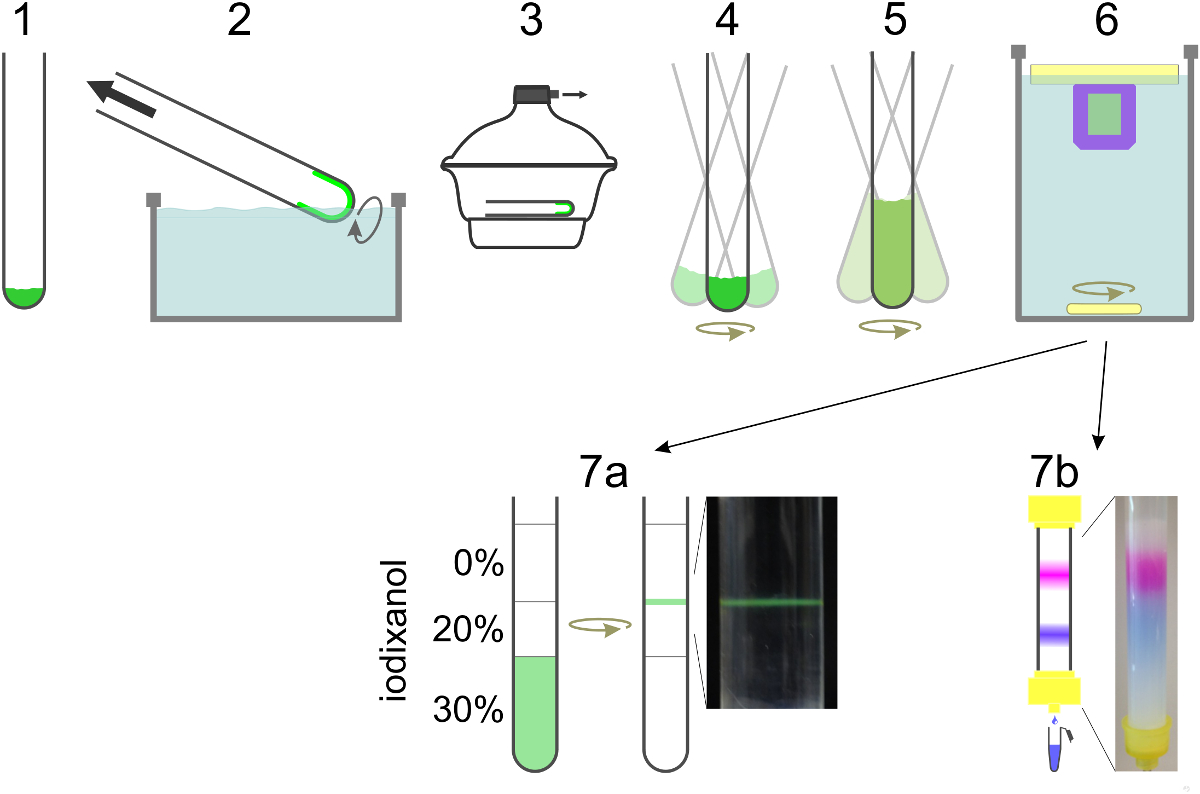{kind=link}
- Préparer 4 litres de tampon de reconstitution, contenant 25 mM de HEPES-KOH, KCl 140 mM, 100 pM d'EGTA et 1 mM de DTT, pH 7,4. Utilisez la plupart du tampon pour dialyse et 100 ml pour les autres étapes.
Note: D' autres tampons peuvent être utilisés, mais il est important de maintenir l'osmolarité du tampon cohérent pendant toute l'expérience et de choisir des conditions dans lesquelles les protéines sont fonctionnelles. Pour la préparation de v-SUV contenant des étiquettes lipidiques seulement et t-VUS, suivez Karatekin et Rothman 25. - Comme les stocks de lipides sont stockés à -20 ° C dans le chloroforme ou le chloroforme: méthanol (2: 1, v / v), soit des fioles à atteindre la température ambiante avant ouvertureéviter la condensation.
- Rinçage d'un tube de verre avec du chloroforme pour éliminer les traces de détergent et mélanger les lipides dans le rapport désiré avec une quantité finale de 1 pmol dans un mélange de chloroforme et de methanol (2: 1, v / v). Utilisez uniquement des seringues en verre / tubes à manipuler des solvants / lipides dissous organiques.
Remarque: Les lipides utilisés ici sont POPC: SAPE: DOPS: Cholestérol: PEG2000-DOPE: DiD à un rapport molaire de 57,4: 15: 12: 10: 4,6: 1 pour v-SNARE contenant des vésicules et POPC: SAPE: DOPS: Cholestérol : cerveau PI (4,5) P 2: PEG2000-DOPE: NBD-PE (54,9: 15: 12: 10: 3: 4,6: 0,5) pour t-SNARE VUS. Voir Matériel pour plus de détails. D'autres compositions lipidiques peuvent être utilisées. - On évapore le solvant sous un léger courant d'azote ou dans un évaporateur rotatif. Immerger l'extrémité du tube dans un bain d'eau (37 ° C) est chauffé au-dessus de la température de fusion la plus élevée des lipides pour donner un film lipidique homogène et à éviter de fortes variations de température tout en évaporant le solvant. Commencez vide à environ300 mbar jusqu'à ce que le film lipidique est formé, puis continuer pendant environ 2 minutes à la plus élevée possible sous vide à l'évaporateur rotatif.
- Retirez le tube de verre et l'envelopper dans du papier d'aluminium pour éviter exposition à la lumière et éliminer les traces restantes de solvant en plaçant le tube dans un dessiccateur sous vide poussé pendant au moins 2 h. Cette étape peut également fonctionner durant la nuit.
- Réhydrater le film lipidique séché, préparer un mélange de détergent (n-octyl-β-D-glucopyranoside, OG), la protéine (v-SNARE) et SRB dans un tampon de reconstitution (volume final 500 ul). Maintenir la concentration finale de détergent ~ 2 fois supérieure à la concentration micellaire critique (CMC, de 20 à 25 mM pour OG) et ajuster le volume final de telle sorte que le détergent taux de lipides est> 10. Dissoudre sulforhodamine B (SRB) de poudre dans la solution de protéine-détergent pour obtenir une concentration finale de mM SRB 10-50 en agitant doucement la solution. Ajuster l'osmolarité du tampon de reconstitution lors de l'ajout de fortes concentrations de SRB en réduisant la concentration de chlorure de potassium en conséquence.
Remarque: le rapport Lipid-à-protéines (LP) pour t-SNAREs est élevé (LP ~ 20.000, ~ 70 t-SNAREs par sq micron 25.). De manière significative des densités t-SNARE dans la SBL peuvent conduire à des agrégats de protéines inactives avec réduit de façon significative les taux de fusion 17,24. LP pour v-SNAREs est beaucoup plus faible (LP ~ 200, ~ 7000 v-SNAREs par um 2), à proximité des densités v-SNARE trouvés sur les vésicules synaptiques 46,47. Pour assurer la fonction des protéines après l' expression recombinant et la purification de v- et t-SNAREs il est utile d'effectuer une simple analyse de fusion en vrac 48 avant de passer par toutes les étapes de l'essai de fusion vésiculaire unique. - Secouer doucement le tube de verre préchauffé contenant le film lipidique séché tout en ajoutant la solution de protéine-détergent SRB à l'étape 3.6. Essayez d'éviter de créer des bulles. Continuer à agiter pendant 15 min à 37 ° C.
- On dilue le détergent quintuple par addition d'une reconstitutiontampon contenant SRB (ajouter 2 ml, 2,5 ml) volume final de tout tourbillonner rapidement pour éviter des gradients de concentration. Continuer à agiter pendant 15 min à 1 h à 37 ° C.
Note: Longer augmentation d'incubation efficacité protéine de reconstitution. La dilution rapide diminue la concentration de détergent en dessous de la CMC et conduit à la formation de petits liposomes. - Dialyser la suspension de vésicules première contre ~ 1 L de tampon de reconstitution pendant 1-2 heures à la température ambiante, puis contre 3 litres de tampon de reconstitution pendant une nuit à 4 ° C avec 4 g de polystyrène adsorbant, en utilisant un tube de dialyse MWCO de 20 000 ou d'une cassette. Utilisez différents béchers pour la dialyse de vésicules avec et sans SRB pour prévenir la contamination croisée.
- Équilibrer une colonne de filtration sur gel avec un tampon de reconstitution. Exécutez la suspension de vésicules à travers la colonne pour séparer SRB libre des v-VUS avec SRB encapsulé. Utiliser un tampon de reconstitution sans SRB comme éluant une fois que l'ensemble de l'échantillon aentrée dans la colonne. Recueillir v-SUVs en fractions de 0,5 ml.
- Vérifier avec succès l' encapsulation SRB à des concentrations d'auto-trempé en mesurant la fluorescence SRB avant et après addition d' un détergent à une aliquote de vésicules 16. En utilisant un spectromètre à fluorescence, exciter l'échantillon à 550 nm et l'émission balayer SRB entre 570 nm et 630 nm. Attendez-vous à une augmentation d'un facteur 4-8 de la fluorescence de SRB selon la quantité encapsulé lors de l'addition de détergent que la membrane est solubilisé et le SRB libéré est dilué.
- Caractériser la récupération des lipides et des protéines en utilisant la spectroscopie de fluorescence 24 et électrophorèse sur gel SDS-PAGE, respectivement. Utilisez une méthode de coloration sensible, en particulier pour les échantillons t-SUV haute LP (voir la liste des matériaux). Typiquement d'environ 50% à la fois des lipides et des protéines d'entrée est perdue lors de la préparation obtenue dans un LP proche de la valeur nominale.
- Caractériser tailles SUV en utilisant la lumière diffusion dynamique de 24 ou électrons microscopy 48. SRB de magasin contenant v-VUS à 4 ° C jusqu'à ~ 3-4 jours. Ne pas congeler, comme la congélation et la décongélation brise la membrane et libère SRB encapsulé.
4. SUV-SBL Fusion Assay pour surveiller Lipid sortie seulement
- La formation de la bicouche supportée attaché à l' intérieur des canaux d'écoulement microfluidique
- Placer le bloc PDMS (étape 1.11) sous vide poussé pendant au moins 20 minutes avant l'essai pour éliminer les gaz dissous. Cela réduit considérablement le risque de bulles d'air dans les canaux microfluidiques au cours de l'expérience de fusion.
- Allumez la configuration du microscope et chauffer la scène et porte-échantillon (figures 3 et 4) à la température désirée.
- Filtrer le tampon de reconstitution utilisée pour diluer les vésicules à travers un filtre de 0,45 um ou plus petite taille des pores.
- Diluer ~ 30 pi du NBD-PE étiquetés t-VUS ou sans protéines (PF-SUV) liposomes témoins (stockssolution 0,5-1 lipidique mM) avec ~ 60 pi de tampon. La concentration finale est pas critique ici.
- Dégazer ce mélange à l'aide d'une seringue de 3 ml. Appuyez sur la majeure partie de l'air au-dessus de l'échantillon tout en tenant la seringue verticalement. Sceller la pointe (sans aiguille) en utilisant un film de paraffine et de créer un vide en tirant sur le piston. Tapoter le corps de seringue afin d'accélérer le dégazage de la solution. Répétez ce processus plusieurs fois jusqu'à ce qu'il n'y a plus de bulles se produisent lorsque le vide est appliqué.
- Utiliser une aiguille hypodermique ayant un diamètre légèrement plus grand que le tube qui est fixé au bloc de PDMS pour percer un trou dans le bouchon d'un tube de microcentrifugation. Assurez-vous que le morceau de matière plastique est découpée pas à l'intérieur du tube, comme il peut obstruer la tubulure reliée à l'entrée du canal microfluidique ultérieurement.
- Remplir la solution SUV dégazé dans le tube à centrifuger ayant un trou dans le bouchon et le placer dans le support sur la platine du microscope à équilibrer à la température de consigne.
- PLace une lamelle préalablement nettoyée (section 2) dans un nettoyeur à plasma et exécuter plasma d'air pendant environ 5 min. Mettez la lamelle traitée au plasma (côté traité vers le haut) sur le dessus de quelques tissus non pelucheux servant de coussin.
- Retirer la feuille d'aluminium à partir du bloc de PDMS dégazé et placez le bloc sur le dessus de la lamelle. Lors de la réutilisation du bloc de PDMS mettre et à détacher un morceau de ruban adhésif à côté du canal pour le nettoyer. Appuyez sur les PDMS bloquer sur la lamelle en utilisant une paire de pinces pour le faire coller, mais ne pas appuyer trop dur que le verre pourrait se briser.
- Placer le tube d'écoulement assemblé sur la platine du microscope et raccorder le tuyau au réservoir de SUV et la pompe à seringue, respectivement. Collez la lamelle à l'étape (figure 3B).
- Commencer à aspirer VUS 3 pl / min jusqu'à ce que la solution remplit les canaux complètement (~ 2,25 mm / s pour le canal de section x 300 um 75 um). Lorsque des solutions pour tous les canaux commencent à se déplacer jusqu'à the tube du côté de la sortie, diminuer le débit à 0,5 ul / min et incuber pendant 30 à 45 min.
Remarque: le temps écoulé entre le traitement au plasma de la lame de verre et l' écoulement du SUV dans le canal ne doit pas dépasser 10 à 20 minutes que l'effet du traitement au plasma est transitoire. - Vérifiez les canaux pour toute fuite. Utilisez un objectif d'air 10-20x pour observer NBD-PE fluorescence du NBD-PE contenant T- ou pf-SUV. Exciter les fluorophores à l'aide du NBD laser à 488 nm. Les fuites sont difficiles à détecter en utilisant un éclairage à fond clair.
- Mettez un tube avec un tampon de reconstitution dégazé dans le support et laissez la température équilibrer les changements rapides que la température pourrait provoquer des défauts dans la SBL. Arrêter l'écoulement et attendre ~ 1 min pour vous assurer que le débit est complètement arrêté et aucune bulle d'air sera aspiré dans le tube avant de passer les tubes d'entrée dans la mémoire tampon dégazé.
- Rincer tous les canaux avec un tampon dégazé pour laver les SUV non liés.
- Basculer vers un higheobjectif de r grossissement FRBR (60X, huile, NA 1,45 à 1,49) et vérifiez que la bicouche semble homogène et est exempt de tout défaut à grande échelle évidents tels que des taches sombres ou tubules lipidiques s'étendant à partir de la SBL.
- Vérifiez la fluidité de la bicouche
- Si une unité de FRAP dédié ne sont pas disponibles, ou si une séquence de FRAP ne peut pas être programmé, qualitativement la fluidité des membranes d'essai comme suit.
- Fermez le diaphragme de champ à une petite taille (~ 40 um de diamètre) et d' ajuster l'intensité de la lumière d'excitation de 488 nm à travers le logiciel (20-80 uW, ou 15-60 nW / um 2) pour blanchir la fluorescence NBD-PE dans l'exposé zone de manière significative, mais pas complètement. Pour une bicouche fluide, à l'état d'équilibre, l'intensité de fluorescence dans le milieu de la zone exposée doit être inférieur au niveau des bords, comme intactes molécules NBD-PE entrent dans la zone exposée et diffusent une certaine distance avant le blanchiment. En revanche, si la surface collée SUVs n'a pas réussi à éclater, ou pour une autre raison de la bicouche supportée est pas fluide, tous les fluorophores dans la zone exposée doit blanchir.
Remarque: Les valeurs d'intensité laser dans ce domaine et plus tard étapes sont donnés à titre de point de départ rugueux et devraient être optimisés pour un ensemble donné de conditions. - Arrêtez l'éclairage et le démarrer à nouveau quelques minutes plus tard pour vérifier les résultats des mesures d'état stable
- Fermez le diaphragme de champ à une petite taille (~ 40 um de diamètre) et d' ajuster l'intensité de la lumière d'excitation de 488 nm à travers le logiciel (20-80 uW, ou 15-60 nW / um 2) pour blanchir la fluorescence NBD-PE dans l'exposé zone de manière significative, mais pas complètement. Pour une bicouche fluide, à l'état d'équilibre, l'intensité de fluorescence dans le milieu de la zone exposée doit être inférieur au niveau des bords, comme intactes molécules NBD-PE entrent dans la zone exposée et diffusent une certaine distance avant le blanchiment. En revanche, si la surface collée SUVs n'a pas réussi à éclater, ou pour une autre raison de la bicouche supportée est pas fluide, tous les fluorophores dans la zone exposée doit blanchir.
- Programmer une séquence de FRAP pour une mesure plus quantitative, si possible. Voir fichiers supplémentaires et la légende correspondante pour les détails.
Note: Parfois , les VUS adhèrent sur la lamelle de verre, mais ne parviennent pas à éclater et former une bicouche fluide. Si cela se produit, rincer les canaux avec un tampon de reconstitution dégazé contenant mM Mg 2+ 10 pour aider à la formation de bicouche pris en charge. Utilisez la fluorescence de blanchiment pour évaluer la fluidité bicouche comme dans 4.2. Une fois une bicouche fluide est formé, rincer avec de Mg 2+ -free recotampon nstitution.
- Si une unité de FRAP dédié ne sont pas disponibles, ou si une séquence de FRAP ne peut pas être programmé, qualitativement la fluidité des membranes d'essai comme suit.
- Présentation de v-VUS dans les canaux d'écoulement microfluidiques
- Degas tampon de reconstitution et de l' utiliser pour diluer la solution mère v-SUV par un facteur d'environ mars 10 à mai 10 en fonction de la concentration des stocks v-SUV. Objectif pour une dilution qui donne lieu à environ 10 à 100 événements de fusion dans les 60 secondes dans le champ de vision.
Remarque: Trop de fusions augmentent la fluorescence de fond (puisque chaque dépôts d'événements LR-PE ou DiD étiquettes lipidiques dans la SBL) et rendent la détection et l' analyse des événements de fusion difficile. En revanche, un taux de fusion qui est trop faible des résultats en mauvais statistiques ou nécessite l'acquisition de beaucoup plus de films. Pour une concentration v-SUV de 0,1 mM de lipide, commencer par dilution 5 pi SUV disponible dans un tampon de reconstitution 995 pi, puis diluer 5-50 pi de cela dans 950-995 ul de tampon de reconstitution. - Laissez la température équilibrer avant d'arrêter l'écoulement et inserting le tube d'entrée dans la solution v-SUV diluée.
- Ajustez l'angle de la FRBR et la polarisation de la manière suivante.
- Après le réglage de la polarisation souhaitée en faisant tourner le faisceau d'excitation, d'ajuster l'inclinaison du miroir, commandant la position du faisceau par l'intermédiaire d'un moteur pas à pas par l'intermédiaire du logiciel. Déplacez lentement la position du faisceau laser du centre de l'objectif au plan focal arrière d'un côté hors-centre. Observez la lumière de l'objectif sur la face avant émergent avec un angle de plus en plus par rapport à l'axe de l'objectif que la position est déplacé plus excentré.
- Noter la position du moteur lorsque le premier faisceau sortant disparaît dans l'objectif, à savoir, lorsque TIR est d' abord atteinte.
- Lentement garder le déplacement de la position de faisceau plus excentré, tout en surveillant la fluorescence de la surface. Notez la position du moteur lorsque la fluorescence de surface disparaît lorsque le faisceau est déplacé trop loin hors-centre.
- Choisissez une positio de faisceau n entre les deux limites déterminées ci-dessus. Pour le meilleur rapport signal à bruit et plus l'amélioration du signal lors de la fusion, pour choisir une profondeur de pénétration peu profonde (position de faisceau plus proche du bord de l'objectif), qui fournit encore un éclairage uniforme de la Viewfield.
Note: Il est préférable de garder la même position de faisceau TIR (même profondeur de pénétration) pour toutes les expériences après les paramètres sont optimisés. Assurez-vous faire tourner la polarisation ne conduit pas à des changements de position du faisceau.
- Écoulement v-SUV dans le canal à un débit de 2 ul / min débit correspondant à une vitesse d'écoulement linéaire moyenne de ~ 1,5 mm / s pour une section de 75 pm x 300 pm. Changer les paramètres d'excitation / émission pour surveiller les lipides de mélange (LR ou DiD seulement).
- Degas tampon de reconstitution et de l' utiliser pour diluer la solution mère v-SUV par un facteur d'environ mars 10 à mai 10 en fonction de la concentration des stocks v-SUV. Objectif pour une dilution qui donne lieu à environ 10 à 100 événements de fusion dans les 60 secondes dans le champ de vision.
- Observant fusion entre v-VUS simples et la SBL
.jpg "/>
Figure 3. Le montage expérimental pTIRF. (A) Représentation schématique d'un contre-SUV et un t-SBL sur un substrat en verre. Faux-images couleur TIRFM de seul événement de fusion SUV-SBL montrant lipides accueil (1) et la libération de lipides colorant dans la SBL (2), suivie par le blanchiment et la diminution de l'intensité de fluorescence (3). L'intensité de fluorescence totale (somme des valeurs de pixels dans les directions x 5,3 um boîte de 5,3 um) de signal est représenté. (B) La lamelle liée au bloc PDMS est enregistrée sur la scène chauffée. Le tube d'entrée pour les canaux microfluidiques prélèvent des échantillons de tube porte-échantillon de métal (à droite) aspiré par la pompe à seringue (à gauche). Sous la pompe est l'unité d'émission double. S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
{kind=link}
oad / 54349 / 54349fig4.jpg "/>
La Figure 4. Représentation schématique du dispositif expérimental. L'onde évanescente est créée à l'interface verre-tampon dans le canal microfluidique. Le SBL est formé sur le verre et v-SUV sont aspirés à partir du porte-échantillon de métal (en haut à droite) à travers le canal dans la seringue (en haut à gauche). M, miroir; DM, miroir dichroïque; L lentille; F, filtre; P, polariseur. S'il vous plaît cliquer ici pour voir une version plus grande de cette figure.
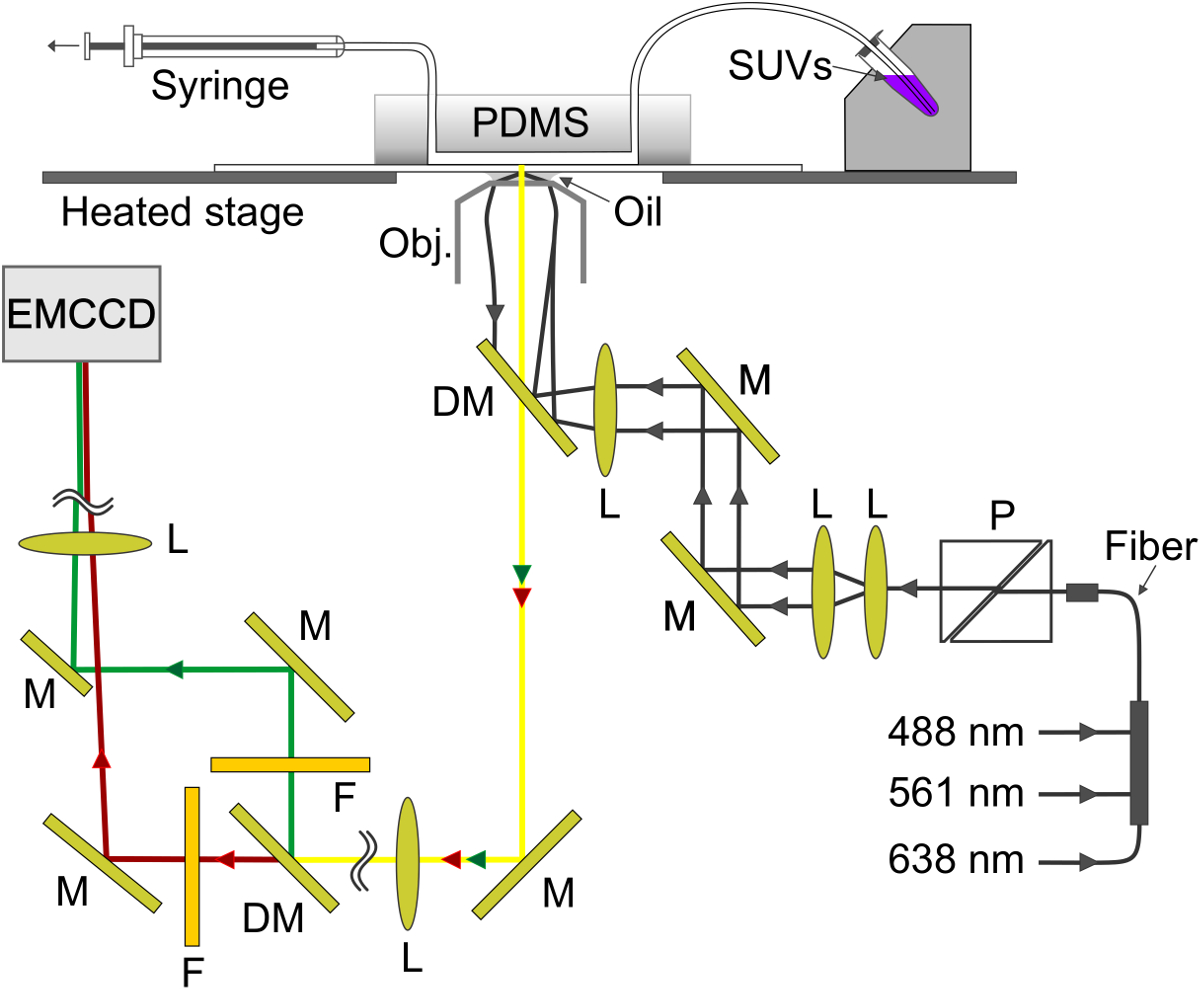{kind=link}
- exciter en continu et de surveiller la fluorescence v-SUV utilisant un 561 nm (pour LR-PE) ou 638 nm (pour DiD) laser en fonction du fluorophore incorporé dans v-SUV. Comme v-VUS atteignent le canal d'écoulement et d'un quai sur et fusible avec le SBL, le signal de fluorescence de fond commence à accumuler.
- Réglez l'intensité du laser d'excitation à travers le logiciel pour blanchir en permanence l'arrière-planfluorescence de telle sorte qu'au état de nouveaux événements d'accueil et de fusion stable peut être facilement observée.
Remarque: Si le blanchiment est trop lent, la fluorescence de fond sera trop élevée. Si le blanchiment est trop rapide, les signaux de VUS à quai et de fluorophores libérés dans l'SBL se fanent rapidement, raccourcissant la fenêtre au cours de laquelle la fusion des VUS amarrés peut être détecté ou un fluorophore peut être suivi. Pour LR-PE excité à 561 nm, 2,5-7,5 puissance mW illuminant un cercle de diamètre de 190 um (100-250 nW / um 2) est une valeur raisonnable pour commencer. Pour 638 nm excitation de DiD, 0,8-1,6 puissance mW sur un cercle de diamètre de 190 um (30-60 nW / um 2) peut être utilisé pour les tests initiaux. - Acquérir plusieurs films à différentes positions dans un canal donné. Vérifier NBD-PE fluorescence pour vérifier que le SBL n'a pas de défauts à ces positions. Acquérir des films full-frame au taux maximal (~ 50 images / sec) ou une région recadrée d'intérêt au taux de trame plus élevée (jusqu'à~ 100 Hz) pendant 60 secondes.
- Déplacer vers un autre canal microfluidique et répéter les enregistrements pour d'autres conditions. Inclure les contrôles négatifs tels qu'un SBL ou VUS exempt de protéines, ajoutez le domaine cytoplasmique soluble du v-SNARE VAMP2 (CDV) comme inhibiteur ou traiter v-SUV avec neurotoxine tétanique (30 min à 37 ° C) dans un ou plusieurs des canaux sur la même lamelle.
- Nettoyer et PDMS recyclage
- Afin de réutiliser le bloc PDMS, rincer les canaux microfluidiques avec ~ 200 ul d'éthanol à 70% à un débit de 5 ul / min. Enfin, aspirer de l'air à travers le tube et les canaux.
- Détacher les PDMS bloquent doucement de la lamelle en serrant légèrement. Placez-le sur un morceau propre de papier d'aluminium. Conserver dans un dessiccateur sous vide. Il peut être réutilisé plusieurs fois.
- Pour un nettoyage plus approfondi rincer les canaux avec une solution d'hydroxyde de détergent ou de sodium avant 70% d'éthanol. Vous pouvez également supprimer tous les tubes fro m PDMS et sonication du bloc pendant 30 minutes dans de l'isopropanol, avant le séchage et l'insertion d'une nouvelle tuyauterie.
5. SUV-SBL Fusion Assay pour surveiller Lipid et contenu de sortie simultanément
- Pour la double surveillance de la couleur de lipides simultanée et contenus solubles libérer, suivez les mêmes étapes que dans l'article 4, à l'exception des liposomes d'utilisation marquées à la fois avec un lipide (DID) et un contenu soluble (SRB) étiquette, comme expliqué dans la section 3. SRB est encapsulé à 10 mM et est initialement très auto-trempé.
- Utilisation de la configuration représentée schématiquement à la figure 4, exciter SRB et DiD fluorescence en utilisant simultanément 561 nm et 638 nm lasers, respectivement. Un miroir dichroïque (640 nm) divise l'émission en deux faisceaux qui traversent un court (595/50 nm) et un long filtre (700/75 nm) de longueur d'onde pour détecter la SRB et DiD émissions, respectivement. Les deux faisceaux d'émission sont projetées côte à côte sur la puce EM-CCD.
- L' analyse des données de FRAP
- Utilisez le programme MATLAB prévu dans le complément d' information pour estimer le coefficient de diffusion des lipides, D. Le programme lit une liste de fichiers OME-TIFF 49, détecte la zone blanchie, trace les valeurs moyennes de pixels dans la zone blanchie en fonction du temps, et correspond à la courbe de récupération résultant d'un modèle par Soumpasis 50 pour extraire le temps de récupération,
 Où w est le rayon du cercle blanchi.
Où w est le rayon du cercle blanchi.
Note: Un programme MATLAB a été fourni précédemment 25 pour l' analyse des films FRAP en utilisant des fichiers Nikon ND2. Le programme actuel lit OME-fichiers TIFF 49, parce que la plupart des formats de fichiers peuvent être facilement convertis en OME-TIFF. Une analyse quantitative des données FRAP est plus facile et plus précis lors du blanchiment est instantané, le bleacrégion hed est un cercle, et le blanchiment au cours de lecture est négligeable. Bien que ces critères ne sont pas strictement respectées dans les simples mesures de FRAP décrites ici, une estimation raisonnable du coefficient de diffusion peut être obtenu. Pour une estimation plus précise, utiliser le suivi des colorants lipidiques simples (section 6.3).
- Utilisez le programme MATLAB prévu dans le complément d' information pour estimer le coefficient de diffusion des lipides, D. Le programme lit une liste de fichiers OME-TIFF 49, détecte la zone blanchie, trace les valeurs moyennes de pixels dans la zone blanchie en fonction du temps, et correspond à la courbe de récupération résultant d'un modèle par Soumpasis 50 pour extraire le temps de récupération,
- Analyse du taux d'accueil, le taux de fusion et les temps docking-à-fusion retard
- Utilisez ImageJ pour ouvrir un film pour analyser et ajuster la luminosité et le contraste pour identifier clairement amarrage vésiculaire et les événements de fusion. Démarrez le plugin SpeckleTrackerJ 26. Voir Smith et al. 26 et la documentation en ligne pour SpeckleTrackerJ pour obtenir des instructions pour l' utilisation.
- Identifier tous les VUS nouvellement amarré à SpeckleTrackerJ. Pour distinguer les VUS qui accostent bien de ceux qui rebondissent sur la SBL, imposer une durée minimale d'accueil de quelques cadres. Enregistrer les pistes, et répéter pour tous les films.
Note: Pour la doctaux roi, tout ce qui importe est d'identifier quand un SUV amarré, de sorte que le premier cadre dans lequel les VUS à quai doit être enregistré dans ces pistes. Le reste des pistes ne sera pas prise en compte dans l'analyse. Cependant, l'auto-suivi de chaque SUV jusqu'à ce qu'il décolore ou fusibles aide VUS de marque qui ont déjà été suivis. - Identifier toutes les vésicules de fusion. Pour ces derniers, les pistes doivent inclure tous les cadres de la première image d'un SUV amarré jusqu'à ce que le premier cadre dans lequel la fusion est évidente par une augmentation soudaine de la fluorescence de la tache suivi. Les durées de ces pistes sont utilisées pour calculer les retards docking-à-fusion. Enregistrer toutes les pistes. Répétez l'opération pour tous les films.
- Pour l' analyse des lots de données d'accueil ou de fusion, de compiler une liste de fichiers de trajectoire des films pertinents et exécuter les programmes MATLAB fournis avec Karatekin et Rothman 25. Suivez les instructions qui sont fournies avec les programmes.
Remarque: Les programmes tracer le spermeaccueil et de fusion des événements ulative en fonction du temps, en fonction des informations extraites des fichiers de trajectoire. Accueil et de fusion des taux sont estimés à partir des pentes de ces parcelles. Pour les données de fusion, les retards à la station d' accueil de fusion sont également calculés et leur répartition tracés en une courbe de survie, à savoir la probabilité que la fusion n'a pas encore eu lieu par un certain retard après l' amarrage.
- Lipid diffusivité
- Pour chaque événement de fusion, de suivre les lipides fluorescents uniques qu'ils deviennent perceptibles quand ils ont diffusé suffisamment éloigné du site de fusion (figure 6). Utilisez SpeckleTrackerJ pour le suivi et enregistrer les pistes pour une analyse plus poussée en utilisant MATLAB. En fonction de la taille de la vésicule de fusion, typiquement 3-30 fluorophores uniques peuvent être suivis. Étant donné que de plus longues pistes sont souhaitables pour le calcul de
 , Essayez de suivre les molécules simples comme à longque possible, en utilisant la correction manuelle des trajectoires si nécessaire.
, Essayez de suivre les molécules simples comme à longque possible, en utilisant la correction manuelle des trajectoires si nécessaire. - Calculer la moyenne au carré déplacement (MSD) pour simples trajectoires de marqueurs lipidiques qui durent> 40-50 cadres (environ ~ 1,5 sec). Utiliser le MSD pour calculer le coefficient de diffusion des lipides, . Voir Smith et al. 26 et Stratton et al. 27 pour plus de détails.
- Pour chaque événement de fusion, de suivre les lipides fluorescents uniques qu'ils deviennent perceptibles quand ils ont diffusé suffisamment éloigné du site de fusion (figure 6). Utilisez SpeckleTrackerJ pour le suivi et enregistrer les pistes pour une analyse plus poussée en utilisant MATLAB. En fonction de la taille de la vésicule de fusion, typiquement 3-30 fluorophores uniques peuvent être suivis. Étant donné que de plus longues pistes sont souhaitables pour le calcul de
- Seule l' intensité de coloration des lipides, SUV SBL facteur de réduction de l' intensité et de la taille des vésicules
- Mesurer la somme des valeurs de pixel d'un marqueur lipidique dans un 3 x 3 pixels (0,8 pm x 0,8 pm) zone autour du marqueur de ~ 15 images avant et après le blanchiment des marqueurs en une seule étape. Soustraire l'intensité de fond en moyenne sur les cadres post-blanchiment de l'intensité pré-blanchiment en moyenne avant blanchiment pour obtenir l'intensité du marqueur lipidique suivi. Répétez la mesure pour autant de marqueurs que pratique pour une donnée film.
- Tracer la distribution des lipides unique intensités d'étiquettes,
 , Et monter une gaussienne pour estimer la moyenne.
, Et monter une gaussienne pour estimer la moyenne. - Calculer le délai entre le moment où la fusion a eu lieu et lorsque la trajectoire d'un marqueur lipidique single sorti dans ce cas, a pris fin dans un blanchiment en une seule étape. Obtenir le temps de blanchiment pour un fluorophore dans le SBL,
 , En traçant la fonction de survie des retards et le raccord à une exponentielle.
, En traçant la fonction de survie des retards et le raccord à une exponentielle.
Remarque: Parce que les couples polarisés de champ d'excitation plus faiblement aux fluorophores sur un SUV, le temps de blanchiment d'un SUV est généralement beaucoup plus lent 27. - Estimation
 Le facteur de réduction de l' intensité d'un colorant lipidique lorsque dans le SUV par rapport au moment où il est dans le SBL, suivant Stratton 27 (tp_upload / 54349 / 54349eq8.jpg "/> est l'intensité d'un seul colorant dans le SUV).
Le facteur de réduction de l' intensité d'un colorant lipidique lorsque dans le SUV par rapport au moment où il est dans le SBL, suivant Stratton 27 (tp_upload / 54349 / 54349eq8.jpg "/> est l'intensité d'un seul colorant dans le SUV).
Remarque: Si le blanchiment étaient négligeables, serait égale à l'intensité amarrée  ( le point (1) sur la figure 3A, panneau de droite), divisé par l'intensité totale atteint après tous les fluorophores sont déposés dans la SBL lors de la fusion (ligne pointillée marquée
( le point (1) sur la figure 3A, panneau de droite), divisé par l'intensité totale atteint après tous les fluorophores sont déposés dans la SBL lors de la fusion (ligne pointillée marquée  sur la figure 3). Cependant, en raison de blanchiment rapide de la SBL, est généralement pas atteint et une estimation précise de nécessite le montage des cinétiques de libération à une expression 27 qui incl udeS fois (Voir 6.4.3) et . Expressions distinctes sont fournies à Stratton et al 27 pour les cas de la cinétique des pores limitée et la diffusion libération limitée. le meilleur des cas en forme varie d'un événement à (voir 6.5.1 pour le cas de la cinétique des pores limitée).
sur la figure 3). Cependant, en raison de blanchiment rapide de la SBL, est généralement pas atteint et une estimation précise de nécessite le montage des cinétiques de libération à une expression 27 qui incl udeS fois (Voir 6.4.3) et . Expressions distinctes sont fournies à Stratton et al 27 pour les cas de la cinétique des pores limitée et la diffusion libération limitée. le meilleur des cas en forme varie d'un événement à (voir 6.5.1 pour le cas de la cinétique des pores limitée). - Calculer la surface des vésicules pour les épreuves individuelles de 27
 , où est l'intensité SUV amarré, est l'intensité d'un seul colorant lipidique signifie dans la SBL, est le facteur de réduction d'intensité pour un colorant lipidique quand il est dans le SUV par rapport au moment où il est dans la SBL, etation 1 "src =" / files / ftp_upload / 54349 / 54349eq12.jpg "/> est la densité surfacique connue des colorants de lipides.
, où est l'intensité SUV amarré, est l'intensité d'un seul colorant lipidique signifie dans la SBL, est le facteur de réduction d'intensité pour un colorant lipidique quand il est dans le SUV par rapport au moment où il est dans la SBL, etation 1 "src =" / files / ftp_upload / 54349 / 54349eq12.jpg "/> est la densité surfacique connue des colorants de lipides.
- Propriétés Fusion des pores
- En supposant que la libération est pore limitée, adapter l'étiquette lipidique cinétique de libération à 27
 , où
, où  est l'intensité SUV amarrée juste avant la fusion et les autres paramètres sont tels que définis précédemment. Utilisez la valeur de obtenu en 6.4.3 comme un paramètre fixe, et extraire les meilleures estimations d'ajustement pour et
est l'intensité SUV amarrée juste avant la fusion et les autres paramètres sont tels que définis précédemment. Utilisez la valeur de obtenu en 6.4.3 comme un paramètre fixe, et extraire les meilleures estimations d'ajustement pour et  .
. - Estimer la fraction du temps le pore est ouvert, P 0, en supposant un retard de lipide libération est due à des pores vacillant 27:60;
 =
=  Où une ESV est la surface de la vésicule (section 6.4.5), b est la hauteur des pores ( en général considérée comme étant ~ 15 nm), r p ≈ 3 nm est le rayon effectif des pores tel que vu par les marqueurs lipidiques diffusantes et comprend la demi l'épaisseur de la double couche (~ 2 nm), est la diffusivité lipidique (calculé en 6.3), et est le temps pour les lipides soient libérés du SUV dans la SBL (de 6.5.1).
Où une ESV est la surface de la vésicule (section 6.4.5), b est la hauteur des pores ( en général considérée comme étant ~ 15 nm), r p ≈ 3 nm est le rayon effectif des pores tel que vu par les marqueurs lipidiques diffusantes et comprend la demi l'épaisseur de la double couche (~ 2 nm), est la diffusivité lipidique (calculé en 6.3), et est le temps pour les lipides soient libérés du SUV dans la SBL (de 6.5.1). - Pour confirmer qu'une P nominale 0> 1 indique un pore complètement ouvert, P 0 = 1, correspond au cours du temps de l' intensité à l' équation 4 de Stratton et al. 27, la cinétique prédite pour un pore ouvert en permanence. Cet ajustement devrait être meilleure que fitting l'expression dans 6.5.1 pour un pore ouvert en permanence.
- En supposant que la libération est pore limitée, adapter l'étiquette lipidique cinétique de libération à 27
Résultats
qualité SBL
Il est crucial de vérifier la qualité et la fluidité du SBL avant l'expérience de fusion. La fluorescence sur le côté inférieur, le verre d'un canal microfluidique doit être uniforme, sans défauts apparents. Si une bulle d'air passe à travers le canal, il laisse généralement des cicatrices visibles sur le SBL. S'il y a de telles grandes cicatrices d'échelle / défa...
Discussion
la mise en œuvre réussie de l'essai de fusion SUV-SBL décrite ici dépend essentiellement de plusieurs étapes clés, telles que la reconstitution fonctionnelle des protéines dans des liposomes, l'obtention de bons LTTS de qualité, et en choisissant les paramètres d'imagerie droite pour détecter des molécules simples. Bien que cela puisse prendre un certain temps et d' efforts pour réussir, une fois que le test est mis en œuvre avec succès, il fournit une foule de renseignements sur le process...
Déclarations de divulgation
Les auteurs déclarent qu'ils ont aucun intérêt financier concurrents.
Remerciements
We thank Vladimir Polejaev (Yale West Campus Imaging Core) for the design and construction of the polarized TIRF microscope, David Baddeley (Yale University) for help with two-color detection instrumentation, and James E. Rothman (Yale University) and Ben O'Shaughnessy (Columbia University) and members of their groups for stimulating discussions. EK is supported by a Kavli Neuroscience Scholar Award from the Kavli Foundation and NIH grant 1R01GM108954.
matériels
| Name | Company | Catalog Number | Comments | |
| Reagents | ||||
| Milli-Q (MQ) water | Millipore | |||
| KOH | J.T. Baker | 3040-05 | ||
| Ethanol 190 Proof | Decon | |||
| Isopropanol | Fisher Chemical | A416P4 | ||
| HEPES | AmericanBio | AB00892 | ||
| Sodium Cholride (KCl) | AB01915 | |||
| Dithiothreitol | AB00490 | |||
| N-[2-hydroxyethyl] piperazine-N'-[2-ethanesulfonic acid] (HEPES) | AmericanBio | AB00892 | ||
| EGTA | Acros Organics | 409911000 | ||
| Buffers | ||||
| HEPES-KOH buffer (pH 7.4) | 25 mM HEPES-KOH, 140 mM KCl, 100 μM EGTA, 1 mM DTT | |||
| Solvents | ||||
| Chloroform | J.T. Baker | 9180-01 | in glass bottle, CAUTION, wear PPE | |
| Methanol | J.T. Baker | 9070-03 | in glass bottle, CAUTION, wear PPE | |
| Liposome preparation | ||||
| Gastight Hamilton syringe | Hamilton | var. sizes | only use glass sringe with solents (Chlorophorm/ Methanon, 2:1, v/v) | http://www.hamiltoncompany.com |
| Glass tubes Pyrex Vista 11 ml, 16x100 mm screw cap culture tube | Pyrex | 70825-16 | clean thoroughly, rinse with chloroform | http://catalog2.corning.com/LifeSciences/ |
| 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine, 16:0-18:1 PC (POPC) | Avanti Polar Lipids | 850457 | Lipids come dissolved in CHCl3 or as lyphilized powder in sealed vials. Aliquot upon opening. Store extra as dried lipid films under inert atmosphere at -20 °C. Keep stocks in CHCl3/MeOH (2:1, v/v) at -20 °C. let come to RT before opening | http://www.avantilipids.com/ |
| 1,2-dioleoyl-sn-glycero-3-phospho-L-serine (sodium salt), 18:1 PS (DOPS) | 840035 | |||
| 1-stearoyl-2-arachidonoyl-sn-glycero-3-phosphoethanolamine, 18:0-20:4 PE (SAPE) | 850804 | |||
| L-α-phosphatidylinositol-4,5-bisphosphate (Brain, Porcine) (ammonium salt), Brain PI(4,5)P2 | 840046 | |||
| 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2-1,3-benzoxadiazol-4-yl) (ammonium salt), 18:1 NBD PE | 810145 | |||
| 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (ammonium salt), 18:1 PEG2000 PE | 880130 | |||
| cholesterol (ovine wool, >98%) | 700000 | |||
| DiD' oil; DiIC18(5) oil (1,1'-Dioctadecyl-3,3,3',3'-Tetramethylindodicarbocyanine Perchlorate) | Molecular Probes | D-307 | https://www.thermofisher.com/ | |
| Rotavapor R-210 | Buchi | R-210 | heat bath above Tm of lipids used | http://www.buchi.com/ |
| OG n-Octyl-β-D-Glucopyranoside | Affymetrix | 0311 | store at -20°C, let come to RT before opening | https://www.anatrace.com/ |
| Shaker - Eppendorf Thermomixer R | Eppendorf | https://www.eppendorf.com/ | ||
| Slide-A-Lyze Dialysis Cassettes, 20K MWCO, 3 mL | life technologies | 66003 | https://www.lifetechnologies.com/ | |
| Bio-Beads SM-2 Adsorbents | Bio-Rad | 1523920 | http://www.bio-rad.com/ | |
| OptiPrep Density Gradient Medium | Sigma-Aldrich | D1556 | http://www.sigmaaldrich.com/ | |
| Ultracentrifugation tube, Thinwall, Ultra-Clear, 13.2 mL, 14 x 89 mm | Beckman Coulter | 41121703 | https://www.beckmancoulter.com/ | |
| Beckman SW41 Ti rotor | ||||
| SuflorhodamineB | Molecular Probes | S-1307 | https://www.thermofisher.com/ | |
| Econo-Column Chromatography Columns, 2.5 × 10 cm | Bio-Rad | 7372512 | http://www.bio-rad.com/ | |
| Sepharose CL-4B | GE Healthcare | 17-0150-01 | http://www.gelifesciences.com/ | |
| SYPRO Orange Protein Gel Stain | Molecular Probes | S-6650 | 5,000X Concentrate in DMSO | https://www.lifetechnologies.com/ |
| PDMS block | ||||
| Sylgard 184 Silicone elastomer kit, PDMS | Dow Corning | 3097358-1004 | http://www.dowcorning.com/ | |
| Pyrex glass petri dish, 150 x 20 mm, complete with cover | Corning | 3160-152 | http://catalog2.corning.com/LifeSciences/ | |
| Hole puncher - Reusable Biopsy Punch, 0.75mm | World Precision Instruments | 504529 | http://www.wpi-europe.com/ | |
| Manual Hole Punching Machine | SYNEO | MHPM-UNV | http://www.syneoco.com/ | |
| Drill .035 x .026 x 1.5 304 SS TiN coated round punch | CR0350265N20R4 | drill diameter: 0.9 mm | ||
| Tygon Microbore tubing, 0.25 mm ID, 0.76 mm OD | Cole-Parmer | 06419-00 | 0.010" ID, 0.030" OD | http://www.coleparmer.com/ |
| Silicone Tubing (0.51 mm ID, 2.1 mm OD | 95802-00 | 0.020" ID, 0.083" OD | ||
| Cover glass - cleanroom cleaned | ||||
| Schott Nexterion cover slip glass D | Schott | 1472305 | http://www.us.schott.com/ | |
| plasma cleaner | Harrick | PDC-32G | http://harrickplasma.com/ | |
| pTIRF setup and accessories | ||||
| IX81 microscope body | Olympus | IX81 | http://www.olympus-lifescience.com/en/ | |
| EM CCD camera | Andor | ixon-ultra-897 | http://www.andor.com/ | |
| Thermo Plate, heated microscope stage | Tokai Hit | MATS-U52RA26 | http://www.tokaihit.com/ | |
| 1 ml hamilton glass syringes (4x) | Hamilton | 81365 | http://www.hamiltoncompany.com | |
| syringe pump | kd Scientific | KDS-230 | http://www.kdscientific.com/ |
Références
- Sudhof, T. C., Rothman, J. E. Membrane fusion: grappling with SNARE and SM proteins. Science. 323, 474-477 (2009).
- Wickner, W., Schekman, R. Membrane fusion. Nat Struct Mol Biol. 15, 658-664 (2008).
- Harrison, S. C. Viral membrane fusion. Nat Struct Mol Biol. 15, 690-698 (2008).
- Jahn, R., Scheller, R. H. SNAREs--engines for membrane fusion. Nat Rev Mol Cell Biol. 7, 631-643 (2006).
- Lindau, M., Alvarez de Toledo, G. The fusion pore. Biochim Biophys Acta. 1641, 167-173 (2003).
- Staal, R. G., Mosharov, E. V., Sulzer, D. Dopamine neurons release transmitter via a flickering fusion pore. Nat Neurosci. 7, 341-346 (2004).
- Wu, Z., et al. Nanodisc-cell fusion: Control of fusion pore nucleation and lifetimes by SNARE protein transmembrane domains. Sci. Rep. 6, 27287 (2016).
- Alabi, A. A., Tsien, R. W. Perspectives on kiss-and-run: role in exocytosis, endocytosis, and neurotransmission. Ann Rev Physiol. 75, 393-422 (2013).
- Rossetto, O., Pirazzini, M., Montecucco, C. Botulinum neurotoxins: genetic, structural and mechanistic insights. Nature Rev Microbiol. 12, 535-549 (2014).
- Weber, T., et al. SNAREpins: minimal machinery for membrane fusion. Cell. 92, 759-772 (1998).
- Nickel, W., et al. Content mixing and membrane integrity during membrane fusion driven by pairing of isolated v-SNAREs and t-SNAREs. Proc Natl Acad Sci U S A. 96, 12571-12576 (1999).
- McNew, J. A., et al. Compartmental specificity of cellular membrane fusion encoded in SNARE proteins. Nature. 407, 153-159 (2000).
- Melia, T. J., You, D. Q., Tareste, D. C., Rothman, J. E. Lipidic antagonists to SNARE-mediated fusion. J Biol Chem. 281, 29597-29605 (2006).
- Hernandez, J. M., et al. Membrane fusion intermediates via directional and full assembly of the SNARE complex. Science. 336, 1581-1584 (2012).
- Fix, M., et al. Imaging single membrane fusion events mediated by SNARE proteins. Proc Natl Acad Sci U S A. 101, 7311-7316 (2004).
- Bowen, M. E., Weninger, K., Brunger, A. T., Chu, S. Single molecule observation of liposome-bilayer fusion thermally induced by soluble N-ethyl maleimide sensitive-factor attachment protein receptors (SNAREs). Biophys J. 87, 3569-3584 (2004).
- Liu, T., Tucker, W. C., Bhalla, A., Chapman, E. R., Weisshaar, J. C. SNARE-driven, 25-millisecond vesicle fusion in vitro. Biophys J. 89, 2458-2472 (2005).
- Yoon, T. Y., Okumus, B., Zhang, F., Shin, Y. K., Ha, T. Multiple intermediates in SNARE-induced membrane fusion. Proc Natl Acad Sci U S A. 103, 19731-19736 (2006).
- Diao, J., et al. A single-vesicle content mixing assay for SNARE-mediated membrane fusion. Nat Commun. 1, 1-6 (2010).
- Kyoung, M., et al. In vitro system capable of differentiating fast Ca2+-triggered content mixing from lipid exchange for mechanistic studies of neurotransmitter release. Proc Natl Acad Sci U S A. 108, E304-E313 (2011).
- Domanska, M. K., Kiessling, V., Stein, A., Fasshauer, D., Tamm, L. K. Single vesicle millisecond fusion kinetics reveals number of SNARE complexes optimal for fast SNARE-mediated membrane fusion. J Biol Chem. 284, 32158-32166 (2009).
- Kreutzberger, A. J., Kiessling, V., Tamm, L. K. High Cholesterol Obviates a Prolonged Hemifusion Intermediate in Fast SNARE-Mediated Membrane Fusion. Biophys J. 109, 319-329 (2015).
- Schwenen, L. L., et al. Resolving single membrane fusion events on planar pore-spanning membranes. Sci Rep. 5, 12006 (2015).
- Karatekin, E., et al. A fast, single-vesicle fusion assay mimics physiological SNARE requirements. Proc Natl Acad Sci U S A. 107, 3517-3521 (2010).
- Karatekin, E., Rothman, J. E. Fusion of single proteoliposomes with planar, cushioned bilayers in microfluidic flow cells. Nat Protoc. 7, 903-920 (2012).
- Smith, M. B., et al. Interactive, computer-assisted tracking of speckle trajectories in fluorescence microscopy: application to actin polymerization and membrane fusion. Biophys J. 101, 1794-1804 (2011).
- Stratton, B. S., et al. Cholesterol Increases the Openness of SNARE-mediated Flickering Fusion Pores. Biophysical journal. 110, (2016).
- Diao, J., et al. Synaptic proteins promote calcium-triggered fast transition from point contact to full fusion. Elife. 1, e00109 (2012).
- Kiessling, V., Domanska, M. K., Tamm, L. K. Single SNARE-mediated vesicle fusion observed in vitro by polarized TIRFM. Biophys J. 99, 4047-4055 (2010).
- Blasi, J., et al. Botulinum neurotoxin A selectively cleaves the synaptic protein SNAP-25. Nature. 365, 160-163 (1993).
- Washbourne, P., et al. Genetic ablation of the t-SNARE SNAP-25 distinguishes mechanisms of neuroexocytosis. Nat Neurosci. 5, 19-26 (2002).
- Diaz, A. J., Albertorio, F., Daniel, S., Cremer, P. S. Double cushions preserve transmembrane protein mobility in supported bilayer systems. Langmuir. 24, 6820-6826 (2008).
- Floyd, D. L., Ragains, J. R., Skehel, J. J., Harrison, S. C., van Oijen, A. M. Single-particle kinetics of influenza virus membrane fusion. Proc Natl Acad Sci U S A. 105, 15382-15387 (2008).
- Albertorio, F., et al. Fluid and air-stable lipopolymer membranes for biosensor applications. Langmuir. 21, 7476-7482 (2005).
- Daniel, S., Albertorio, F., Cremer, P. S. Making lipid membranes rough, tough, and ready to hit the road. Mrs Bulletin. 31, 536-540 (2006).
- Gao, Y., et al. Single reconstituted neuronal SNARE complexes zipper in three distinct stages. Science. 337, 1340-1343 (2012).
- Kenworthy, A. K., Hristova, K., Needham, D., Mcintosh, T. J. Range and Magnitude of the Steric Pressure between Bilayers Containing Phospholipids with Covalently Attached Poly(Ethylene Glycol). Biophys J. 68, 1921-1936 (1995).
- Knoll, W., et al. Solid supported lipid membranes: New concepts for the biomimetic functionalization of solid surfaces. Biointerphases. 3, Fa125-Fa135 (2008).
- Quinn, P., Griffiths, G., Warren, G. Density of newly synthesized plasma membrane proteins in intracellular membranes II. Biochemical studies. J Cell Biol. 98, 2142-2147 (1984).
- Sund, S. E., Swanson, J. A., Axelrod, D. Cell membrane orientation visualized by polarized total internal reflection fluorescence. Biophys J. 77, 2266-2283 (1999).
- Johnson, D. S., Toledo-Crow, R., Mattheyses, A. L., Simon, S. M. Polarization-controlled TIRFM with focal drift and spatial field intensity correction. Biophys J. 106, 1008-1019 (2014).
- Anantharam, A., Onoa, B., Edwards, R. H., Holz, R. W., Axelrod, D. Localized topological changes of the plasma membrane upon exocytosis visualized by polarized TIRFM. J Cell Biol. 188, 415-428 (2010).
- Axelrod, D. Carbocyanine dye orientation in red cell membrane studied by microscopic fluorescence polarization. Biophys J. 26, 557-573 (1979).
- Wang, T., Smith, E. A., Chapman, E. R., Weisshaar, J. C. Lipid mixing and content release in single-vesicle, SNARE-driven fusion assay with 1-5 msec resolution. Biophys J. 96, 4122-4131 (2009).
- Chernomordik, L. V., Frolov, V. A., Leikina, E., Bronk, P., Zimmerberg, J. The pathway of membrane fusion catalyzed by influenza hemagglutinin: restriction of lipids, hemifusion, and lipidic fusion pore formation. J Cell Biol. 140, 1369-1382 (1998).
- Takamori, S., et al. Molecular anatomy of a trafficking organelle. Cell. 127, 831-846 (2006).
- Wilhelm, B. G., et al. Composition of isolated synaptic boutons reveals the amounts of vesicle trafficking proteins. Science. 344, 1023-1028 (2014).
- Scott, B. L., et al. Liposome fusion assay to monitor intracellular membrane fusion machines. Methods Enzymol. 372, 274-300 (2003).
- Linkert, M., et al. Metadata matters: access to image data in the real world. J Cell Biol. 189, 777-782 (2010).
- Soumpasis, D. M. Theoretical analysis of fluorescence photobleaching recovery experiments. Biophys J. 41, 95-97 (1983).
- Ohki, S. A mechanism of divalent ion-induced phosphatidylserine membrane fusion. Biochim Biophys Acta. 689, 1-11 (1982).
- Berquand, A., et al. Two-step formation of streptavidin-supported lipid bilayers by PEG-triggered vesicle fusion. Fluorescence and atomic force microscopy characterization. Langmuir. 19, 1700-1707 (2003).
- Tamm, L. K., McConnell, H. M. Supported phospholipid bilayers. Biophys J. 47, 105-113 (1985).
- Rawle, R. J., van Lengerich, B., Chung, M., Bendix, P. M., Boxer, S. G. Vesicle fusion observed by content transfer across a tethered lipid bilayer. Biophys J. 101, L37-L39 (2011).
- Wagner, M. L., Tamm, L. K. Reconstituted syntaxin1a/SNAP25 interacts with negatively charged lipids as measured by lateral diffusion in planar supported bilayers. Biophys J. 81, 266-275 (2001).
- Kalb, E., Frey, S., Tamm, L. K. Formation of Supported Planar Bilayers by Fusion of Vesicles to Supported Phospholipid Monolayers. Biochimica Et Biophysica Acta. 1103, 307-316 (1992).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.