É necessária uma assinatura da JoVE para visualizar este conteúdo. Faça login ou comece sua avaliação gratuita.
Method Article
Fusão SNARE mediada por uma única proteolipossomos com Bicamadas suportados Tethered em uma célula Microfluidic fluxo monitorado por polarizada TIRF microscopia
Neste Artigo
Resumo
Aqui, apresentamos um protocolo para detectar eventos individuais, mediada por SNARE fusão entre lipossomas e bicamadas suportados em canais microfluídicos usando TIRFM polarizada, com sensibilidade única molécula e ~ 15 resolução de tempo ms. Lipídico e liberação de cargas solúveis podem ser detectados simultaneamente. o tamanho do lipossoma, de difusividade lípido, e propriedades de fusão dos poros são medidos.
Resumo
No processo de fusão membranar omnipresente a abertura de um poro de fusão estabelece a primeira ligação entre dois compartimentos anteriormente separadas. Durante neurotransmissor ou a liberação de hormônio através de exocitose, o poro de fusão podem transitoriamente abrir e fechar repetidamente, regulando cinética de liberação de carga. dinâmica dos poros também determinam o modo de reciclagem de vesícula; irreversíveis revedação resulta na fusão transitória, "beije-and-run", enquanto a dilatação leva a fusão completa. Para entender melhor quais os fatores que regem a dinâmica dos poros, desenvolvemos um ensaio para monitorar a fusão da membrana utilizando microscopia polarizada total de fluorescência de reflexão interna (TIRF), com sensibilidade única molécula e ~ 15 resolução de tempo ms em um bioquimicamente bem definida sistema in vitro. Fusão de fluorescente etiquetado pequenas vesículas unilamelares contendo proteínas v-SNARE (V-SUVs) com um planar bicamada rolamento t-SNAREs, apoiada sobre uma almofada de polímero macio (t-SBL, bicamada suportada-t), É monitorizada. O ensaio utiliza canais de fluxo de microfluidos que asseguram o consumo da amostra mínima, enquanto o fornecimento de uma densidade constante de SUVs. Explorando o reforço de sinal rápida mediante a transferência de etiquetas de lipídios do SUV à SBL durante a fusão, cinética de transferência de corante de lípidos é monitorada. A sensibilidade da microscopia TIRF permite rastrear etiquetas lipídicas fluorescentes individuais, a partir do qual diffusivity lipídico e tamanho SUV podem ser deduzidas para cada evento de fusão. tempos de liberação corante de lípidos pode ser muito maior do que o esperado para a passagem desimpedida através de poros abertos em permanência. Usando um modelo que assume retardamento da liberação de lipídios é devido a pore cintilação, um poro "abertura", a fração de tempo do poro permanece aberta durante a fusão, pode ser estimado. Um marcador solúvel pode ser encapsulado nas SUVs de monitorização simultânea de lípido e de libertação de carga solúvel. Tais medições indicam alguns poros pode selar novamente depois de perder uma fracção da carga solúvel.
Introdução
A fusão da membrana é um processo biológico universal necessário para o tráfico intracelular de lípidos e proteínas, secreção, fertilização, desenvolvimento, e envolveu a entrada do vírus em organismos hospedeiros 1-3. Para a maioria das reacções de fusão intracelular incluindo libertação de hormonas e de neurotransmissores via exocitose, a energia para fundir duas bicamadas lipídicas é proporcionado pela formação de um feixe de quatro hélices entre proteínas de receptor cognato solúvel N-etilmaleimida-sensível proteína factor de fixação (SNARE), ancorada na a vesícula (v-SNARE) e da membrana alvo (T-SNARE) 4, respectivamente. Synaptic exocitose da vesícula é a reação de fusão mais fortemente regulada e ocorre dentro de um milésimo de segundo depois da chegada de um 1,4,5 potencial de ação. O poro de fusão, a ligação inicial entre os dois compartimentos de fusão, pode piscar aberta e fechada várias vezes antes de selar ou expandir irreversivelmente 5-7. Os resultados anterioresem transitória, "Kiss & run" fusion, enquanto o segundo leva a fusão completa. Fatores que regem o equilíbrio entre estes dois modos de fusão e mecanismos que regulam cintilação poros não são bem compreendidos 5,8.
SNARE proteínas são necessárias para a exocitose; fusão das vesículas sinápticas é abolida por quebra de SNAREs por neurotoxinas 9. Experiências de fusão em massa usando pequenas vesículas unilamelares (SUVs) mostrou que SNAREs não só necessário, mas também suficiente para conduzir a fusão da membrana 10. Neste ensaio a granel, SUVs reconstituído com v-SNARE (V-SUV) foram dopados com fosfolípidos fluorescentes (N - (7-nitro-2-1,3-benzoxadiazol-4-il) -phosphoethanolamine (NBD-PE) e ( N -. (lisamina rodamina B sulfonil) -phosphoethanolamine (LR-PE) e misturado com vesículas não marcados contendo t-SNAREs (t-SUV) Inicialmente, a fluorescência do NBD-PE no v-SUVs é extinta por Förster transferência de energia de ressonância (FRET ) para LR-PE. Como laboratórioELED v-SUVs fundir-se com t-SUVs não marcados, a densidade da superfície fluoróforo na membrana agora combinada é reduzido e o aumento resultante na fluorescência de NBD-PE relata a extensão de lípido de mistura 10. Como o ensaio é fácil de grandes quantidades definir-se e analisar, que tem sido amplamente utilizada para estudar os mecanismos de fusão mediada por SNARE 10-14. No entanto, tem várias limitações, como a baixa sensibilidade e baixa resolução tempo. O mais importante, como um conjunto de medição, ela é em média resultados de todos os eventos que fazem a discriminação entre o encaixe e de fusão, bem como a detecção de intermediários hemifusão difíceis.
Durante a última década vários grupos, incluindo o nosso, desenvolveram novos ensaios para monitorar eventos de fusão no único nível vesícula 15-27. Ha e seus colegas usaram v-SUVs amarrados em uma superfície e monitorados sua fusão com livre t-SUVs 18,19. mistura lipídica foi monitorizada utilizando FRET entre um par de fluoróforos ligados a lípidos inalojado no V e t-SUVs, respectivamente, através de microscopia de fluorescência de reflexão total interna (TIRF) 18. Mais tarde, o laboratório de Brunger utilizada uma única espécie de rótulo de lípidos juntamente com um marcador de conteúdos para a detecção simultânea de lípido de mistura e os conteúdos 20,28. Tanto o lipídico e os marcadores de conteúdo foram incluídos em concentrações elevadas, auto-extingue; fusão com SUVs não marcados resultou em fluorescência dequenching 20,28.
Outros têm fundido v-SUVs para bicamadas planar reconstituídos com t-SNAREs 15-17,21-27,29. A geometria planar do alvo (T-SNARE contendo) bicamada imita melhor o processo de fusão fisiológica de pequenas vesículas, altamente curvadas com uma membrana de plasma plana. O grupo Steinem empregada membranas de abrangência dos poros reconstituídos com t-SNAREs, suspensas sobre um substrato de nitreto de silício poroso e detectado fusão com v-SUVs individuais usando microscopia confocal de varrimento laser 23. outros fusados v-SUVs para bicamadas planar reconstituídos com t-SNAREs, apoiada sobre um substrato de vidro 15-17,21,22,24-27,29. A grande vantagem do uso de camadas duplas suportados (SBLs) é que a microscopia TIRF pode ser usado para detectar atracação e de fusão eventos com excelente relação sinal-ruído e sem interferência de V-SUVs livres, embora usando microfluídica também fornece resolução single-evento usando microscopia de epifluorescência-campo distante padrão de 24.
Uma preocupação importante é se, e como interacções substrato-bicamada suportada afectar a qualidade da bicamada e o processo de fusão. Os primeiros trabalhos feito uso de SBLs planares que foram suportados directamente sobre um substrato de vidro ou de quartzo 15-17. Estes SBLs foram feitas por adsorção, a rebentar, espalhando e fusão de membranas t-SUV sobre o substrato. Foi logo percebeu, porém, que omitir um componente-chave t-SNARE, SNAP25, de SBLs preparados desta maneira resultou em v-SUV de atracação e de fusão cinética indistinguishable daqueles obtidos utilizando a completa t-SNAREs 17. Porque SNAP25 é absolutamente necessária para a fusão in vivo 30,31, a relevância fisiológica desses primeiras tentativas foi posta em causa. O grupo de Tamm superou este desafio usando a formação de bicamada melhor controlada suportado 21. Usou-se a deposição de Langmuir-Blodgett, pela primeira folheto livre de proteínas de SBL, seguido por fusão de que monocamada com t-SUVs 21. Isto resultou na fusão dependente de SNAP25.
Para evitar artefactos potenciais associados a uma bicamada suportada directamente sobre um substrato de vidro, sem necessidade de utilizar métodos de Langmuir-Blodgett, Karatekin et ai. Introduziu um poli hidratado (etileno glicol) de apoio macio, (PEG) entre a bicamada e o substrato 24. Esta modificação também resultou na fusão dependente da SNAP25 24. Bicamadas almofadadas em uma camada de polímero macio tinha sido conhecido para melhor preservar transmembranamobilidade e função das proteínas de 32, e tinha sido utilizado em estudos de fusão com vírus 33. Além disso, bicamadas PEGilados parecem manter alguma capacidade de se auto-curar e são muito robustos 34,35. Em primeiro lugar, uma fracção de cadeias de PEG disponíveis comercialmente, ligados em lípidos incluídos na membrana t-SUV. Quando estes t-SUVs rebentar e formar uma bicamada planar num substrato de vidro, uma escova de PEG abrange ambos os folhetos da bicamada planar. Como a formação de bicamada planar é impulsionado por intermédio de adesão das cadeias de PEG circundantes t-SUVs sobre a superfície de vidro hidrófilo, rebentamento de lipossomas e formação em bicamada planar são relativamente insensíveis à composição de lípido utilizado. No entanto, quando grandes quantidades de colesterol estão incluídos, aumentando as propriedades coesivas dos SUVs, as SUVs não pode rebentar espontaneamente. Se este for o caso, os iões divalentes ou choque osmótico pode ser empregado para ajudar a formação planar de duas camadas 25.
Como mencionado acima, nesta approach uma escova de PEG cobre ambos os lados do cartão planar, suportado bicamada. A escova de frente para o canal de fluxo de microfluidos ajuda a evitar a aderência não específica da v-SUVs de entrada que também são geralmente cobertos com uma camada de PEG. A formação de complexos e t-V- SNARE começa a partir do terminal N da membrana distal e procede em etapas para com os domínios membrana proximal 36. Para os v-SUVs para interagir com o t-SBL, a necessidade e v- t-SNARE terminais N a projectar-se acima das escovas de PEG, o que parece ser o caso nas condições do ensaio. Altura da escova pode ser adaptada para estudar outras proteínas que SNARE, variando a densidade de lipidos PEGilados e o comprimento da cadeia de PEG 37,38. Um outro benefício das escovas de PEG que cobre as superfícies proximais das bicamadas de fusão é que eles imitam o ambiente de aglomerado de membranas biológicas, que são embalados com 30.000-40.000 proteínas de membrana integrais por quadrado 39 micron. Assim como as cadeias de PEG Neste ensaio, o repucamada de proteína lsive abrangendo membranas biológicas tem de ser empurrado de lado para permitir o contacto entre as duas bicamadas de fosfolípidos para a fusão ocorresse.
canais de microfluidos de fluxo são utilizados neste ensaio, uma vez que oferecem vantagens únicas. Primeiro, o fluxo microfluídico permite a deposição uniforme mais de t-SUVs para espalhar e se fundem para formar o t-SBL. Em segundo lugar, o volume do canal de pequeno (<1 ul) minimiza o consumo da amostra. Em terceiro lugar, as pequenas quantidades necessárias de permitir que todo o experimento ser conduzida sob fluxo constante. Fluxo remove fracamente, presumivelmente não-especificamente, aderiram v-SUVs da SBL 16. Ele também mantém uma densidade constante de v-SUVs acima do t-SBL, simplificando a análise cinética 17. Finalmente, vesículas ancoradas são facilmente distinguidos dos mais livre transportados pelo fluxo 25. Em quarto lugar, vários canais de microfluidos pode ser utilizado na mesma lamela, cada sondagem de uma condição diferente. Isto permite a comparação das condições de during mesma corrida experimental. Uma abordagem semelhante foi usada pelo grupo van Oijen para estudar fusão entre vírus influenza e SBLs almofadadas 33.
Em microscopia TIRF, o decaimento exponencial do campo evanescente (com um decaimento constante de ~ 100 nm) limites de excitação de fluorescência para aquelas moléculas que são muito próximo da interface vidro-tampão. Isso minimiza a contribuição de moléculas fluorescentes que estão mais longe, aumenta a relação sinal-ruído, e permite sensibilidade única molécula com tempos de exposição de quadro de 10-40 ms. O campo evanescente também leva a um aumento de sinal em cima de fusão: como a transferência de lípidos marcados a partir do SUV para o SBL, encontram-se, em média, em um campo de excitação mais forte. Este aumento na fluorescência é mais forte para os lipossomas maiores.
Se a luz polarizada é usada para gerar o campo evanescente, efeitos adicionais contribuir para alterações na fluorescência após transferência de etiquetas do SUV na SBL. Alguns corantes de lípidos tem um dipolo de transição orientada com um ângulo médio preferido em relação à camada dupla em que eles são incorporados. Isto cria uma diferença de a quantidade de fluorescência emitida pelos fluoroforos quando estão no SUV versus a SBL, uma vez que o feixe polarizado vai excitar corantes nas duas membranas de forma diferente. Para o primeiro, o feixe de excitação vai interagir com dipolos orientados de transição em torno do SUV esférica, ao passo que para o segundo, orientações dipolo será confinado pela geometria plana SBL. Por exemplo, quando é usado s-luz polarizada incidente (polarizado normal ao plano de incidência), a excitação é mais eficiente quando o corante é no SBL do que no SUV por um corante de lípidos transição dipolo orientado paralelamente à membrana 29,40 (como a de DII ou fez 41-43). Um SUV dopado com tal fluoróforo aparece dim quando se dirige para o SBL (Figura 7, Representat ive Resultados). Como um poro de fusão abre e liga as membranas SUV e SBL, sondas fluorescentes difundem-se para a SBL e tornar-se mais susceptível de ser excitado pelo campo evanescente s-polarizados 25,27,29. Por conseguinte, o sinal de fluorescência integrada em torno do local de fusão aumenta acentuadamente durante a transferência de corantes a partir de SUV para a SBL 27 (Figura 3 e Figura 7). Um factor adicional que contribui para sinalizar mudanças que acompanham fusão é dequenching de etiquetas fluorescentes, como eles são diluídos quando transferido para a SBL. A contribuição de dequenching é geralmente menor em comparação com efeitos evanescentes decaem campo e polarização no ensaio descrito aqui, porque apenas uma pequena fração (  ) Dos lípidos são rotulados.
) Dos lípidos são rotulados.
O aumento do sinal mediante a fusão pode ser explorada para deduzir propriedades de poros de fusão através da comparação do tempo,1 "src =" / files / ftp_upload / 54349 / 54349eq2.jpg "/>, necessário para um lipídio de escapar através de um poro que é livremente permeável à lipídios com o tempo de lançamento real,  . Se as duas escalas de tempo são comparáveis, ao que se concluir que a poro apresenta pouca resistência ao fluxo de lípido. No entanto, se o tempo efectivo de libertação é significativamente mais longo do que o tempo para a libertação limitada por difusão, isso indicaria um processo, tal como tremulação de poro, retardador de libertação de lípidos. O tempo de liberação limitada por difusão,
. Se as duas escalas de tempo são comparáveis, ao que se concluir que a poro apresenta pouca resistência ao fluxo de lípido. No entanto, se o tempo efectivo de libertação é significativamente mais longo do que o tempo para a libertação limitada por difusão, isso indicaria um processo, tal como tremulação de poro, retardador de libertação de lípidos. O tempo de liberação limitada por difusão,  , Depende do tamanho do lipossoma de fusão e de lípidos-difusividade; sua estimativa exige estes dois parâmetros para ser quantificado. A sensibilidade única molécula do ensaio permite diffusivity lipídica a ser medido através do rastreamento vários fluoróforos única de lipídios após a sua libertação no SBL para cada evento de fusão 26. O tamanho de cada vesícula fusãopode ser cerca de 27 por combinação de (I) a intensidade de um único corante lípido, (ii) a alteração na fluorescência total em torno de um local de ancoragem depois de todos os fluoróforos são transferidos para a SBL mediante fusão, (iii) a densidade de rotulagem conhecidos de SUV lípidos, e (iv) a superfície por lípido. Para muitos eventos de fusão, os tempos de libertação de lípidos reais foram encontrados para ser muito mais lento que o esperado pela libertação de difusão controlada 27, como foi observado anteriormente assumindo SUV tamanho uniforme 44. Assumindo retardamento da liberação de lipídios é devido a pore cintilação, um modelo quantitativo permite a estimativa de "abertura dos poros", a fração de tempo do poro permanece aberta durante a fusão 27.
, Depende do tamanho do lipossoma de fusão e de lípidos-difusividade; sua estimativa exige estes dois parâmetros para ser quantificado. A sensibilidade única molécula do ensaio permite diffusivity lipídica a ser medido através do rastreamento vários fluoróforos única de lipídios após a sua libertação no SBL para cada evento de fusão 26. O tamanho de cada vesícula fusãopode ser cerca de 27 por combinação de (I) a intensidade de um único corante lípido, (ii) a alteração na fluorescência total em torno de um local de ancoragem depois de todos os fluoróforos são transferidos para a SBL mediante fusão, (iii) a densidade de rotulagem conhecidos de SUV lípidos, e (iv) a superfície por lípido. Para muitos eventos de fusão, os tempos de libertação de lípidos reais foram encontrados para ser muito mais lento que o esperado pela libertação de difusão controlada 27, como foi observado anteriormente assumindo SUV tamanho uniforme 44. Assumindo retardamento da liberação de lipídios é devido a pore cintilação, um modelo quantitativo permite a estimativa de "abertura dos poros", a fração de tempo do poro permanece aberta durante a fusão 27.
Sempre que possível, é importante para testar mecanismos de fusão usando tanto conteúdo etiquetas solúveis lipídico e. Por exemplo, a liberação de lipídios pode ser retardado por outros que cintilação poros processos, como a restrição da difusão de lípidos pelas proteínas SNARE que cercam tele poros. Se este fosse o caso, em seguida, solte do conteúdo precederia de suporte de etiquetas de lipídios, desde o poro é grande o suficiente para permitir a passagem de sondas solúveis. Uma falha mais fundamental na abordagem poderia ser sob a suposição de que a transferência de lípidos marcados para a SBL ocorre através de um poro de fusão estreita que liga a SBL a uma vesícula que manteve a sua forma, em grande parte de pré-fusão. Transferência de lípides para a SBL também pode resultar de uma rápida dilatação dos poros de fusão com uma concomitante, extremamente rápido colapso do SUV na membrana SBL, como previamente sugerido com base em dados de libertação de lípidos sozinho 29. Monitoramento tanto lipídica e conteúdo liberar simultaneamente, verificou-se que muitos poros fechados de novo depois de libertar todos os seus rótulos de lipídios, mas manteve alguma da sua carga solúvel 27. Isto indica que, pelo menos alguns lipossomas não colapsam para a SBL após a fusão, e que a transferência de corante de lípidos no SBL ocorre através de um poro de fusão. Além disso, lIPID e conteúdo de libertação ocorreu simultaneamente 27, tornando improvável que retardamento da libertação de lípidos era devido ao impedimento de difusão de lípidos pelas proteínas SNARE circundantes do poro 45.
Um protocolo de fusão SUV-SBL que não monitorar liberação conteúdos solúveis foi publicado anteriormente pelo Karatekin e Rothman 25. Aqui, os desenvolvimentos mais recentes são incluídos, monitoramento ou seja simultânea de lipídios e conteúdo liberar e estimativa de propriedades SUV, lipídios, e de poros de fusão 27. O protocolo começa com instruções para preparar as células de microfluidos, realizadas por ligação de um poli (dimetilsiloxano) (PDMS) elastómero de blocos contendo ranhuras com uma lamela de vidro de 25. Em seguida, preparação de v-SUVs com tanto lipídica e conteúdo marcadores é explicado. Secções 4 e 5 fornecem instruções para a montagem das células microfluídicos, formando os SBLs in situ e verificar se há defeitos e fluidez, introdução de vSUVS para as células de fluxo e detecção de eventos de fusão. Secção 6 fornece instruções para a análise de dados.
Protocolo
1. Preparação de um bloco de PDMS para formar o canal microfluídicos

Figura 1. Microfabricação de modelo de célula de fluxo e preparação de blocos PDMS. (A) Projeto de uma célula de fluxo de quatro canais que se encaixa em uma lamela de vidro 24 x 60 mm (inferior). Seis modelos idênticos estão dispostas para se encaixar sobre uma bolacha de silício de 10 cm (topo). (B) Cortar bloco de PDMS espessura de aproximadamente 5-8 mm em um furador. (C) A inserção de tubos para o buraco perfurado usando um par de pinças. Por favor clique aqui para ver uma versão maior desta figura.
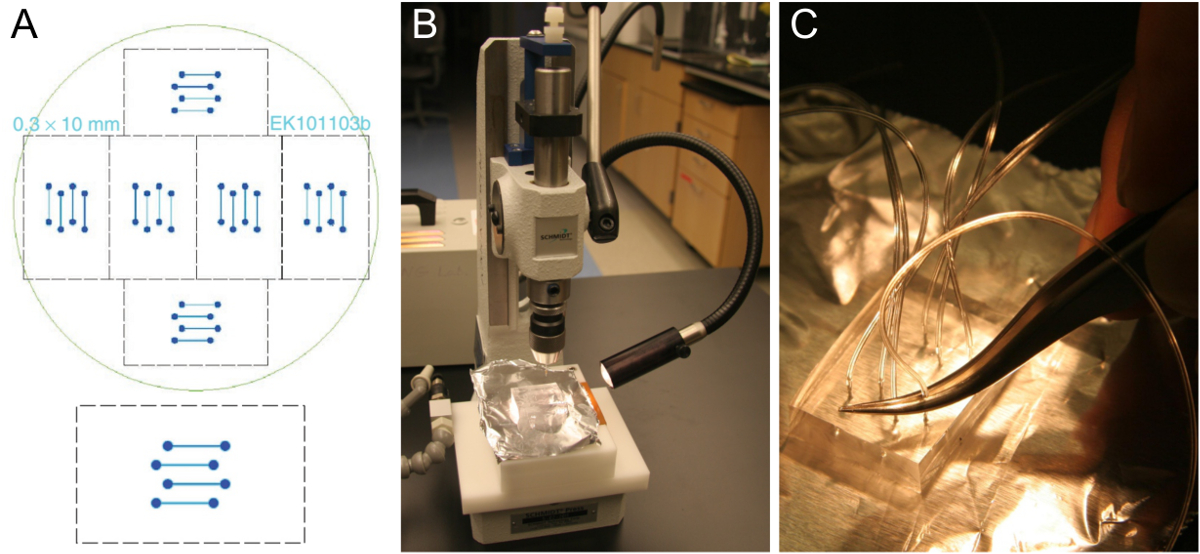{kind=link}
- Obter um modelo de célula de fluxo, tais como o mostrado na Figura 1A. canais típicos são largas 0,3-1 mm, 70-100 mm de altura e 1-2 cm de comprimento. Fabricar um modelo de nósing técnicas de fotolitografia padrão em uma instalação de sala limpa 25 ou ordem se o acesso de sala limpa não está disponível. Alternativamente, a máquina de um modelo de maiores dimensões a partir de um material adequado.
Nota: o pessoal da sala de limpeza pode treinar e orientar os usuários inexperientes (acesso a uma instalação de sala limpa é sempre restrito a usuários com formação adequada) na concepção e ordenação de uma máscara, limpeza de wafer e fotolitografia. Uma vez que um molde é obtido, ele pode ser usado fora da sala limpa repetidamente, desde que seja mantida em uma cápsula fechada e se o cuidado de excluir poeira. - Prepare uma mistura de ~ 100 ml PDMS (silicone base de elastômero) e ~ 10 ml de agente de ligação cruzada (agente de cura) em um copo de plástico descartável. Parta a ponta de uma pipeta descartável para lidar com o PDMS viscosos mais facilmente. Pese os PDMS no copo de plástico como pipetagem não é preciso.
- Mexa bem a mistura. Retirar as bolhas de ar por muitos desgaseificação num excicador sob vácuo (cerca de 20min). À medida que a taça é colocada sob vácuo, as bolhas serão inicialmente crescer em tamanho, aumentando o volume da mistura grandemente. Controlar o vácuo que é aplicado e certifique-se o cálice é profundo o suficiente para evitar um derramamento.
- Pour uma grande queda da mistura desgaseificada PDMS em uma placa de Petri de vidro (150 mm x 20 mm) e pressione a bolacha para o PDMS com o molde virado para cima. Isto evita bolhas sendo preso sob a hóstia. de bolhas de ar expande-se e pode inclinar a bolacha quando o prato é colocado no forno. Agora despeje PDMS desgaseificados para o molde no topo da bolacha até que seja coberta por cerca de 5-8 mm de EPM. Se uma bolha de ar ocorre suavemente removê-lo com uma ponta de pipeta.
- Coza os PMDS num forno a 60 ° C durante 3 horas. Certifique-se de que o prato está nivelado.
- Use uma nova lâmina de bisturi para cortar um bloco de PDMS contendo as estruturas de canais moldados. O bloco de cortar precisa caber em uma lamela (veja o passo 4.1.9).
- Descasque o bloco cortado do prato e place-lo em um pedaço de papel alumínio limpo.
Nota: Os blocos PMDS reticulados podem ser mantidos por alguns meses. Em uma única bolacha, existem 6 conjuntos de canais de fluxo (Figura 1a), de modo 6 PDMS blocos podem ser produzidos numa única moldagem PDMS. Depois de cortar todos os 6 dos blocos de PDMS, peças soltas limpas de PDMS, mas caso contrário, não retire as PDMS restantes antes de derramar e cross-linking o próximo lote, o que exigirá apenas cerca de metade dos PDMS usados para o primeiro lote. Um bom modelo pode durar alguns anos. - Use um furador para perfurar através do bloco de PDMS em um movimento em linha reta (Figura 1B). Iniciar no lado das ranhuras de canal. Certifique-se de remover o pedaço perfurado fora do PDMS. Repita esse procedimento para todos os oito furos do design de quatro canais.
- Coloque o lado do canal do bloco do PDMS para baixo em uma peça nova e livre de rugas de papel alumínio. Armazenar o bloco de até alguns meses numa caixa seca, de preferência em um exsicador.
- Empurrar o tubo (0,25 mm de diâmetro, 0,76 mm de diâmetro externo) cerca de um terço para o buraco perfurado usando um par de pinças (Figura 1C). Corte o tubo inclinado para facilitar a inserção. Deixar tubos de tempo suficiente para atingir o reservatório de SUV e a bomba de seringa, respectivamente. Depois de colocar o chip montado no microscópio, cortar os tubos novamente se eles são muito longos.
- Para conectar-se as seringas da bomba, cortar um pequeno pedaço de tubo de silicone maior (0,51 mm de diâmetro, 2,1 mm de diâmetro externo) e inserir o tubo mais fino para um lado. Este bloco de pronto-para-utilização dos PDMS pode ser mantido durante alguns meses.
2. Limpeza Coverslip
- Lamelas limpo de acordo com o protocolo descrito em Karatekin Rothman e 25 utilizando uma mistura oxidante forte de ácido sulfúrico (H 2 SO 4) e peróxido de hidrogénio (H 2 O 2). Executar este procedimento em uma sala limpa, e observar as precauções de segurança adequadas. Como alternativa, use uma sala limpa limpa coverslip que está comercialmente disponível (ver Lista de Materiais).
3. Preparação de v-SUVS contendo tanto lipídico e marcadores de conteúdo

Figura 2. Diagrama esquemático da preparação de SUV. Os lípidos são misturados num tubo de vidro (1) e o solvente é evaporado para formar uma película de lípido, rodando o tubo num banho de água (2). Restantes vestígios de solvente são removidos sob alto vácuo (3). O filme de lípido é hidratado em tampão contendo detergente e reconstituição da proteína enquanto vórtice (4). Se o conteúdo de corante é para ser encapsulada, está incluído neste passo, bem como na etapa de diluição (5). A diluição da concentração de detergente abaixo da sua concentração micelar crítica leva à formação de lipossomas. O detergente é dialisado durante a noite distância (6). Para t-SUVs para a formação de SBL incluindo NBD-PE (verde), vesículas estão flutuando em um gradiente de densidade e recolhida naa interface entre duas camadas (7a). Para separar v-SUVs com marcador conteúdos encapsulados de corante livre da amostra é executado através de uma coluna de exclusão de tamanho e recolhidos em fracções de 0,5 ml (7b). Por favor clique aqui para ver uma versão maior desta figura.
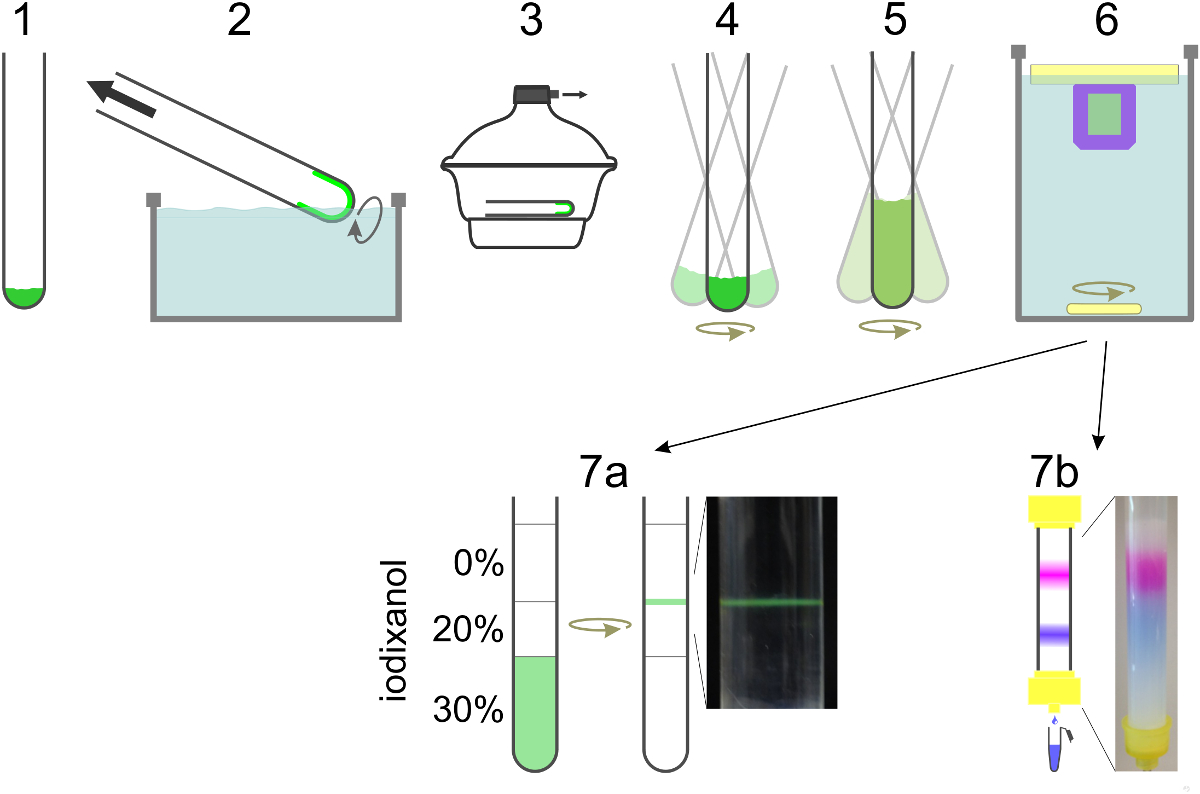{kind=link}
- Prepare a 4 L de tampão de reconstituição, que contém 25 mM de HEPES-KOH, KCl 140 mM, 100 ^ M EGTA e DTT 1 mM, pH 7,4. Usar a maioria do tampão de diálise e 100 ml de outras fases.
Nota: podem ser utilizados outros tampões, mas é importante para manter a osmolaridade tampão consistente ao longo da experiência e escolher as condições em que as proteínas são funcionais. Para a preparação de v-SUVs contendo apenas rótulos de lipídios e t-SUVs, siga Karatekin e Rothman 25. - Os estoques de lípidos são armazenadas a -20 ° C em clorofórmio ou clorofórmio: metanol (2: 1, v / v), deixe os frascos atingir a temperatura ambiente antes de abrir aevitar a condensação.
- Lavar um tubo de vidro com clorofórmio para remover vestígios de detergentes e misturar os lípidos na proporção desejada com um valor final de 1? Mol de uma mistura de clorofórmio e metanol (2: 1, v / v). Utilize apenas seringas de vidro / tubos para lidar com solventes orgânicos / lipídios dissolvidos.
Nota: Os lipídios usados aqui são POPC: SAPE: DOPS: Colesterol: PEG2000-DOPE: Será que a uma razão molar de 57,4: 15: 12: 10: 4,6: 1 para v-SNARE contendo vesículas e POPC: SAPE: DOPS: Colesterol : PI cérebro (4,5) P 2: DOPE-PEG2000: NBD-PE (54,9: 15: 12: 10: 3: 4,6: 0,5) para o t-SNARE SUVs. Veja material para mais detalhes. Outras composições de lipidos podem ser usados. - Evapora-se o solvente sob uma corrente suave de azoto, ou em um evaporador rotativo. Imergir a ponta do tubo em banho-maria (37 ° C), aquecido acima da mais elevada temperatura de fusão dos lipidos, para se obter uma película de lípido homogéneo e para evitar grandes alterações na temperatura enquanto que a evaporação do solvente. Iniciar vácuo em torno300 mbar, até que o filme de lípidos é formada, em seguida, continuar durante ~ 2 min a mais elevada possível a vácuo no evaporador rotativo.
- Remover o tubo de vidro e envolvê-la em papel de alumínio para evitar a exposição à luz e remover os vestígios restantes de solvente, colocando o tubo num exsicador sob alto vácuo durante pelo menos 2 h. Esta etapa também pode ser executado durante a noite.
- Para hidratar a película de lípido seca, preparar uma mistura de detergente (N-octil-β-D-glucopiranósido, OG), proteínas (v-SNARE), e de SRB em tampão de reconstituição (volume final 500 ul). Manter a concentração de detergente final de ~ 2 vezes acima da concentração crítica de micelas (CMC, 20-25 mM de OG) e ajustar o volume final de tal modo que o detergente para lípido é> 10. Dissolve-se Sulforodamina B em pó (SRB) na solução detergente-proteína para se obter uma concentração final de SRB 10-50 mM, por agitação suave da solução. Ajustar a osmolaridade do tampão de reconstituição quando a adição de elevadas concentrações de SRB, reduzindo o concentration de cloreto de potássio em conformidade.
Nota: lípido-proteína (LP) para o t-SNARE é alta (LP 20.000 ~, ~ 70 t-SNARE por quadrado 25 micron.). Significativamente maiores densidades t-laço no SBL pode levar a agregados de proteínas inativas com reduziu significativamente as taxas de fusão 17,24. LP para v-SNAREs é muito mais baixa (LP ~ 200, ~ 7.000 v-SNAREs por mm 2), próximo às densidades v-SNARE encontrados em vesículas sinápticas 46,47. Para garantir a função da proteína depois de expressão recombinante e purificação de V e t-SNAREs é útil para realizar um ensaio de fusão em massa simples 48 antes de passar por todas as etapas do ensaio de fusão de vesícula única. - Agite suavemente o tubo de vidro pré-aquecido contendo o filme lipídico seco, enquanto a adição da solução de proteína-detergente-SRB a partir do passo 3.6. Tente evitar a criação de bolhas. Continue a agitar durante 15 min a 37 ° C.
- Dilui-se a cinco vezes por adição de detergente reconstituiçãotampão contendo SRB (2 ml, adicionar 2,5 ml de volume final), enquanto vortex rapidamente para evitar gradientes de concentração. Continue a agitar durante 15 min a 1 h a 37 ° C.
Nota: Maior aumento de incubação eficiência reconstituição proteína. A diluição rápida diminui a concentração de detergente abaixo da CMC e conduz à formação de pequenos lipossomas. - Dializar a suspensão vesícula primeiro contra ~ 1 L de tampão de reconstituição durante 1-2 horas à temperatura ambiente e, em seguida, contra 3 L de tampão de reconstituição durante a noite a 4 ° C com 4 g de adsorvente de poliestireno, usando um tubo de 20,000 diálise MWCO ou cassete. Use diferentes taças de diálise de vesículas com e sem SRB para evitar a contaminação cruzada.
- Equilibrar a coluna de filtração através de gel com tampão de reconstituição. Executar a suspensão de vesículas através da coluna para separar SRB livre do V-SUVs com SRB encapsulado. Use buffer de reconstituição sem SRB como eluente uma vez toda a amostra tementrou na coluna. Colete V-SUVs em fracções de 0,5 ml.
- Verifique sucesso encapsulamento SRB em concentrações auto-extinguiu-se por medição da fluorescência de SRB antes e após a adição de detergente a uma alíquota de 16 vesículas. Usando um espectrômetro de fluorescência, excitar a amostra a 550 nm e digitalizar a emissão SRB entre 570 nm e 630 nm. Esperar um aumento de 4-8 vezes na fluorescência SRB dependendo da quantidade encapsulada após a adição de detergente como a membrana é solubilizado e a SRB libertado é diluída.
- Caracterizar a recuperação de lípido e proteína usando espectroscopia de fluorescência 24 e electroforese em gel de SDS-PAGE, respectivamente. Use um método de coloração sensível, especialmente para as amostras t-SUV alta LP (veja Lista de Materiais). Tipicamente, cerca de 50% de ambas as entradas de lípidos e de proteínas é perdido durante a preparação resultante numa LP perto do valor nominal.
- Caracterizar tamanhos SUV usando luz dinâmica de espalhamento 24 ou elétrons microscopy 48. SRB loja contendo V-SUVs a 4 ° C até ~ 3-4 dias. Não congelar, como congelamento e descongelamento rompe a membrana e libera SRB encapsulado.
4. SUV-SBL Fusão Ensaio para monitorar Lipid Apenas lançamento
- A formação da bicamada suportada presa dentro dos canais de fluxo de microfluidos
- Colocar o bloco de PDMS (passo 1.11) sob alto vácuo durante pelo menos 20 minutos antes da experiência para remover gases dissolvidos. Isto reduz grandemente o risco de bolhas de ar nos canais de microfluidos durante a experiência de fusão.
- Ligue a configuração microscópio e aquecer a fase e suporte de amostras (Figuras 3 e 4) para a temperatura desejada.
- Filtra-se o tampão de reconstituição utilizada para diluir as vesículas através de um filtro com 0,45? M ou menor tamanho de poro.
- Diluir ~ 30 ul da NBD-PE marcado t-SUVs ou (PF-SUVs) livres de proteínas lipossomas de controlo (estoque0,5-1 solução lipídica mM) com ~ 60 mL de tampão. A concentração final não é critica aqui.
- Desgaseifica esta mistura utilizando uma seringa de 3 ml. Pressionar a maior parte do ar por cima da amostra, mantendo a seringa verticalmente. Selar a ponta (sem agulhas) usando filme de parafina e criar um vácuo, puxando para baixo o êmbolo. Toque o tambor da seringa para acelerar a desgaseificação da solução. Repita esse processo algumas vezes até que não haja mais bolhas ocorrer quando o vácuo é aplicado.
- Utilize uma agulha hipodérmica com um diâmetro ligeiramente maior do que o tubo que está ligado ao bloco de PDMS de perfurar um furo na tampa de um tubo de microcentrífuga. Certifique-se de que a peça perfurada de plástico não é no interior do tubo, uma vez que pode entupir o tubo ligado à entrada do canal microfluídico mais tarde.
- Encha a solução SUV desgaseificado para dentro do tubo de microcentrífuga de ter um furo na tampa e coloque-a no suporte no palco microscópio para equilibrar a temperatura definida.
- Place uma lamela previamente limpas (seção 2) em um aspirador de plasma e executar plasma do ar por cerca de 5 min. Coloque o plasma lamela tratado (lado tratado para cima) no topo de alguns tecidos que não soltem fiapos que servem como uma almofada.
- Remover a película de alumínio a partir do bloco de PDMS desgaseif içada e colocar o bloco em cima da lamela. Ao reutilizar o bloco PDMS colocar e retirar um pedaço de fita adesiva para o lado do canal para limpá-lo. Pressione para baixo as PDMS bloquear sobre a lamela usando um par de pinças para fazê-lo ficar, mas não pressione muito duro como o vidro pode quebrar.
- Coloque a célula de fluxo montada na platina do microscópio e ligar a tubagem para o reservatório SUV e a bomba de seringa, respectivamente. Tape o lamela para a fase (Figura 3B).
- Iniciar aspiração SUVs em 3 mL / min, até a solução enche os canais completamente (~ 2,25 milímetros / seg para a secção transversal do canal x 300 mm 75 mm). Quando as soluções para todos os canais começam a se mover para cima the tubos no lado de saída, para reduzir o fluxo de 0,5 mL / min e incubar durante 30-45 min.
Nota: O tempo de tratamento de plasma entre a lâmina de vidro e as SUVs que flui para dentro do canal não deve exceder 10-20 min como o efeito do tratamento com plasma é transiente. - Verifique os canais para qualquer vazamento. Use um objetivo ar 10-20X observar NBD-PE fluorescência do NBD-PE contendo T ou PF-SUVs. Excite NBD fluoróforos utilizando o laser de 488 nm. Vazamentos são difíceis de detectar usando brilhante iluminação de campo.
- Coloque um tubo com tampão de reconstituição desgaseificado no suporte e deixe a temperatura equilibrar mudanças o mais rápido na temperatura pode causar defeitos no SBL. Pare o fluxo e esperar ~ 1 min para garantir que o fluxo parou completamente e nenhuma bolha de ar será aspirado para dentro do tubo antes de ligar os tubos de entrada para a memória intermédia desgaseificado.
- Lavar todos os canais com tampão desgaseificado para lavar SUVs não ligados.
- Mudar para um higher objetivo de ampliação TIRF (60X, óleo, NA 1,45-1,49) e verificar se a bicamada parece homogênea e é livre de quaisquer defeitos de grande escala óbvios, tais como manchas escuras ou túbulos lipídicas que se estendem para fora da SBL.
- Verifique a fluidez da bicamada
- Se uma unidade FRAP dedicado não está disponível, ou se uma sequência FRAP não pode ser programado, a fluidez da membrana teste, então qualitativamente como se segue.
- Feche o diafragma de campo para um tamanho pequeno (~ 40 um de diâmetro) e ajustar a 488 nm, a intensidade da luz de excitação através do software (20-80 mW, ou 15-60 NW / uM 2) para branquear a fluorescência de NBD-PE na exposta área significativamente, mas não completamente. Para uma bicamada de fluido, no estado estacionário, a intensidade de fluorescência no meio da área exposta deve ser menor do que nas arestas, como moléculas de NBD-PE intactas entrar na área exposta e difundir uma certa distância antes do branqueamento. Em contraste, se o S aderida à superfícieUVs não conseguiu estourar, ou por algum outro motivo bicamada suportada não é fluido, todos os fluoróforos na área exposta deve branquear.
Nota: os valores de intensidade de laser da e posteriores passos são dados como ponto de partida áspera e devem ser optimizados para um determinado conjunto de condições. - Pare a iluminação e iniciá-lo novamente alguns minutos mais tarde para verificar os resultados das medições de estado estacionário
- Feche o diafragma de campo para um tamanho pequeno (~ 40 um de diâmetro) e ajustar a 488 nm, a intensidade da luz de excitação através do software (20-80 mW, ou 15-60 NW / uM 2) para branquear a fluorescência de NBD-PE na exposta área significativamente, mas não completamente. Para uma bicamada de fluido, no estado estacionário, a intensidade de fluorescência no meio da área exposta deve ser menor do que nas arestas, como moléculas de NBD-PE intactas entrar na área exposta e difundir uma certa distância antes do branqueamento. Em contraste, se o S aderida à superfícieUVs não conseguiu estourar, ou por algum outro motivo bicamada suportada não é fluido, todos os fluoróforos na área exposta deve branquear.
- Programar uma sequência FRAP para uma medida mais quantitativa, se possível. Veja arquivos complementares e a legenda correspondente para mais detalhes.
Nota: Às vezes SUVs irá aderir para a lamela de vidro, mas não estourar e formar uma bicamada fluido. Se isto ocorrer, lavar os canais com tampão de reconstituição desgaseificada contendo 10 mM de Mg2 + para ajudar a formação de bicamada suportada. Use branqueamento de fluorescência para avaliar a fluidez da bicamada como em 4.2. Uma vez que uma bicamada de fluido é formada, enxaguar com Mg 2 + livre recotampão nstitution.
- Se uma unidade FRAP dedicado não está disponível, ou se uma sequência FRAP não pode ser programado, a fluidez da membrana teste, então qualitativamente como se segue.
- Apresentando v-SUVs para os canais de fluxo de microfluidos
- Desgaseifica reconstituição tampão e usá-lo para diluir a solução estoque V-SUV por um factor de cerca de marco 10-maio 10, dependendo da concentração da v-SUV. Alvo para uma diluição que resulta em cerca de 10-100 eventos de fusão dentro de 60 seg, no campo de visão.
Nota: Muitas fusões aumentar a fluorescência de fundo (uma vez que cada depósitos de eventos LR-PE ou fez rótulos de lipídios no SBL) e fazer detecção e análise de eventos de fusão difícil. Em contraste, uma taxa de fusão que é demasiado baixa resulta em mau estatísticas ou exige a aquisição de muitos mais filmes. Para uma concentração de v-SUV de lípido de 0,1 mM, começar por diluição da SUV 5 ul em tampão de reconstituição 995 ul e depois dilui-se 5-50 uL desta em 950-995 ul de tampão de reconstituição. - Deixe a temperatura equilibrar antes de parar o fluxo e inserTing o tubo de entrada para dentro da solução V-SUV diluída.
- Ajuste o ângulo TIRF e polarização da seguinte forma.
- Depois de ajustar a polarização desejado, rodando o feixe de excitação, ajustar a inclinação do espelho comandar a posição do feixe por meio de um motor passo a passo através do software. mover-se lentamente a posição do feixe de laser a partir do centro da objectiva no plano focal posterior de um dos lados fora do centro. Observar a luz da objectiva no lado frontal emergem com um ângulo crescente em relação ao eixo objectivo que a posição é movido ainda mais para fora do centro.
- Observe a posição do motor quando o feixe de saída desaparece primeiro no objetivo, isto é, quando TIR é primeiro atingido.
- Lentamente manter em movimento a posição do feixe ainda mais para fora do centro enquanto se monitoriza a fluorescência da superfície. Observe a posição do motor quando a fluorescência de superfície desaparece quando o feixe passou muito longe do centro.
- Escolha um positio feixe n entre os dois limites acima determinado. Para uma melhor relação sinal-ruído e mais realce sinal em cima de fusão, escolha uma profundidade de penetração rasa (posição do feixe mais perto da borda do objectivo) que ainda proporciona uma iluminação uniforme da viewfield.
Nota: É melhor manter a mesma posição do feixe TIR (mesma profundidade de penetração) para todos os experimentos após as configurações são otimizadas. Certifique-se de rodar a polarização não resulta em mudanças na posição do feixe.
- Fluxo V-SUVs para dentro do canal, a uma taxa de fluxo de 2 mL / min, correspondendo a uma velocidade de fluxo linear média de ~ a 1,5 mm / seg para uma secção transversal de 75 um x 300 fim. Mudar para as definições de excitação / emissão para monitorar lipídica de mistura (LR ou só fez).
- Desgaseifica reconstituição tampão e usá-lo para diluir a solução estoque V-SUV por um factor de cerca de marco 10-maio 10, dependendo da concentração da v-SUV. Alvo para uma diluição que resulta em cerca de 10-100 eventos de fusão dentro de 60 seg, no campo de visão.
- Observando fusão entre v-SUVs individuais e da SBL
.jpg "/>
Figura 3. A configuração experimental pTIRF. (A) Representação esquemática de um v-SUV e um t-SBL sobre um substrato de vidro. De cores falsas imagens TIRFM de fusão único evento SUV-SBL mostrando ancoragem lípido (1) e a libertação de corante de lípidos no SBL (2) seguido por branqueamento e diminuição da intensidade de fluorescência (3). A intensidade de fluorescência total (soma dos valores de pixel no 5,3 mm x 5,3 caixa de um) sinal é mostrado. (B) A lamela ligado ao bloco de PDMS é gravado na fase aquecida. tubo de entrada para os canais de microfluidos extrair amostras a partir de tubo de amostra no suporte de metal (direita) aspirada pela bomba de seringa (esquerda). Abaixo da bomba é a unidade de emissão dupla. Por favor clique aqui para ver uma versão maior desta figura.
{kind=link}
oad / 54349 / 54349fig4.jpg "/>
Figura 4. Representação esquemática da montagem experimental. A onda evanescente é criado na interface vidro-tampão no canal microfluídico. O SBL é formado sobre o vidro e v-SUV são aspirados a partir de o suporte de amostra de metal (parte superior direita) através do canal para a seringa (parte superior esquerda). M, espelho; DM, espelho dicróico; G, lente; F, filtro; P, polarizador. Por favor clique aqui para ver uma versão maior desta figura.
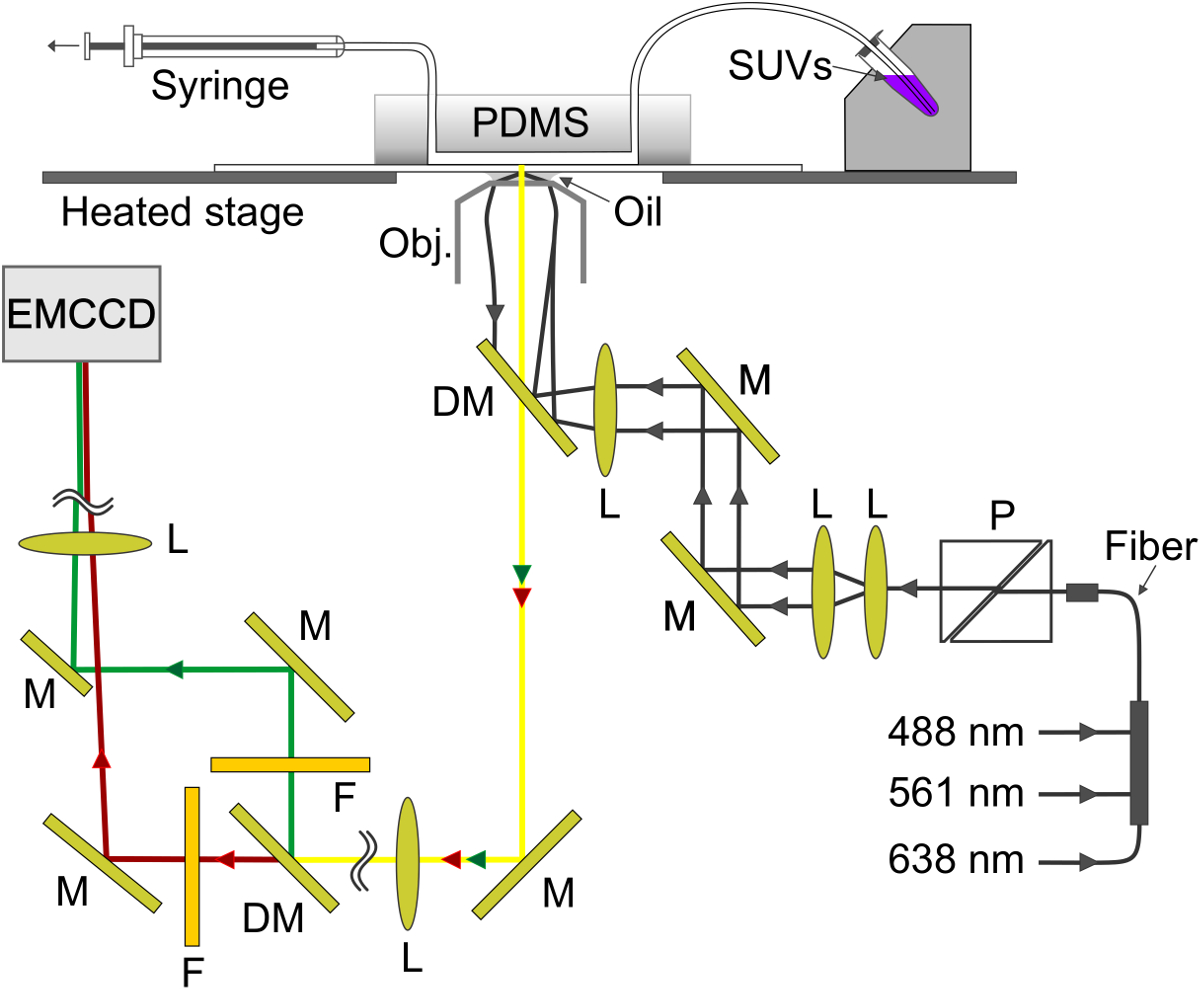{kind=link}
- Continuamente estimular e monitorar a fluorescência v-SUV usando um 561 nm (para LR-PE) ou 638 nm (para DID) a laser, dependendo do fluoróforo incorporada em v-SUVs. Como v-SUVs chegar ao canal de fluxo e cais para e se fundem com o SBL, o sinal de fluorescência de fundo começa a se acumular.
- Ajuste a intensidade do laser de excitação através do software para branquear continuamente o fundofluorescência tal que pelo novos eventos de encaixe e de fusão de estado estacionário pode ser facilmente observado.
Nota: Se o branqueamento é muito lenta, a fluorescência de fundo será demasiado elevada. Se o branqueamento é muito rápido, os sinais de SUVs encaixado e de fluoróforos liberados no SBL vai desaparecer rapidamente, diminuindo a janela durante a qual fusão de SUVs ancorados pode ser detectada ou um fluoróforo pode ser rastreado. Para LR-PE excitado a 561 nm, 2,5-7,5 mW de potência iluminando um círculo de 190 mm de diâmetro (100-250 NW / mm 2) é um valor razoável para começar. Para 638 nm de excitação de DID, 0,8-1,6 mW de potência ao longo de um diâmetro do círculo de 190 mm (30-60 NW / mm 2) pode ser usado para testes iniciais. - Adquirir vários filmes em posições diferentes em um determinado canal. Verificar NBD-PE fluorescência para verificar que a SBL não tem defeitos nestas posições. Adquirir filmes full-frame, à taxa máxima (~ 50 frames / seg) ou de uma região recortada de interesse em maior taxa de quadros (até~ 100 Hz), durante 60 seg.
- Mover para outro canal microfluídico e repetir as gravações para outras condições. Incluem controlos negativos, tais como um SBL ou SUVs livre de proteína, adicionar o domínio citoplásmico solúvel do v-SNARE VAMP2 (CDV) como um inibidor ou tratar V-SUV com neurotoxina tetânica (30 min a 37 ° C) em um ou mais dos canais no mesmo lamela.
- Limpar e PDMS reciclagem
- A fim de reutilizar o bloco de PDMS, lavar os canais de microfluidos com ~ 200 mL de etanol a 70% a uma taxa de fluxo de 5 ul / min. Finalmente, aspirar ar através da tubagem e canais.
- Retire os PDMS bloquear suavemente do lamela, apertando-o levemente. Coloque-o em um pedaço limpo de papel alumínio. Armazenar num exsicador de vácuo. Ele pode ser reutilizado várias vezes.
- Para uma limpeza mais profunda lavar os canais com uma solução de hidróxido de sódio ou de detergente antes de 70% de etanol. Como alternativa, remova todos os tubos fro M, o PDMS e sonicate o bloco de 30 minutos em isopropanol, antes da secagem e inserção de nova tubulação.
5. SUV-SBL Fusão Ensaio para monitorar lipídico e conteúdo lançamento Simultaneamente
- Para o monitoramento cor dupla de lípidos simultânea e conteúdo solúveis liberar, siga os mesmos passos na seção 4, excepto lipossomas uso marcadas com tanto um lipídio (DID) e um conteúdo solúveis (SRB) etiqueta, conforme explicado na secção 3. SRB é encapsulado a 10 mM e é inicialmente altamente auto-extinguiu.
- Usando a configuração representada esquematicamente na Figura 4, e fez SRB excita a fluorescência simultaneamente utilizando 561 nm e 638 nm lasers, respectivamente. Um espelho dicróico (640 nm) divide a emissão em dois feixes que percorrem uma curta (595/50 nm) e um filtro de comprimento (700/75 nm) comprimento de onda para detectar a SRB e fez emissões, respectivamente. Os dois feixes de emissão são projectadas lado-a-lado no chip EM-CCD.
- Análise de dados FRAP
- Use o programa MATLAB fornecida nas informações complementares para estimar o coeficiente de difusão de lipídios, D. O programa de ler uma lista de ficheiros OME-TIFF 49, detecta a zona branqueada, representa graficamente os valores de pixel significativo na área branqueada como uma função do tempo, e se ajusta a curva de recuperação resultante para um modelo por Soumpasis 50 para extrair o tempo de recuperação,
 , Em que W é o raio do círculo branqueada.
, Em que W é o raio do círculo branqueada.
Nota: Um programa MATLAB foi fornecido anteriormente 25 para análise de filmes FRAP usando arquivos Nikon ND2. O programa actual lê OME-TIFF arquivos de 49, porque a maioria dos formatos de arquivo podem ser facilmente convertidos para OME-TIFF. A análise quantitativa dos dados FRAP é mais fácil e mais preciso quando o branqueamento é instantânea, o bleacregião HED é um círculo, e de branqueamento durante read-out é negligenciável. Embora estes critérios não são estritamente satisfeito nas medições FRAP simples descritas aqui, pode ser obtida uma estimativa razoável do coeficiente de difusão. Para uma estimativa mais precisa, use o acompanhamento de corantes lipídicas única (Ponto 6.3).
- Use o programa MATLAB fornecida nas informações complementares para estimar o coeficiente de difusão de lipídios, D. O programa de ler uma lista de ficheiros OME-TIFF 49, detecta a zona branqueada, representa graficamente os valores de pixel significativo na área branqueada como uma função do tempo, e se ajusta a curva de recuperação resultante para um modelo por Soumpasis 50 para extrair o tempo de recuperação,
- Análise da taxa de encaixe, taxa de fusão e tempos de atraso de encaixe-a-fusion
- Use ImageJ para abrir um filme para analisar e ajustar o brilho eo contraste para identificar claramente encaixe da vesícula e eventos de fusão. Comece o plugin SpeckleTrackerJ 26. Veja Smith et al. 26 e online documentação para SpeckleTrackerJ para obter instruções para o uso.
- Identificar todos os SUVs recém-entrados em SpeckleTrackerJ. Para distinguir SUVs que doca firmemente daqueles que saltam fora da SBL, impor uma duração mínima de ancoragem de alguns quadros. Guarde as faixas, e repita para todos os filmes.
Nota: Para o doctaxa de rei, tudo o que importa é identificar quando um SUV ancorado, portanto, apenas o primeiro quadro em que os SUVs ancorado precisa ser gravados nestas faixas. O resto das faixas não serão tidas em conta na análise. No entanto, auto-tracking cada SUV até que alvejantes ou fusíveis ajuda SUVs da marca que já foram rastreados. - Identificar todas as vesículas de fusão. Para estes, as faixas devem incluir todos os quadros a partir de primeiro quadro um SUV ancorado até que o primeiro quadro no qual a fusão é evidente por um aumento súbito da fluorescência da mancha rastreados. As durações de estas faixas são utilizados para calcular os atrasos de encaixe-a-fusão. Salve todas as faixas. Repita o procedimento para todos os filmes.
- Para a análise de série de dados para conexão de aparelhos ou de fusão, compilar uma lista de arquivos de trajetória dos filmes relevantes e executar os programas MATLAB fornecidos com Karatekin e Rothman 25. Siga as instruções que são fornecidos juntamente com os programas.
Nota: Os programas de traçar o cumeventos de encaixe e de fusão ulative como uma função do tempo, com base na informação extraída a partir dos ficheiros de trajectória. taxas de atracação e de fusão são estimados a partir das pistas dessas parcelas. Para os dados de fusão, os atrasos de encaixe-a-fusão são também calculados e sua distribuição representada graficamente como uma trama de sobrevivência, ou seja, a probabilidade de que a fusão ainda não tiver ocorrido um determinado atraso após o acoplamento.
- diffusivity Lipid
- Para cada evento de fusão, pista única lípidos tornam-se fluorescentes como discernível quando eles tenham difundido suficientemente longe do local de fusão (Figura 6). Use SpeckleTrackerJ para rastreamento e salvar as faixas para uma análise mais aprofundada usando MATLAB. Dependendo do tamanho da vesícula de fusão, tipicamente 3-30 fluoróforos individuais podem ser rastreados. Uma vez que as faixas mais longos são desejáveis para o cálculo de
 , Tentam detectar moléculas individuais como longquanto possível, utilizando a correcção manual das trajectórias se necessário.
, Tentam detectar moléculas individuais como longquanto possível, utilizando a correcção manual das trajectórias se necessário. - Calcular o quadrado médio de deslocamento (MSD) para únicos trajetórias de marcadores lipídicos que duram> 40-50 quadros (cerca de ~ 1,5 seg). Utilizar a MSD para calcular o coeficiente de difusão de lípidos, . Veja Smith et al. 26 e Stratton et al. 27 para mais detalhes.
- Para cada evento de fusão, pista única lípidos tornam-se fluorescentes como discernível quando eles tenham difundido suficientemente longe do local de fusão (Figura 6). Use SpeckleTrackerJ para rastreamento e salvar as faixas para uma análise mais aprofundada usando MATLAB. Dependendo do tamanho da vesícula de fusão, tipicamente 3-30 fluoróforos individuais podem ser rastreados. Uma vez que as faixas mais longos são desejáveis para o cálculo de
- Lipídica única intensidade corante, fator de redução da intensidade SUV-SBL, eo tamanho da vesícula
- Medir a soma dos valores de pixel de um marcador lipídico em um pixel de 3 x 3 (0,8 um x 0,8 um) em torno da área de marcador para ~ 15 quadros antes e após os branqueamentos marcador numa única etapa. Subtrair a média da intensidade de fundo ao longo dos quadros pós-branqueamento da intensidade pré-lixívia média antes do branqueamento para obter a intensidade do marcador de lípido rastreados. Repetir a medição para tantos marcadores quanto prático para um dado filme.
- Traçar a distribuição de intensidades rótulo único lipídicas,
 E coloque um Gaussian para estimar a média.
E coloque um Gaussian para estimar a média. - Calcule o atraso entre quando a fusão ocorreu e quando a trajetória de um único marcador lipídico lançado nesse caso terminou em um de branqueamento de etapa única. Obter o tempo de branqueamento de um fluoróforo na SBL,
 , Traçando a função sobrevivente dos atrasos e de montagem a um exponencial.
, Traçando a função sobrevivente dos atrasos e de montagem a um exponencial.
Nota: Como as polarizadas casais de excitação mais fracamente aos fluoróforos em um SUV, o tempo de branqueamento de um SUV é geralmente muito mais lentos 27. - Estimativa
 , O factor de redução de intensidade para um corante de lípidos quando no SUV relação a quando está na SBL, seguindo Stratton 27 (tp_upload / 54349 / 54349eq8.jpg "/> é a intensidade de um único corante no SUV).
, O factor de redução de intensidade para um corante de lípidos quando no SUV relação a quando está na SBL, seguindo Stratton 27 (tp_upload / 54349 / 54349eq8.jpg "/> é a intensidade de um único corante no SUV).
Nota: Se o branqueamento eram insignificantes, seria igual à intensidade encaixado  (ponto (1) na Figura 3A, painel da direita), dividida pela intensidade total atingiu após todos os fluoróforos são depositados sobre a SBL fusão (linha tracejada marcado
(ponto (1) na Figura 3A, painel da direita), dividida pela intensidade total atingiu após todos os fluoróforos são depositados sobre a SBL fusão (linha tracejada marcado  na Figura 3). No entanto, devido ao rápido branqueamento na SBL, normalmente não é alcançado e uma estimativa precisa da requer montagem da cinética de liberação para uma expressão 27 que incl udes tanto (Ver 6.4.3) e . Expressões separadas são fornecidas em Stratton et al 27 para os casos de cinética limitado de poros e difusão de libertação limitada.; o melhor caso ajuste varia de caso para caso (ver 6.5.1 para o caso de cinética limitado de poros).
na Figura 3). No entanto, devido ao rápido branqueamento na SBL, normalmente não é alcançado e uma estimativa precisa da requer montagem da cinética de liberação para uma expressão 27 que incl udes tanto (Ver 6.4.3) e . Expressões separadas são fornecidas em Stratton et al 27 para os casos de cinética limitado de poros e difusão de libertação limitada.; o melhor caso ajuste varia de caso para caso (ver 6.5.1 para o caso de cinética limitado de poros). - Calcular a área da vesícula para os eventos individuais a partir de 27
 , Onde é a intensidade SUV encaixado, é a intensidade de um único corante em lípido significa a SBL, é o factor de redução de intensidade para um corante de lípidos quando está no SUV relação a quando está na SBL, eção 1 "src =" / files / ftp_upload / 54349 / 54349eq12.jpg "/> é a densidade de área conhecida de corantes lipídicas.
, Onde é a intensidade SUV encaixado, é a intensidade de um único corante em lípido significa a SBL, é o factor de redução de intensidade para um corante de lípidos quando está no SUV relação a quando está na SBL, eção 1 "src =" / files / ftp_upload / 54349 / 54349eq12.jpg "/> é a densidade de área conhecida de corantes lipídicas.
- Propriedades de fusão dos poros
- Assumindo lançamento é limitado de poros, se encaixam a cinética de libertação rótulo lipídica a 27
 , Onde
, Onde  é a intensidade SUV encaixado apenas antes da fusão e os outros parâmetros são como definidos anteriormente. Use o valor de 6.4.3 obtido no como um parâmetro fixo, e extrair os melhores estimativas para ajuste e
é a intensidade SUV encaixado apenas antes da fusão e os outros parâmetros são como definidos anteriormente. Use o valor de 6.4.3 obtido no como um parâmetro fixo, e extrair os melhores estimativas para ajuste e  .
. - Estimar a fração de tempo que o poro está aberto, P 0, assumindo retardamento da liberação de lipídios é devido a pore piscando 27:60;
 =
=  , Onde uma ves é a área da vesícula (secção 6.4.5), b representa a altura dos poros (normalmente levado para ser ~ 15 nm), r p ≈ 3 nm é o raio do poro eficaz como pode ser visto pelas etiquetas de lípidos e de difusão inclui uma meia a espessura da bicamada (~ 2 nm), é a difusividade lípido (calculado em 6,3), e é o momento para os lipídios para ser lançado a partir do SUV para o SBL (de 6.5.1).
, Onde uma ves é a área da vesícula (secção 6.4.5), b representa a altura dos poros (normalmente levado para ser ~ 15 nm), r p ≈ 3 nm é o raio do poro eficaz como pode ser visto pelas etiquetas de lípidos e de difusão inclui uma meia a espessura da bicamada (~ 2 nm), é a difusividade lípido (calculado em 6,3), e é o momento para os lipídios para ser lançado a partir do SUV para o SBL (de 6.5.1). - Para confirmar que um P nominal 0> 1 indica uma poro totalmente aberta, P 0 = 1, ajustar o curso de tempo da intensidade com a equação 4 de Stratton et ai. 27, a cinética preditos para uma poro permanentemente aberta. Este ajuste deve ser melhor do que fitting a expressão em 6.5.1 para um poro permanentemente aberta.
- Assumindo lançamento é limitado de poros, se encaixam a cinética de libertação rótulo lipídica a 27
Resultados
Qualidade SBL
É crucial para verificar a qualidade e a fluidez do SBL antes da experiência da fusão. A fluorescência no lado de baixo, de vidro de um canal microfluídico deve ser uniforme, sem quaisquer defeitos óbvios. Se uma bolha de ar passa através do canal, ele geralmente deixa cicatrizes visíveis na SBL. Se existem tais grandes cicatrizes escala / defeitos, não utilize esse canal. Às vezes SUVs...
Discussão
A implementação bem sucedida do ensaio de fusão SUV-SBL aqui descrita depende criticamente vários passos-chave, tais como a reconstituição funcional de proteínas em lipossomas, obtendo SBLs de boa qualidade, e escolher os parâmetros de imagem para a direita para detectar moléculas individuais. Embora possa demorar algum tempo e esforço para ter sucesso, uma vez que o ensaio é executado com sucesso, ele oferece uma riqueza de informações sobre o processo de fusão não disponível a partir de qualquer outro ...
Divulgações
Os autores declaram que não têm interesses financeiros concorrentes.
Agradecimentos
We thank Vladimir Polejaev (Yale West Campus Imaging Core) for the design and construction of the polarized TIRF microscope, David Baddeley (Yale University) for help with two-color detection instrumentation, and James E. Rothman (Yale University) and Ben O'Shaughnessy (Columbia University) and members of their groups for stimulating discussions. EK is supported by a Kavli Neuroscience Scholar Award from the Kavli Foundation and NIH grant 1R01GM108954.
Materiais
| Name | Company | Catalog Number | Comments | |
| Reagents | ||||
| Milli-Q (MQ) water | Millipore | |||
| KOH | J.T. Baker | 3040-05 | ||
| Ethanol 190 Proof | Decon | |||
| Isopropanol | Fisher Chemical | A416P4 | ||
| HEPES | AmericanBio | AB00892 | ||
| Sodium Cholride (KCl) | AB01915 | |||
| Dithiothreitol | AB00490 | |||
| N-[2-hydroxyethyl] piperazine-N'-[2-ethanesulfonic acid] (HEPES) | AmericanBio | AB00892 | ||
| EGTA | Acros Organics | 409911000 | ||
| Buffers | ||||
| HEPES-KOH buffer (pH 7.4) | 25 mM HEPES-KOH, 140 mM KCl, 100 μM EGTA, 1 mM DTT | |||
| Solvents | ||||
| Chloroform | J.T. Baker | 9180-01 | in glass bottle, CAUTION, wear PPE | |
| Methanol | J.T. Baker | 9070-03 | in glass bottle, CAUTION, wear PPE | |
| Liposome preparation | ||||
| Gastight Hamilton syringe | Hamilton | var. sizes | only use glass sringe with solents (Chlorophorm/ Methanon, 2:1, v/v) | http://www.hamiltoncompany.com |
| Glass tubes Pyrex Vista 11 ml, 16x100 mm screw cap culture tube | Pyrex | 70825-16 | clean thoroughly, rinse with chloroform | http://catalog2.corning.com/LifeSciences/ |
| 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine, 16:0-18:1 PC (POPC) | Avanti Polar Lipids | 850457 | Lipids come dissolved in CHCl3 or as lyphilized powder in sealed vials. Aliquot upon opening. Store extra as dried lipid films under inert atmosphere at -20 °C. Keep stocks in CHCl3/MeOH (2:1, v/v) at -20 °C. let come to RT before opening | http://www.avantilipids.com/ |
| 1,2-dioleoyl-sn-glycero-3-phospho-L-serine (sodium salt), 18:1 PS (DOPS) | 840035 | |||
| 1-stearoyl-2-arachidonoyl-sn-glycero-3-phosphoethanolamine, 18:0-20:4 PE (SAPE) | 850804 | |||
| L-α-phosphatidylinositol-4,5-bisphosphate (Brain, Porcine) (ammonium salt), Brain PI(4,5)P2 | 840046 | |||
| 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitro-2-1,3-benzoxadiazol-4-yl) (ammonium salt), 18:1 NBD PE | 810145 | |||
| 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (ammonium salt), 18:1 PEG2000 PE | 880130 | |||
| cholesterol (ovine wool, >98%) | 700000 | |||
| DiD' oil; DiIC18(5) oil (1,1'-Dioctadecyl-3,3,3',3'-Tetramethylindodicarbocyanine Perchlorate) | Molecular Probes | D-307 | https://www.thermofisher.com/ | |
| Rotavapor R-210 | Buchi | R-210 | heat bath above Tm of lipids used | http://www.buchi.com/ |
| OG n-Octyl-β-D-Glucopyranoside | Affymetrix | 0311 | store at -20°C, let come to RT before opening | https://www.anatrace.com/ |
| Shaker - Eppendorf Thermomixer R | Eppendorf | https://www.eppendorf.com/ | ||
| Slide-A-Lyze Dialysis Cassettes, 20K MWCO, 3 mL | life technologies | 66003 | https://www.lifetechnologies.com/ | |
| Bio-Beads SM-2 Adsorbents | Bio-Rad | 1523920 | http://www.bio-rad.com/ | |
| OptiPrep Density Gradient Medium | Sigma-Aldrich | D1556 | http://www.sigmaaldrich.com/ | |
| Ultracentrifugation tube, Thinwall, Ultra-Clear, 13.2 mL, 14 x 89 mm | Beckman Coulter | 41121703 | https://www.beckmancoulter.com/ | |
| Beckman SW41 Ti rotor | ||||
| SuflorhodamineB | Molecular Probes | S-1307 | https://www.thermofisher.com/ | |
| Econo-Column Chromatography Columns, 2.5 × 10 cm | Bio-Rad | 7372512 | http://www.bio-rad.com/ | |
| Sepharose CL-4B | GE Healthcare | 17-0150-01 | http://www.gelifesciences.com/ | |
| SYPRO Orange Protein Gel Stain | Molecular Probes | S-6650 | 5,000X Concentrate in DMSO | https://www.lifetechnologies.com/ |
| PDMS block | ||||
| Sylgard 184 Silicone elastomer kit, PDMS | Dow Corning | 3097358-1004 | http://www.dowcorning.com/ | |
| Pyrex glass petri dish, 150 x 20 mm, complete with cover | Corning | 3160-152 | http://catalog2.corning.com/LifeSciences/ | |
| Hole puncher - Reusable Biopsy Punch, 0.75mm | World Precision Instruments | 504529 | http://www.wpi-europe.com/ | |
| Manual Hole Punching Machine | SYNEO | MHPM-UNV | http://www.syneoco.com/ | |
| Drill .035 x .026 x 1.5 304 SS TiN coated round punch | CR0350265N20R4 | drill diameter: 0.9 mm | ||
| Tygon Microbore tubing, 0.25 mm ID, 0.76 mm OD | Cole-Parmer | 06419-00 | 0.010" ID, 0.030" OD | http://www.coleparmer.com/ |
| Silicone Tubing (0.51 mm ID, 2.1 mm OD | 95802-00 | 0.020" ID, 0.083" OD | ||
| Cover glass - cleanroom cleaned | ||||
| Schott Nexterion cover slip glass D | Schott | 1472305 | http://www.us.schott.com/ | |
| plasma cleaner | Harrick | PDC-32G | http://harrickplasma.com/ | |
| pTIRF setup and accessories | ||||
| IX81 microscope body | Olympus | IX81 | http://www.olympus-lifescience.com/en/ | |
| EM CCD camera | Andor | ixon-ultra-897 | http://www.andor.com/ | |
| Thermo Plate, heated microscope stage | Tokai Hit | MATS-U52RA26 | http://www.tokaihit.com/ | |
| 1 ml hamilton glass syringes (4x) | Hamilton | 81365 | http://www.hamiltoncompany.com | |
| syringe pump | kd Scientific | KDS-230 | http://www.kdscientific.com/ |
Referências
- Sudhof, T. C., Rothman, J. E. Membrane fusion: grappling with SNARE and SM proteins. Science. 323, 474-477 (2009).
- Wickner, W., Schekman, R. Membrane fusion. Nat Struct Mol Biol. 15, 658-664 (2008).
- Harrison, S. C. Viral membrane fusion. Nat Struct Mol Biol. 15, 690-698 (2008).
- Jahn, R., Scheller, R. H. SNAREs--engines for membrane fusion. Nat Rev Mol Cell Biol. 7, 631-643 (2006).
- Lindau, M., Alvarez de Toledo, G. The fusion pore. Biochim Biophys Acta. 1641, 167-173 (2003).
- Staal, R. G., Mosharov, E. V., Sulzer, D. Dopamine neurons release transmitter via a flickering fusion pore. Nat Neurosci. 7, 341-346 (2004).
- Wu, Z., et al. Nanodisc-cell fusion: Control of fusion pore nucleation and lifetimes by SNARE protein transmembrane domains. Sci. Rep. 6, 27287 (2016).
- Alabi, A. A., Tsien, R. W. Perspectives on kiss-and-run: role in exocytosis, endocytosis, and neurotransmission. Ann Rev Physiol. 75, 393-422 (2013).
- Rossetto, O., Pirazzini, M., Montecucco, C. Botulinum neurotoxins: genetic, structural and mechanistic insights. Nature Rev Microbiol. 12, 535-549 (2014).
- Weber, T., et al. SNAREpins: minimal machinery for membrane fusion. Cell. 92, 759-772 (1998).
- Nickel, W., et al. Content mixing and membrane integrity during membrane fusion driven by pairing of isolated v-SNAREs and t-SNAREs. Proc Natl Acad Sci U S A. 96, 12571-12576 (1999).
- McNew, J. A., et al. Compartmental specificity of cellular membrane fusion encoded in SNARE proteins. Nature. 407, 153-159 (2000).
- Melia, T. J., You, D. Q., Tareste, D. C., Rothman, J. E. Lipidic antagonists to SNARE-mediated fusion. J Biol Chem. 281, 29597-29605 (2006).
- Hernandez, J. M., et al. Membrane fusion intermediates via directional and full assembly of the SNARE complex. Science. 336, 1581-1584 (2012).
- Fix, M., et al. Imaging single membrane fusion events mediated by SNARE proteins. Proc Natl Acad Sci U S A. 101, 7311-7316 (2004).
- Bowen, M. E., Weninger, K., Brunger, A. T., Chu, S. Single molecule observation of liposome-bilayer fusion thermally induced by soluble N-ethyl maleimide sensitive-factor attachment protein receptors (SNAREs). Biophys J. 87, 3569-3584 (2004).
- Liu, T., Tucker, W. C., Bhalla, A., Chapman, E. R., Weisshaar, J. C. SNARE-driven, 25-millisecond vesicle fusion in vitro. Biophys J. 89, 2458-2472 (2005).
- Yoon, T. Y., Okumus, B., Zhang, F., Shin, Y. K., Ha, T. Multiple intermediates in SNARE-induced membrane fusion. Proc Natl Acad Sci U S A. 103, 19731-19736 (2006).
- Diao, J., et al. A single-vesicle content mixing assay for SNARE-mediated membrane fusion. Nat Commun. 1, 1-6 (2010).
- Kyoung, M., et al. In vitro system capable of differentiating fast Ca2+-triggered content mixing from lipid exchange for mechanistic studies of neurotransmitter release. Proc Natl Acad Sci U S A. 108, E304-E313 (2011).
- Domanska, M. K., Kiessling, V., Stein, A., Fasshauer, D., Tamm, L. K. Single vesicle millisecond fusion kinetics reveals number of SNARE complexes optimal for fast SNARE-mediated membrane fusion. J Biol Chem. 284, 32158-32166 (2009).
- Kreutzberger, A. J., Kiessling, V., Tamm, L. K. High Cholesterol Obviates a Prolonged Hemifusion Intermediate in Fast SNARE-Mediated Membrane Fusion. Biophys J. 109, 319-329 (2015).
- Schwenen, L. L., et al. Resolving single membrane fusion events on planar pore-spanning membranes. Sci Rep. 5, 12006 (2015).
- Karatekin, E., et al. A fast, single-vesicle fusion assay mimics physiological SNARE requirements. Proc Natl Acad Sci U S A. 107, 3517-3521 (2010).
- Karatekin, E., Rothman, J. E. Fusion of single proteoliposomes with planar, cushioned bilayers in microfluidic flow cells. Nat Protoc. 7, 903-920 (2012).
- Smith, M. B., et al. Interactive, computer-assisted tracking of speckle trajectories in fluorescence microscopy: application to actin polymerization and membrane fusion. Biophys J. 101, 1794-1804 (2011).
- Stratton, B. S., et al. Cholesterol Increases the Openness of SNARE-mediated Flickering Fusion Pores. Biophysical journal. 110, (2016).
- Diao, J., et al. Synaptic proteins promote calcium-triggered fast transition from point contact to full fusion. Elife. 1, e00109 (2012).
- Kiessling, V., Domanska, M. K., Tamm, L. K. Single SNARE-mediated vesicle fusion observed in vitro by polarized TIRFM. Biophys J. 99, 4047-4055 (2010).
- Blasi, J., et al. Botulinum neurotoxin A selectively cleaves the synaptic protein SNAP-25. Nature. 365, 160-163 (1993).
- Washbourne, P., et al. Genetic ablation of the t-SNARE SNAP-25 distinguishes mechanisms of neuroexocytosis. Nat Neurosci. 5, 19-26 (2002).
- Diaz, A. J., Albertorio, F., Daniel, S., Cremer, P. S. Double cushions preserve transmembrane protein mobility in supported bilayer systems. Langmuir. 24, 6820-6826 (2008).
- Floyd, D. L., Ragains, J. R., Skehel, J. J., Harrison, S. C., van Oijen, A. M. Single-particle kinetics of influenza virus membrane fusion. Proc Natl Acad Sci U S A. 105, 15382-15387 (2008).
- Albertorio, F., et al. Fluid and air-stable lipopolymer membranes for biosensor applications. Langmuir. 21, 7476-7482 (2005).
- Daniel, S., Albertorio, F., Cremer, P. S. Making lipid membranes rough, tough, and ready to hit the road. Mrs Bulletin. 31, 536-540 (2006).
- Gao, Y., et al. Single reconstituted neuronal SNARE complexes zipper in three distinct stages. Science. 337, 1340-1343 (2012).
- Kenworthy, A. K., Hristova, K., Needham, D., Mcintosh, T. J. Range and Magnitude of the Steric Pressure between Bilayers Containing Phospholipids with Covalently Attached Poly(Ethylene Glycol). Biophys J. 68, 1921-1936 (1995).
- Knoll, W., et al. Solid supported lipid membranes: New concepts for the biomimetic functionalization of solid surfaces. Biointerphases. 3, Fa125-Fa135 (2008).
- Quinn, P., Griffiths, G., Warren, G. Density of newly synthesized plasma membrane proteins in intracellular membranes II. Biochemical studies. J Cell Biol. 98, 2142-2147 (1984).
- Sund, S. E., Swanson, J. A., Axelrod, D. Cell membrane orientation visualized by polarized total internal reflection fluorescence. Biophys J. 77, 2266-2283 (1999).
- Johnson, D. S., Toledo-Crow, R., Mattheyses, A. L., Simon, S. M. Polarization-controlled TIRFM with focal drift and spatial field intensity correction. Biophys J. 106, 1008-1019 (2014).
- Anantharam, A., Onoa, B., Edwards, R. H., Holz, R. W., Axelrod, D. Localized topological changes of the plasma membrane upon exocytosis visualized by polarized TIRFM. J Cell Biol. 188, 415-428 (2010).
- Axelrod, D. Carbocyanine dye orientation in red cell membrane studied by microscopic fluorescence polarization. Biophys J. 26, 557-573 (1979).
- Wang, T., Smith, E. A., Chapman, E. R., Weisshaar, J. C. Lipid mixing and content release in single-vesicle, SNARE-driven fusion assay with 1-5 msec resolution. Biophys J. 96, 4122-4131 (2009).
- Chernomordik, L. V., Frolov, V. A., Leikina, E., Bronk, P., Zimmerberg, J. The pathway of membrane fusion catalyzed by influenza hemagglutinin: restriction of lipids, hemifusion, and lipidic fusion pore formation. J Cell Biol. 140, 1369-1382 (1998).
- Takamori, S., et al. Molecular anatomy of a trafficking organelle. Cell. 127, 831-846 (2006).
- Wilhelm, B. G., et al. Composition of isolated synaptic boutons reveals the amounts of vesicle trafficking proteins. Science. 344, 1023-1028 (2014).
- Scott, B. L., et al. Liposome fusion assay to monitor intracellular membrane fusion machines. Methods Enzymol. 372, 274-300 (2003).
- Linkert, M., et al. Metadata matters: access to image data in the real world. J Cell Biol. 189, 777-782 (2010).
- Soumpasis, D. M. Theoretical analysis of fluorescence photobleaching recovery experiments. Biophys J. 41, 95-97 (1983).
- Ohki, S. A mechanism of divalent ion-induced phosphatidylserine membrane fusion. Biochim Biophys Acta. 689, 1-11 (1982).
- Berquand, A., et al. Two-step formation of streptavidin-supported lipid bilayers by PEG-triggered vesicle fusion. Fluorescence and atomic force microscopy characterization. Langmuir. 19, 1700-1707 (2003).
- Tamm, L. K., McConnell, H. M. Supported phospholipid bilayers. Biophys J. 47, 105-113 (1985).
- Rawle, R. J., van Lengerich, B., Chung, M., Bendix, P. M., Boxer, S. G. Vesicle fusion observed by content transfer across a tethered lipid bilayer. Biophys J. 101, L37-L39 (2011).
- Wagner, M. L., Tamm, L. K. Reconstituted syntaxin1a/SNAP25 interacts with negatively charged lipids as measured by lateral diffusion in planar supported bilayers. Biophys J. 81, 266-275 (2001).
- Kalb, E., Frey, S., Tamm, L. K. Formation of Supported Planar Bilayers by Fusion of Vesicles to Supported Phospholipid Monolayers. Biochimica Et Biophysica Acta. 1103, 307-316 (1992).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados