
Method Article
Administration exogène d’Alpha-synucléine Microsomes associées aux agrégats de neurones primaires comme un modèle de cellulaire puissant de la Formation de fibrilles
Dans cet article
Résumé
Le but du présent protocole est de fournir un système de base de cellules qui réplique la formation d’alpha-synucléine aggregates in vivo. Des inclusions intracellulaires alpha-synucléine sont ensemencées dans des neurones primaires par l’internalisation et propagation d’exogène administrée natif associées aux microsomes alpha-synucléine agrégats isolés de souris transgénique alpha-synucléine malades.
Résumé
Pendant des années, l’incapacité de reproduire la formation d’inclusions insoluble alpha-synucléine (αS) dans des cultures cellulaires a été une grande limitation dans l’étude d’agrégation αS dans la maladie de Parkinson (MP). Récemment, le développement de nouveaux modèles animaux par le biais de l’inoculation exogène d’extraits de cerveau de souris transgéniques αS malades ou patients Parkinsoniens a donné de nouveaux espoirs à la possibilité de créer des modèles de cellules plus adéquats d’agrégation αS. Malheureusement, quand il s’agit de cellules en culture, administration d’extraits de cerveau raw n’a pas prouvé aussi bons souris et la source de choix d’agrégats exogènes est encore in vitro préformées des fibrilles αS.
Nous avons développé une méthode pour induire la formation d’inclusions intracellulaires αS dans des neurones primaires par l’intermédiaire de l’administration exogène d’agrégats native αS associées aux microsomes, une espèce hautement toxiques αS isolée des zones malades de souris transgéniques. Cette fraction des agrégats αS qui est associée avec les vésicules de microsomes, est internalisé efficacement et induit la formation d’inclusions intracellulaires positives pour αS agrégées et phosphorylée. Par rapport à in vitro-fibrilles préformées qui sont fabriqués à partir de recombinant αS, notre méthode est plus rapide et garantit que l’ensemencement pathogène est faite avec des agrégats αS authentique extraites des modèles animaux malades du Parkinson, imitant plus étroitement du type de inclusions obtient in vivo. En conséquence, la disponibilité des tissus riches en inclusions αS est obligatoire.
Nous croyons que cette méthode fournira un modèle polyvalent à base de cellules afin d’étudier les aspects microscopiques de αS agrégation et le connexe physiopathologie cellulaire in vivo et qu’il sera un point de départ pour la création d’une cellule plus précise et sophistiquée paradigme de PD.
Introduction
L’accumulation d’alpha-synucléine (αS) inclusions protéiques est une caractéristique importante et importante de Parkinson Disease (DP) et alpha-synulceinopathies1. Malheureusement, alors que les modèles animaux sont capables de fournir un environnement cellulaire et biochimique suffisant pour induire les étapes d’agrégation nécessaires pour la formation des fibrilles de protéine2, reproduisant la formation de corps de Lewy complexes (LB)-comme des agrégats dans des cultures cellulaires sont difficile et exigeante.
Nous décrivons ici une méthode pour induire la formation d’inclusions αS, semblables aux agrégats de protéines obtenus dans des modèles animaux et les patients Parkinsoniens, dans des cellules cultivées, à l’aide de neurones primaires de souris cerveau isolé. Notre protocole est basé sur l’administration exogène d’agrégats αS associées aux microsomes isolés de αS symptomatique transgéniques (Tg) souris à souris hippocampe ou cortical neurones primaires. Cette méthode tire parti de la capacité de diffusion et de propagation des αS espèces toxiques qui, une fois ajoutée au milieu de culture, sont capables de devenir intériorisé et induisent la formation de mature αS séropositifs agrégats3,4, 5,6,7,8.
A l’origine, les méthodes standard pour obtenir la formation de fibrilles αS dans des cultures cellulaires reposaient sur la surexpression de l’ADNc αS correspondant via les protocoles de transfection régulière ou infection virale véhiculée par9. Alors que dans le premier cas obtenir αS LB-comme agrégats ont été fortuites, a montré une efficacité faible et dépend du type de cellule, le deuxième protocole a conduit à la formation de fibrilles insolubles, y compris les espèces de poids moléculaire élevé (HMW) en 24-48 h de l’infection 10. dans ces méthodes, la formation d’agrégats est probablement due à une excessive et asymétrique en quantité de protéines αS qui devient insoluble plutôt qu’une conversion pathologique de la conformation αS qui dicte l’agrégation. Au lieu de cela, la technique que nous présentons ici ne modifie pas le niveau d’expression αS mais provoque l’agrégation des protéines très répandue en raison de l’internalisation des fibrilles exogènes. En outre, la formation d’agrégats αS en administrant des fibrilles exogènes est un long processus qui nécessite des jours ou des semaines pour devenir exhaustive nous permettant d’étudier les étapes intermédiaires et début de la formation d’inclusions αS dans un mode Time-lapse et pour elle en corrélation avec les changements biochimiques cellulaires. Ainsi, notre méthode est une application utile pour créer des modèles d’agrégation αS cellulaires qui sont utiles pour étudier la formation de fibrilles αS au microscope en ce qui concerne la physiopathologie cellulaire.
En outre bien qu’administration de cerveau brut extrait de souris transgéniques (Tg) malades αS11,12 ou humaine PD cerveaux6,13 est capable d’induire des dépôts αS Tg ou animaux de type sauvage (WT), application de la même façon pour les cultures de cellules n’a pas été prouvé d’être aussi franc succès, peut-être à cause de la faible quantité de granulats dans les échantillons utilisés et l’absence d’une procédure standard pour isoler αS native espèces toxiques14. Pour cette raison, en vitro préformé fibrilles (FFP) de αS ont été la source d’agrégats de choix jusqu'à maintenant pour l’induction des inclusions αS dans les cellules et modèles animaux3,4,6,7 ,15,16. Avec notre protocole, cependant, nous montrent que les espèces agrégées αS associées aux microsomes isolés de souris Tg αS peuvent induire efficacement accumulation intracellulaire inclusions de αS LB-comme dans des neurones primaires.
Dans notre laboratoire, les agrégés espèces associées aux microsomes αS sont isolés du tissu médullaire (SpC) des souris malades Tg exprimant le gène humain de αS A53T sous le contrôle du promoteur souris prion (PrP) de protéines [Prp humain A53T αS Tg souris, ligne G2-3,17]. Ces souris montrent un phénotype de neurodegenerative âge-dépendante qui inclut dysfonctionnement moteur robuste et formation d’inclusions dans le système nerveux central fait de phosphorylée, ubiquitinées et αS insoluble, commençant après 9 mois d’âge. Une fois que le dysfonctionnement moteur apparaît, le phénotype évolue rapidement dans la paralysie, à partir des membres postérieurs, qui mène à la mort en 2 à 3 semaines. Accumulation des agrégats αS parallels manifestation de la maladie. Souris sacrifiées à l’apparition du dysfonctionnement moteur présentent une agrégation αS robuste dans le CPS, le tronc cérébral et le cervelet. Il n’y a pas besoin d’attendre jusqu'à ce que la paralysie affecte à sacrifier la souris. Présymptomatiques souris sont prises à 9 mois-vieux animaux qui n’affichent pas de troubles moteurs.
Protocole
L’utilisation d’animaux WT et Tg a été approuvée et respectée dans leur intégralité par le national et les lois internationales de bien-être animal de laboratoire et d’expérimentation (CEE du Conseil directive 86/609, 12 décembre 1987 et Directive 2010/63/UE, 22 septembre 2010). Tous les protocoles décrits dans cet article suivent les directives de protection des animaux de notre institution.
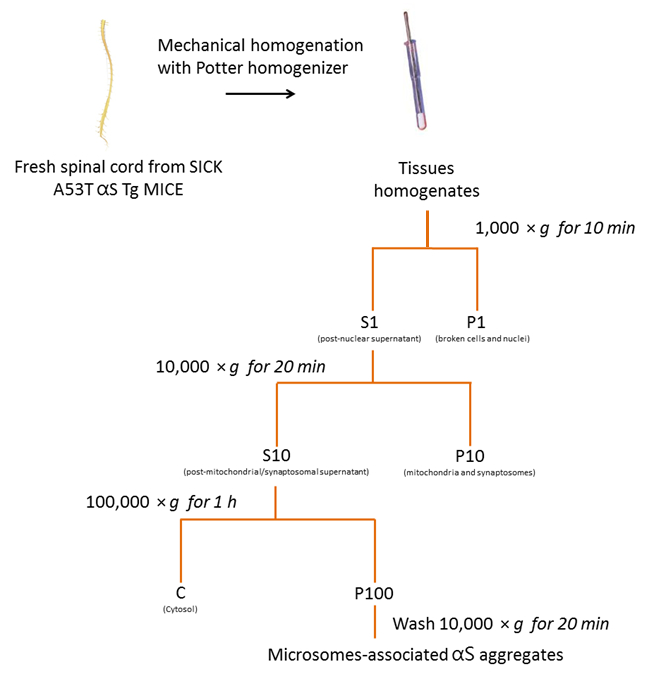
1. isolement des agrégats αS Microsomes associées à des malades A53T αS Tg souris
- Préparer le tampon d’homogénéisation qui est composé de saccharose de 250 mM, 20 mM HEPES, KCl 10 mM, 1,5 mM MgCl2, 2 mM EDTA et 1 x phosphatase /-inhibiteurs de la protéase. Garder le tampon sur la glace.
- Homogénéiser les frais ou congelés de tissu dans un 01:10 volume (p/v) de tampon d’homogénéisation glacee utilisant un téflon Pilon homogénéisateur, avec 10 à 15 coups.
- Transférer l’homogénat initial (1 à 2 mL) dans un tube de microcentrifuge et centrifuger à 1 000 × g pendant 10 min à 4 ° C à l’aide d’une centrifugeuse réfrigérée afin d’enlever les noyaux et ininterrompu de cellules dans le culot qui en résulte (P1). Jeter le P1.
- Transférer le surnageant (S1) dans un tube de microcentrifuge propre et centrifuger S1 à 10 000 x g pendant 20 min à 4 ° C à l’aide d’une centrifugeuse réfrigérée afin d’obtenir le deuxième surnageant (S10) et le culot (P10).
NOTE : P10 est une boulette de membrane brut qui contient des mitochondries et des synaptosomes. Jeter les P10. - Transférer le surnageant (S10) dans une bouteille de polycarbonate (> 1 mL) et centrifuger S10 à 100 000 × g, pendant 1 h à 4 ° C à l’aide d’une ultracentrifugeuse et un rotor à angle fixe (90 Ti).
Remarque : Le surnageant est la fraction pure cytosol tandis que le culot, P100, contient agrégats αS microsomes-associés. - Resuspendre le culot P100 avec 500 µL de tampon de l’homogénéisation. Transférer P100 dans un tube propre microcentrifuge et centrifuger à 10 000 x g pendant 20 min à 4 ° C dans une centrifugeuse réfrigérée.
- Jeter le surnageant et remettre en suspension P100 avec 100 µL de tampon de l’homogénéisation.
Remarque : Cette fraction est les agrégats αS microsomes-associés. - Soniquer échantillons pour 2 s sur la glace [sortie puissance 1 watt (RMS)]. Conserver les échantillons à-80 ° C.
- Le lendemain, déterminer la quantité de protéines en utilisant l’analyse de la BCA.
2. Western Blot
Remarque : La caractérisation biochimique des agrégats αS associées aux microsomes est évaluée par Western Blot.
- Monter un appareil d’électrophorèse verticale à un gel de polyacrylamide de Tris-glycine dégradé 4 à 20 %.
- Charge 1 µg de fractions αS associées aux microsomes, dissous dans la solution tampon dénaturation.
- Dans un autre puits, charger 5 µL de marqueur standard de protéines.
- Exécutez le gel à 100 V en un Tris/glycine/SDS exécutant tampon marqueur protéique jusqu'à la fin du gel.
- Protéines de transfert sur une membrane de nitrocellulose, à l’aide d’un tampon de carbonate basique (10 mM de l’OCNA3, 3 mM NaHCO3, 20 % méthanol) O/N à 4 ° C, à raison de 200 mA, constante.
- Bloquer la membrane avec PBS-non ioniques tensioactif 0,05 % (PBS-T) avec 5 % non-lait écrémé dans un agitateur orbital pendant 30 min à température ambiante.
- Rincez brièvement, la membrane avec PBS-T.
- Incuber la membrane avec Syn-1 (1:5 000) ou anticorps pSer129-αS (1:5, 000) à 2,5 % non-lait écrémé en PBS-T, O/N à 4 ° C dans un agitateur orbital.
- Laver la membrane pendant 10 min à RT avec PBS-T dans un agitateur orbital.
- Incuber la membrane avec anti-souris ou anti-lapin HRP-conjugués anticorps secondaire (1:3, 000) à 2,5 % non-lait écrémé en PBS-T pendant 1 h à température ambiante.
- Laver la membrane pendant 10 min à RT avec PBS-T dans un agitateur orbital. Répéter 3 x.
- Obtenir le signal à travers les kits de chimiluminescence ordinaire.
3. principales Cultures de neurones
Remarque : Les cultures primaires de neurones ont été préparés de la WT nouveau-né (P0) souris hippocampe ou cortex (ligne C57BL/6). L’ensemble de la procédure, moins les étapes de centrifugation, est effectuée sous une hotte de culture cellulaire, dans des conditions stériles.
- Traiter des lamelles couvre-objet (Ø 18 mm) avec la solution d’acide nitrique 65 % pendant au moins 12 h à température ambiante.
- Retirer l’acide nitrique. Rincer deux fois avec du PBS 10 x et plusieurs fois avec de l’eau distillée lamelles couvre-objet.
- Insérer les lamelles couvre-objet en 24 paraboloïdes et enduisez-les avec poly-D-lysine (0,1 mg/mL dans l’eau distillée stérile ou PBS 1 x) pendant 1 h à 37 ° C.
- Enlever poly-D-lysine et laver les lamelles couvre-objet trois fois avec de l’eau distillée. Stocker à 4 ° C jusqu'à ce que nécessaire.
- Euthanasier les souriceaux par décapitation et séparer les têtes des corps. Placez les têtes sur la vaisselle et disséquer doucement la peau.
- À l’aide des ciseaux, ouvrir le crâne en faisant une incision à la base du cerveau. Séparer les deux moitiés des crânes et retirez-les avec précaution.
- À l’aide de pinces, pincez les cerveaux des bases et isoler le cortex et l’hippocampe. Transférez-les dans deux plats séparés contenant Balanced Salt Solution (HBSS) un milieu de Hank contenant 1 % pénicilline/streptomycine.
- Répétez les étapes 3.6 et 3.7 pour chaque animal. Sachez que l’ensemble du processus ne devrait pas prendre plus de 30-45 min.
- Émincer les tissus prélevés et transférez-les dans deux tubes conique de 50 mL (un pour l’hippocampe) et l’autre pour cortex avec 10 mL de milieu HBSS contenant 0,1 % de trypsine. Incuber dans un bain-marie pendant 7 min à 37 ° C.
Remarque : Aucune agitation n’est nécessaire. - Ajouter 1 mL de DMEM contenant 10 % sérum fœtal (SVF) et 10 µg/mL DNase à l’homogénat pour bloquer l’activité de la trypsine.
- Centrifuger le tissu résultant dissocié à 200 × g pendant 5 min à RT sur une centrifugeuse et éliminer le surnageant.
Remarque : Le pellet représente les cellules disséqués par le tissu. - Remettre en suspension les cellules dans le milieu de l’électrodéposition, contenant 2 % B27, 2 mM de glutamine, glucose 6 mg/mL, 10 % FBS, 12,5 µM glutamate et la gentamicine 10 µg/mL.
- Plaque dissociée des neurones sur lamelles de poly-D-Lysine dans 24 plaques bien selon le ratio : puits de cortex 1/12 et 1 hippocampe/6 puits, entraînant environ 150 000/200 000 cellules par puits. Maintenir les cellules dans un incubateur à 37 ° C.
- Le lendemain (jour in vitro, 1 DIV), remplacer le support de placage avec le milieu contenant 2 % B27, 10 gentamicine µg/mL et 2 mM de glutamine.
- DIV 2, enlever 1/3 du milieu et ajouter 1/3 du milieu frais contenant 2,5 µM cytosine arabinoside pendant 48 h afin de réduire la contamination gliale.
- Maintenir les neurones à 37 ° C et remplacer la moitié de la moyenne tous les trois jours.
4. les neurones traitement
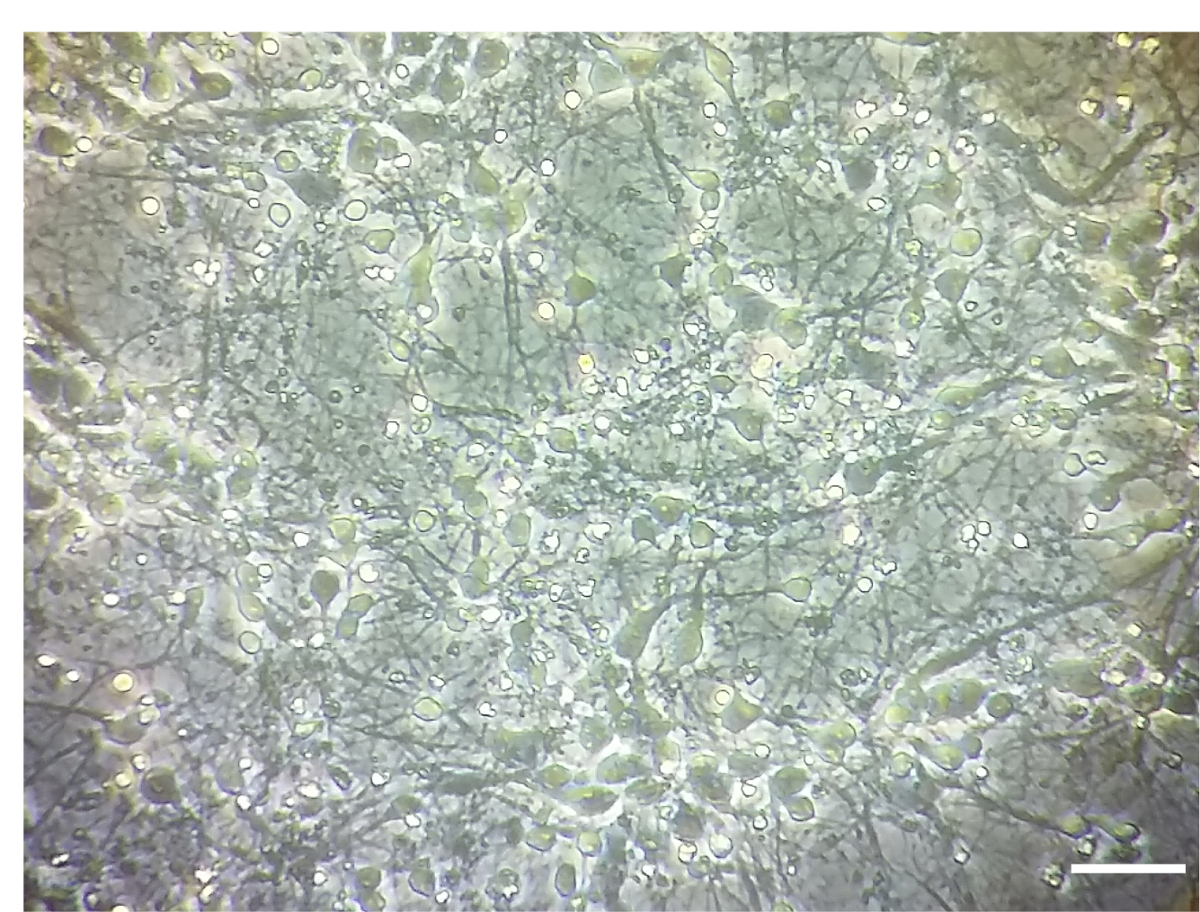
Remarque : Le traitement a été joué dans DIV 7. Toutes les étapes sont exercées sous une hotte de culture de cellules dans des conditions stériles. Un exemple de la densité neuronale corticale de la culture à la DIV 7 est illustré à la Figure 1.
- Agrégats αS associées aux microsomes de piscine provenant de la moelle épinière des trois souris Tg malades différentes afin d’avoir un 1 µg/µL de solution, à l’aide de la mémoire tampon d’homogénéisation original pour la dilution.
- Retirez 1/3 du milieu et doucement le remplacer par du milieu frais contenant 2 % B27, 1 x gentamicine et 2 mM de glutamine.
- Ajouter 1 µg d’agrégats communs associés aux microsomes αS dans le milieu cellulaire. Retourner les neurones dans l’incubateur à 37 ° C.
- Tous les 3 jours pendant 1 semaine, ajouter 1/3 du milieu frais. Ne remplacez pas le milieu. Juste l’ajouter.
- Après 1 semaine de traitement, enlever 1/3 du milieu et doucement le remplacer par du milieu frais contenant 2 % B27, 10 gentamicine µg/mL et 2 mM de glutamine. Répéter tous les 3 jours.
- Difficulté les neurones après 2 semaines de traitement (DIV 21).
5. immunofluorescence
- Difficulté des neurones avec paraformaldéhyde à 2 % dans du PBS 1 x et solution de sucrose de 5 % pendant 15 min à RT sans agitation, en vertu d’un produit chimique des fumées hotte.
- Enlever la solution de fixation. En bref, laver avec du PBS 1 x, 3 fois.
Remarque : Effectuez toutes les étapes de lavage doucement car les neurones primaires ne collent pas fermement à lamelles poly-lysine. - Permeabilize des neurones avec 0,3 % de tensioactifs non ioniques dans du PBS 1 x pendant 5 min à température ambiante.
- En bref, laver avec du PBS 1 x, 3 fois.
- Incuber les neurones avec 3 % FBS dans du PBS 1 x pendant 30 min à RT, de bloquer les sites de liaison non-spécifique, dans un agitateur orbital.
- Incuber les neurones avec des anticorps primaire approprié dissous dans 3 % FBS dans du PBS 1 x, O/N à 4 ° C dans un agitateur orbital.
NOTE : syn303 (1:1 000), la souris αS (1 : 200), pser129-αS (1:1, 000) et le Tau (01:10, 000) anticorps ont été utilisés. - Supprimer la solution d’anticorps et de lavage, brièvement, avec du PBS 1 x, 3 fois.
- Incuber les neurones avec des anticorps secondaire fluorescent approprié dissous dans 3 % FBS dans du PBS, pendant 1 h à RT dans l’obscurité dans un agitateur orbital.
- Supprimer la solution d’anticorps et de lavage, brièvement, avec du PBS 1 x, 3 fois.
- Colorer les neurones avec solution DAPI (0,1 µg/mL dans du PBS 1 x) pendant 15 min à ta dans le noir dans un agitateur orbital.
- Supprimer la solution d’anticorps et de lavage, brièvement, avec du PBS 1 x, 3 fois.
- Monter les lamelles couvre-objet sur une diapositive à l’aide du milieu de montage antifade.
Résultats
Suivant le protocole décrit ci-dessus et résumées dans la Figure 2, nous avons purifié agrégats αS associées aux microsomes de trois malades A53T les souris αS Tg (Figure 3). Les microsomes sont des fractions de pellet membranaire brute qui contiennent le réticulum endoplasmique, appareil de Golgi et petites vésicules synaptiques. Le degré de pureté de la pastille microsomique par rapport aux autres fractions a été évalué précédemment à l’aide de marqueurs organite spécifique18.
Une fois isolés, caractérisation biochimique des agrégats αS est évaluée au moyen de dénaturant SDS-Page, suivie d’une incubation avec Syn-1 ou αS phosphorylés à sérine 129 (pSer129-αS) anticorps. Comparativement à présymptomatique (PreS) et appariés selon l’âge des souris-Tg (nTg), microsomes isolés de souris malades précipité Co avec des agrégats αS. Les espèces associées aux microsomes αS montrent les caractéristiques typiques des agrégats αS, tels que l’accumulation des espèces résistant aux détergents HMW, phosphorylation de sérine 12919 et C-N-terminal écimée fragments (pour une caractérisation complète de agrégats de microsomes-associated αS voir référence18,20,21). Ce sont des exigences fondamentales car αS monomère ne pas obtenir efficacement internalisé et n’induit pas de αS dépôt7,15,22. Il est important de ne pas utiliser n’importe quel détergent (ionique ou non ioniques) pour remettre en suspension les granulés P100 puisqu’il peut être nocif pour les cellules. Aussi, afin d’éviter la variabilité de l’échantillon, agrégats αS associées aux microsomes de trois différentes souris malades vont être regroupés pour traitement neuronal.
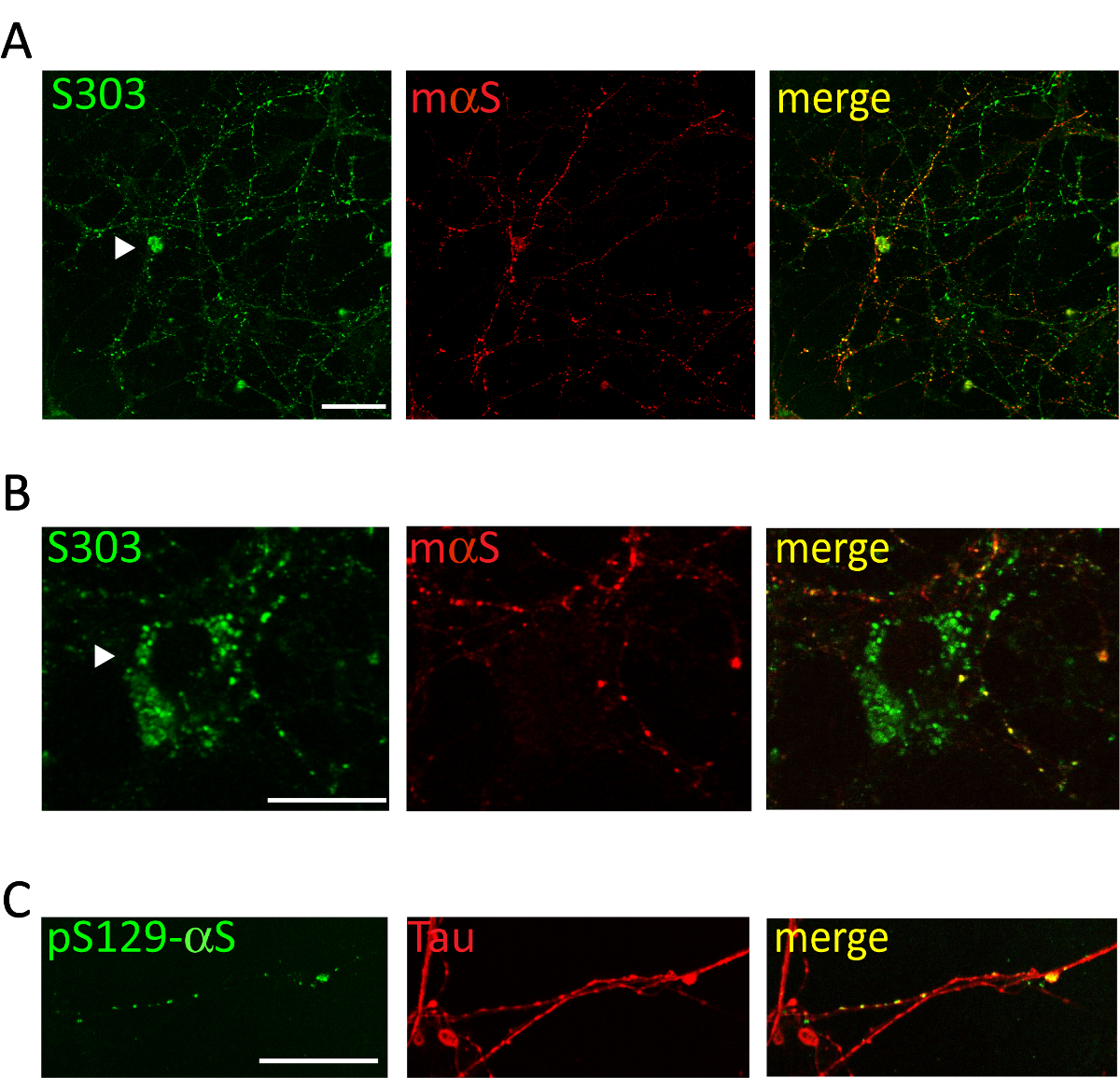
Administration de 1 µg d’agrégats communs associés aux microsomes αS des malades A53T souris au milieu de culture de neurones corticaux ou hippocampe induit la formation d’une dépendant du temps des inclusions αS, positives pour les anticorps αS agrégats spécifiques tels que Syn303 (Figure 4, 5) ou pser129-αS (Figure 5). Après deux jours (2d) de ces agrégats apparaissent comme des punctums petits, épars qui deviendront plus abondantes aux périodes ultérieures de traitement. Après deux semaines, inclusions αS ressemblent à longs et mûrissent structures ressemblant à des perles, largement répartis dans les cultures de neurones, suivant un modèle de neurites et partiellement co localisation avec présynaptique et marqueurs de neurites (Figure 5). Occasionnellement, αS nouvellement formé des inclusions sont visibles à localiser conjointement et tacher le soma de cellule ou couvrir l’ensemble du processus, ressemblant à des neurites nécrotiques.
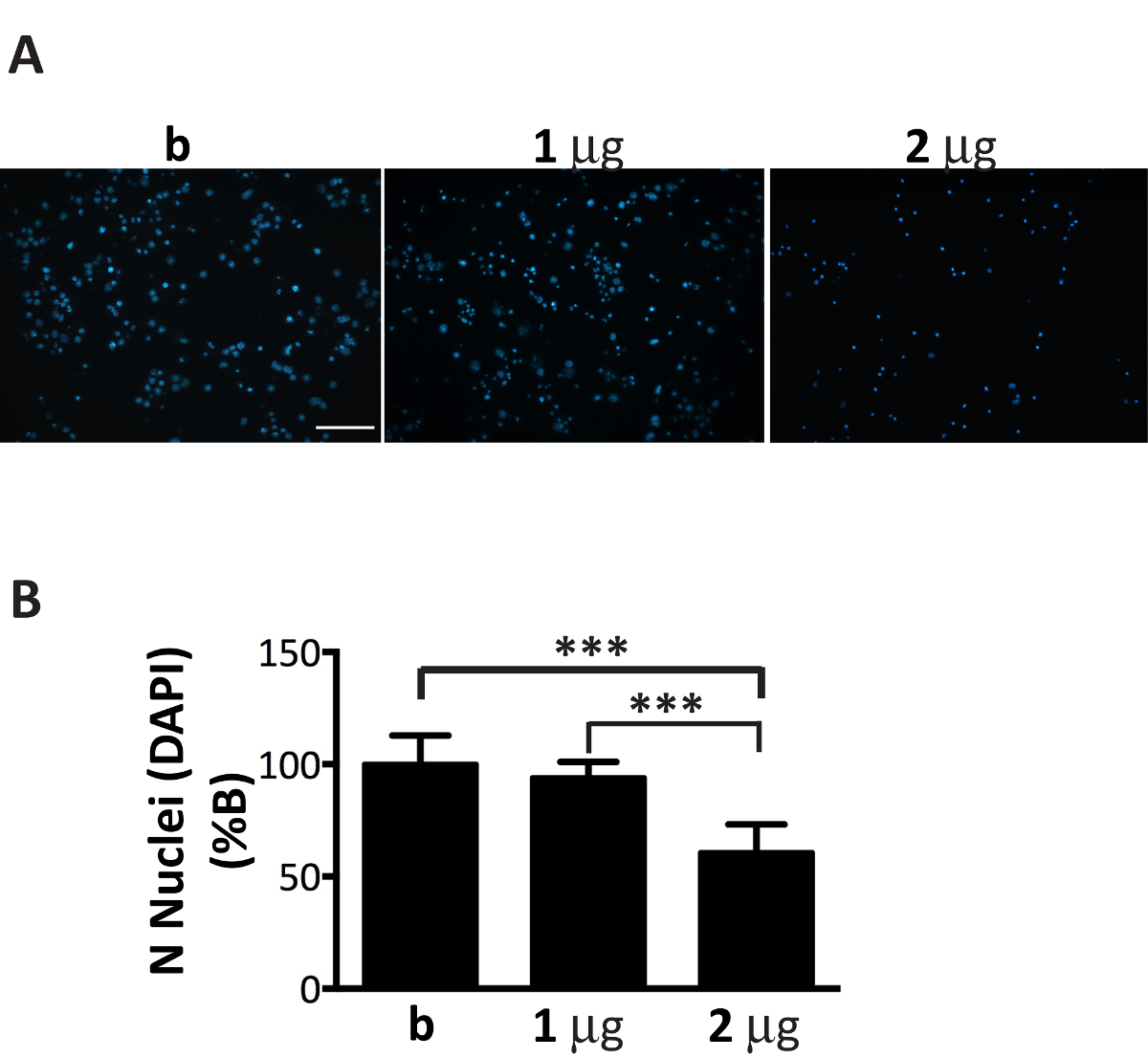
Bien qu’associés aux microsomes αS agrégats fractions peuvent se propager efficacement dans les cultures de neurones, leur montant doit être finement réglé pour le nombre de neurones plaqué (Figure 1). Dépassant en fait, le ratio recommandé, µg de microsomes associées à des agrégats : nombre de neurones, va entraîner la mort prématurée des cellules dans les prochains jours (Figure 6), alors qu’une quantité insuffisante d’agrégats associées aux microsomes αS conduira à une rare et réduit nombre d’inclusions après deux semaines de traitement, similaire à ce qui a été obtenue aux périodes précédentes (Figure 4A).

Figure 1 . Cultures de neurones corticaux. L’image représentative de densité DIV 7 des cultures de neurones corticaux. Des images ont été prises avec un microscope inversé léger, objectif 10 X. Echelle = 100 µm. s’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 2 . Isolement des microsomes-associated αS agrège de souris SpC. Organigramme du protocole de purification des agrégats αS associées aux microsomes de SpC de souris malades. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 3 . Isolement des fractions de microsomes de malades, présymptomatiques A53T αS Tg nTg souris et. Affichage de l’analyse par transfert Western purifiée agrégats αS associées aux microsomes isolés de trois malades (malade), présymptomatiques (PreS) et des souris nTg appariés selon l’âge. Les souris malades sont souris de Tg αS A53T qui montrent le dysfonctionnement moteur et neurologique, y compris l’accumulation des inclusions αS, tandis que présymptomatique animaux est en bonne santé A53T αS Tgs de 9 mois d’âge qui ne présentent pas encore de toute pathologie αS associés phénotype. nTg souris sont ceux des souris malades qui ne portent pas le transgène αS et donc ne se développent pas αS induits par la pathologie. 1 µg de chaque fractions purifiées ont été exécutées sur un dénaturant SDS-Page, transféré sur une membrane de nitrocellulose et effacé avec Syn-1 ou pSer129-αS anticorps. Seulement des fractions de microsomes isolement de souris malades des agrégats de détersif-résistante αS HMW confinées phosphorylés à sérine 129 et a montré C- et N-terminale troncation. Ce chiffre a été adapté de Colla et al. 18. s’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 4 . Induction dépendant du temps des dépôts αS après administration d’agrégats αS microsomes-associés. (A) Immunofluorescence des neurones de l’hippocampe primaires traités avec 1 µg de microsomes-associated αS agrège les fractions regroupées à partir de trois différentes de souris malades. Les neurones ont été fixés à 2 jours (2d), 1 semaine (1w) ou 2 semaines (2w) de traitement et immunomarquage avec syn303 (S303, 1/1000), un anticorps spécifique pour oxydé et agrégées αS. Les cellules étaient Eosine au DAPI. Images confocales ont été prises à l’aide d’un laser scanning microscope confocal, 63 X, objectif. Echelle = 50 µm. (B) analyse Quantitative de la fluorescence totale, après soustraction du fond, a été fait en utilisant le plugin de comptage de particules du logiciel Image J. Les valeurs ont été normalisées pour le nombre de noyaux par champ (DAPI count) et exprimées en pourcentage du signal de fluorescence S303 à 2D. Les valeurs sont indiquées comme la moyenne ± écart-type (n = 5). ** p < 0,001, *** p < 0,00001, unidirectionnel ANOVA, suivie de LSD posteriori Fisher. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 5 . Des inclusions intracellulaires αS sont colocalisées avec réseau cortical neurites. Images confocales représentatifs des neurones corticaux traitement avec agrégats αS associées aux microsomes provenant de souris malades de Tg. Après que 2 semaines de neurones de traitement étaient fixes et double colorées avec des anticorps spécifiques agrégats [S303 (A, B) ou pser129-αS, 1 : 1 000 (C)] et marqueurs neurites [souris αS, 1 : 200 (A, B) ou Tau, 1/10 000 (C)]. Co, étiquetage des signaux fluorescents démontré co-localisation partielle des structures ressemblant à des perles αS nouvellement formé avec le réseau des neurites. Parfois des inclusions αS s’accumulent dans le soma neuronal (A, B, pointes de flèches). Images superposées ont été acquises avec un laser scanning microscope confocal, 63 X, objectif. Echelle = 50 µm. Ce chiffre a été modifié par Colla et al. 20. s’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 6 . Addition d’une quantité sous-optimal des agrégats αS associées aux microsomes est toxique pour les neurones. Coloration DAPI des neurones traités avec augmentation de la concentration des agrégats associées aux microsomes αS mène à la mort cellulaire. (A) les neurones corticaux ont été traités avec 1, 2 µg d’agrégats associées aux microsomes αS extraite de souris malades ou tampon seul qui ne contient-elle pas d’agrégats (B) et colorées au DAPI. Les images fluorescentes ont été acquises avec un microscope épifluorescente en utilisant un objectif 20 X. Echelle = 100 µm.()B) cellules DAPI-positives ont été dénombrées à l’aide du logiciel Image J. Le graphique montre une réduction du nombre des noyaux avec augmentation de la concentration des agrégats associées aux microsomes αS ajouté aux médias neuronales. Les valeurs sont exprimées en % de b et sont donnés comme la moyenne ± écart-type (n = 5), *** p < 0,0001, unidirectionnel ANOVA, suivie de LSD posteriori Fisher. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}
Discussion
Nous avons décrit une méthode pour obtenir formation d’inclusions αS dans les cultures primaires de neurones encéphales de souris WT, grâce à l’ajout des agrégats purifiée αS associées aux microsomes isolés des modèles animaux de αS Tg.
Les étapes critiques du présent protocole sont les suivants : le ratio de µg de microsomes-associated αS agrégats/neurones et la source des agrégats αS. Comme indiqué dans la session de résultats, il est essentiel optimiser le ratio de µg de microsomes-associated αS agrégats/nombre de neurones puisque travaillant dans des conditions sous-optimales peut conduire à la mort prématurée des cellules ou les trop rare agrégation intracellulaire (voir le Section résultats représentatifs). Pour cette raison, il est très important d’évaluer la densité de la culture neuronale à 7 DIV (illustré à la Figure 1) avant de commencer le traitement. En outre, agrégats αS doivent être purifiés de souris de Tg αS malades, c’est à dire. des modèles animaux qui s’accumulent inclusions LB-comme caractérisent par phosphorylées et détergent αS insolubles de fibrilles HMW.
Les modifications éventuelles au présent protocole considèrent le tissu, congelé ou fraîche, les microsomes peuvent être isolés et le montant de départ. Alors que nous avons utilisé des souris SpC de Tg en raison de la forte teneur des agrégats insolubles αS, le protocole est conçu pour isoler les microsomes associées à des agrégats de n’importe quel tissus, pourvu que la région a une teneur élevée en agrégats αS. Les échantillons congelés peuvent également servir comme gel n’affecte pas les étapes de purification des microsomes associées à des agrégats ou des agrégats en soi. Alors que le poids de départ recommandé pour tissus est environ 100-150 mg, ce protocole est convenable pour l’obtention de microsomes d’aussi faibles que 50 mg de matière première (pas de limite de poids maximum). Dans le cas de montant inférieur à 100 mg, cependant, le ratio approprié d’homogénéisation sera 01:20 (p/v) afin d’avoir au moins 1 mL du surnageant S10 à charger sur la bouteille en polycarbonate pour la précipitation ultracentrifugeuse. En fait, chargement des volumes inférieurs à 1 mL peut entraîner l’effondrement de la perte de tube et échantillon. Augmentation du volume d’homogénéisation conduira à un surnageant plus dilué, mais la concentration de la pastille microsomique restera inchangée.
Une limitation du présent protocole concerne l’ensemencement des croix inefficace dans la formation d’inclusions αS qui ont été récemment signalés dans l’administration des litiges de FFP αS d’origine humaine à des cultures de neurones murins par opposition aux souris αS FFP24. Étant donné que la hausse du montant des fibrilles exogène αS donné aux cultures murines peut contourner ce problème, nous vous recommandons de régler finement la quantité de microsomes associées à des agrégats dans le cas de l’administration de fibrilles issues d’autres variantes αS ou d’autre espèces de cultures de neurones de souris que ce que nous avons décrit.
Agrégats αS exogènes ajoutés pour les milieux de culture peuvent être de différentes sources. In vitro αS FFP ont été précédemment utilisés comme modèle de semis d’agrégats αS intracellulaire dans les cultures de cellules, neurones primaires et des modèles animaux3,7,8,15. Par rapport à notre méthode où les agrégats αS associées aux microsomes peuvent être isolés en quelques heures, la formation de FFP est longue et laborieuse, nécessitant plusieurs étapes de purifications, suivies d’essais supplémentaires pour vérifier αS agrégats confomations25 . En outre, FFP étant obtenu de homme exprimés par les bactéries ou les souris αS, c'est-à-dire sans modifications post-traductionnelles typiques des eucaryotes, peuvent présenter des conformations différentes, avec ensemencement sélectif et propriétés pathogènes, conformément à les protocoles de nucléation suivi (p. ex. rubans vs fibrilles)5,8 , conduisant à des conclusions et des résultats différents. Au lieu de cela, l’administration unique de in vivo purifiée αS agrégats garantit la transmission des modèles de pathogènes plus authentiques, imitant étroitement le processus de formation d’inclusions αS dans des modèles animaux et les patients Parkinsoniens.
Comme une application future de cette technique, nous croyons que ce protocole peut être avec succès utilisé pour isoler les graines pathogènes αS le cerveau de patients Parkinsoniens ou autres modèles animaux de la Tg αS, pourvu que les malade zone d'où les microsomes sont isolés sont riches en αS inclusions.
À notre connaissance, c’est la première méthode qui permet la purification des espèces toxiques indigènes de αS en vivo modèles PD pour servir de modèle pour obtenir la formation d’inclusions αS dans des neurones primaires de l’ensemencement.
Nous croyons que cette méthode est extrêmement polyvalente et peut fournir un modèle exceptionnel à base de cellules pour étudier les différents aspects de l’agrégation αS et son influence sur la physiopathologie de la cellule. Parce que la formation d’inclusions αS représentent un processus complexe qui a été difficile de reproduire dans les cellules cultivées, nous avons bon espoir que ce modèle offrira de grandes intuitions dans les mécanismes pathogéniques aiguës, difficiles à identifier dans les chroniques et plus élaborées les systèmes sont telles que les modèles animaux.
Déclarations de divulgation
Les auteurs n’ont rien à divulguer.
Remerciements
Ce travail a été soutenu par le ministère italien de l’Université et la recherche (MIUR) à travers le programme de subventions de carrière réinsertion (programme RLM pour jeune chercheur) et de la Scuola Normale Superiore. Nous remercions Prof Michael Lee de l’Université du Minnesota, é.-u., pour fournir le αS Prp humain A53T souris Tg, d'où les agrégats sont isolés.
matériels
| Name | Company | Catalog Number | Comments |
| Sucrose | Sigma-Aldrich | 84097-1KG | |
| Hepes | Sigma-Aldrich | H0887-100ML | 1M pH=7-7.6 |
| EDTA | Sigma-Aldrich | 0390-100ml | pH=8 0.5M |
| MgCl2 | Sigma-Aldrich | M8266-100G | |
| NaCO3 | Sigma-Aldrich | S7795-500G | |
| NAHCO3 | Sigma-Aldrich | S5761-500G | |
| Methanol | Sigma-Aldrich | 322415-6X1L | |
| KCl | Sigma-Aldrich | P9541-500G | |
| cOmplete Mini | Roche | 11836170001 | protease inhibitor |
| PhosStop | Roche | 4906837001 | phosphatase inhibitor |
| BCA Protein Assay Kit | Euroclone | EMPO14500 | |
| Criterion TGX 4-20% Stain Free, 18 wells | Biorad | 5678094 | |
| Supported Nitrocellulose membrane | Biorad | 1620097 | 0.2 μm |
| Blotting-Grade Blocker | Biorad | 1706404 | Non-fat dry milk |
| SuperSignal West Pico Chemiluminescent Substrate | Termo Fisher Scientific | 34077 | |
| Nitric acid | Sigma-Aldrich | 1004411000 | 65% |
| Glass Coverslips | Termo Fisher Scientific | 1014355118NR1 | 18 mm x |
| Poly-D-Lysine | Sigma-Aldrich | P7280 | |
| Hank's Balanced Salt Solution | Termo Fisher Scientific | 14170-500 mL | |
| Penicillin/Streptomycin | Termo Fisher Scientific | 15140122 | 10,000 U/mL, 100 mL |
| Dulbecco’s Modified Eagle’s Medium | Termo Fisher Scientific | D5796-500 mL | |
| Trypsin-EDTA | Termo Fisher Scientific | 15400054 | 0.50% |
| B27 Supplement | Termo Fisher Scientific | 17504044 | 50X |
| Glutamax | Termo Fisher Scientific | 35050-038 | 100x |
| DNAse | Sigma-Aldrich | D5025 | |
| Fetal bovine serum | Euroclone | EC50182L | |
| Glutamate | Sigma-Aldrich | 1446600-1G | |
| Gentamicin | Termo Fisher Scientific | 15710 | 10 mg/ml |
| Neurobasal Medium | Termo Fisher Scientific | 10888-022 | |
| Cytosine arabinoside (AraC) | Sigma-Aldrich | C3350000 | |
| VECTASHIELD antifade mounting medium | Vector Laboratories | H-1000 | |
| DAPI | Termo Fisher Scientific | 62247 | |
| 90 Ti rotor | Beckman | N/A | Ultracentrifuge rotor |
| Optima L-90K Ultracentrifuge | Beckman | N/A | |
| Syn-1 antibody, clone 42 | BD Biosciences | 610786 | anti-mouse WB: 1:5000 |
| Syn303 antibody | BioLegend | 824301 | anti-mouse IF: 1:1000 |
| Tau antibody | Synaptic Systems | 314 002 | anti-rabbit IF: 1:10,000 |
| pser129-αS antibody | A gift from Fujiwara et al, reference 19 | anti-rabbit WB: 1:5000 | |
| pser129-αS antibody | Abcam | ab51253 | anti-rabbit IF: 1:1000 |
| Mouse αS (D37A6) XP | Cell Signaling | 4179 | anti-rabbit IF 1:200 |
| Alexa fluor 555-conjugated anti-rabbit antibody | Termo Fisher Scientific | A27039 | |
| Alexa fluor 488-conjugated anti-mouse antibody | Termo Fisher Scientific | A-11029 | |
| Microson XL-2000 | Misonix | Sonicator | |
| Ultra Bottles (Oakridge Bottles), PCB, 16x76mm, Assembly, Noryl Cap, Beckman-type | Science Service EU | S4484 | Ultracentrifuge tubes |
| AXIO Observer Inverted Light Microscope | Zeiss | N/A | |
| TCS SP2 laser scanning confocal microscope | Leica | N/A | |
| Inverted epi-fluorescence microscope | Nikon | N/A | |
| Triton x-100 | Sigma-Aldrich | X100-500ML | Nonionic surfactant |
Références
- Goedert, M., Spillantini, M. G., Del Tredici, K., Braak, H. 100 years of Lewy pathology. Nat. Rev. Neurol. 9, 13-24 (2012).
- Visanji, N. P., et al. α-Synuclein-Based Animal Models of Parkinson's Disease: Challenges and Opportunities in a New Era. Trends Neurosci. 39, 750-762 (2016).
- Luk, K. C., et al. Pathological α-Synuclein Transmission Initiates Parkinson-like Neurodegeneration in Nontransgenic Mice. Science. 338, 949-953 (2012).
- Rey, N. L., Petit, G. H., Bousset, L., Melki, R., Brundin, P. Transfer of human α-synuclein from the olfactory bulb to interconnected brain regions in mice. Acta Neuropathol. (Berl). 126, 555-573 (2013).
- Guo, J. L., et al. Distinct α-Synuclein Strains Differentially Promote Tau Inclusions in Neurons. Cell. 154, 103-117 (2013).
- Masuda-Suzukake, M., et al. Prion-like spreading of pathological α-synuclein in brain. Brain. 136, 1128-1138 (2013).
- Volpicelli-Daley, L. A., et al. Exogenous α-Synuclein Fibrils Induce Lewy Body Pathology Leading to Synaptic Dysfunction and Neuron Death. Neuron. 72, 57-71 (2011).
- Peelaerts, W., et al. α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature. 522, 340-344 (2015).
- Lázaro, D. F., Pavlou, M. A. S., Outeiro, T. F. Cellular models as tools for the study of the role of alpha-synuclein in Parkinson's disease. Exp. Neurol. 298, 162-171 (2017).
- Lee, H. -. J., Patel, S., Lee, S. -. J. Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J. Neurosci. Off. J. Soc. Neurosci. 25, 6016-6024 (2005).
- Luk, K. C., et al. Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegenerative α-synucleinopathy in mice. J. Exp. Med. 209, 975-986 (2012).
- Mougenot, A. -. L., et al. Prion-like acceleration of a synucleinopathy in a transgenic mouse model. Neurobiol. Aging. 33, 2225-2228 (2012).
- Recasens, A., et al. Lewy body extracts from Parkinson disease brains trigger α-synuclein pathology and neurodegeneration in mice and monkeys: LB-Induced Pathology. Ann. Neurol. 75, 351-362 (2014).
- Woerman, A. L., et al. Propagation of prions causing synucleinopathies in cultured cells. Proc. Natl. Acad. Sci. 112, 4949-4958 (2015).
- Luk, K. C., et al. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc. Natl. Acad. Sci. U. S. A. 106, 20051-20056 (2009).
- Sacino, A. N., et al. Intramuscular injection of α-synuclein induces CNS α-synuclein pathology and a rapid-onset motor phenotype in transgenic mice. Proc. Natl. Acad. Sci. U. S. A. 111, 10732-10737 (2014).
- Lee, M. K., et al. Human alpha-synuclein-harboring familial Parkinson's disease-linked Ala-53 --> Thr mutation causes neurodegenerative disease with alpha-synuclein aggregation in transgenic mice. Proc. Natl. Acad. Sci. U. S. A. 99, 8968-8973 (2002).
- Colla, E., et al. Endoplasmic Reticulum Stress Is Important for the Manifestations of -Synucleinopathy In Vivo. J. Neurosci. 32, 3306-3320 (2012).
- Fujiwara, H., et al. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 4, 160-164 (2002).
- Colla, E., et al. Toxic properties of microsome-associated alpha-synuclein species in mouse primary neurons. Neurobiol. Dis. 111, 36-47 (2018).
- Colla, E., et al. Accumulation of Toxic -Synuclein Oligomer within Endoplasmic Reticulum Occurs in -Synucleinopathy In Vivo. J. Neurosci. 32, 3301-3305 (2012).
- Volpicelli-Daley, L. A., Luk, K. C., Lee, V. M. -. Y. Addition of exogenous α-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous α-synuclein to Lewy body and Lewy neurite-like aggregates. Nat. Protoc. 9, 2135-2146 (2014).
- Li, W., et al. Aggregation promoting C-terminal truncation of alpha-synuclein is a normal cellular process and is enhanced by the familial Parkinson's disease-linked mutations. Proc. Natl. Acad. Sci. U. S. A. 102, 2162-2167 (2005).
- Luk, K. C., et al. Molecular and Biological Compatibility with Host Alpha-Synuclein Influences Fibril Pathogenicity. Cell Rep. 16, 3373-3387 (2016).
- Volpicelli-Daley, L. A., Kirik, D., Stoyka, L. E., Standaert, D. G., Harms, A. S. How can rAAV-α-synuclein and the fibril α-synuclein models advance our understanding of Parkinson's disease. J. Neurochem. 139, 131-155 (2016).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.