
Method Article
Analyse des Complexes protéiques dynamique assemblés sur et libéré de Biolayer interférométrie biocapteur à l’aide de la spectrométrie de masse et de microscopie électronique
Dans cet article
Résumé
Nous présentons ici un protocole visant à contrôler le montage et le démontage de la toxine du charbon à l’aide de biolayer l’interférométrie (BLI). Suite de montage/démontage sur la surface du biocapteur, les complexes de protéine importante sont dispensés de la surface pour la visualisation et l’identification des composantes des complexes à l’aide de la microscopie électronique et microanalyse, respectivement.
Résumé
Des protéines in vivo, font souvent partie de grands complexes macromoléculaires où spécificité de liaison et de la dynamique en fin de compte dicte les résultats fonctionnels. Dans cet ouvrage, la toxine du charbon de pre-endosomes est Assemblée et passée dans les endosomes complexes. Tout d’abord, le domaine N-terminal d’un facteur létal mutant de cystéine (LFN) est attaché à un biocapteur d’interférométrie (BLI) biolayer par le biais de disulfure de couplage dans une orientation optimale, permettant prepore antigène protecteur (AP) lier (Kd 1 nM). La LFN-PA idéalement orientéprepore complexe lie ensuite à capillaire morphogène gène-2 (CMG2) cell surface récepteur soluble (Kd 170 MP), résultant en un complexe de pre-endosomes anthrax représentatif, stable à pH 7.5. Ce complexe assemblé est ensuite soumis au représentant de l’acidification (pH 5,0) de l’environnement endosome tardif à la PAprepore de transition dans l’état de pore membranaire inséré. Cet état depore PA se traduit par un affaiblissement obligatoire entre le récepteur CMG2 et le LFN-PAdes pores et une dissociation importante du CMG2 de la pore de transition. La thio-saisie des LFN à la surface du biocapteur est facilement renversée par le dithiothréitol. Réduction sur la surface du biocapteur BLI libère le LFN-PAprepore-CMG2 complexe ternaire ou l’acide passés complexes depore LFN-PA dans les volumes de microlitre. Libéré des complexes sont ensuite visualisées et identifié à l’aide de la microscopie électronique et microanalyse. Ces expériences démontrent comment suivre la cinétique de montage/démontage des complexes de protéine spécifique à l’aide de méthodologies BLI exempte d’étiquette et d’évaluer la structure et l’identité de ces BLI assemblé complexes par microscopie électronique et de la masse spectrométrie, respectivement, à l’aide de procédures séquentielles facile à reproduire.
Introduction
Identifier et comprendre la spécificité régissant les protéines complexes d’assemblage en vivo sont d’un intérêt extrême pour les chercheurs biochimiques. Les assemblys de grosse protéine hétérogènes sont la norme plutôt que l’exception. Cette notion est pris en charge par surveillance spectroscopiques en vivo Assemblée, isoler les complexes à l’aide de techniques de désorganisation cellulaire plus douces, évaluation des produits, des méthodes de purification d’affinité-basé et visualiser les à l’aide de haute résolution microscopie cryogénique (cryo-EM). Pour comprendre le contrôle de la spécificité de l’Assemblée au sein de la cellule, chercheurs doivent systématiquement isoler, identifier et caractériser en fin de compte ces structures dynamiques de montage/démontage. L’outil moléculaire plus abondamment utilisé pour identifier les composants de ces assemblys fréquemment exige immunoprécipitation fondée sur les anticorps, qui repose sur la stabilité des complexes au cours de la désorganisation de la cellule. Différentes techniques analytiques couplées ont été récemment mis au point pour capturer des complexes des échantillons cellulaires en utilisant des approches de microfluidique basée, comme la résonance plasmonique de surface (SPR). Après le retrait de la surface de la SPR, ces échantillons ont été analysés par temps de désorption-ionisation laser assistée par matrice de vol (MALDI-TOF)1,2. Faire progresser cette méthodologie en utilisant les protocoles plus faciles permettra aux chercheurs de visualiser et de valider les prévisions complexes qui se produisent dans le milieu cellulaire. Étant donné que le SPR est un système microfluidique, problèmes découlent souvent de formation globale. Contourner ce problème exige la dilution de l’échantillon, qui à son tour, peut diminuer l’intégrité de complexes biologiques susceptibles de concentration.
Un avancement relativement récent en technologies sans étiquette est le développement de l' biolayer système d’interférométrie (BLI)3. Ces particulière axée sur la lumière réflectance émulent des systèmes identiques ou meilleures, un coincement de la SPR et résultats cinétiques à une fraction du coût3,4 surtout si monocanal unités sont utilisées. BLI mesure les changements dans les modèles d’interférence de lumière réfléchie entre une couche de référence (contrôle) et la surface biolayer (expérimentale). Le changement de phase en résultant est mesuré en temps réel comme une lecture cinétiques et quantitatives5. La surface du biocapteur, contenant des chimies d’immobilisation spécifique, est physiquement transférée entre les solutions, par opposition aux fluctuations de la mémoire tampon par des approches de microfluidique en SPR, pour la mesure par l’intermédiaire de déflexions de phase de longueur d’onde. Les effets de transfert de masse ne peuvent pas en agitant la solution. Contrairement à la SPR, ces systèmes sont très utiles dans l’évaluation des complexes à partir d’échantillons biologiques bruts. Le paramètre physique mesuré au cours d’expériences BLI principalement dépend d’un changement dans la masse ou l’épaisseur à la surface du biocapteur qui résulte de l’assemblage complexe de protéine ou de démontage.
Ces biocapteurs optiques de fibre sont faciles à utiliser et relativement peu coûteux. L’un des nouveaux aspects utiles de BLI est l’élimination facile des complexes de protéines nouvellement assemblés de la surface. Une application récente de cette méthode a permis à notre laboratoire pour observer la cinétique en temps réel du pH à grande échelle induit par réarrangement de structure de protéine du composant anthrax toxine antigène protecteur (AP) que la prepore (PAprepore) passés à son forme de pores (PAdes pores). Cette transition sur la pointe de biocapteur a été vérifiée à l’aide de la microscopie électronique (me)6. Enlèvement des complexes des surfaces de biocapteur évite des effets de dilution volume plus fréquemment rencontrés lors de la publication des complexes des surfaces puce lors de l’utilisation des systèmes microfluidiques.
Dans les travaux en cours, complexes de toxine d’anthrax sont assemblés et démontés sur la surface du biocapteur et puis relâchés en volumes de microlitre. Les composantes complexes qui en résultent sont validés orthogonalement à l’aide de EM et spectrométrie de masse (MS).
Protocole
1. assemblage des Complexes macromoléculaires définies sur des Surfaces de biocapteur Biolayer interférométrie (BLI)
-
Assemblage de la toxine d’anthrax prepore complexe sur une surface de biocapteur réactive amine PDEA-modifié
- Hydrater une amine réactive astuce de le biocapteur BLI de deuxième génération (AR2G) dans 250 µL d’eau pendant dix minutes.
- Fois de l’étape, répertoriés dans le tableau 1, sur le logiciel contrôlant l’unité BLI de programme (voir Table des matières).
- Démarrer le BLI géré par immersion de la pointe du biocapteur en 250 µL d’eau pendant 30 s pour mesurer un niveau de référence initial de biocapteur épaisseur et la densité.
- Activer le biocapteur en plongeant la pointe en 250 µL 50 NHS (N-hydroxysuccinimide) et 200 mM EDC (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide)) pendant 7 minutes.
- Immerger le biocapteur activé en 50 mM PDEA (2-(2-pyridinyldithio)ethanamine) dissous dans le tampon de borate 0,1 M (pH 8,5) pendant 5 minutes générer une surface thiol-réactive activée.
- Immerger le biocapteur thiol réactif activé dans 250 µL de solution contenant 100 nM E126C LFN 10 mM acétate de sodium pH 5.0, 100 mM NaCl, mettre en mémoire tampon pendant 5 minutes.
- Plongez la pointe LFN en 50 mM L-cystéine, 1 M NaCl, acétate de sodium 0,1 M, pH 5.0, pendant 4 minutes, pour étancher tout groupes thiol réactif gratuits réactifs restants.
- Plongez la pointe LFN trempée dans 50 mM Tris, 50 mM NaCl pendant 2 minutes pour établir la base de la mémoire tampon.
- Plongez la pointe LFN trempée dans prepore d’antigène protecteur 0,5 µM (PAprepore), 50 mM Tris, 50 mM NaCl pendant 5 minutes pour créer le LFN-PAprepore complexes.
- Une fois PAprepore est associé, retirez l’embout de la solution deprepore PA et plonger l’extrémité dans 50 mM Tris, 50 mM NaCl pendant 30 secondes évacuer toute non spécifiquement liée PAprepore.
- Immerger le LFN-PAprepore complexes en récepteur de CMG2 0,5 µM (sans le domaine transmembranaire), 50 mM Tris, 50 mM NaCl, pendant 5 minutes.
- Plongez le LFN-PAprepore -CMG2 complexes dans 50 mM Tris, 50 mM NaCl pendant 30 secondes pour évacuer toute CMG2 non lié au formulaire pré-endosomes complexe.
- Pour l’analyse de l’EM, libérer le LFN-PAprepore-CMG2 complexes de l’extrémité de biocapteurs par immersion de la pointe dans 5 µL de 50 mM DTT, 50 mM Tris, 50 mM NaCl, à l’intérieur d’un tube de PCR.
- Pour l’analyse du tandem MS des peptides du complexe, libérer le LFN-PAprepore-CMG2 complexes de l’extrémité du biocapteur en immergeant le biocapteur dans 5 µL 50 mM DTT, 6 M GuHCl (kératine-libre), 25 mM de bicarbonate d’ammonium pH 8.0 à l’intérieur d’une PCR tube . Ceci est exécuté sur un biocapteur différent que celle utilisée pour l’analyse de l’EM.
-
Assemblage de la toxine du charbon de pore complexe sur surface réactive biocapteur amine PDEA-modifiée
- Pour consulter le complexe après la transition de pH, effectuez les étapes 1.1.1-1.1.12 dans la section précédente pour générer la LFN-PAprepore-CMG2 complexe.
- Plonger l’extrémité du biocapteur dans un acétate pH 5.0 pendant 5 minutes pour initier la transition de la PAprepore transitionpore de PA de 10 mM. Cette transition est indiquée par l’augmentation de l’amplitude (environ 0,2 nm) suivie d’une baisse d’amplitude plus grande qui fait l’hypothèse d’être dissociation récepteur important ou complet dû à diminué affinité.
- Plonger le cône de LFN-PAdes pores en 50 mM Tris, 50 mM NaCl, pH 8,0, pendant 30 secondes à laver tampon acide.
- Pour l’échantillon pour analyse EM, plonger la pointe du biocapteur dans une solution contenant des micelles de 1,25 mM (2,5 mM MSP1D1, 25 mM Na-cholate, 162,5 mM POPC) pendant 5 minutes afin d’empêcher l’agrégation en solution après disulfure de sortie.
- Pour l’analyse de l’EM, libérer le LFN-PApore -Micelle complexe de l’extrémité du biocapteur par immersion de la pointe dans 5 µL de 50 mM DTT, 50 mM Tris, 50 mM NaCl à l’intérieur d’un tube de PCR.
- Pour l’analyse en tandem MS des peptides du complexe, release le LFN-PApoore complexe (pas de micelles) depuis le biocapteur tip en immergeant le biocapteur dans 5 µL 50 mM DTT, 6 M GuHCl (kératine-libre), 25 mM de bicarbonate d’ammonium pH 8.0 à l’intérieur d’une PCR tube. Ceci est exécuté sur un biocapteur différent que celle utilisée pour l’analyse de l’EM.
2. visualisation et validation sorti des assemblages macromoléculaires de biocapteurs BLI par microscopie électronique à colorant négatif
- Une grille de Cu 300 carbone enduit à décharge luminescente. Paramètres de décharge typique lueur sont 0,38 mBar pression de l’atmosphère stable, négatifs 15 mAmps, 20 secondes, puis évacuation d’air.
- Grille sécurisée entre une paire de pinces à épiler propre.
- Pipetter 4 µL d’échantillon complexe libéré dans le tube PCR sur la grille et laisser adsorption pendant 60 s.
- Mèche loin restant liquide avec un coin de papier filtre.
- Détachant la grille par pipetage 5 µL de formiate d’uranyle de 0,75 % 0,02 micron filtrée et mèche tache trop loin après 5 secondes. Permettre la grille à sécher à température ambiante.
- Découvre les grilles échantillon teinté à l’aide d’un microscope électronique à transmission.
3. identification des complet pré-endosomes Anthrax toxine complexes (LFN-Paprepore-CMG2) et spectrométrie de masse à l’aide de transition complexes (LFN-Pades pores sans CMG2).
- Diluer les échantillons libérés à 20 µL et incuber pendant 1 heure.
- Ajoutez 2 µL de 55 mM iodoacétamide, bicarbonate d’ammonium 25 mM, pH 8,0 et incuber pendant 1 heure à température ambiante dans l’obscurité (recouverte de papier d’aluminium).
- Diluer l’échantillon avec 100 µL de bicarbonate d’ammonium 25 mM, pH 8,0, pour réduire la concentration de chlorhydrate de guanidine inférieure à 1 M.
- Ajouter 5 µL de séquencer la trypsine grade modifié à 20 ng/µL et incuber à 37 ° C pendant la nuit.
- Ajouter l’acide acétique glacial à une concentration finale de 5 % pour réduire le pH à < 3, puis réduire le volume à 10 µL au concentrateur sous vide.
- Transférer la solution de peptide à la plaque de l’échantillon sur l’échantillonneur automatique de la BNC 1200 uHPLC.
- Charger 5 µL de la solution de peptide sur une colonne en phase inversée uHPLC montée sur la scène de l’ionisation de la SP.
- Laver la colonne avec 15 µL de 0,1 % d’acide formique à un débit maximal de 5 µL/min / pression maximale de 800 lb/po2.
- Éluer les peptides de la colonne C18 à phase inverse à un débit de 350 nL/min sur une période de 90 min à l’aide d’un dégradé linéaire de 5 à 40 % de solvant B à A + B (solvant A: 0,1 % d’acide formique ; solvant b : 95 % d’acétonitrile avec 0,1 % d’acide formique).
- Analyser des peptides élution en ligne à l’aide de tandem MS.
- Définissez la source d’ionisation à 2500 volts et température de transfert ionique à 250 ° C.
- Fonctionner en mode automatique pour effectuer en permanence une analyse MS suivie autant tandem MSMS que possible dans une période de 3 s, le spectromètre de masse à l’aide de CID et une énergie de collision normalisée de 35.
- Identifier les composants de peptides et des protéines à l’aide de méthodes standard. Pour ce travail, deux ensembles de peptide et protéine d’analyse ont été effectuées.
Résultats
La capacité de contrôler et de valider l’Assemblée des grands complexes macromoléculaires est une étape cruciale vers la compréhension de la spécificité et la fonctionnalité de biomoléculaire de grandes assemblées. Les résultats des méthodes présentées ci-dessus démontrent la facilité avec laquelle complexes grosse protéine (> 150 kDa masse) peuvent être assemblés à l’aide de biolayer l’interférométrie, tout en écoutant la cinétique et l’amplitude de l’Assemblée. La nature unique et compacte de la surface du biocapteur permet une analyse Assemblée prolongée, libérant des complexes assemblés dans petite périfusion. Ces périfusion permet de visualiser la structure physique initiale des complexes à l’aide de EM et de vérifier l’identité des composants complexes en utilisant MS. Un aperçu schématique de tout ce processus est illustré à la Figure 1.
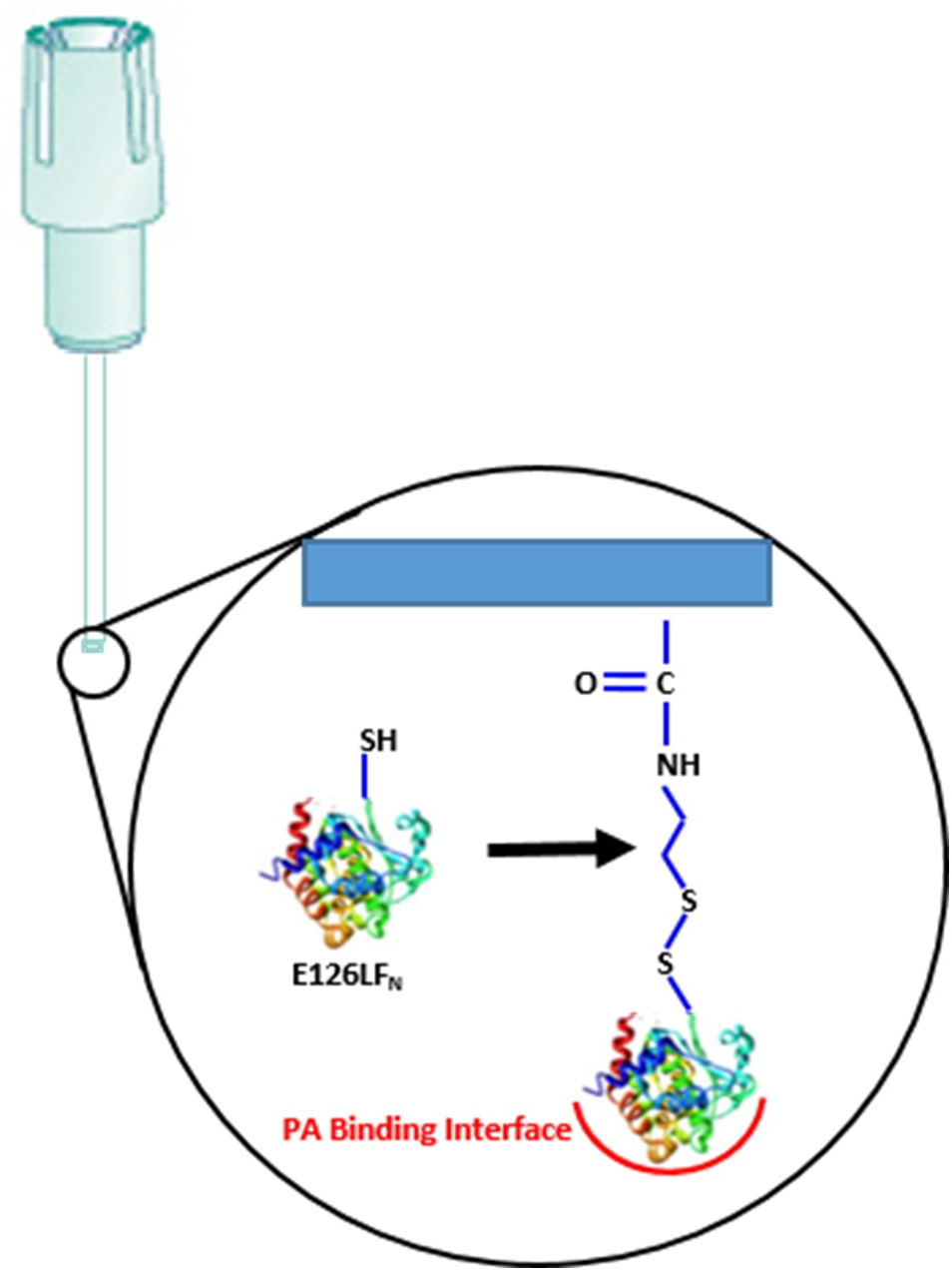
Un élément clé pour le succès de l’Assemblée et de la vérification du complexe macromoléculaire sur la surface du biocapteur implique l’orientation correcte de la protéine de la graine initiale. Cela garantit que les sites d’interaction de protéine-protéine sont accessibles, pas stériquement bloqués et idéalement placé loin de la surface du biocapteur. Comme illustré à la Figure 2, l’orientation correcte de la toxine du charbon complexe est obtenue en utilisant un fragment N-terminal spécifiquement machiné de facteur létal (LFN) donc le site de liaisonprepore LFN-PA est toujours positionné en face du biocapteur covalente site6,7. L’accumulation subséquente du complexe avec liaison du PA au biocapteur LFN BLI suivie soluble CMG2 liaison vers PA crée finalement une toxine d’anthrax compétente de translocation complexe.
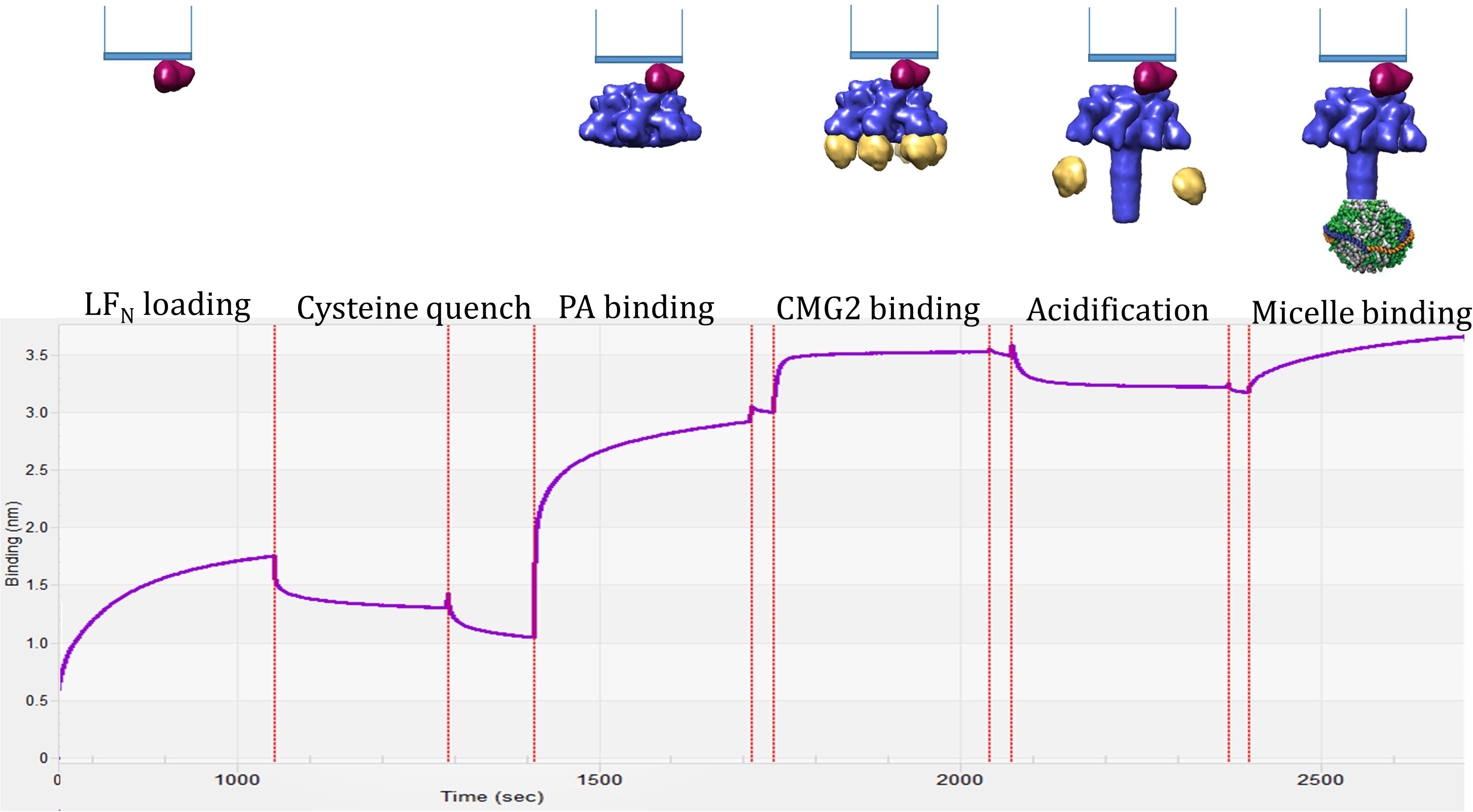
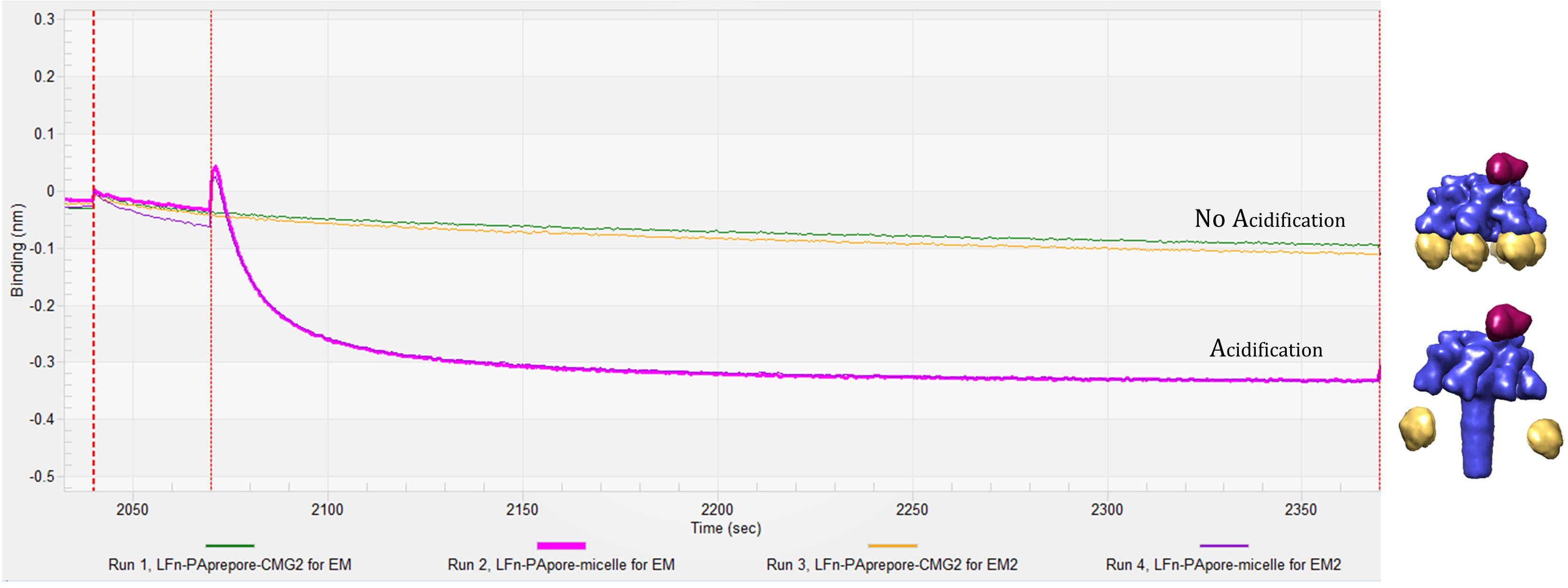
La trace de sensogram BLI est une lecture en temps réel sur les changements d’amplitude en raison de l’ajout spécifique des composants anthrax toxine lorsqu’elles sont ajoutées sur le biocapteur. La figure 3 montre avec qu'une trace représentative jumelée un modèle du complexe prédit pour former à cette étape du processus. La première montée est LFN chargement sur la pointe. Après trempe et baseline, PAprepore lie ensuite à LFN suivie de l’addition du récepteur soluble de CMG2 résultant dans le complexe de pré assemblé-endosomes de LFN-PAprepore-CMG2. Pour progresser vers l’environnement endosome tardif, l’ensemble du complexe est soumis à une impulsion de faible pH (pH 5.0) qui affaiblit le récepteur contraignant, permettant la prepore pour passer dans sa conformation de pores de membrane étendue insérée. 6 Sensogram traces de l’étape de l’acidification sont indiquées à la Figure 4. L’augmentation initiale ou « pic » en amplitude est probablement le pore extension6 qui doit se produire avant la diminution de la fixation aux récepteurs CMG2. La diminution d’amplitude plus grande est plus probable dissociation récepteur important ou complet en raison de la diminution d’affinité. Des travaux antérieurs dans ce laboratoire a indiqué CMG2 liaison à la pleine extension PApore est négligeable par rapport à l’interaction deprepore CMG2-PA6. En outre, la trace de cinétique sensogram, observé dans toutes les sensograms de LFN-PAprepore-CMG2 transitions vers LFN-PAdes pores avec une baisse de pH, est reproductible en cours d’essais multiples.
Avant et après l’acidification, les complexes de biocapteur attaché sortent facilement pour visualisation par colorant négatif EM et l’identification par SM (Figure 5). Représentant complexes à partir des résultats de l’EM sont indiquées à la Figure 6. Grilles d’échantillon pré-endosomes, présentent des densités en accord avec intacts complexes ternaires consistant en LFN-PAprepore-CMG2. Grilles de complexes après acidification, montrer PA passés pour des pores et solubilisée par inclusion de micelle avec aucune densité CMG2 évidente.
Les identités de l’avant et les complexes après acidification ont été vérifiées par SM. Une base de données où les séquences de PA, P13423 ; LFN, P15917 ; et CMG2, P58335 ont été inclus dans un contexte d’une base de données de protéines souris dérivé du référentiel NCBInr. Seules les protéines d’intérêt ont été extraites de cette première recherche de la base de données, avec les garanties suivantes d’acides aminés pour les complexes ternaires et binaires : 54 % et 22 % pour les PA, 36 % et 6 % pour les LF et 43 % pour CMG2, respectivement (CMG2 n’était pas détecté sur le complexe binaire). Afin de maximiser la couverture de l’acide aminé de protéine, une deuxième identification de peptides/protéines a été réalisée à l’aide d’une base de données de protéine contenant seulement les trois protéines d’intérêt. Pre-endosomes MS échantillons contenaient des peptides de tous les composants de trois toxines 60.46 % 67,97 % et une couverture de 54,15 % LFN, PA et CMG2, respectivement (Figure 7). Résultats post-endosomes, illustrés à la Figure 8, ne contenaient que les peptides de LFN et PA (couverture 57.41 et 67.79 %, respectivement). Le manque de CMG2 dans les échantillons post-endosomes sont compatibles avec la diminution de nm BLI observée au cours de la formation de pores.

Figure 1. Aperçu schématique pour l’analyse des complexes protéiques assemblés sur et isolée du biocapteur BLI utilisant EM et Mme s’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 2. Activation de biocapteurs : première étape dans l’assembly spécifique d’orientation sur le biocapteur BLI. Le domaine E126C facteur létal N-terminal, (LFN) est lié par une liaison thio création de l’interface de liaisonprepore PA correctement orienté. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 3. Surveillance d’anthrax toxine montage et démontage avec BLI : le sensogram présente variations d’amplitude en fonction du temps en raison de l’ajout spécifique de l’anthrax toxine composants lorsqu’elles sont ajoutées sur le biocapteur commençant par le chargement de LFN (étape 4 de la Le tableau 1). PAprepore est ensuite chargé sur la surface, suivie par l’addition du récepteur soluble de CMG2. Le complexe pré assemblé-endosomes se compose d’un LF-PAprepore- CMG2. Pour progresser vers l’environnement endosome fin, toute l’assembler anthrax fonctionnelle toxine est soumise à une impulsion de faible pH (pH 5.0) qui affaiblit le récepteur contraignant, permettant la prepore pour passer dans sa conformation de pores de membrane étendue insérée. Exposition de la membrane couvrant le pore est confirmée et solubilisée par addition de micelles. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 4. BLI sensogram de libération CMG2 : Traces de l’étape de l’acidification montrent une augmentation initiale ou « spike » amplitude suivie d’une baisse d’amplitude plus grande qui risquent des pores formation et dissociation de récepteur, respectivement. Lignes rouges en pointillés verticaux indiquent le début d’une nouvelle étape. Sensograms correspondent à la ligne rouge en pointillé apparaît en caractères gras. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 5. Analyse des complexes protéiques assemblés sur et isolée du biocapteur BLI utilisant EM et MS : biocapteur ci-joint complexes sont facilement libérés dans 5 µL de tampon contenant du TNT pour visualisation par colorant négatif EM et identification par MS. s’il vous plaît cliquez ici pour obtenir une version agrandie de cette figure.
{kind=link}

Figure 6. Visualisation des complexes de protéine avec EM : grilles d’échantillon pré-endosomes, présentent des densités en accord avec intacts complexes ternaires consistant en LFN-PAprepore-CMG2. Post-endosomes grilles complexes, voir la PA fait la transition pour des pores et solubilisée par micelle avec aucune densité CMG2 évidente. Modèles de prévisions complexes (côté gauche) sont à la même échelle que les particules individuelles illustré. Particules colorisés avec protéine (basé sur la taille de la masse volumique EM) sont indiqués ci-dessous chaque particule. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 7. Vérification de complexes protéiques avec MS : Pre-endosomes MS échantillons contenaient des peptides de tous les composants de trois toxines 60.46 % 67,97 % et une couverture de 54,15 % LFN, PA et CMG2, respectivement. Peptides détectés à un taux de fausse découverte (FDR) égal ou supérieur à 5 % indiqué en jaune, FDR égal ou supérieur à 1 % en vert. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 8. Vérification de complexes protéiques avec MS : peptides échantillons contenues Post-endosomes de LFN et PA (couverture 57.41 et 67.79 %, respectivement), mais pas CMG2. FDR égal ou supérieur à 5 % indiqué en jaune, FDR égal ou supérieur à 1 % en vert. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}
| Étape | Heure (s) | Description |
| 1 | 30 | Niveau de référence initial |
| 2 | 420 | Activation |
| 3 | 300 | Activation |
| 4 | 300 | ChargementN LF |
| 5 | 240 | Cystéine quench |
| 6 | 120 | Ligne de base |
| 7 | 300 | Associationprepore PA |
| 8 | 30 | Ligne de base |
| 9 | 300 | CMG2 association |
| 10 | 30 | Ligne de base |
| 11 | 300 | Transition acide |
| 12 | 30 | Ligne de base |
| 13 | 300 | Association de micelle |
Table 1. Fois d’étape simple BLI exécuter à la toxine du charbon de montage complexe. Notez que les lignes de base sont utilisées pour laver la protéine non lié à l’étape précédente et/ou de proposer une nouvelle base de tampon.
Discussion
Cette démonstration illustre comment la formation d’un complexe macromoléculaire peut être facilement contrôlée à l’aide de biolayer interférométrie, visualisé à l’aide de EM et vérifié avec MS, climatisées et dotées de microvolumes dans un court laps de temps. Ensemble structurel et complexes observés suivent les prédictions biologiquement pertinentes, en plus de valider cette méthodologie combinée. Comme mentionné dans la section résultats, l’élément clé pour le succès de l’Assemblée exige la mutagénèse cystéine rationnellement machiné pour s’assurer que les interfaces complexes protéine-protéine sont correctement orientés de la surface du biocapteur.
Les systèmes précédents ont utilisé des techniques BLI et MS pour évaluer la liaison aux protéines de deux et trois systèmes de composants, ainsi que l’intégrité de la liaison aux récepteurs de la protéine exprimée, mais, dans les deux cas, les méthodes n’a été élaborés pour profiter du tandem EM/MS approche de8,9. Le seuls autre interférométrie système combinant MS analyse afin de caractériser les interactions était une double polarisation interférométrie10. Malheureusement, ce système n’est plus disponible pour une utilisation générale. Tel que mentionné dans l’introduction, on a un certain nombre d’études achevées où les échantillons ont été formés sur des surfaces de biocapteur SPR-like et retirés de MS analyse. Aucun de ces exemples n’a révélé complexes visualisées à l’aide de EM.
Les limitations de cette méthode séquentielle peuvent être nombreuses, mais peuvent être résolubles. Pour l’immobilisation initiale et donc pas de fondation de l’Assemblée, un manque de connaissances sur les surfaces structurelles interaction gênerait certainement progrès associées au contrôle des phases de montage initial. Le manque d’information sur la structure peut être résolu en concevant un machinés Fondation construire où les chimies de pièce jointe (p. ex. sulfhydryle portion pour disulfures / His-tag positionnement) peut être déplacé vers diverses régions au sein de la base système d’assemblage. Dans le cas de la toxine du charbon complexe, il est heureux que la structure du LFN lié à PAprepore est disponible11. Placement rationnel de la cystéine machinée a été localisé aux régions loin de la face de liaisonprepore PA. Chimies de linkage de cystéine, il est préférable qu’aucun autres cystéines réactives ne sont présents sur la surface de la protéine.
Il y a différentes chimies de fixation qui permet à l’ingénieur site d’attachement spécifique sur les surfaces de la protéine. Un des plus populaires accessoires spécifiques implique une portion de la biotine de positionnement à un emplacement défini spécifiquement sur la protéine de surface12. Malheureusement, la liaison biotine à streptavidin ou avidine enduit surfaces est assez serré. Renversement de la réaction de liaison n’est pas simple. L’utilisation d’une ingénierie His-balise à l’extrémité N - ou C-terminale et la facilité ultérieure de l’attachement aux surfaces Ni-NTA est une application plus universelle de l’immobilisation de l’affinité. Bien sûr, d'entre les avertissements relatifs à ingénierie sites d’attachement de l’Assemblée avec les systèmes de son-le tag est l’exigence que les N - et C-terminus de la protéine Assemblée restent exposés et séparés de sorte que la pièce jointe est facile. Comme avec tous les processus d’assemblage, l’interface d’interaction de la protéine Assemblée doit rester disponible en cours de montage complexe.
Peut-être le souci plus courant d’utiliser chimies surface biocapteur est non-spécifiques. Streptavidine conseils sont souvent une source d’effets significatifs non-spécifiques. Disulfure lié biotine peut servir à libérer les complexes très spécifiques, laissant derrière lui le lien S-biotine réduit, étroitement lié à la Streptavidine immobilisée biocapteurs13. Il existe d’autres chimies réversibles deviennent disponibles, comme l’iminoboronates et à un moindre degré CETOAMIDE14. Ce champ est actuellement sous-développés, mais il n’y a grand intérêt à développer des protocoles covalentes réversibles pour éviter des effets de toxicité des médicaments hors-cible qui accompagnent généralement l’utilisation de développement de médicaments ciblés covalente.
Une limitation d’utilisation EM pour visualiser des complexes est interprétation, en particulier dans les cas où les structures des complexes montés ne sont pas connus au départ. La localisation spatiale des composants d’un complexe macromoléculaire assemblé indéfini peut être identifiée en utilisant des anticorps monoclonaux (ACM) comme des marqueurs spécifiques de cinétiques et structurales. Par exemple, une fois qu’un complexe est formé, anticorps monoclonaux peuvent être ajoutées qui se lient à des composants spécifiques. Cette méthode est fréquemment utilisée en EM pour identifier des composants spécifiques au sein de grandes assemblées15. Une autre limitation est proportionnelle à la taille du complexe, bien qu’il y a eu des cas où les assemblys symétriques définies aussi petit que 70 kDa (GroES PA63) sont facilement résolus à l’aide négatif tache EM. Assemblé des complexes qui sont analysés par EM sont typiquement dans la gamme de taille de ~ 100 Å de diamètre ou plus. Récemment cependant, protéines aussi petites que 20 kDa ont été résolus et structures de basse résolution ont été obtenues lors de l’utilisation supérieure coloration méthodologies16.
Pour l’analyse de MS, la sensibilité accrue de l’instrumentation actuelle de MS jusqu’au niveau de femtomolar (attomoles) peut, dans certains cas, augmenter la sensibilité de détection de BLI. Il est très concevable que des signaux de protéine qui montrent une augmentation minime mais répétable amplitudes seront traduira par identification de la protéine en question. En outre, sondant les interactions protéine-protéine avec un des partenaires a été affectés le biocapteur et l’autre dans un milieu cellulaire en vigueur entraînera une purification et détection par la suite plus facile du complexe nouvellement formé. Une des limitations qui peuvent être observées avec les systèmes actuels de MS très sensibles est que la protéine d’intérêt peut ne pas être dans la base de données, mais cette observation est rare (par exemple, les protéomes d’espèces rares). Si les séquences des protéines d’intérêt sont connus, ce problème est facilement résolu en incluant la séquence d’acides aminés ou des protéines dans une base de données de protéine fond (tel que décrit dans ce travail dans la section protocole). Une autre limitation potentielle de la méthodologie résulte de la résistance d’une protéine à trypsinolysis. La digestion trypsique est généralement la méthode par défaut de bas en haut l’identification des protéines. Cependant, les protéines peuvent être résistants à la trypsine si ils manquent de résidus Arg et Lys ou l’accès à ces résidus sont restreints par la structure plissée. Ces limitations sont résolues, respectivement, à l’aide de rechange ou une combinaison des protéases ou incluant un réactif qui se déroule (urée ou guanidine HCl, comme indiqué dans 1.1.14 et 1.2.6) avant la digestion enzymatique.
Les expansions possibles de cette méthode incluent permettant à l’utilisateur de suivre et d’identifier les complexes cellulaires Assemblée de lysats cellulaires bruts. Il s’avère que tests de composants d’assemblage dans des extraits concentrés de cellulaires est facile à réaliser en utilisant les procédures de l’interférométrie biolayer. Contrairement aux méthodes plus couramment utilisés microfluidique basé qui a tendance à encrasser et sensibles à l’agrégation, l’approche BLI peut être utilisé pour plonger directement biolayer extrémités du capteur dans les extraits bruts potentiellement assembler des complexes spécifiques directement ces échantillons impurs concentré. Une fois assemblé, il est tout à fait envisageable d’utiliser des sondes de l’anticorps spécifique faisant suite à la système BLI à mieux identifier et à quantifier même soupçonnée de composants dans des extraits cellulaires qui ont été identifiés à l’aide de la méthode microvolume MS. Encore une fois, la clé ici est d’utiliser des protéines essentielles définies et correctement orientés comme sondes d’affinité spécifique.
La possibilité de visualiser le prepore pour le processus de transition cinétiquement avec BLI des pores sera très utile pour identifier les éventuels « anti-toxine » petites molécules inhibitrices des transitions protéines spécifiquement fonctionnant sous fin endosomale, un pH faible (5.0) conditions. Cette prepore pH induite à pore de transition est inhibée en présence de pliage stabilisateur (osmolytes) tels que le glycérol ou le saccharose et donc prête appui solide de développement spécifiques ciblés stabilisateurs pliants qui empêchent les PA formationdes pores . Cette approche spécifique évite et remplace bruts tests basés sur l’agrégation où pH chute de plomb à la précipitation des protéines. Cette dernière méthode, même si c’est bon pour les principales méthodes de présélection, conduit souvent à des résultats faussement positifs où certains composés inhibent l’agrégation plutôt que les transitions moléculaires réelles.
Les observations en aval de la structure et l’identification des différents composants assemblés dans des échantillons de petite microvolume peuvent être également utiles pour valider le potentiel des composés de plomb. Ceci peut être appliqué dans les cas où la stabilisation assembly spécifique ou déstabilisation est le résultat de la cible. Cette approche parallèle cinétique/structure/identification est utile pour confirmer directement la validité des effecteurs composé principal suspect de l’Assemblée et sert comme étape de dépistage secondaire raisonnables ou approche de débit moyen.
Cryo-EM est une technique utile pour étudier les détails atomiques des complexes de la macromolécule dans différents États de l’Assemblée. Avant de préparer des échantillons de cryo-EM, il est important de vérifier tout d’abord qu'une préparation contient raisonnablement purs complexes homogènes avec colorant négatif EM. Le travail présenté ci-après illustre Assemblée de complexes protéiques sur des surfaces de biocapteur BLI, libération de ces complexes pour la visualisation de l’EM et l’identification de ces composants à l’aide de MS. Cette méthode particulière d’Assemblée contrôlée et la libération peut être utile dans la génération des protocoles très spécifiques qui améliorent la préparation de l’échantillon séquentielle homogène pour une coloration négative EM, une étape nécessaire qui doit être démontrée avant de passer à Cryo-EM. Pour obtenir la structure 3D de basse résolution, seules les particules de 30-50 du complexe seraient nécessaire pour effectuer une série d’inclinaison conique (70 vues différentes image 2D par particule) autant il y a diversité de l’orientation (plusieurs vues différentes).
En ce qui concerne l’amélioration des méthodes de MS, progrès de la sensibilité et réduction du volume de l’échantillon de continuent à améliorer. Flux de nano et chromatographie liquide à ultra haute pression ainsi que le développement de spectromètres de masse avec un service rapide du cycle, une sensibilité accrue et pouvoir de résolution. L’introduction récente du spectromètre de masse Orbitrap valant, en particulier la dernière version (Orbitrap valant Fusion Lumos et son successeur attendu le Lumos de Fusion Orbitrap valant 1M), mais aussi les algorithmes de recherche facilitent considérablement ce processus.
La méthodologie actuelle surveille la cinétique montage et le démontage des composants de toxine d’anthrax conformément aux méthodes exempte d’étiquette BLI et évalue la structure et l’identité de ces composants à l’aide de EM et MS, respectivement. L’utilisation d’un canal unique simple système BLI couplé avec routine analyse EM de coloration négative et techniques élémentaires de MS sont plus que suffisantes pour caractériser un processus d’assemblage.
Déclarations de divulgation
Aucune divulgation en ce moment.
Remerciements
Ce travail a été soutenu par le Madison et Lila Self Graduate Fellowship (AJM), NIH 5T32GM008359 (PTO), KUMC Biomedical Research Training programme (PTO), NIH R01AI090085 (MTF), KU fonds de transition et le centre de Technologies des interactions biomoléculaires (BITC) Grant ( CT et MTF). Les auteurs tiens à remercier Barbara Fegley à base de microscopie électronique de KUMC pour l’aide à l’acquisition d’images TEM.
matériels
| Name | Company | Catalog Number | Comments |
| (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide)) (EDC) | Thermo Scientific | 22980 | |
| (2-(2-pyridinyldithio)ethanamine) (PDEA) | GE Lifesciences | BR100058 | |
| 0.5 mL black microtubes | Sigma-Aldrich | Z688304-500EA | |
| 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) | Avanti | 850457C-25mg | |
| Acclaim PepMap 50 μm x 150mm, C18, 2 μm, 100 Å | ThermoFisher Scientific | 164561 | |
| Acetic acid glacial | Fisher | A38SL-212 | |
| Acetonitrile | Fisher | A955-4 | |
| Amine reactive 2nd generation (AR2G) tips | Forte Bio | 18-5092 | |
| Ammonium bicarbonate | Fisher | A643-500 | |
| Anotop 10 syringe filter, 0.02um | GE Lifesciences | 6809-1002 | |
| BLItz System | Pall Forte Bio | 45-5000 | |
| Boric acid | Fisher Scientific | 10043-35-3 | |
| Capillary Morphogenic Gene-2 | Protein produced and purified in-house | ||
| Carbon coated Cu 300 grid | Electron Microscopy Sciences | CF300-CU-50 | |
| Dithiothreitol (DTT) | GoldBio | DTT25 | |
| Formic acids | JT Baker | 0129-01 | |
| Guanidine hydrochloride (GuHCl) | Sigma-Aldrich | G4505-100G | |
| Iodoacetamide | MP-BioMedicals,LLC | 100-351 | |
| JEM-1400 Transmission Electron Microscope | JEOL | ||
| L-cystiene hydrochloride hydrate | Sigma-Aldrich | C121800-5G | |
| Lethal Factor N-terminal domain | Protein produced and purified in-house | ||
| MS software version 2.2 | ThermoFisher Scientific | ||
| N-hydroxysuccinimide (NHS) | Sigma-Aldrich | 090M14531V | |
| Orbitrap Fusion Lumos | ThermoFisher | ||
| PCR tubes | ThermoFisher Scientific | AB-0620 | |
| Pelco easiGlow glow discharge cleaning system | Ted Pella, Inc | 91000 | |
| Protective Antigen prepore | Protein produced and purified in-house | ||
| Qualitative filter paper circles | Fisher Scientific | 09-795C 5 | |
| Sequencing grade modified trypsin | Promega | V5111 | |
| Sequest HT search engine | ThermoFisher Scientific | ||
| Sodium acetate trihydrate | Fisher Scientific | BP334-500 | |
| Sodium chloride | Fisher Scientific | S271-10 | |
| Sodium cholate | Sigma-Aldrich | C1254-100G | |
| Tris base | Fisher Scientific | BP152-10 | |
| Uranyl formate (UF) | Electron Microscopy Sciences | 22450 | |
| Xcalibur | ThermoFisher Scientific | OPTON-30487 |
Références
- Bellon, S., Buchmann, W., Gonnet, F., Jarroux, N., Anger-Leroy, M., Guillonneau, F., Daniel, R. Hyphenation of surface plasmon resonance imaging to matrix-assisted laser desorption ionization mass spectrometry by on-chip mass spectrometry and tandem mass spectrometry analysis. Analytical Chemistry. 81, 7695-7702 (2009).
- Kim, Y. E., Yi, S. Y., Lee, C. S., Jung, Y., Chung, B. H. Gold patterned biochips for on-chip immuno-MALDI-TOF MS: SPR imaging coupled multi-protein MS analysis. The Analyst. 137, 386-392 (2012).
- Abdiche, Y., Malashock, D., Pinkerton, A., Pons, J. Determining kinetics and affinities of protein interactions using a parallel real-time label-free biosensor, the Octet. Analytical Biochemistry. 377, 209-217 (2008).
- Abdiche, Y. N., Malashock, D. S., Pinkerton, A., Pons, J. Exploring blocking assays using Octet, ProteOn, and Biacore biosensors. Analytical Biochemistry. , 172-180 (2009).
- Naik, S., Brock, S., Akkaladevi, N., Tally, J., McGinn-Straub, W., Zhang, N., Gao, P., Gogol, E. P., Pentelute, B. L., Collier, R. J., Fisher, M. T. Monitoring the kinetics of the pH-driven transition of the anthrax toxin prepore to the pore by biolayer interferometry and surface plasmon resonance. Biochemistry. , (2013).
- Akkaladevi, N., Hinton-Chollet, L., Katayama, H., Mitchell, J., Szerszen, L., Mukherjee, S., Gogol, E., Pentelute, B., Collier, R., Fisher, M. Assembly of anthrax toxin pore: Lethal-factor complexes into lipid nanodiscs. Protein Science. 22, 492-501 (2013).
- Jin, H., Cantin, G. T., Maki, S., Chew, L. C., Resnick, S. M., Ngai, J., Retallack, D. M. Soluble periplasmic production of human granulocyte colony-stimulating factor (G-CSF) in Pseudomonas fluorescens. Protein Expression and Purification. 78, 69-77 (2011).
- Yamniuk, A. P., Edavettal, S. C., Bergqvist, S., Yadav, S. P., Doyle, M. L., Calabrese, K., Parsons, J. F., Eisenstein, E. ABRF-MIRG benchmark study: Molecular interactions in a three-component system. Journal of Biomolecular Techniques. , 101-114 (2012).
- Moore, J. D., Perez-Pardo, M. A., Popplewell, J. F., Spencer, S. J., Ray, S., Swann, M. J., Shard, A. G., Jones, W., Hills, A., Bracewell, D. G. Chemical and biological characterisation of a sensor surface for bioprocess monitoring. Biosensors & Bioelectronics. 26, 2940-2947 (2011).
- Feld, G. K., Thoren, K. L., Kintzer, A. F., Sterling, H. J., Tang, I. I., Greenberg, S. G., Williams, E. R., Krantz, B. A. Structural basis for the unfolding of anthrax lethal factor by protective antigen oligomers. Nature Structural & Molecular Biology. 17, 1383-1390 (2010).
- Fairhead, M., Howarth, M. Site-specific biotinylation of purified proteins using BirA. Site-Specific Protein Labeling: Methods and Protocols. , 171-184 (2015).
- Naik, S., Kumru, O. S., Cullom, M., Telikepalli, S. N., Lindboe, E., Roop, T. L., Joshi, S. B., Amin, D., Gao, P., Middaugh, C. R. Probing structurally altered and aggregated states of therapeutically relevant proteins using GroEL coupled to bio-layer interferometry. Protein Science. 23, 1461-1478 (2014).
- Bandyopadhyay, A., Gao, J. Iminoboronate-based peptide cyclization that responds to pH, oxidation, and small molecule modulators. Journal of the American Chemical Society. 138, 2098-2101 (2016).
- Grantham, J., Llorca, O., Valpuesta, J. M., Willison, K. R. Partial occlusion of both cavities of the eukaryotic chaperonin with antibody has no effect upon the rates of beta-actin or alpha-tubulin folding. The Journal of Biological Chemistry. 275, 4587-4591 (2000).
- Ercius, P., Alaidi, O., Rames, M. J., Ren, G. Electron Tomography: A Three-Dimensional Analytic Tool for Hard and Soft Materials Research. Advanced Materials. 27, 5638-5663 (2015).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.