
Method Article
Isolement et analyse des communautés microbiennes dans le sol, rhizosphère et racines dans des expériences de graminée vivace
Dans cet article
Résumé
Excavation des racines des plantes sur le terrain, ainsi que traitement d’échantillons dans la rhizosphère, endosphère et le sol sont décrites en détail, y compris les méthodes d’analyse de données et extraction de l’ADN. Cet article est conçu pour permettre aux autres laboratoires d’utiliser ces techniques pour l’étude du sol, endosphère et rhizosphère microbiomes.
Résumé
Plante et sol études microbiome gagnent en importance pour comprendre que les rôles les microorganismes jouent dans la productivité agricole. Le but de ce manuscrit est pour fournir des détails sur comment rapidement des échantillons de sol, rhizosphère et endosphère des essais répliqués sur le terrain et analyser les changements qui peuvent survenir dans les communautés microbiennes grâce à type d’échantillon, traitement et le génotype de la plante. L’expérience permettant de démontrer ces méthodes compose de parcelles de terrain répliquée contenant deux, pure, graminées de saison chaude (Panicum virgatum et Andropogon gerardii) et un mélange de graminées de faible diversité (a. gerardii, Sorghastrum nutans, et Bouteloua curtipendula). Brièvement, excavation des plantes, des racines très sont coupé et placé dans un tampon phosphate et puis secoué pour recueillir la rhizosphère. Racines sont présentées au laboratoire sur la glace et surface stérilisé à l’eau de Javel et de l’éthanol (EtOH). La rhizosphère est filtrée et concentrée par centrifugation. Déblais d’autour de la Motte est placé dans des sacs en plastique et apporté au laboratoire où une petite quantité de sol est prise pour les extractions d’ADN. L’ADN est extrait des racines, le sol et la rhizosphère et ensuite amplifié avec des amorces pour la région de la V4 du gène de l’ARNr 16 s. Amplicons sont séquencés, puis analysés avec outils bioinformatiques de libre accès. Ces méthodes permettent aux chercheurs de tester comment la diversité des communautés microbiennes et la composition varie en raison du type d’échantillon, traitement et plant le génotype. En utilisant ces méthodes ainsi que des modèles statistiques, les résultats représentatifs démontrent il y a des différences significatives dans les communautés microbiennes des racines rhizosphère et sol. Méthodes présentées ici fournissent un ensemble complet des étapes sur la façon de recueillir des échantillons sur le terrain, isoler, extraire, quantifier, amplifier et séquence ADN et analysent la diversité des communautés microbiennes et la composition des essais de terrain répliqué.
Introduction
Recherche de microbiome a des implications importantes pour comprendre et manipuler des processus écosystémiques comme nutriments cyclisme, chiffre d’affaires de matière organique et le développement ou l’inhibition de sol pathogènes1,2. Ce domaine de recherche a également un grand potentiel pour comprendre les impacts des microbes du sol sur la productivité des communautés végétales naturelles et des écosystèmes agricoles. Bien qu’il existe de nombreuses études qui ont mis l’accent sur le microbiome du sol dans les écosystèmes naturels, moins ont mis l’accent sur les microbes de la rhizosphère et endosphère plante dans les agroécosystèmes3. Dans le Nebraska, l’agriculture domine le paysage dans une grande partie de l’État, qui fait les études de ces sols où les cultures agricoles importantes sont cultivés un sujet essentiel pour la recherche. Le but de cet article des méthodes consiste à fournir aux chercheurs un ensemble standard de protocoles pour décrire les microbes présents dans les agroécosystèmes, afin de déterminer comment les racines des plantes modifient les communautés microbiennes dans la rhizosphère et endosphère et à terme comprendre les fonctions de que ces microbes jouent dans la productivité du sol sanitaire et phytosanitaire.
La méthode présentée ici diffère légèrement de méthodes utilisées par les autres4,5 dans que cet article vise à apprendre quels microbes sont exclusivement à l’intérieur de la racine et comment elles diffèrent des microbes immédiatement en dehors de la racine dans le rhizosphère. Le séquençage de l’amplicon utilisé dans cette étude identifie les microbiennes taxons présents dans l’échantillon d’ADN et permet aux chercheurs de déterminer comment les communautés changent selon le type d’échantillon ou de traitement. Une des principales différences entre ce protocole et un protocole très similaire utilisé par Lundberg et al. 6 , c’est qu’au lieu de la sonication, ce protocole utilise stérilisation superficielle avec eau de Javel et de l’éthanol pour retirer les racines de la rhizosphère. D’autres ont utilisé également stérilisation superficielle effectivement7,8,9,10. Ces méthodes ne sont pas plus avantageuses que d’autres méthodes, mais légèrement différent. Ces méthodes sont particulièrement bien adaptés pour des expériences de grand champ car avec assez de gens, il est possible de traiter plus de 150 par jour, ce qui ajoute jusqu'à environ 450 échantillons lorsque divisé en endosphère, rhizosphère et le sol des parcelles de terrain. Ce manuscrit décrit en détail les méthodes utilisées pour l’échantillon dans le domaine, de traiter le matériel dans le laboratoire, d’extraire et de séquencer l’ADN et donne un bref aperçu des étapes pour analyser les données de séquençage qui en résulte.
Protocole
1. Description du Site champ
- Décrire les sites expérimentales sur le terrain pendant les périodes de collecte. Déterminer l’emplacement du champ (latitude, longitude et altitude) à l’aide d’un GPS.
- Décrire la profondeur d’échantillonnage, le temps d’échantillonnage et la texture du sol.
- Les facteurs environnementaux jouent un rôle important dans le façonnement des communautés microbiennes. Enregistrer les informations climatiques tels que la température moyenne annuelle, les précipitations annuelles, la rotation des cultures des exercices antérieurs, pratiques culturales, méthode de fertilisation et l’histoire de l’emplacement du champ. Stations météorologiques automatisées ou autres dispositifs sont utiles pour enregistrer la pluie quotidienne et la température pendant une saison de croissance.
2. collecte et traitement des échantillons de sol, rhizosphère et racine domaine
- Excavation de plantes.
- Étiqueter un pan de lavage et un seau avec un pense-bête contenant des informations sur le matériel végétal à échantillonner. Inclure des informations comme numéro de parcelle, génotype des plantes et espèces végétales. Porter le seau étiqueté à l’intrigue et laisser la cuve de lavage à la station de travail établie dans le domaine.
- Percer le sol avec une pelle à une profondeur de 30 cm pour couper toutes les racines latérales tenant la plante dans le sol. Le volume approximatif est de 18 cm3. Au hasard choisir et de recueillir deux plantes par parcelle provenant de régions différentes au sein de l’intrigue.
- Creuser les racines des plantes en s’appuyant sur la pelle et placer la motte dans le seau étiqueté. Ramener le seau étiqueté avec la Motte excavé à la station de travail dans le domaine. Couper et jeter la biomasse au-dessus du sol.
- Enlèvement du sol de racines et de la perception du sol en vrac.
- Secouer les racines pour éliminer les salissures ou utiliser une bêche ou un motoculteur à main pour enlever la terre sur les racines manuellement. Secouer les racines est suffisante pour enlever la saleté dans les sols très sablonneux. Porter des gants et placez les racines près de la station de traitement.
- Après avoir secoué les racines, la plus grande partie du sol sera dans la cuve de lavage. Mélanger le sol dans la cuve de lavage et briser les mottes de n’importe quel sol avec un cultivateur à main. Placez un échantillon du sol qui est exempt de débris dans un étiquetées, sac de rangement 17,7 x 19,5 cm fermeture à glissière et placez dans un endroit frais ou sur glace.
- Collection de racines et de la rhizosphère.
- À l’aide de ciseaux élagage stérilisé dans 70 % EtOH, exciser une variété de racines, environ 4 à 6 racines par plante et chaque racine autour de 9 à 12 cm de longueur. Placez les racines excisées (coupe si nécessaire pour s’adapter) dans un tube marqué 50 mL contenant 35 mL de tampon de phosphate autoclavé, (6,33 g/L NaH2PO4, 8,5 g/L de Na2HPO4 anhydre, pH = 6,5, agent de surface 200 µl/L).
Note : L’agent de surface (voir Table des matières) a été ajouté après la stérilisation le tampon phosphate. Le volume de la Motte et la longueur des racines varient selon l’âge de la plante et les plantes. - Secouer les tubes pendant 2 min libérer la rhizosphère de la surface des racines. Avec une pince stérilisée dans 70 % EtOH, enlever les racines du tube, tache brièvement sur papier absorbant et placer dans un nouveau étiquetés tube de 50 mL. Placez les deux le tube contenant la rhizosphère et celui contenant les racines sur la glace.
- À l’aide de ciseaux élagage stérilisé dans 70 % EtOH, exciser une variété de racines, environ 4 à 6 racines par plante et chaque racine autour de 9 à 12 cm de longueur. Placez les racines excisées (coupe si nécessaire pour s’adapter) dans un tube marqué 50 mL contenant 35 mL de tampon de phosphate autoclavé, (6,33 g/L NaH2PO4, 8,5 g/L de Na2HPO4 anhydre, pH = 6,5, agent de surface 200 µl/L).
3. le traitement du champ des échantillons en laboratoire
- Stérilisation en surface des racines après collecte sur le terrain.
Remarque : Effectuez la stérilisation en surface dès que possible après son retour sur le terrain. S’il n’est pas possible à la surface de stériliser les racines dans la même journée, stocker des racines à 4 ° C jusqu'à ce que les traités.- Verser environ 35 mL d’eau de Javel 50 % + 0.01 % Tween-20 à 50 tubes de racine mL recueillies sur le terrain. Agiter à centrifuger de 50 mL pendant 30 à 60 secondes. Décanter 50 % eau de Javel et d’ajouter 35 mL de 70 % EtOH. Agiter pendant encore 30 à 60 secondes.
- Décanter 70 % EtOH et ajouter 35 mL d’eau ultrapure stérile. Agiter pendant 1 minute. Répéter le lavage à l’eau deux fois plus.
Remarque : Pour garantir que notre traitement de stérilisation en surface est suffisant, nous plaqué des échantillons d’eau provenant du dernier rinçage et n’observé aucune croissance de bactéries (P. Wang, données non publiées, 2014). Autres chercheurs ont testé pour l’efficacité de stérilisation de racine à l’aide de semblables méthodes9,10. - Racines sécher sur un essuie-tout propre. Utilisez une serviette en papier propre pour chaque échantillon.
Remarque : Les serviettes en papier peuvent être stérilisés avant usage. Nous ne pas stériliser nos serviettes en papier. Cependant, nous utilisons 1 serviette en papier par exemple et garder les serviettes enveloppés jusqu'à utilisation. - À l’aide de la pince stérile et ciseaux élagage, couper les racines en morceaux de 5 mm environ et le lieu coupe les racines dans un tube conique propre, étiqueté 15 mL. Conserver les échantillons à-80 ° C jusqu'à être traitées ultérieurement.
- Traitement des échantillons de la rhizosphère.
- Secouer les tubes de 50 mL contenant des échantillons de la rhizosphère du champ pour remettre en suspension l’ensemble de l’échantillon. À l’aide d’une passoire de cellules stériles, maille de 100 µm (voir Table des matières), filtre l’échantillon remis en suspension dans un nouveau tube de 50 mL.
- Centrifuger les tubes à 3000 g pendant 5 min à température ambiante. Immédiatement, décanter et éliminer le surnageant.
- Placer les boulettes rhizosphère dans les tubes de 50 mL sur la glace. Ajouter 1,5 mL de tampon phosphate stérile (sans agent tensio-actif) à granulés de la rhizosphère et vortex à suspendre.
- Pipette de suspension liquide dans un tube à centrifuger propre, étiqueté, 2 mL. Tourner les tubes à 15 871 x g pendant 2 min à température ambiante. Immédiatement, décanter le liquide surnageant et égoutter les tubes sur des essuie-tout propre.
- Stocker les pellets à-20 ° C jusqu'à être traitées ultérieurement.
Remarque : Les étapes 3.2.2 - 3.2.4 sont faites pour réduire la taille de tube d’échantillon pour le stockage. Il est plus efficace de stocker les petits tubes de 2 mL par rapport aux tubes de 50 mL d’espace.
- Traitement des échantillons de sol pour l’analyse de l’ADN d’extraction et le sol.
- À l’aide d’une spatule métallique stérile, remplir un tube propre, étiqueté 2 mL avec environ 3 g de sol pour l’extraction de l’ADN. Éviter les pièces de petite racine et les débris. Stocker cet échantillon de sol à-20 ° C. Spatule métallique rinçage dans 70 % EtOH entre chaque échantillon.
- Dans une casserole propre lavage, vider le sac de sol en tamis superposés (voir Table des matières), plus grand tamis au-dessus du tamis plus petit et tamiser manuellement le sol par les deux tamis. Utilisez une brosse pour nettoyer soigneusement les tamis entre les échantillons.
- Mettre de côté 100 à 125 g de sol tamisé dans un sac à fermeture à glissière de 17,7 x 19,5 cm pour futur sol physico-chimique et analyse de la texture. Placer les sacs de terre à 4 ° C pour le stockage à court terme.
- Traitement des échantillons de sol pour l’humidité du sol.
- Un sac de papier brun marqué sur une échelle à zéro. Mesure 40 à 45 g de sol tamisé dans le sac de papier brun. Noter le poids du sac en papier brun et du sol sur une feuille de données et de placer des sacs dans une étuve réglé à 55-60 ° C.
- Après 72 heures, retirez les sacs de l’étuve. Laisser les sacs de sol refroidir pendant au moins 30 min et puis peser.
- Pour enregistrer le poids, tarer la balance à zéro et placer les sacs de papier brun sur la balance. Calculer le pourcentage d’humidité du sol pour chaque échantillon à l’aide de la formule :

Remarque : La méthode de l’humidité du sol est de Kellogg Biological Station Long terme Ecological Research (LTER KBS). KBS LTER fournit un large éventail de protocoles établis pour les chercheurs sur leur site Internet (https://Lter.kbs.msu.edu/).
4. préparation des racines transformées échantillons pour les Extractions d’ADN.
- Moudre le matériel racine congelés afin que l’échantillon est homogène.
- Versez de l’azote liquide dans un bécher en plastique avec des spatules propres et dans un propre mortier et un pilon pour conserver les échantillons congelés pendant tout le processus de broyage.
- Placez le tissu congelé dans le mortier et broyer avec le pilon en une fine poudre. Continuellement ajouter azote liquide tout au long de meulage pour conserver les échantillons congelés.
- Utilisez une spatule pour placer le tissu fondamental dans un tube propre, étiqueté 2 mL. Conserver à-80 ° C.
5. extraction de l’ADN du sol et la rhizosphère échantillons au Format 96 puits
- Chargement des échantillons de sol en plaques 96 puits.
- Essuyer la zone de travail avec 70 % EtOH et 1 % eau de Javel. Porter des gants de laboratoire au cours de ces étapes. Enlever les échantillons de sol de stockage de-20 ° C et laisser pour décongeler dans un bac à glaçons.
- Retirer la housse de matelas étanchéité une plaque 96 puits d’extraction qui est fournie avec le kit d’extraction d’ADN. Placer la housse de matelas étanchéité entre 2 lingettes en papier pour garder propre tandis que ne l’utilisez pas. Pour éviter toute contamination, utilisez adhésive 8 puits PCR bandes pour couvrir les 12 colonnes de la plaque de 96 puits d’extraction.
- Tare d’un pesée stérile-entonnoir (taille SM) sur une échelle et peser de 200 à 250 mg de sol.
Remarque : Ces entonnoirs stériles sont aplatis sur une face, donc l’entonnoir repose à plat sur une échelle. L’entonnoir est rempli de terre et placé directement dans un puits. Cette technique permet d’éviter la perte des échantillons, minimise les fuites et empêche la contamination croisée. - Pour découvrir le premier puits de la plaque d’extraction, soulever délicatement la bande adhésive place, Eh bien le goulot de l’entonnoir-pesée remplie dans le cas échéant et guidez l’échantillon de sol dans le puits correspondant. Remplacer la bande adhésive pour couvrir le puits.
- Répétez ce processus pour chaque puits de la plaque, avec un entonnoir nouvel, stérile pour chaque échantillon, jusqu'à ce que la plaque est remplie. Laisser un bien vide comme un contrôle vide d’extraction.
Remarque : Un bien est laissé vide sur chaque plaque d’extraction pour servir de témoin négatif (sans indication). Ce contrôle des contaminants qui peuvent être présents dans les réactifs de kit11. - Remplacer la housse de matelas étanchéité sur la plaque d’extraction et de stocker la plaque à-20 ° C jusqu’au moment de l’extraction d’ADN.
- Chargement d’échantillons de la rhizosphère en plaques 96 puits.
- Enlever les échantillons de la rhizosphère de stockage-20 ° C et laisser pour décongeler dans un bac à glaçons.
- Suivez l’étape 5.1.2 ci-dessus pour préparer la plaque d’extraction de l’ADN.
- Placer un chiffon de papier propre sur une échelle, puis une spatule métallique stérile sur la balance à zéro. Utilisez la spatule pour évider soigneusement les certains de la pastille de la rhizosphère d’un tube à essais. Retourner la spatule à l’échelle et pèsent entre 200 et 250 mg de l’échantillon de la rhizosphère.
- Soulever délicatement la bande adhésive pour découvrir le premier puits de la plaque d’extraction, la spatule remplie d’angle dans le puits et gratter le matériau de la rhizosphère dans le puits correspondant avec un cure-dent stérile.
- Rincer la spatule métallique dans l’eau, suivi par 70 % EtOH entre les échantillons. Répétez cette procédure pour chaque puits de la plaque jusqu'à ce que la plaque est remplie, laissant un bien vide comme un contrôle vide d’extraction.
- Extraction de l’ADN provenant d’échantillons de la rhizosphère et sol en format 96 puits.
- Extrait du sol et la rhizosphère ADN en utilisant un kit optimisé pour le sol (voir Table des matières), suivant le protocole du fabricant.
Remarque : Nous utilisons ce kit de fixation spécifique pour isoler le sol et la rhizosphère ADN en raison de la capacité des propriétaires réactifs pour éliminer l’acide humique et autres puissants inhibiteurs de PCR trouvés dans le sol.
- Extrait du sol et la rhizosphère ADN en utilisant un kit optimisé pour le sol (voir Table des matières), suivant le protocole du fabricant.
- Quantification de l’ADN.
- Quantifier les 92 échantillons et 4 concentrations de normes à l’aide d’un kit (voir Table des matières; normes sont inclus), selon le protocole du fabricant.
- Quantifier les 4 échantillons restants qui ont été retirés de la plaque pour accueillir les puits pour les quatre normes à l’aide d’un kit (voir Table des matières), selon le protocole du fabricant.
6. extraction de l’ADN de racine d’échantillons au Format 96 puits.
- Chargement d’échantillons de racines dans des plaques de 96 puits.
- Essuyer la zone de travail avec 70 % EtOH et 1 % eau de Javel. Porter des gants lors de ces étapes.
- Conserver des échantillons de racines de terre congelés à tout moment en plaçant les échantillons dans un seau avec la glace sèche.
- Remplir un gobelet en plastique avec de l’azote liquide et placer microspatulas antistatique et stériles peser-entonnoirs (taille XSM) dans le bécher en plastique se refroidir.
- Retirer la housse de matelas étanchéité de la plaque de perle de 96 puits d’extraction qui est fournie avec le kit et le placer entre 2 lingettes en papier pour garder propre tandis que ne l’utilisez pas.
Remarque : La version plus récente de ce kit, libéré à la suite de l’époque, que nous avons effectué ces extractions d’ADN, oblige l’utilisateur à fournir les plaques de perle d’extraction. Dans la Table des matières, nous avons listé les informations du catalogue fournisseur requises pour commander ces articles. - Pour éviter toute contamination, utilisez adhésive 8 puits PCR bandes pour couvrir les 12 colonnes de la plaque de 96 puits d’extraction.
- Place la plaque d’extraction perle sur la glace sèche pour conserver les échantillons dans les puits congelés.
- Soulevez soigneusement les bandes adhésives pour découvrir le premier puits de la plaque d’extraction, placer le goulot de l’entonnoir-poids rempli dans le puits correspondant et ajoutez 3 mesurettes de spatule de tissus racinaires au sol. Remplacer la bande adhésive pour couvrir le puits.
Remarque : Le sol et la rhizosphère sont décongelés sur la glace avant le pesage, tandis que le matériel végétal est pesé, puis congelé. Des tissus végétaux gelés, surtout de petites quantités, sont difficile à évaluer sur une échelle sans décongélation. Peser tests ont été effectués sur des tissus racinaires au sol afin de déterminer combien de boules spatule suffisaient. Notez que le fabricant du kit ne nécessite pas une quantité exacte de tissu mais recommande environ 50 mg. types d’échantillons de plantes différentes variera et l’utilisateur devra déterminer le montant approprié. - Répétez cette procédure pour chaque puits de la plaque jusqu'à ce que la plaque est remplie.
- Stocker les plaques à-20 ° C jusqu’au moment de l’extraction d’ADN.
- Extraction de l’ADN des tissus racinaires.
- Extraire l’ADN à l’aide d’un kit optimisé pour plantes (voir la Table des matières) suivant le protocole du fabricant.
Remarque : Nous utilisons ce kit qui est spécialement conçu pour les tissus végétaux pour obtenir des rendements maximaux des échantillons de racines. À la différence du sol et la rhizosphère, les acides humiques et autres contaminants sont moins problématique pour les tissus de la racine.
- Extraire l’ADN à l’aide d’un kit optimisé pour plantes (voir la Table des matières) suivant le protocole du fabricant.
- Quantifier l’ADN comme au point 5.4.
7. amplification et séquençage de l’ADN isolé.
- Amplifier la région V4 du gène 16 s avec une preuve lecture polymérase (voir Table des matières), comme décrit dans Gohl et al. 12 les échantillons de code à barres avec différentes amorces d’indexation et de la piscine avant de séquençage. Séquence en utilisant les méthodes décrites par Gohl et coll.12 en utilisant un processus en deux étapes PCR avec des amorces V4 (supplémentaire 1 fichier).
Remarque : Certaines des méthodes pour ces étapes sont décrites en détaillent ailleurs12,13,14 et ne seront donc pas être décrit ici. Pour les échantillons de racines, bloqueurs de l’ANP ont été ajoutés afin de réduire la quantité d’ADN de plastes amplifié dans les tissus végétaux, qui a été précédemment décrit précisément15. Deux contrôles ont été utilisés dans le séquençage, un témoin négatif qui comprenait l’extraction plaque vierge contrôles (voir la note après l’étape 5.1.5), et une communauté de simulacre d’une population connue de l’ADN bactérien (voir Table des matières) qui a servi comme positif contrôle.
Remarque : Dans la plupart des cas, les trousses de réactifs MiSeq v3 sont utilisés dans le mode de base fin paires 2 X 300. Pour les échantillons dans ce manuscrit, le Illumina HiSeq 2500 a été utilisé en mode rapide avec 250 mode jumelés-fin (2 x 250). Tous les échantillons ont été séquencés dans la même voie. - Traiter les données de séquençage à travers un pipeline pour les analyses de la communauté microbienne (USEARCH v9.2.64, QIIME v1.9.1 et RStudio v3.4.3 16).
- Préparer les données de séquençage en utilisant USEARCH17.
Remarque : USEARCH est disponible en ligne avec des instructions complètes (https://www.drive5.com/usearch/). - Démultiplexer les données de séquençage à l’aide de lectures d’index ou de codes à barres pour assigner Illumina lectures aux échantillons.
- Fusionner les lectures jumelé-fin pour obtenir des séquences consensus. Utilisez la commande : usearch-fastq_mergepairs * R1*.fastq-relabel @ - fastq_maxdiffs 10 - fastq_minmergelen - 230 fastq_maxmergelen 320 - fastq_pctid 80 - fastqout merged.fq.
Remarque : Les paramètres sont réglés par le biais de référencement manuel USEARCH. - Retirez les amorces les données de séquençage afin d’éviter les substitutions dans les séquences d’amorces, qui peuvent être causées par la réaction de PCR. Utilisez la commande : usearch-fastx_truncate merge.fq - stripleft 19 - stripright 20 - fastqout stripped.fq.
- Filtrer les données de séquençage pour supprimer les lectures de faible qualité et garder les séquences d’unité taxonomique opérationnelle (UTO) de haute qualité. Utilisez la commande : usearch-fastq_filter stripped.fq-filtered.fa - fastaout fastq_maxee 1.0.
- Préparer les données de séquençage en utilisant USEARCH17.
- OTUs génère USEARCH.
- Effectuer la duplication pour identifier l’ensemble des séquences OTU uniques. Utilisez la commande : usearch-fastx_uniques filtered.fa - fastaout uniques.fa - sizeout-relabel Uniq.
- OTUs cluster avec 97 – 100 % de similarité de séquence pour désigner l’OTUs uniques. Utilisez la commande : usearch-cluster_otus uniques.fa - minsize 2 - otus otus.fa-relabel Otu.
Remarque : Cette étape intègre également la suppression des singletons de l’OTUs en cluster et l’enlèvement des chimères de données de séquençage. - Créer une table de l’OTU dans USEARCH. Utilisez la commande : usearch-usearch_global stripped.fq - db otus.fa-chapelet plus - otutable.txt - otutabout id 0,97.
Remarque : Cette commande génère un tableau avec le nombre de lectures (chefs) de OTUs tous pour chaque échantillon. La table OTU est utilisée pour les étapes en aval, y compris les analyses différentielles de l’abondance et la diversité microbienne. Exemple de table de l’OTU est montré dans la figure complémentaire 2. - Procéder à une analyse de la raréfaction dans QIIME v1.9.1 18.
Remarque : La courbe de raréfaction est calculée à l’aide de la table de l’OTU pour déterminer si la profondeur du séquençage des échantillons correctement la communauté microbienne. Exemple de courbes de raréfaction apparaissent complémentaires Figure3. - Procéder à une analyse de la diversité alpha18. Utiliser alpha_rarefaction.py dans QIIME v1.9.1 pour calculer la diversité de la communauté microbienne au sein de chaque échantillon.
Remarque : Cette analyse calcule les indices de diversité comme Shannon19, Simpson20et Chao121. - Mener une analyse la diversité des bêta18,22. Utiliser le script Python : beta_diversity_through_plots.py dans la matrice de dissimilarité QIIME v1.9.1 Bray-Curtis.
Remarque : Cette analyse compare la composition de la communauté microbienne entre les échantillons. - Effectuer des analyses statistiques entre les groupes. Utiliser les matrices de distance calculées pour PERMONOVA en utilisant la fonction d’adonis et d’analyse de la variance dans le végétalien paquet23 v2.4.5 RStudio16. Procéder à une analyse canonique d’analyse de coordonnées principales (CAP) à l’aide de la fonction capscale dans le package végétalien. Visualiser les données à l’aide de ggplot2 paquet24 v2.2.1 dans RStudio.
Résultats
Les résultats représentatifs présentés dans ce manuscrit proviennent d’un site mis en place en 2012 à l’Université de Nebraska-Lincoln Agriculture Research Division ferme près de Mead, né avant l’expérience, le site avait été géré comme une rotation maïs-soya . Le site d’étude est situé sur trois différents types de sols, mais les données ont été analysées comme si toutes les modifications dans les propriétés du sol mesurées étaient dus à des traitements imposées.
Le site du champ contenait deux, pure, stands de switchgrass (P. virgatum cv Liberty) et barbon (a) ainsi qu’un mélange de faible diversité herbe contenant barbon, Sorghastrum (S. nutans) et « Butte » boutelou grama ( B. curtipendula). Les trois parcelles de graminées de saison chaude étaient dans une conception en blocs aléatoires complets qui ont été répliquée trois fois. Imbriqué dans trois parcelles différentes herbes étaient deux traitements de fertilisation d’azote (N), qui comptait 56 (N1) et 112 (N2) kg N ha-1 d’urée appliquée. Au moment de l’échantillonnage microbiome à la fin de la saison de croissance, le sol contenu 8,0 ± 1.1 (moyenne ± écart type) ppm nitrate dans les parcelles fertilisées avec 112 kg N ha-1 et 6,8 ± 0,7 (moyenne + SD) nitrate ppm dans les parcelles fertilisées avec 56 kg N ha-1. Les parcelles avaient été fertilisés une fois par an. L’herbe de saison chaude, les parcelles ont été désignés comme les principaux emplacements (8000 m2) et les traitements N étaient les parcelles de split (4000 m2). Barbon a été classée comme un mélange de 50/50 de « Bonanza » et « Mine d’or » et Sorghastrum a été classée comme un mélange de 50/50 de « Éclaireur » et de « Guerrier ». Les parcelles ont été plantées en 2012, et la première application de N s’est produite au printemps 2013.
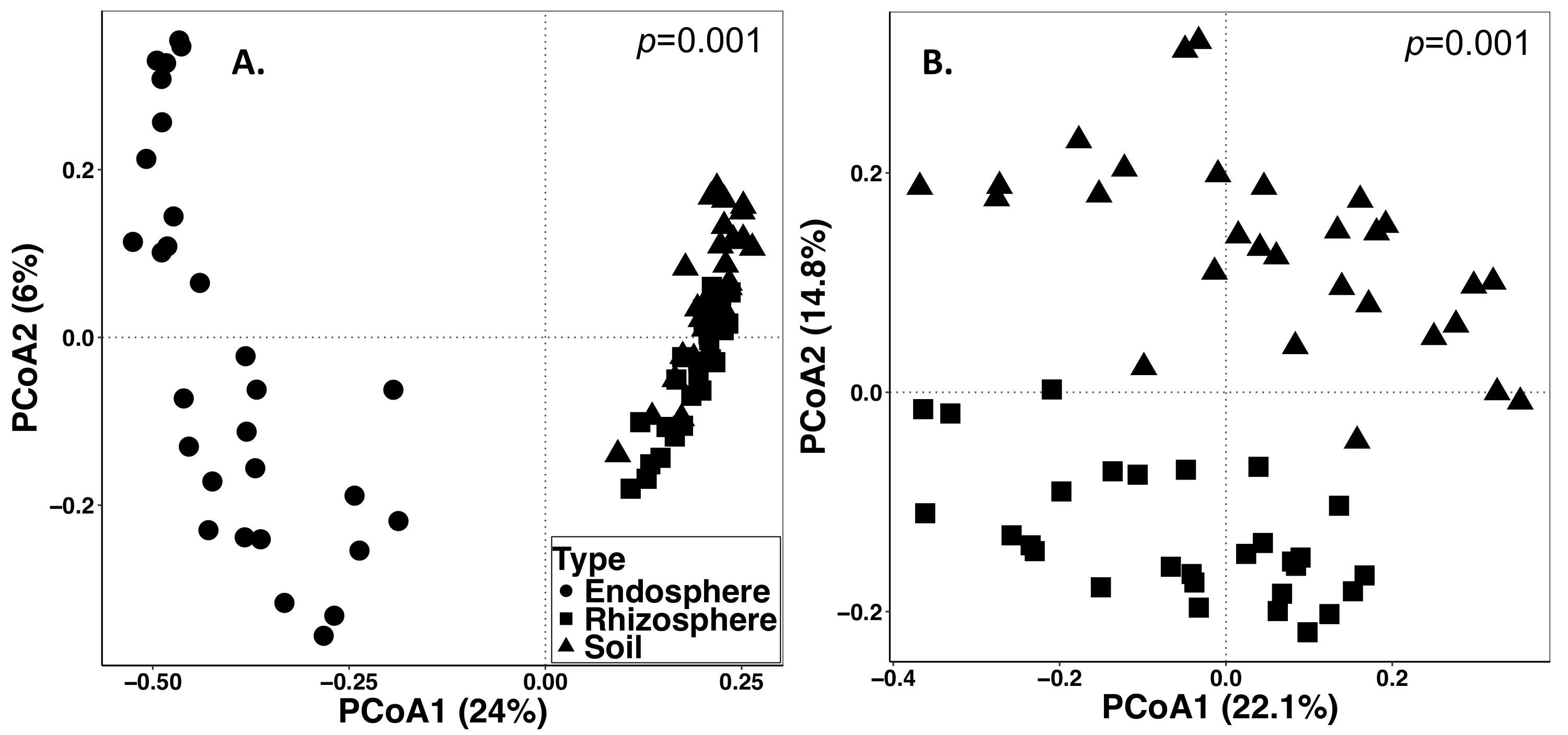
Le sol et l’échantillonnage de la racine a été menée sur 15 septembre 2014. L’ouvrage décrit ci-dessous a été réalisée sur un champ qui a été défini comme un design aléatoire de parcelles divisées avec trois répétitions (Figure 1). La profondeur moyenne de séquençage de tous les échantillons d’endosphère ont été comme suit : 4871 ± 5711 (moyenne ± écart-type), rhizosphère : 40726 ± 14684, sol : 38184 ± 9043. Parmi les principales sources de variation dans ces expériences, en utilisant les méthodes décrites, est la différence dans les communautés microbiennes trouvée entre les types d’échantillons (Figure 2). Dans ce jeu de données représentatif, la rhizosphère et les sols semblent être plus une composition semblable à l’autre que l’endosphère (Figure 2 a). Cependant, il y avait aussi hautement significative (p = 0,001) différences dans la composition des communautés microbiennes rhizosphère et le sol (Figure 2 b). La variation totale pris en compte dans ces expériences analysées par type d’échantillon était de 26 %.
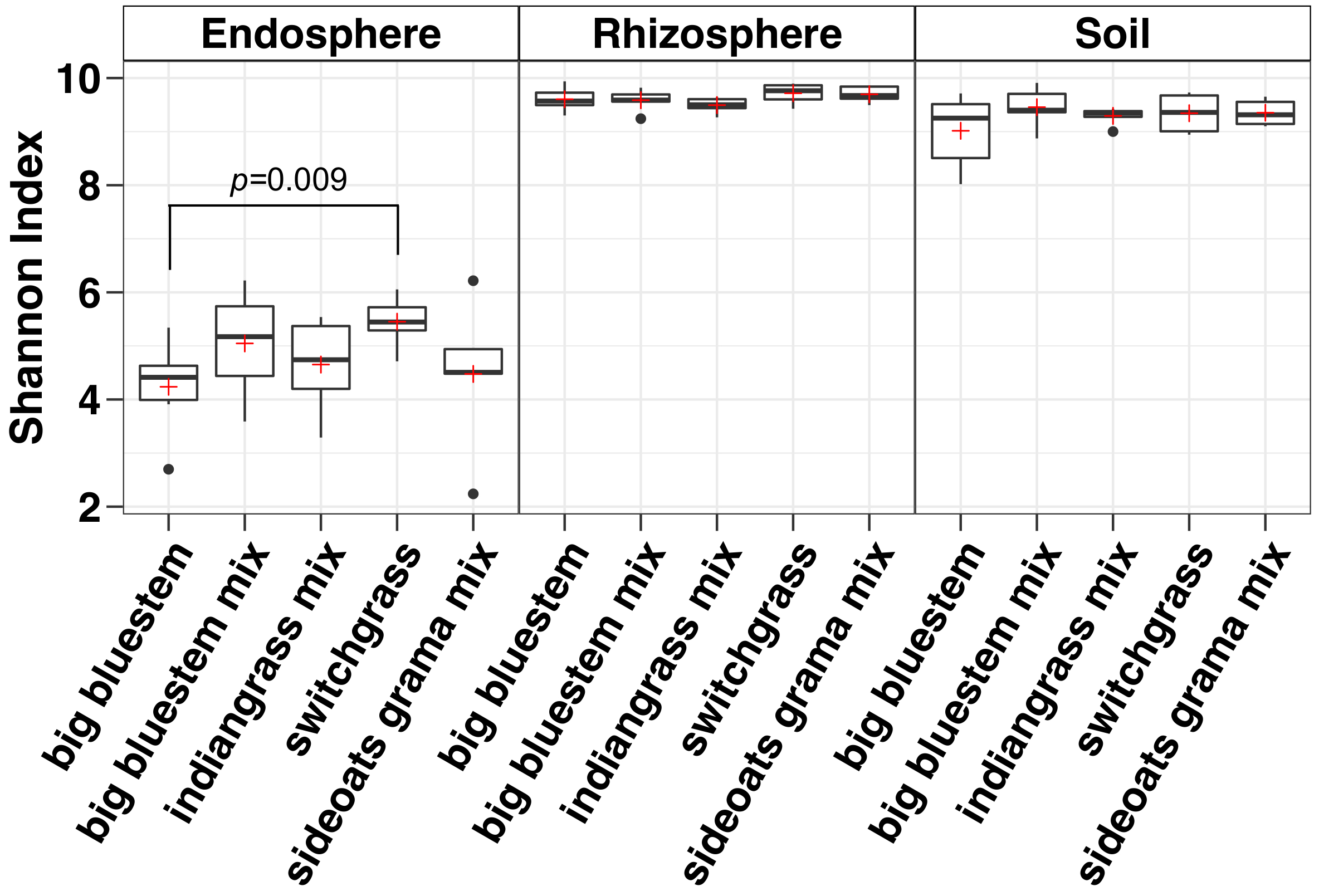
Analyse de la diversité alpha a montré que les communautés microbiennes dans l’endosphère étaient inférieures dans la diversité d’échantillon par rapport au sol et la rhizosphère (Figure 3). Les seules différences significatives dans la diversité entre les espèces de graminées dans un compartiment quelconque variaient entre les échantillons endosphère de barbon et le panic raide, avec le panic raide, vu la diversité des espèces microbiennes significativement plus élevée (Figure 3). L’analyse de l’abondance relative (Figure 4) met en évidence la prédominance des protéobactéries suivie Actinobacteria dans tous les types d’échantillons. Sol et la rhizosphère sont également dominées par Acidobacteria et Chloroflexi tandis que l’endosphère avait une plus grande abondance relative des Bacteriodetes.
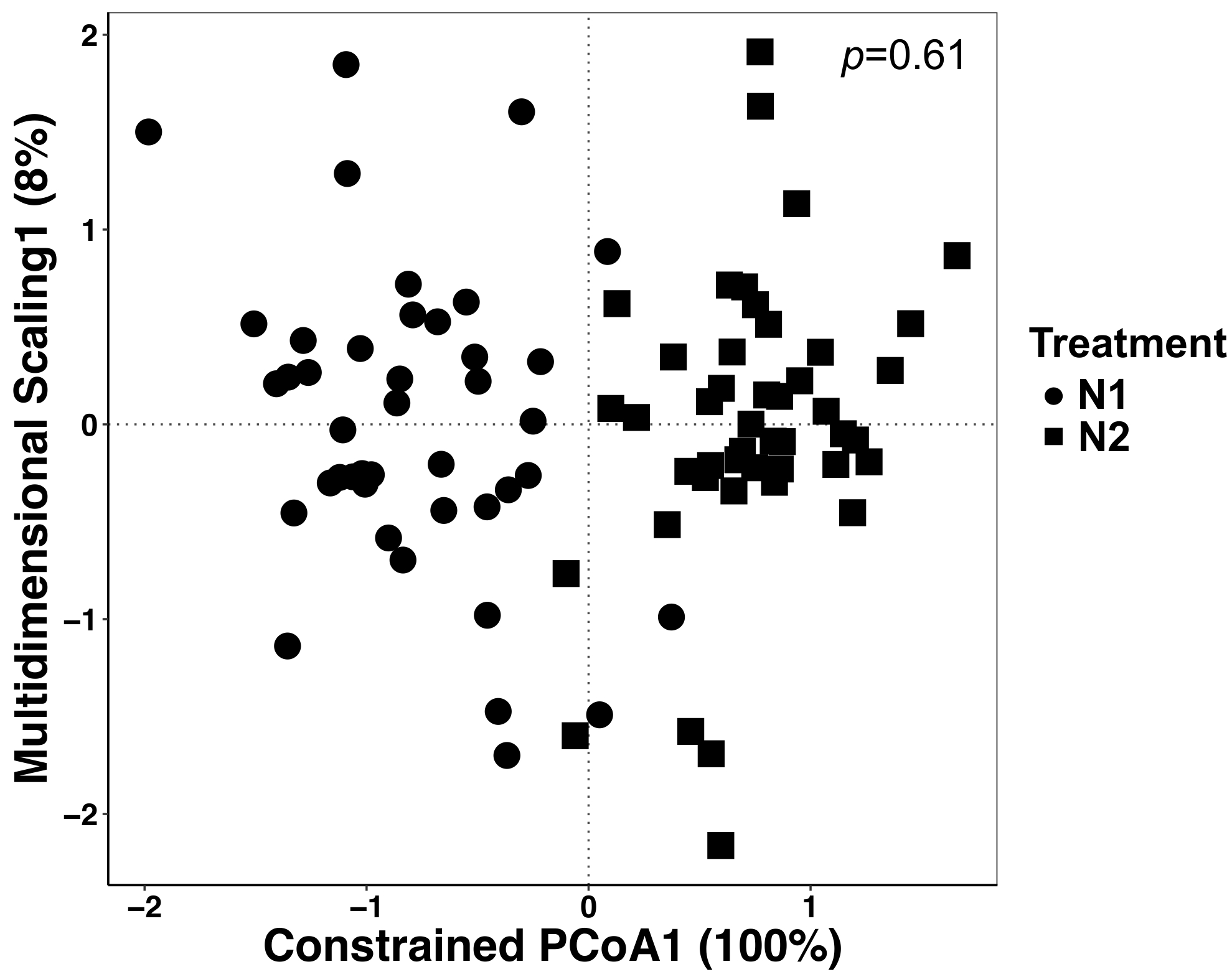
Dans cette expérience, les plantes ont été cultivées avec deux différentes quantités de fertilisation azotée et c’est pourquoi nous avons analysé les données afin de déterminer s’il y a des effets du traitement. Effets du traitement ont représenté 12 % de la variation totale mais n’étaient pas significativement différentes, bien qu’à l’ordination les deux traitements semblent différentes (Figure 5). Cela met en évidence l’importance des analyses statistiques pour ces ensembles de données plutôt que des inspection visuelle ou des jugements qualitatifs.
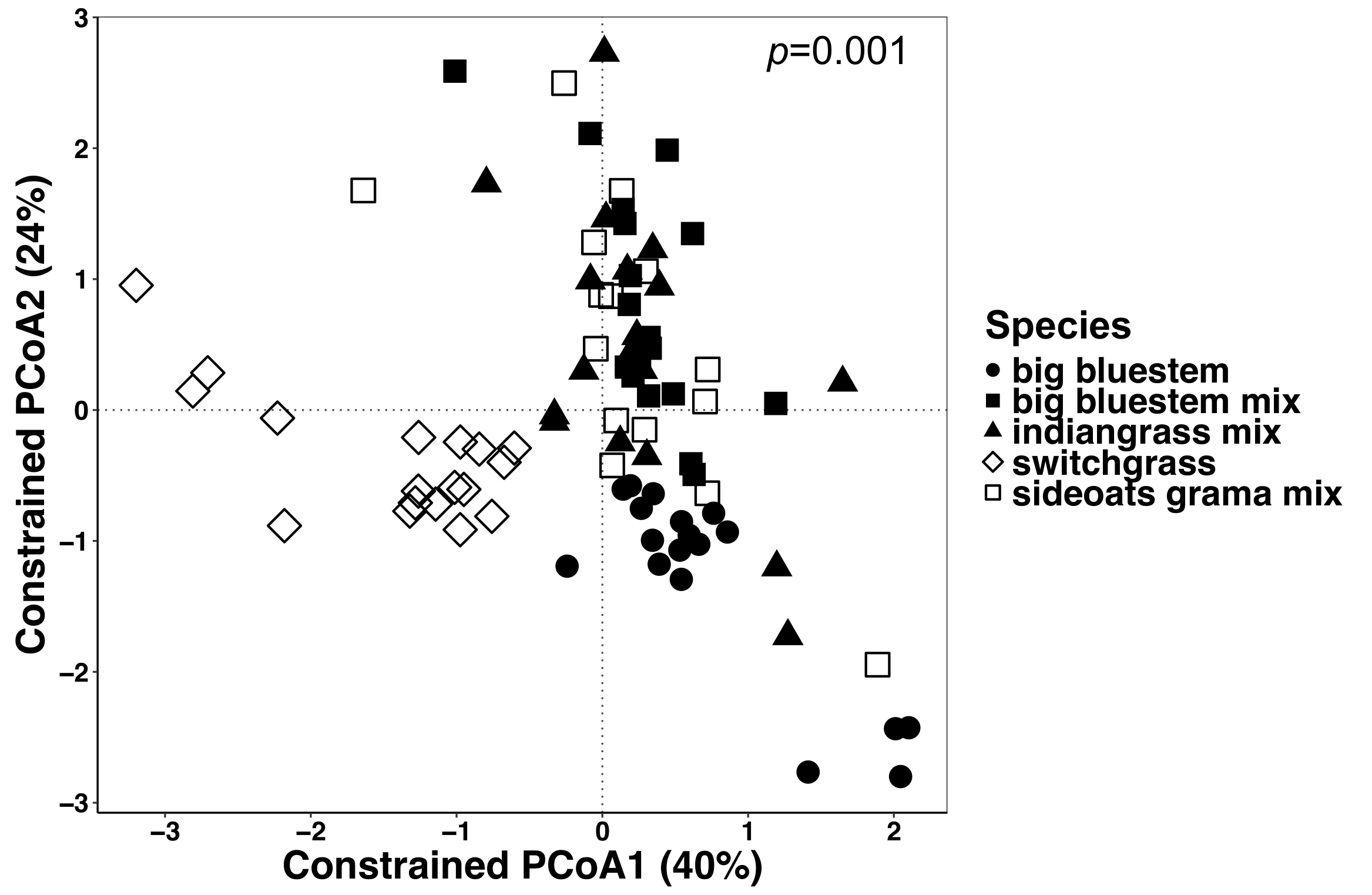
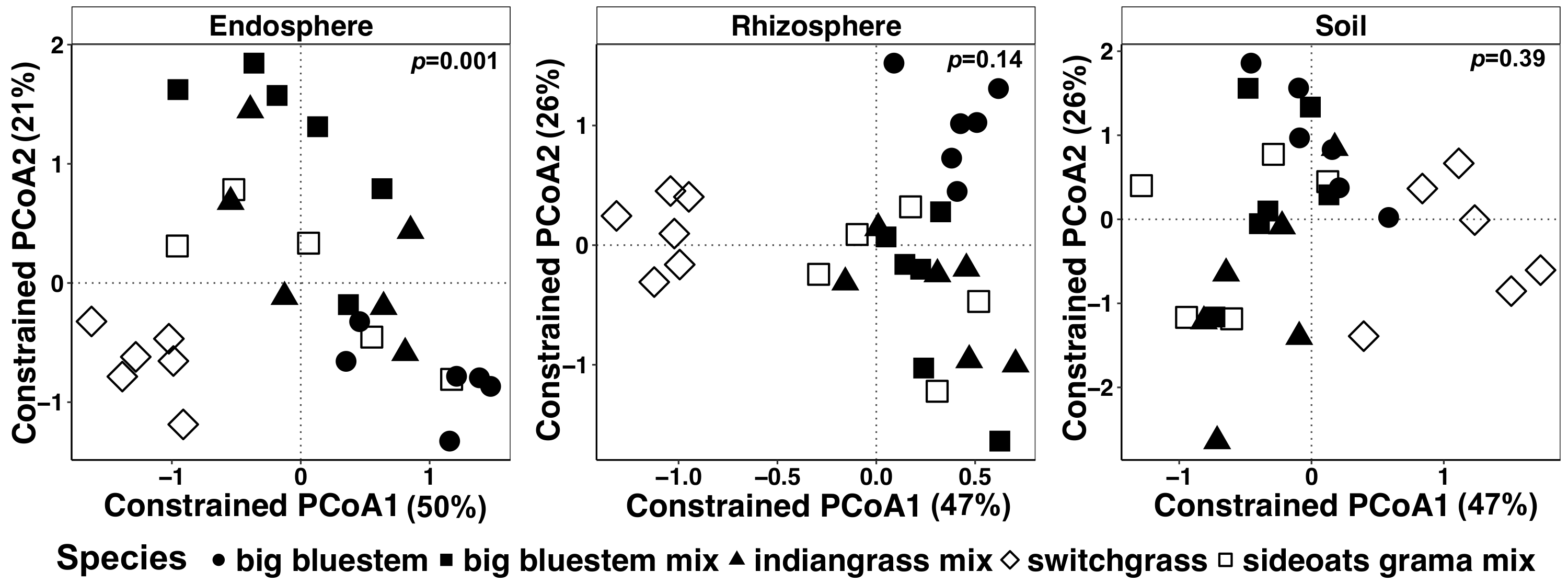
Plante-influencé les différences dans le microbiome des tissus végétaux et du sol ont été visualisées à l’aide d’une méthode de contrainte de l’ordination. Des différences statistiques ont été déterminées à l’aide d’une analyse PERMANOVA pour vérifier si les variables spécifiques, telles que les espèces, entraînent dans la composition de la communauté microbienne significativement différente entre les échantillons. Lorsque tous les types d’échantillons ont été analysés ensemble, une différence hautement significative a été trouvée dans la composition des communautés microbiennes dues aux espèces de végétaux (Figure 6). Dans cette expérience, la quantité de variation expliquée par les espèces végétales était de 6,7 %. Enfin, chaque type d’échantillon a été analysé individuellement afin de déterminer lequel des types échantillon pourrait être conduite l’effet d’espèces végétales importantes. Seulement dans l’endosphère était là une différence hautement significative (p = 0,001) entre les compositions de la communauté microbienne des espèces de plantes différentes (Figure 7). Dans les autres types d’échantillons, l’effet de l’espèce n’était pas significative lorsque analysés individuellement. Dans l’endosphère, la variation en pourcentage en raison de l’espèce était de 27 %, alors qu’il était plus faible dans la rhizosphère (18 %) et le sol (15 %). De plus, cela met en évidence l’importance d’analyser chaque type de tissu individuellement.

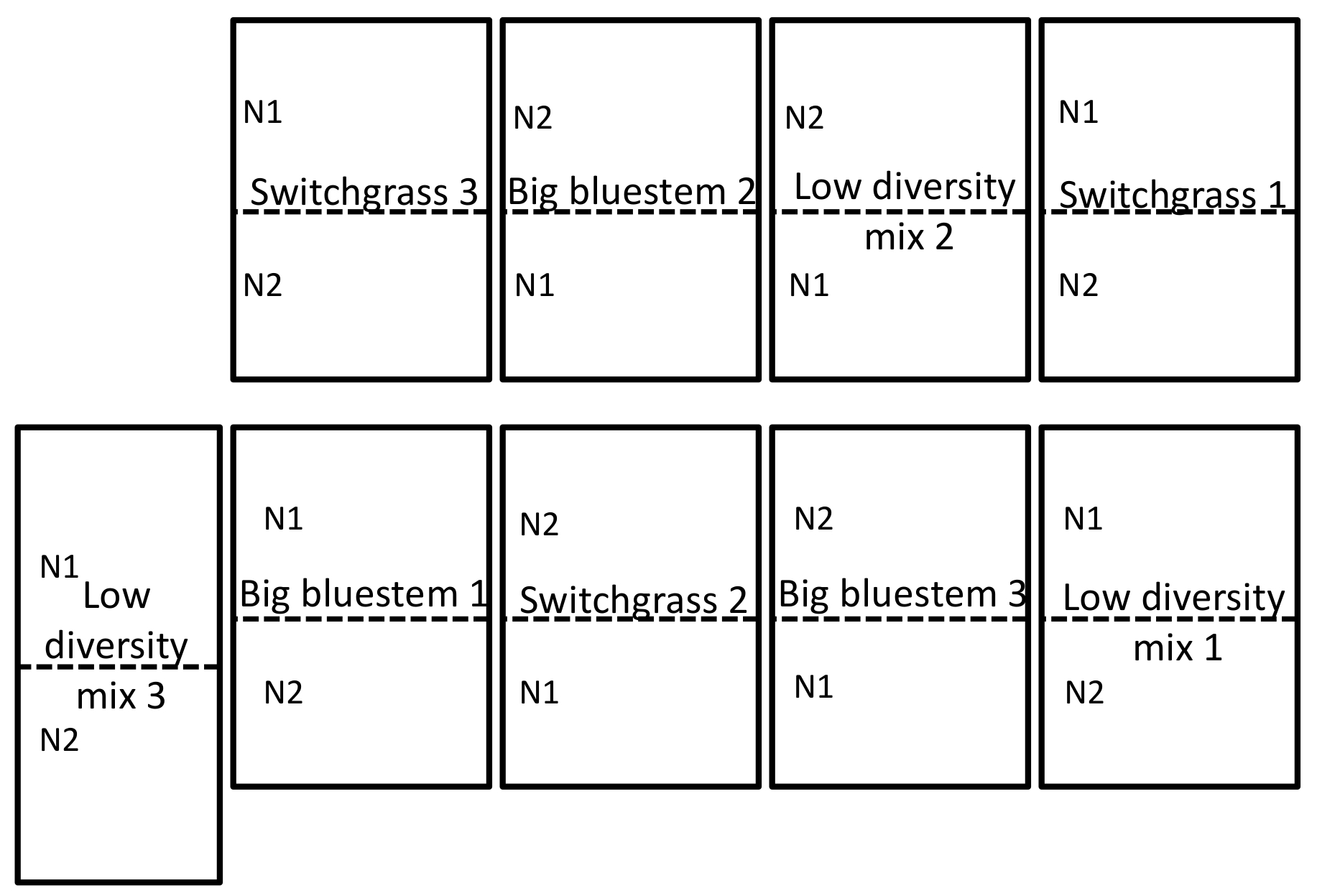
Figure 1 : exemple de la conception expérimentale. Conception de domaine expérimental illustrant une conception en blocs aléatoires complets en triple exemplaire du site champ situé au University of Nebraska-Lincoln Eastern Nebraska Research and Extension Cetres Mead, NE. Pour une description complète du site, voir la section résultats. N1 est le faible (56 kg N ha-1 urée) et N2 (112 kg N ha-1 urée) est le plus haut taux d’azote qui a été appliqué. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 2 : analyse de la diversité bêta en comparant la composition microbienne dans les types d’échantillons différents y compris endosphère, rhizosphère et le sol de l’échantillonnage de graminée vivace en 2014. L’analyse a été réalisée à l’aide d’un script Python dans QIIME1.9.1 pour produire la matrice de dissimilitude de Bray-Curtis. Analyse de coordonnées principales (PCoA) basé sur la matrice de dissimilitude de Bray-Curtis a été visualisée dans RStudio. PCoA1 et PCoA2 indiquent la variance de première et deuxième expliquée par l’analyse PCoA. PERMANOVA analyse statistique a été réalisée pour déterminer l’importance entre les types d’échantillons, et la valeur de p est affichée dans le coin supérieur droit. Chaque symbole dans les figures représentent l’ensemble de la communauté microbienne pour chaque échantillon. (A) endosphère, la rhizosphère et sol types d’échantillons ont été analysés ensemble. Tous les 87 échantillons ont raréfié à 486 séquences par échantillon. (B) les échantillons rhizosphère et de sol ont été analysés ensemble. Toutes les 59 échantillons ont raréfié avec 8231 séquences. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 3 : analyse de la diversité Alpha utilisant l’indice de Shannon pour chaque espèce dans l’endosphère, la rhizosphère et le sol. L’analyse a été réalisée à l’aide d’un script Python dans le QIIME1.9.1. Raréfaction a été faite pour l’endosphère, la rhizosphère et les types d’échantillons de sol respectivement avec des séquences 486, 17154 et 8231 par échantillon. Cases indiquent les 25e et 75e centiles (premier et troisième quartiles). La ligne horizontale au sein de la zone désigne la médiane et le rouge plus montre la moyenne. Moustaches afficher l’étendue des données à l’exclusion des valeurs aberrantes (qui apparaissent comme des points noirs) qui a chuté de plus de 1,5 fois l’écart interquartile (n = 6 pour chaque échantillon sauf pour boutelou grama mélanger où n = 5). L’indice de Shannon de cinq espèces dans l’endosphère ont été inférieurs de la rhizosphère et le sol. Non paramétriques test de Wilcoxon somme a servi à déterminer l’importance entre l’espèce et seulement des différences significatives entre les espèces ont été montrés sur le dessus les cases. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 4 : abondance Relative au niveau de l’embranchement dans l’endosphère, la rhizosphère et sol. Des échantillons ont été analysés afin de comparer l’abondance des phylums microbiennes entre types différents échantillons (n = 29, pour chaque type d’échantillon). L’analyse a été réalisée à l’aide d’un script Python dans QIIME1.9.1 de la table de l’OTU. Les différentes couleurs à l’intérieur du graphique à secteurs indiquent les embranchements. Le pourcentage indique l’abondance relative de chaque phylum dans chaque type d’échantillon. Les informations de l’embranchement a été annotées en utilisant le projet de base de données Ribosomal classificateur (RDP)25. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 5 : analyse à l’aide de traitement comme facteur contraignant entre tous les types d’échantillons. L’analyse canonique de l’analyse de coordonnées principales (PAC) a été réalisée afin de déterminer s’il y a des différences dans la composition de la communauté microbienne entre les traitements. Pour chaque traitement de N, n = 42 pour N1 (56 kg N ha-1) et n = 45 pour N2 (112 kg N ha-1). La matrice de dissimilitude de Bray-Curtis a été générée à l’aide d’un script python dans le QIIME1.9.1. Analyse de la calotte basé sur la matrice de dissimilitude de Bray-Curtis a été fait en limitant le traitement comme étant le facteur RStudio. PERMANOVA analyse a été effectuée pour déterminer si les différences de traitement sont significatives, et la valeur de p est affichée dans le coin supérieur droit. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 6 : analyse utilisant des espèces végétales comme facteur contraignant entre tous les types d’échantillons. L’analyse a été réalisée afin de déterminer s’il y a des différences dans la composition de la communauté microbienne entre des espèces de plantes dans tous les types d’échantillons. Ordination de coordonnées principales et analyse de la calotte de tous types d’échantillons (endosphère, rhizosphère et le sol) ont été réalisées à l’aide d’une matrice de dissimilitude de Bray-Curtis. La matrice de dissimilitude de Bray-Curtis a été générée à l’aide du script Python dans QIIME1.9.1. Analyse de la calotte basé sur la matrice de dissimilitude de Bray-Curtis a été fait en contraignant les espèces végétales comme étant le facteur RStudio. PERMANOVA analyse statistique a été réalisée pour déterminer l’importance entre les espèces végétales, et la valeur de P est affichée dans le coin supérieur droit. Chaque symbole dans les chiffres représente l’ensemble de la communauté microbienne pour cet échantillon. n = 18 pour chacune des espèces dans tous les types d’échantillons sauf n = 15 pour le mélange de grama boutelou. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 7 : exemple d’analyse de la calotte espèces en utilisant comme facteur contraignant pour chaque type d’échantillon individuellement. Ordination de coordination principale et analyse de la calotte de chaque type d’échantillon (endosphère, rhizosphère et le sol) à l’aide de la matrice de dissimilitude de Bray-Curtis. Chaque type d’échantillon s’est raréfié à 486, 17154 et 8231 lectures par exemple respectivement à endosphère, la rhizosphère et sol. Espèces a été utilisé comme le facteur pour contraindre l’ordination. PERMANOVA analyse statistique a été réalisée pour déterminer l’importance entre les espèces de plantes dans chaque type d’échantillon, et la valeur de p est affichée dans le coin supérieur droit. Chaque symbole dans la figure représente l’ensemble de la communauté microbienne pour chaque échantillon. Taille de l’échantillon est n = 29, pour chaque type d’échantillon, n = 6 pour chacune des espèces végétales dans chaque type d’échantillon à l’exception de la combinaison de grama boutelou (n = 5). S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}
Discussion
Les méthodes décrites dans ce manuscrit doivent aux scientifiques d’entrer facilement dans le domaine du sol et plante métagénomique. Au cours des années, nous avons raffiné nos méthodes depuis mène l’expérience décrite dans ce manuscrit. Un seul changement est que nous maintenant avant l’étiquette des tubes avant de sortir sur le terrain pour l’échantillon. Notre laboratoire utilise un système de codes à barres et une imprimante. L’imprimante d’étiquettes non seulement gagner du temps quand l’étiquetage tubes, mais aussi rend tout plus facile de suivre et d’identifier correctement les échantillons sans les aléas de l’écriture de la main de l’homme. Un autre point critique est que nous essayons de traiter le matériel après avoir porté de retour sur le terrain dès que possible. Nous visons à geler le sol utilisé pour l’analyse de l’ADN, stériliser et geler les racines et filtrer et geler la rhizosphère dans les 12 à 36 heures après son retour sur le terrain. Les procédures d’extraction de l’ADN sont longs avec nombreuses étapes, notamment pour le sol et la rhizosphère, donc nous avons acheté un robot (Kingfisher Flex, ThermoFisher) qui minimise les mains à l’heure pour les protocoles d’extraction d’ADN, réduit les erreurs humaines qui peuvent être introduites, et améliore la cohérence dans la manière différents lots de terre, racines ou rhizosphère sont traités. Lorsque vous travaillez avec le matériel végétal, il est important de décider du type de racine d’étudier ou de prendre une variété de types de racines pour obtenir un « échantillon représentatif ». Maintien de racines et les feuilles à l’état congelé quand effectuer les extractions d’ADN est important, comme est de s’assurer il n’y a aucune contamination croisée entre les échantillons lors du remplissage des plaques 96 puits d’extraction des ADN. Un autre facteur important à considérer est le nombre de répétitions à utiliser pour concevoir des expériences sur le terrain et en utilisant un complet randomisé conception si possible26. En raison de la variabilité du champ élevé, il peut être nécessaire d’avoir un grand nombre de répétitions pour détecter de petites différences. Enfin, d’après notre expérience, il est essentiel pour s’assurer que les sols ne sont pas trop humides, lorsque les racines de l’excavation. Si les sols sont saturés d’eau n’est pas seulement salissant à travailler avec, mais il est aussi très difficile de définir la rhizosphère et de retirer du sol les racines.
Au lieu de secouer les tubes à la main pour libérer la rhizosphère, que nous sommes passés à vortexers alimenté par un générateur de gaz pour faciliter le travail dans le domaine et plus encore, une modification qui a été faite très tôt durant le développement de ces méthodes a été normalisée en termes de le temps et la manière que chaque tube a été agité. Une des limites de l’approche du séquençage de l’amplicon sont que la résolution taxonomique des résultats est souvent limitée et nombreux OTUs sont connus inconnus ou seulement au niveau de la famille ou du genre. Ce domaine de recherche est en évolution rapide, il est donc important d’être conscient des approches nouvelles ou en développement, en particulier pour l’analyse des données susceptibles de renforcer la résolution des résultats.
Ces protocoles sont seulement pour l’étude des bactéries et archaea, pas de champignons. L’utilisation de différentes amorces pour l’amplification permettra l’étude des communautés de champignons à l’aide de la même ADN échantillons27,28. Ces méthodes ne requièrent pas l’achat de grandes quantités de matériel parce que les méthodes peuvent être simplifiées. Les méthodes que nous décrivons ici sont généralement pour déterminer « qui est là », mais le domaine évolue rapidement en posant des questions importantes sur la fonction, qui peut-être être résolue en utilisant les méthodes de séquençage de fusil de chasse, d’isolation et de tester les fonctionnalités de microbes, ou génomes microbiens ensemble de séquençage.
Les résultats représentatifs soulignent les différences entre les communautés microbiennes qui peuvent être identifiées à l’aide des méthodes décrites. En utilisant une approche de la diversité bêta à l' analyse de données22, les différences de composition apparaissaient entre les types d’échantillons. Ces différence ont été clairement observées dans la plupart des autres études où endosphère, rhizosphère et le sol contiennent des communautés microbiennes unique3. L’indice de diversité de Shannon a été calculée pour déterminer l’abondance et la régularité des espèces microbiennes présentes au sein de chacune des espèces végétales dans l’endosphère, la rhizosphère et sol. Comme indiqué dans cette étude et dans beaucoup d’autres, diversité alpha est plus élevée dans le sol, diminuant légèrement dans la rhizosphère et puis diminue significativement dans l’endosphère3,5,29. Ces résultats indiquent que les méthodes décrites ici sont adaptés pour identifier les changements de composition dans l’endosphère, rhizosphère et le sol.
La domination des protéobactéries est commun à la conclusion des études sur l’endosphère et le sol30,31,32. Endosphère a généralement une plus faible diversité d’espèces microbiennes avec une plus grande abondance relative des Proteobacteria. Encore une fois, cela met en évidence que les résultats présentés ici sont représentatifs des autres résultats dans la littérature. Les effets du traitement dans cette étude n’étaient pas significativement différents et deux principales raisons pour cela peut-être que les différences imposées par les traitements n’étaient pas assez grands pour générer une variation suffisante pour détecter et que cet échantillonnage a été effectué à la fin de la saison, quand les champs pourraient avoir suffisamment de temps pour tirer vers le bas de l’azote à des niveaux similaires, qui est ce qui a été mesuré à la fin de la saison de croissance. Dans une autre étude utilisant des taux de fécondation similaires sur une longue période de temps, seulement relativement petits changements dans la composition de la microbiome ont été mesurées33. Autres études ont montré des changements dans les communautés en raison de l’azote engrais34,35fongiques et bactériennes.
Espèces de plantes sont connus pour jouer un rôle dans la détermination de leur microbiomes3,32,36 et même de petites différences dans la variation de la population microbienne ont été démontrées entre les génotypes des plantes différentes dans un 37de même espèce. Dans cette étude, une différence significative dans la composition de la communauté microbienne a été trouvée entre les espèces végétales. Dans tous les types d’échantillon, il est apparu que Panic avait la composition microbienne plus distincte, mais les différences entre les espèces seulement statistiquement sont dans l’endosphère. Composition de la communauté rhizosphère peut sont devenus importante si plusieurs répétitions étaient disponibles pour l’analyse.
Le champ combiné, lab et les protocoles analytiques décrits ici fournissent une méthode puissante pour étudier comment les différents facteurs influence la composition des communautés microbiennes dans les sols, la rhizosphère et l’endosphère de racines36. Il y a beaucoup de travail à faire dans le domaine de l’étude microbiomes, particulièrement dans les champs agricoles. Questions importantes à propos de comment les rendements sont altérées par le microbiome du sol n’ont pas encore entièrement élucidée. Même les questions plus fondamentales au sujet de l’influence de la rotation des cultures le microbiome du sol, comment le calendrier altère le microbiome, stress abiotique comment modifie le microbiome, comment le type de sol interagit avec ces facteurs afin d’altérer le microbiome et s’il existe des microbes universels dans certaines cultures ou des régions des USA sont toutes les questions en suspens. Ces méthodes seront également utiles pour les études épidémiologiques d’identifier la présence et la persistance de bactéries pathogènes et bénéfiques. Un autre horizon futur pour ces méthodes sera de commencer à intégrer les méthodes de l’ADN décrites ici avec plante et microbe RNA et données de métabolite. Amélioration supplémentaire et l’essai de plusieurs variables sera importants pour l’optimisation supplémentaire de ces protocoles.
Déclarations de divulgation
Les auteurs n’ont rien à divulguer.
Remerciements
Le développement de ce manuscrit est pris en charge par la National Science Foundation EPSCoR Center pour Root et Rhizobiome Innovation Award OIA-1557417. La collecte des données a été soutenue par des fonds de l’Université de Nebraska-Lincoln, Agricultural Research and Development et par une subvention de la trappe de l’USDA. Nous remercions également le soutien de l’USDA-ARS et appui a été fourni par l’Agriculture and Food Research Initiative compétitive Grant no 2011-68005-30411 de l’USDA National Institute of Food et de l’Agriculture d’établir et de gérer ces domaines.
matériels
| Name | Company | Catalog Number | Comments |
| Dneasy PowerSoil HTP 96 Kit | Qiagen/MoBio | 12955-4 | Extraction kit for soil and rhizosphere |
| Dneasy PowerPlant HTP 96 Kit | Qiagen/MoBio | 13496-4 | Extraction kit for roots |
| D-Handle Digging shovel, 101 cm L | Fiskars | 9669 | |
| Rapid Tiller, 40 cm L | Truper | 34316 | |
| Ziploc Bags, 17.7 cm x 19.5 cm | Ziploc | NA | |
| Cooler | Any | NA | |
| Wash pan | Any | NA | |
| Plastic bucket | Any | NA | |
| Gloves (work and lab) | Any | NA | |
| 20 cm diameter Soil sieve #8, 2360 μm mesh size | Dual Manufacturing Co., Chicago IL | US8-8FS | |
| 20 cm diameter Soil Sieve #4, 4750 μm mesh size | Dual Manufacturing Co., Chicago IL | US8-4FS | |
| portable generator | Honda brand works well | NA | |
| Sterile cell strainers 100 μm mesh size | Fisher Scientific | 22-363-549 | |
| NaH2PO4·H2O | VWR | 0823 | |
| Na2HPO4 | VWR | 0404 | |
| Silwet L-77 | Lehman Seeds | VIS-30 | Surfactant |
| Autoclaves | Any | NA | |
| Drying Oven | Any | NA | |
| Scale | Any | Any | |
| Bleach | CLOROX - household strength | NA | |
| Tween 20 | Any | NA | |
| Liquid Nitrogen | Any | NA | |
| Dry Ice pellets | Any | NA | |
| Ethanol | Any | NA | |
| 11 cm precision fine point tweezers | Fisher | 17456209 | |
| 18 cm Straight point specimen forceps | VWR | 82027-436 | |
| 13.5 cm Pruning Scissors | Fiskars | 9921 | |
| 2 mL tube | Any | NA | |
| 15 mL PP conical tube | MIDSCI | C15B | |
| 50 mL PP conical tube | MIDSCI | C50B | |
| Ultrapure water | Millipore-sigma | Milli-Q Integral, Q-POD | |
| Qubit 2.0 fluorometer | Invitrogen | Q32866 | |
| Qubit dsDNA HS Assay Kit | Invitrogen | Q32854 | For DNA quantification of removed samples |
| QuantiFluor dsDNA System | Promega | E2670 | For DNA quantification |
| 96-Well Black with Clear Flat Bottom Plates | Corning | 3631 | |
| pPNA PCR Blocker | PNA Bio | PP01-50 | |
| mPNA PCR Blocker | PNA Bio | MP01-50 | |
| Genomic DNA from Microbial Mock Community B (Even Low Concentration) v5.1L, for 16s rRNA Gene sequencing | BEI Resources | HM782D | |
| Adhesive 8 well-strips for plates | VWR | 89134-434 | |
| Stainless steel beads, 3.2 mm dia | Next Advance | SSB32 | |
| 1 ml Assay block (DNA extraction plate for the Qiagen/MoBio Dneasy PowerPlant HTP Kit) | CoStar | 3959 | |
| antistatic PP weighing funnel, size small for soil/rhizosphere | TWD Tradewinds, INC | ASWF1SPK | |
| antistatic PP weighing funnel, size x-small for root/leaf | TWD Tradewinds, INC | ASWFXSCS | |
| Genie 2 Digital Vortex | Scientific Industries | SI-0236 | |
| Vortex adapter for 50 mL tubes | Scientific Industries | SI-H506 | |
| Mortar (100 mL) and pestle | Any | NA | |
| Metal micro-spatula | VWR | 80071-672 | |
| Disposable antistatic microspatulas | VWR | 231-0106 | |
| Brown Paper bag 2# (10.95 cm x 6.19 cm x 20 cm) | Duro | 18402 | |
| 5424 Centrifuge for 2 mL tube | Eppendorf | 22620461 | |
| Centrifuge for 96-well plate | Sigma4-16S | 81510 | |
| Centrifuge rotor for 50 mL tubes | Sigma4-16S | 12269 - Biosafe | |
| KAPA HiFi DNA polymerase | Kapa Biosystems |
Références
- Philippot, L., Raaijmakers, J. M., Lemanceau, P., vander Putten, W. H. Going back to the roots: the microbial ecology of the rhizosphere. Nature Reviews Microbiology. 11, 789-799 (2013).
- Fierer, N. Embracing the unknown: disentangling the complexities of the soil microbiome. Nature Review Microbiology. 15 (10), 579-590 (2017).
- Wang, P., et al. Shifts in microbial communities in soil, rhizosphere and roots of two major crop systems under elevated CO2 and O3. Scientific Reports. 7 (1), 15019(2017).
- White, L. J., Jothibasu, K., Reese, R. N., Brözel, V. S., Subramanian, S. Spatio Temporal influence of isoflavonoids on bacterial diversity in the soybean Rhizosphere. Molecular Plant-Microbe Interactions. 28 (1), 22-29 (2015).
- Edwards, J., et al. Structure, variation, and assembly of the root-associated microbiomes of rice. Proceedings of the National Academy of Science of the United States of America. 112 (8), E911-E920 (2015).
- Lundberg, D. S., et al. Defining the core Arabidopsis thaliana root microbiome. Nature. 488, 86-90 (2012).
- Bougoure, D. S., Cairney, J. W. Assemblages of ericoid mycorrhizal and other root-associated fungi from Epacris pulchella (Ericaceae) as determined by culturing and direct DNA extraction from roots. Environmental Microbiology. 7 (6), 819-827 (2005).
- Doty, S. L., et al. Diazotrophic endophytes of native black cottonwood and willow. Symbiosis. 47 (1), 23-33 (2009).
- Gottel, N. R., et al. Distinct microbial communities within the endosphere and rhizosphere of populus deltoides roots across contrasting soil types. Applied and Environmental Microbiology. 77 (17), 5934-5944 (2011).
- Xin, G., Glawe, D., Doty, S. L. Characterization of three endophytic, indole-3-acetic acid-producing yeasts occurring in Populus trees. Mycological Research. 113, 973-980 (2009).
- Salter, S. J., et al. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biolology. 12, 87(2014).
- Gohl, D. M., et al. Systematic improvement of amplicon marker gene methods for increased accuracy in microbiome studies. Nature Biotechnology. 34 (9), 942-949 (2016).
- Kozich, J. J., Westcott, S. L., Baxter, N. T., Highlander, S. K., Schloss, P. D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Applied Environmental Microbiology. 79 (17), 5112-5120 (2013).
- Caporaso, J. G., et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME Journal. 6 (8), 1621-1624 (2012).
- Lundberg, D. S., Yourstone, S., Mieczkowski, P., Jones, C. D., Dangl, J. L. Practical innovations for high-throughput amplicon sequencing. Nature Methods. 10 (10), 999-1002 (2013).
- RStudio, T. RStudio: Integrated Development for R. RStudio, Inc. , Available from: http://www.rstudio.com/ (2015).
- Edgar, R. C. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nature Methods. 10 (10), 996-998 (2013).
- Caporaso, J. G., et al. QIIME allows analysis of high-throughput community sequencing data. Nature Methods. 7 (5), 335-336 (2010).
- Shannon, C. A mathematical theory of communication. Bell System Technical Journal. 27, 379-423 (1948).
- Simpson, E. H. Measurement of diversity. Nature. 163, 688(1949).
- Chao, A. Non-parametric estimation of the number of classes in a population. Scandanavian Journal of Statistics. 11, 265-270 (1984).
- Legendre, P. Studying beta diversity: Ecological variation partitioning by multiple regression and canonical analysis. Journal of Plant Ecology. 1 (1), 3-8 (2008).
- Oksanen, J., et al. The vegan package. Community Ecology Package. 10, 631-637 (2007).
- Wickham, H. ggplot2: Elegant graphics for data analysis. Ggplot2: Elegant Graphics for Data Analysis. , Springer Nature. Switzerland. 1-212 (2009).
- Cole, J. R., et al. The Ribosomal Database Project (RDP-II): sequences and tools for high-throughput rRNA analysis. Nucleic Acids Research. 33, D294-D296 (2006).
- Wagner, M. R., et al. Host genotype and age shape the leaf and root microbiomes of a wild perennial plant. Nature Communications. 7, 12151(2016).
- O'Brien, H. E., Parrent, J. L., Jackson, J. A., Moncalvo, J. M., Vilgalys, R. Fungal community analysis by large-scale sequencing of environmental samples. Applied and Environmental Microbiology. 71 (9), 5544-5550 (2005).
- Taylor, D. L., et al. A first comprehensive census of fungi in soil reveals both hyperdiversity and fine-scale niche partitioning. Ecological Monographs. 84 (1), 3-20 (2014).
- Lebeis, S. L., et al. Salicylic acid modulates colonization of the root microbiome by specific bacterial taxa. Science. 349 (6250), 860-864 (2015).
- Ofek-Lalzar, M., et al. Niche and host-associated functional signatures of the root surface microbiome. Nature Commununications. 5, 4950(2014).
- Niu, B., Paulson, J. N., Zheng, X., Kolter, R. Simplified and representative bacterial community of maize roots. Proceedings of the National Academy of Science of the United States of America. 114 (12), E2450-E2459 (2017).
- Fitzpatrick, C. R., et al. Assembly and ecological function of the root microbiome across angiosperm plant species. Proceedings of the National Academy of Science of the United States of America. 115 (6), E1157-E1165 (2018).
- Ramirez, K. S., Lauber, C. L., Knight, R., Bradford, M. A., Fierer, N. Consistent effects of nitrogen fertilization on soil bacterial communities in contrasting systems. Ecology. 91 (12), 3463-3470 (2010).
- Paungfoo-Lonhienne, C., et al. Turning the table: Plants consume microbes as a source of nutrients. PLoS One. 5 (7), e11915(2010).
- Yeoh, Y. K., et al. The core root microbiome of sugarcanes cultivated under varying nitrogen fertilizer application. Environmental Microbiology. 18 (5), 1338-1351 (2016).
- Bulgarelli, D., Schlaeppi, K., Spaepen, S., Ver Loren van Themaat, E., Schulze-Lefert, P. Structure and functions of the bacterial microbiota of plants. Annual Review of Plant Biology. 64, 807-838 (2013).
- Peiffer, J. A., et al. Diversity and heritability of the maize rhizosphere microbiome under field conditions. Proceedings of the National Academy of Science of the United States of America. 110 (16), 6548-6553 (2013).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.