Method Article
Un écran de suppresseur de Deep-séquençage-assisted, spontanée chez la levure Scissipare Schizosaccharomyces pombe
Dans cet article
Résumé
Nous présentons un protocole simple suppresseur écran fission levure. Cette méthode est efficace, sans mutagène et sélective pour les mutations qui se produisent souvent à un seul locus génomique. Le protocole est adapté pour isoler les suppresseurs qu’atténuer les défauts de croissance dans un milieu liquide qui sont causées par une mutation ou d’une drogue.
Résumé
Un dépistage génétique des allèles mutants qui suppriment phénotypiques défauts dus à une mutation est une approche puissante pour identifier les gènes qui appartiennent à des voies biochimiques étroitement apparentées. Méthodes précédentes telles que l’analyse de tableau génétique synthétique (SGA) et les techniques de mutagénèse aléatoire à l’aide de rayons ultraviolets (UV) ou produits chimiques comme le méthanesulfonate d’éthyle (EMS) ou N-éthyl-N-nitrosourée (ENU), ont été largement utilisés, mais sont souvent coûteuses et laborieux. En outre, ces méthodes de dépistage basé sur mutagène sont fréquemment associées à des effets secondaires graves sur l’organisme, induisant des mutations multiples qui ajoutent à la complexité d’isoler les suppresseurs. Nous présentons ici un protocole simple et efficace pour identifier les mutations chez les mutants qui confèrent un défaut de croissance chez Schizosaccharomyces pombesuppresseur. La remise en forme des cellules avec un déficit de croissance en milieu liquide riche standard ou un milieu synthétique liquide peut être surveillé pour récupération à l’aide d’un lecteur de plaque à 96 puits automatisé sur une longue période. Une fois qu’une cellule acquiert une mutation de suppresseur de la culture, ses descendants supplanter celles des cellules parentales. Les cellules récupérées qui présentent un avantage de croissance compétitive sur les cellules parentales peuvent alors être isolés et rétrocroisés avec les cellules parentales. Les mutations de l’éliminateur sont identifiées puis en utilisant le séquençage du génome entier. En utilisant cette approche, nous avons isolé avec succès plusieurs suppresseurs qu’atténuer les défauts de croissance sévère causées par la perte de Elf1, une famille de AAA + ATPase qui est important dans le transport nucléaire de mRNA et de maintien de la stabilité génomique. On compte actuellement plus de 400 gènes chez S. pombe avec des mutants conférant un défaut de croissance. Comme beaucoup de ces gènes sont indéterminés, nous proposons que notre méthode accélérera l’identification de nouvelles interactions fonctionnelles grâce à cette approche conviviale et à haut débit.
Introduction
La base de la compréhension des liens fonctionnels entre les gènes s’appuie sur la capacité d’identifier l’ou les mécanismes moléculaire par lesquels les traits génétiques complexes divergent pour produire des phénotypes divers1. Dans la levure de fission, Schizosaccharomyces pombe (S. pombe), la majorité des gènes codant pour des protéines est indispensable pour viabilité2. Ce résultat ne parle pas de l’insignifiance de ces gènes, mais plutôt les mécanismes compensatoires complexes qui sous-tendent les voies biochimiques à laquelle appartiennent ces gènes. Ces mécanismes compensatoires de dissection a généré des cartes d’épistasie, qui ont découvert les interactions génétiques complètes et élargi notre compréhension des voies biochimiques fonctionnelles3,4.
Méthodes de haut-débit (p. ex., analyse génétique synthétique ou SGA) ont été développées afin d’identifier les interactions génétiques tout le génome de la levure de bière et ont été développés pour une utilisation en fission levure5,6. Ces approches reposent souvent sur une bibliothèque de souches contenant tous les viable unique codant pour des protéines gène destructions (environ 3 300 mutants de délétion haploïde couvrant plus de 92 % du génome de levure de fission) et nécessitent un bras robotisé pour effectuer des croisements génétiques entre les souche d’intérêt et toutes les souches possibles dans la library6. De plus, les techniques de SGA dépendent de la capacité des souches de bibliothèque d’avoir un accouplement correct et efficace, un phénotype qui est anormal au 444, actuellement caractérisées gènes chez S. pombe2.
Malgré la complexité des interactions génétiques, comparant le phénotype d’une souche portant des mutations dans deux gènes au phénotype de deux souches porteuses de mutations individuelles de chaque gène peut avoir l’un des deux résultats notables : 1) est le phénotype mutant double pire que les phénotypes parents multiplicatifs attendues sous la forme de la maladie ou, dans le cas le plus extrême, la létalité. Ceci est appelé une interaction génétique négative et est généralement un signe que les deux gènes agissent en parallèles voies biologiques. 2) le phénotype mutant double est mieux que la combinaison attendue des phénotypes parents, également connu sous le nom d’une interaction génétique positive. Une interaction positive génétique est particulièrement intéressante car il indique que ces gènes fonctionnent dans le même processus. Deux gènes interagissants positivement ont trois relations possibles : un gène mutant peut up-réguler l’expression de l’autre gène dans une voie parallèle, les deux gènes peuvent travailler de concert dans la même voie en aval de l’autre ou les deux gènes codent pour protéines qui interagissent directement avec les autres. Par conséquent, des interactions génétiques positives peuvent être utilisées pour mapper les nœuds de régulation génique et classer des gènes non caractérisés dans des voies biochimiques7,8.
Un suppresseur est une mutation qui peut alléger le phénotype de la maladie de la mutation d’un autre gène, représentant généralement une interaction positive génétique entre les deux gènes9,10. Des mutations sur un locus différent de celui de la mutation qu'ils répriment suppresseur sont appelées extragéniques suppresseurs. Ils sont particulièrement utiles dans l’étude des mutations génétiques non viables en sauvant synthétiquement le phénotype létal (également connu sous l’effet de Lazarus)11. Ils ont aussi des applications thérapeutiques potentielles dans le traitement de maladies héréditaires12,13.
Pour toutes ces raisons, l’identification de mutations suppresseur dans divers organismes modèles a été largement utilisée pour faciliter la compréhension des diverses voies biochimiques14,15,16. Dépistage des suppresseurs repose généralement sur le phénotype de la mutation en question et exige la tenue de mutagénèse aléatoire pour isoler les mutations qui permettrait d’atténuer le phénotype. Presque tous les organismes modèles ont établi des méthodes de mutagénèse aléatoire. Par exemple, N-éthyl-N-nitrosourée (ENU) et éthyle (EMS), deux agents mutagènes qui sont capables d’induire des mutations ponctuelles dans l’ADN, sont largement employées dans divers modèles de bactéries à souris17,18,19 . En outre, le chlorure de manganèse a longtemps été utilisé dans les levures pour la capacité du cation manganèse d’inhiber l’ADN réparation voies20. Une autre approche commune est mutagenèse induite sur les UV, ce qui génère des dimères de pyrimidine mutagènes pangénomique21,22.
Bien que l’utilisation de mutagenèse chimique afin d’identifier les mutations suppresseur a été populaire, la méthode a beaucoup d’inconvénients, y compris l’utilisation de produits chimiques dangereux, les taux de succès très variable et l’introduction de variables confusionnelles extra présenté par les effets secondaires négatifs de l’agent mutagène sur plusieurs processus cellulaires23,24. En outre, la mutagenèse chimique induit souvent plusieurs mutations dans le génome qui ajoute à la complexité de l’utilisation génétique et les techniques de séquençage d’identifier la mutation exacte qui confère le phénotype suppresseur dans l’organisme des25.
Pour combler les lacunes des méthodes actuelles de mutagenèse, nous présentons une méthode à l’écran pour les mutations spontanées suppresseur chez la levure qui ne repose pas sur n’importe quel mutagènes ou une bibliothèque de suppression. La méthode isolats suppresseurs grâce à un test de sélection positive. Le principe de cette méthode repose sur l’avantage de la croissance de la sous-population de suppresseur muté dans la culture liquide, qui peut être surveillée par un lecteur de plaque automatisé. L’accouplement et la méiose sont utilisés uniquement si l'on souhaite nettoyer le bagage génétique ou de confirmer la présence d’allèles monogéniques de suppresseurs avant le séquençage du génome entier. Si le phénotype de la répression est causé par une mutation, le phénotype suppresseur va séparer 2:2 après croisement avec les souches parentales. Les mutations de suppresseur peuvent ensuite être identifiées au moyen du séquençage de génomes complets. Nous proposons que cette méthode est applicable pour le dépistage des suppresseurs dans tous les microorganismes qui peuvent atteindre une grande population dans des cultures liquides.
Protocole
1. construction et préparation de la souche
- Générer une mutation ou une délétion du gène (yfm, votre mutation préférée) à l’aide de standard mutagénèse dirigée (SDM) comme décrit plus haut26.
- Avant de commencer l’écran, (optimale) rétrocroisement souches mutantes avec une souche sauvage pour nettoyer le bagage génétique et générer des frais-né des cellules mutantes comme les souches parentales. Ensemencer la souche parentale aux colonies individuelles sur des plaques standard de média enrichi. Choisir au hasard huit à seize colonies indépendantes (réplique biologique) avec les mutations souhaitées pour le dosage de lecteur de plaque (voir 3.1).
Remarque : Ce protocole est efficace seulement quand les souches parentales ont une croissance défectuosité dans les milieux liquides (minime ou riches, avec ou sans médicaments ou avec des changements de température qui provoquent le défaut de croissance). Toutes les souches parentales devraient être haploïdes et donc en mesure d’être génétiquement croisé avec d’autres souches haploïdes avec un type accouplement complémentaire.
2. essai lecteur de plaque
- Avec un applicateur stérile, prendre une petite quantité de chacune des colonies préparées à l’étape 1.1 (aucune quantité exacte nécessaire pour ensemencer le démarrage ne culture) et placez-les dans une microplaque 96 puits en polystyrène. Chacune des colonies dans 200 µL de milieux liquide appropriés (riche ou minime, avec ou sans drogue) suspendre. Inclure un puits blanc pour tous les rangs sur la plaque contenant 200 µL de ces mêmes médias (pas de cellules).
- Exécuter le protocole suivant sur un logiciel de détection de lecteur de plaque connecté à un lecteur de microplaques automatisé : définir un programme cinétique pour 24h et la température à 30 ° C, avec une agitation rapide orbital continue (425 cpm, amplitude de 3 mm). Affectez les lectures optiques pour mesurer la dispersion lumineuse à une longueur d’onde de 600nm de densité optique et la lumière pour lire en bas de la plaque à une fréquence de lecture de 2 min (721 lectures totales sur une période de 24 h par puits).
- Après 24h, enregistrer les lectures de densité optique occultées finale (DO600 occultées) et utiliser la formule suivante pour déterminer le volume nécessaire pour diluer chacun des échantillons jusqu'à O.D. = 0,1 :

NOTE : Exporter les données depuis le logiciel de lecteur de plaque et utiliser un tableur pour insérer la formule ci-dessus comme une fonction à lot de traiter le volume de dilution à utiliser dans chaque puits expérimental. - Toutes les 24 h, diluer chacun des échantillons à l’aide de ces mêmes médias comme jour 0 à O.D. = 0,1 (environ 1. 5 x 106 cellules/mL) à l’aide de la formule indiquée à l’étape 2.3. Enregistrez toute croissance courbes générées quotidiennement et notez toute colonie individuel qui montre un taux de croissance accrue, jugé par un D.E. final qui est significativement plus élevé que le reste de la cohorte avec le même bagage génétique ou par une courbe de croissance semblable à celle de colonies de type sauvage.
Remarque : Ce test prend habituellement environ de 7 à 14 jours. Effectuez toutes les opérations dans des conditions stériles.
3. sélection des colonies suppresseur et confirmation du phénotype.
- Depuis le dernier jour de l’essai lecteur de plaque (étape 2.4), sauver les cultures liquides qui ont un taux de croissance nettement récupéré, sans doute en acquérant une mutation suppresseur qui peut alléger le phénotype de la mutation parental. Transfert et mélanger 250 µL culture liquide à un cryotube contenant 250 µL de glycérol à 50 %. Flash geler les cellules dans l’azote liquide et sauver les souches in French - 80oC indéfiniment.
- Pour confirmer que la mutation du suppresseur est un élément génétiquement héritable, utiliser des méthodes de croisement génétique standard pour traverser yfm P (pour la souche parentale, utilisé au début de l’essai lecteur de plaque) avec yfm S (pour suppresseur, la souche sauvée à la fin de l’essai lecteur de plaque). Si la mutation du suppresseur est en effet un élément génétiquement héritable, yfm P × yfm S devrait donner des tétrades dans lequel deux colonies ont le phénotype de la maladie de la souche parentale et deux colonies ont le taux de croissance récupérés de l’éliminateur souche.
- Issue du croisement de l’étape 3.2, prélever des trois colonies avec le phénotype suppresseur (souche S) et trois colonies avec le phénotype parental (souche P) de la même génétique croisent (3 réplicats biologiques pour chacun) et procéder à l’extraction d’ADN génomique et étapes de séquençage ci-dessous.
NOTE : Les étapes 3.2 et 3.3 sont fortement recommandés, mais ne sont pas nécessaires. Alternativement, on peut étendre récupérée culture liquide prélevée 3.1 sur un milieu enrichi en colonies individuelles, puis choisir au hasard trois colonies comme biologiques réanalysés pour le séquençage de génomes complets sans une confirmation génétique supplémentaire. Dans ce cas, trois réanalysés biologiques de souche parentale devraient être utilisés pour la comparaison de séquençage génomique.
4. genomic DNA extraction, production de bibliothèque et séquençage.
- Extraction, préparation de la bibliothèque et le séquençage, ADN au hasard trois réplicats biologiques par souche yfm P , et biologique trois réplicats de chaque individuellement mastiquer yfm S souches issues des croisements génétiques (étape 3.2) ou de la plaques qui ont été diffusées pour obtenir des colonies individuelles de la souche S (étape 3.3 remarque).
- Cultiver des souches cultivées dans des milieux riches en 10 mL à phase logarithmique (OD = 0,5 à 0,8, de 0,75 à 1,2 x 107 cellules/mL) et un incubateur à agitation permet de cultiver des cultures en milieu liquide à 30 ° C sous agitation continue à 250 tr/min. Prélever des cellules par centrifugation à 4 ° C pendant 5 min à 1000 x g.
- Suspendre les granulés cellules de 400 µL de tampon d’extraction de l’ADN (2 % Triton X-100, 1 % SDS, 100 mM NaCl, 10 mM Tris-Cl (pH 8,0), 1 mM Na2-EDTA), puis ajouter 400 µL de perles de verre et de 400 µL de 25:24:1 phénol : chloroforme : isoamylique alcool. Vortex vigoureusement pendant 2 min à 4 ° C.
- Ajouter une supplémentaire 200 µL du tampon d’extraction de l’ADN et les mélanger en retournant plusieurs fois. Centrifuger 5 min à 4 ° C à 20 000 x g.
- Transférer la phase aqueuse dans un tube propre, ajouter 20 µg de mélange de RNase A/T1 et incuber à 37 ° C pendant 15 min.
- Ajouter un volume égal de 25:24:1 phénol : chloroforme : isoamylique alcool, essorage pendant 5 min à 4 ° C à 20 000 x g, puis transférer la phase aqueuse dans un tube propre.
- Ajouter un volume égal de chloroforme, mélanger en retournant plusieurs fois, puis tournez pendant 5 min à 4 ° C à 20 000 x g, puis transférer la phase aqueuse dans un tube propre.
- ADN précipité avec deux volumes d’éthanol à 100 % plus de 10 % en volume de 3 M Acona (pH 4,3) à-20 ° C pendant au moins 2 h, puis tourner pendant 5 min à 4 ° C à 20 000 x g et collecter le culot.
- Laver le culot (ADN précipité) deux fois avec l’éthanol à 70 % réfrigérés (centrifuger à 20 000 x g, 5 min, 4 ° C) et suspendre le culot dans 50 µL de 10 mM Tris tamponné (pH 7,4).
- Utiliser une préparation de bibliothèque kit (voir Table des matières) par les recommandations du fabricant pour préparer la bibliothèque de séquençage de génomes complets.
NOTE: nous recommandons le kit énuméré dans la Table des matières , car il permet la construction de la banque génomique sans amplification par PCR, ce qui minimise les mutations d’erreur générées lors de l’amplification par PCR. En outre, lors de la préparation de la banque génomique, n’autorisent pas les billes pour séchage complet en raccourcissant le talon à 1 – 2 min le temps de séchage. - Pour la tonte paramètres lors de la préparation de la bibliothèque, utilisez un sonicateur ciblé (voir Table des matières) et régler le facteur d’utilisation à 20 %, puissance de crête de 175 W, avec 200 cycles / rafale, et le mode balayage de fréquence à 5.5oC à 6 ° C pendant 45 s. alternativement, utilisez un ADN et chomatin système de cisaillement (voir Table des matières) avec les paramètres suivants : amplitude de 50 % à 4 ° C en mode impulsion, 15 s sur et 15 s éteint pendant 10 min, avec un temps total de traitement de 20 min.
- Il est essentiel de traiter les matériaux dangereux utilisés dans cette étape avec soin. Consulter l’approprié Material Safety Data Sheets et d’institution santé environnementale et bureau de la sécurité pour la manipulation de NaOAC, éthanol, 25:24:1 phénol : chloroforme : isoamylique alcool et le chloroforme.
- Séquencer les banques génomiques qui en résulte. Les lectures de séquençage de l’ensemble devraient couvrir au moins trois fois de tout le génome avec la résolution à une gamme de nucléotide. Jumelé-terminé le séquençage (ou dernière technologie) est recommandée.
5. bioinformatique analyse pour l’identification des mutations suppresseur
- Effectuer l’analyse bio-informatique de se concentrer sur les changements génomiques qui sont systématiquement identifiés entre parents et supprimés yfm souches biologique tous les réplique.
Remarque : Le processus de pipeline complet est décrit ci-dessous, mais, en outre, les deux fichiers de script BASH de texte brut, fastq_to_vcf.sh et vcfprocess.sh, figurent parmi les matériaux supplémentaires pour montrer des exemples de flux de travail de transformation de lectures à la variante VCF fichiers et de traitement et d’intersection de la VCF fichiers, respectivement. - Trim court-lectures à l’aide de cisaillement (https://github.com/jbpease/shear) en suivant les lignes de commande (toutes les autre options par défaut) :
Shear.py--fq1 $FASTQ1--fq2 $FASTQ2--out1 $OUTFQ1--out2 $OUTFQ2 \
--barcodes1 $BARCODE--TruSeq--trimqual de plate-forme 20:20 \
trimpolyat--0--trimambig filterlength--50--filterunpaired - Carte : lit à la v2.30 de génome de référence S. pombe provenant de PomBase (ftp://ftp.ebi.ac.uk/pub/databases/pombase/pombe/Chromosome_Dumps/fasta/) à l’aide de BWA v0.7.1527. Utilisez la ligne de commande suivante (tous les autre options par défaut) :
BWA mem -t 8 $GENOME $OUTFQ1 $OUTFQ2 > $SAM1 - Placez les fichiers de l’alignement SAM par GATK conseillées pipeline28 afin d’appeler variante utilisant GATK v3.629, PicardTools v2.5.0 (http://broadinstitute.github.io/picard) et SAMtools v1.3.130. Utilisez les paramètres (toutes les autre options par défaut) et les lignes de commande suivantes :
Java-Xmx30g-jar picard.jar AddOrReplaceReadGroups entrée = $SAM1 \
SORTIE = $BAMMARKED RGID = 1 RGLB = lib01 RGPL = illumina \
RGPU = $BARCODE RGSM = $SAMPLENUMBER
SamTools fixmate bam - O $BAMMARKED $BAMFIXED
SamTools trier - O bam -o $BAMSORTED -T /home/peasejb/tmp $BAMFIXED
SamTools index $BAMSORTED
Java-Xmx30g-jar GenomeAnalysisTK.jar -T HaplotypeCaller \
-R $GENOME-je $BAMSORTED--genotyping_mode découverte \
-stand_emit_conf 10 - stand_call_conf 30 -o $VCFRAW - Compresser et indexer les fichiers VCF utilisant tabix :
bgzip $VCFRAW.vcf
tabix $VCFRAW.vcf.gz - Comparer les fichiers VCF chez les parents et suppresseur de souches réplicats de séquençage à l’aide de BCFtools v1.3.127. Utilisez les paramètres (toutes les autre options gauche par défaut) et les lignes de commande suivantes :
bcftools isec - n + 1 $VCFPARENTAL1.gz $VCFPARENTAL2.gz $VCFPARENTAL3.gz \
$VCFMUTANT1.gz $VCFMUTANT2.gz $VCFMUTANT3.gz > common_variants.list
Remarque : Cette commande produit un fichier encodé avec schémas binaires où les variantes de séquence figurant dans le premier mutant seulement serait binaires codés comme « 000100 », le deuxième mutant seulement comme « 000010 », tous trois mutants comme « 000111, » etc.. Ces fichiers ont été générés pour chaque ensemble de l’autorité parentale et mutant répliquer les fichiers VCF. - Compiler les fichiers de liste de variantes intersection ainsi que le nom de fichier à chaque ligne à l’aide de la commande grep UNIX :
grep «. » *.list > all.list - Renvoyer à la liste complète variant avec le fichier d’annotation GFF3 actuel (ftp://ftp.ebi.ac.uk/pub/databases/pombase/pombe/Chromosome_Dumps/gff3/schizosacchar omyces_pombe.chr.gff3) à l’aide d’un script personnalisé de Python (variant_characterize.py) pour recenser les sites compatibles de SNP en régions (synonymes et non-synonymes) codant pour des protéines, 5′ et 3′ RTNs et Arnnc.
python3 variant_characterize.py--liste common_variants.list \
--gff schizosaccharomyces_pombe.chr.gff3 \
--fasta Schizosaccharomyces_pombe. ASM294v2.30.DNA.Genome.fa \
--modèle 000100--sur all.list.filter.000100
Répétez cette modification de script--modèle tandis que le suffixe du fichier en sortie (--out) en utilisant le binaire
modèles : 000010 000001, 000110, 000011, 000101 et 000111 - Combiner le résultat de toutes ces courses de script dans un fichier séparé par des tabulations, pour être considéré comme une feuille de calcul. La table annotée des variantes comprend celles qui apparaissent dans une ou les deux mutants souche (s) par rapport à l’arrière-plan. Le champ indicateur binaire désigne une apparition dans une seule souche mutante (000100, 000010, 000001), deux souches mutantes (000011, 000101, 000110), ou tous les trois souches mutantes (000111).
- Analyser les listes de sortie annoté des variantes non trouvé dans les échantillons parentales, mais trouvé dans un, deux ou tous les trois échantillons de mutant. L’annotation désigne aussi bien l’emplacement génomique et la classe de variante (synonyme/non-synonymes dans une région codante 3' / 5' UTR, non codante, etc..). Dans cette liste des mutations du candidat, un exemple d’un candidats fortement pertinentes pourrait être une variante de codage non-synonymes apparaissant régulièrement dans les trois souches. Un autre type de candidat solide serait une accumulation de différentes mutations réglementaires non-synonymes ou putatives dans les souches mutantes apparaissant rapprochés ou à l’intérieur du même gène.
Résultats
Croissance lente des mutants montrent phénotypique reprise en milieu liquide
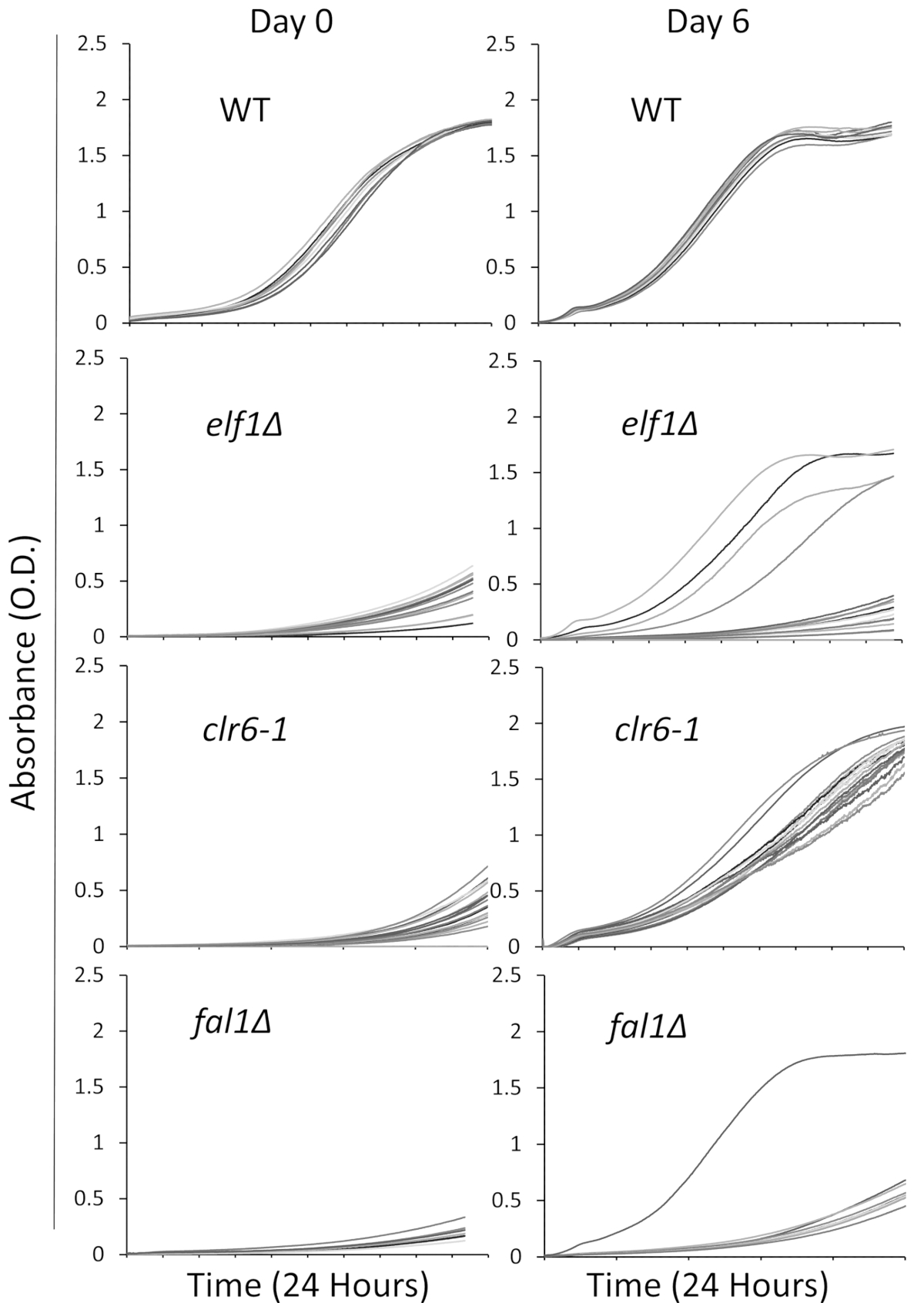
Nous avons sélectionné trois mutants impliqués dans une variété de voies biologiques avec un phénotype malade, croissance lente, : AAA famille ATPase Elf1, Histone Deacetylase Clr6 et complexe de jonction Exon composant Fal1. Une souche sauvage et porteuses de mutations de ces trois gènes qui avaient été rétrocroisés avec les souches sauvages des souches étaient striées de colonies individuelles et 16 colonies individuelles ont été choisis au hasard pour être mis en culture dans un milieu liquide riche en utilisant la plaque 96 puits décrites ci-dessus. Courbes de croissance des colonies individuelles ont été enregistrés à l’instant initial (jour 0) et pour 6 jours avec une surveillance continue en utilisant le lecteur de plaque. Comme prévu, colonies de type sauvage ne montrent aucun changement notable dans leurs courbes de croissance tout au long de l’expérience31 (Figure 1). Notamment, quatre colonies avec le fond de elf1∆ et une colonie de fal1∆ montrent un changement dramatique dans la croissance de croissance lente à des niveaux variables de croissance similaire ou proche de celui des colonies de type sauvage. Dramatiquement, tous les mutants de clr6-1 montrent une reprise phénotypique cohérents, croît à un rythme plus rapide à la fin de l’essai31 (Figure 1). Afin de caractériser les différents phénotypes, nous renvoyons à l’original les souches à croissance lente comme les « Souches P » (ou souches parentales) et les souches montrant phénotypique rétablissement comme « Souches S » (ou souches supprimés). Veuillez noter que la Figure 1 est un exemple d’un tour de l’expérience de dépistage et ne représente pas les totales suppresseurs non complémentaires identifiés et séquencés dans les résultats représentatifs suivants.
Récupération phénotypique est attribuée à caractères héréditaires
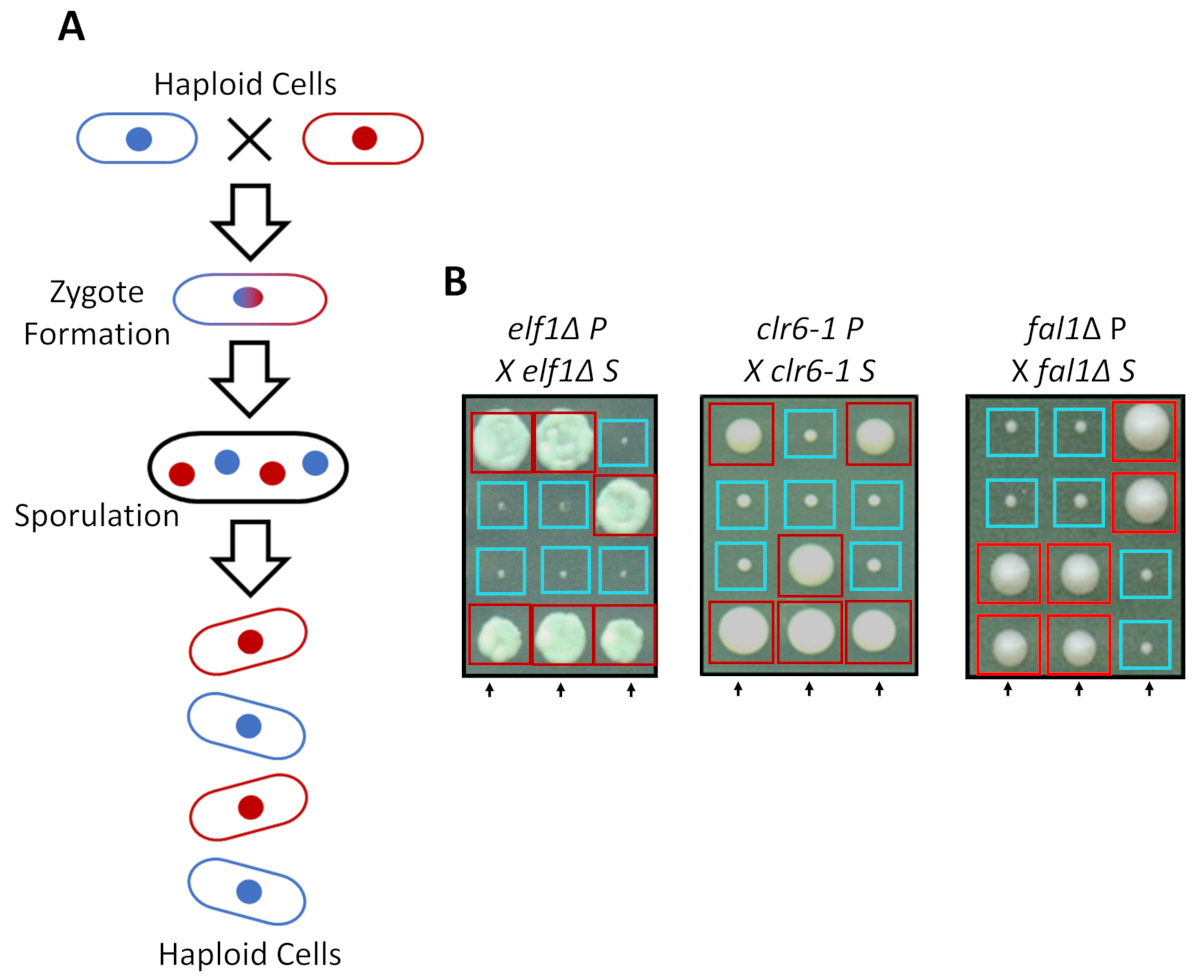
S. pombe peuvent se développer comme un haploïde dans des milieux riches, mais deux lignées haploïdes avec complémentaires accouplement types mate sous azotée. La méiose chez la levure comporte un tour de duplication, suivie de deux cycles de division cellulaire. Le cycle sexuel conduit à la formation de quatre spores haploïdes transportant le matériel génétique de la souche parentale avec 2,2 ségrégation des traits génétiques suivant les règles de la génétique mendélienne classique (Figure 2 a). Lorsqu’il est cultivé sur la même plaque pour le même laps de temps, nous avons confirmé la 2,2 ségrégation lors du rétrocroisement tous supprimés souches (les souches S) avec leurs souches parentales (souches de P), qui a fait 2 petits (défaut de croissance) et 2 grands (phénotype suppresseur) colonies. Des exemples individuels pour les cellules elf1∆, clr6-1 et fal1∆ supprimés sont indiquées dans la Figure 2 b. Nous avons confirmé que toutes les souches isolées de S transporter un élément génétique monogénique qui supprime le phénotype de croissance lente de leurs souches P (données non présentées).
Le séquençage du génome entier identifie correctement les mutations de l’éliminateur
À titre d’exemple, nous avons utilisé le séquençage du génome entier jumelés-fin d’identifier les éléments génétiques responsables du recouvrement phénotypique chez les souches de S elf1∆ . Une description plus complète de l’analyse des données est disponible en ligne31. En bref, nous avons utilisé réanalysés biologiques de deux souches de elf1∆ P indépendamment généré et doublons biologiques de cinq groupes non complémentaires des souches de S elf1∆ , dont chacun contient différents suppresseurs. Après que nous avons obtenu une liste de variantes annotées de bioinformatique analyse (6.1-10), nous avons priorisé certaines classes de variants qui présentaient un intérêt pour notre analyse. Nous nous sommes concentrés sur l’identification des changements génomiques cohérente qui étaient identiques dans tous les réplicats biologiques de souches elf1∆ S individuelles par rapport à leurs souches parentales de elf1∆ P (Figure 3 et supplémentaire tableaux 1 α 4 ). Nous avons identifié cinq changements non-synonymes dans les régions de CD chez toutes les souches elf1∆ S différents cinq, y compris rli1 +, SPBPJ4664.02, cue2 + et rpl2702 +. S-A1 et contiennent tous deux S-A2 mutée SPBPJ4664.02, bien que les mutations se produisent à différents acides aminés. SPBPJ4664.02 étant un gène long (11 916 nucléotides) avec des centaines de répétitions, les mutations n’ont pas être confirmées en effectuant des PCR suivie par séquençage. S-A3 contient un mutant de délétion dans rli1 qui est compatible dans les deux doublons biologiques. Cependant, le mutant ne pas co séparer avec le phénotype S en fond de elf1∆ . Nous avons identifié un mutant cue2 (cue2-1) en S-B1, avec les acides aminés 396-400 disparus. S-B2 contient un mutant rpl2702 (rpl2702-1), qui change des acides aminés en position 45 de Glycine en Aspartate31. Suppresseurs de elf1∆ les cue2-1 et rpl2702-1 ont été confirmés comme indiqué ci-dessous.
Confirmation génétique des mutations identifiées suppresseur vérifie l’héritabilité du phénotype récupération
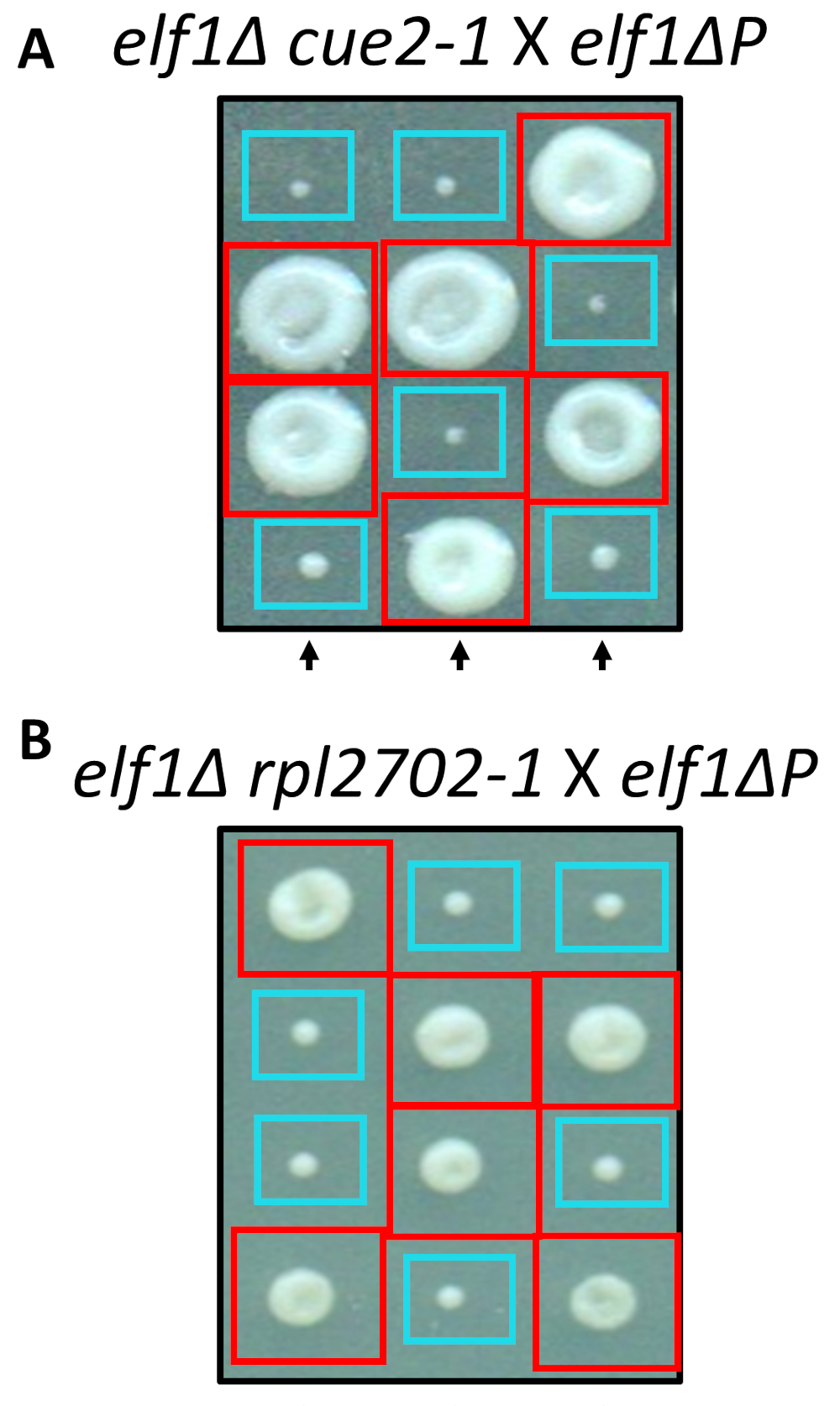
Parmi les modifications identifiées de non-synonymes, cue2-1 et rpl2702-1, a été reconstitué en laboratoire en utilisant des protocoles standard pour la mutagénèse dirigée. Double mutant souches cue2-1 P elf1∆ et rpl2702-1 elf1∆ P ont été croisés avec le gratuit elf1∆ P souche31 (Figure 4). Si les mutations non-synonymes, identifiées par le biais de cet écran, étaient suffisantes pour supprimer elf1∆ P, puis les tétrades résultantes afficherait un 2:2 petit à grand rapport dans les colonies résultant des 4 spores dans chaque tétrade. En effet, croisement génétique a montré que les mutations identifiées suppresseur réussissent en supprimant le phénotype de croissance lente de elf1∆ P et sont héréditaires.

Figure 1 : récupération phénotypique peut être surveillée par des courbes de croissance d’enregistrement dans un lecteur de plaque. Seize des colonies individuelles de type sauvage (WT), elf1∆, clr6-1et fal1∆ ont été placés dans une plaque à 96 puits. Courbes de croissance ont été enregistrées sur une période de 24 h et les colonies ont été re-dilués quotidiennement dans les médias enrichis. Le défaut de croissance est évident par la faible absorption (O.D.) à la fin de la période de 24h au jour 0. Souches phénotypiquement récupérés sont celles qui affichent une courbe de croissance semblable, ou à proximité, celle de type sauvage pendant la période de 24 heures le jour 6. Quatre colonies de elf1∆, une colonie de fal1∆et de toutes les colonies de clr6-1 a montré divers niveaux de reprise phénotypique après 6 jours. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 2 : croisement génétique peut-elle confirmer que la récupération phénotypique est attribuée à un seul allèle héritable. (A) lorsque la levure cellules subissent azotée, deux cellules haploïdes avec un type accouplement complémentaire peut générer un zygote qui sporule pour générer une tétrade de 4 spores. Le matériel génétique parental se séparer pendant la méiose suivant les règles de la génétique mendélienne. (B) récupéré phénotypiquement colonies (marqué S, car supprimée) avec indiqué génotypes parentaux ont été rétrocroisée avec leur colonie parental gratuit (qui montre sans récupération phénotypique, marquée P, pour parental). Croisements génétiques montrant 2:2 petits (mauvaise adaptation) dans des colonies de grand (fitness récupérés) démontrent que la récupération phénotypique est héréditaire et peut être attribuée à un seul élément génétique. Boîtes rouges sont porteurs de l’allèle suppresseur de colonies, et boîtes bleues sont des colonies portant les allèles parentaux. Ce chiffre a été modifié d’Al Marayati, 201831. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 3 : analyse des données de séquençage de génome-large d’identifier des éléments génétiques responsables du recouvrement phénotypique. Trois biologiques réplique des deux souches parentales de « P » (P-A et P-B), et deux réplicats biologiques de cinq phénotypiquement récupéré commuté souches « S » (S-A1, S-A2 et A3 S - récupérés de P-A ; S-B1 et S-B2 de P-B), ont été séquencés et les mutations ont été organisées sous forme de liste de chaque mutation chez la souche récupérée par rapport à du génome de la souche parentale il était dérivé (par exemple, p-a vs S-A1, etc.). Le nombre total des mutations détectées au sein du génome entier de toutes ces comparaisons par paires était 660. Un total de 44 mutations ont été identifiées lorsque seules des mutations qui se produisent dans les deux réplicats biologiques de la même souche « S » ont été sélectionnées. Sur les 44 mutations, 12 mutations étaient insertion/délétion (INDEL) ou non - des mutations synonymes. Sur les 12 INDEL ou mutations non-synonymes, cinq ont eu lieu dans la protéine séquence codante. Les cinq mutations potentiellement mettre en corrélation avec le seul élément génétique responsable par les souches phénotypiquement récupérés : une mutation non-synonyme de SPBPJ4664.02 trouvé en S-A1 et A2-S, INDEL dans rli1 en S-A3, INDEL dans cue2 dans S-B1 et des mutations non-synonymes rpl2702 dans S-B2. Séquence détaillée des informations sur les mutations et le fond filtré sont incluses dans supplémentaire tableaux 1 α 4. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}

Figure 4 : Confirmation des suppresseurs identifiés par séquençage de génome entier. Résultats de séquençage de génomes complets ont été confirmés en générant les mutations indépendamment et en effectuant des croisements génétiques pour confirmer la récupération phénotypique en croisant une souche elf1∆ cue2-1 avec une souche de P elf1∆ et elf1∆ rpl2702-1 avec la souche elf1∆ P . Trois tétrades verticales représentant sont indiqués. Boîtes rouges sont double-mutant colonies (elf1 cue2-1, ou elf1 rpl2702-1) ; boîtes bleues sont des colonies de elf1∆ . Ce chiffre a été modifié d’Al Marayati, 201831. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}
La Table 1. S’il vous plaît cliquez ici pour télécharger ce tableau.
La Table 2 . S’il vous plaît cliquez ici pour télécharger ce tableau.
Supplemental tableau 3 . S’il vous plaît cliquez ici pour télécharger ce tableau.
Supplemental tableau 4 . S’il vous plaît cliquez ici pour télécharger ce tableau.
Codage de fichiers supplémentaires . S’il vous plaît cliquez ici pour télécharger les fichiers.
Discussion
Le protocole décrit ici représente un roman et un simple écran pour les mutations spontanées suppresseur détectables grâce à la récupération phénotypique des mutations conférant une croissance lente chez la levure, un phénotype caractéristique de plus de 400 gènes chez S. pombe, le fonction d’un grand nombre qui reste inconnu2,32. Méthodes précédentes ont pris d’autres approches pour dépister les mutations de suppresseur de micro-organismes, y compris l’utilisation d’agents mutagènes21, ou l’application d’un changement de température en milieux mutant sensible à la température de33. En revanche, ce protocole indique que phénotypique récupération se produit sans interférence d’environnement/produit chimique supplémentaire et met en évidence l’avantage de remise en forme de la montée des mutations de répression éventuellement prise en charge des ressources disponibles dans un liquide culture. Cet écran permet l’isolement des deux suppresseurs de dérivation ou de suppresseurs d’interaction parce qu’elle est efficace pour les deux mutations perte de fonction telles que elf1∆ ou fal1∆ et des mutations ponctuelles telles que clr6-1, aussi longtemps que les mutants démontrer des défauts de conditionnement physique en milieu liquide.
Jusqu’ici, toutes les souches de S récupérés que nous avons étudiés ont montré des degrés de récupération phénotypique. Tel que détecté par le biais de croisement génétique, le phénotype récupéré est attribuable à un seul élément génétique et héréditaires (exemples présentés dans la Figure 2). C’est l’un des plus importants avantages de cette méthode par rapport aux écrans de suppresseur axée sur les produits chimiques ou à base de rayons UV, qui ciblent souvent plusieurs locus génomiques. Il est fréquent d’observer une ou deux colonies récupérés hors 16 colonies/souche (environ 10 %) moins d’une semaine. Cependant, nous avons remarqué que certains mutants, tels que la perte de fonction de Rrp6, la sous-unité de l’exosome nucléaires spécifiques, jamais retrouvé le taux de croissance presque sauvage observé dans les cellules de elf1Δ 31. Il est probable que la fonction de Rrp6 ne peut être partiellement compensée par des suppresseurs, contrairement à la fonction des autres mutants testés, y compris fal1∆, qui a été montré pour causer une malformation grave méiotique à travers sa fonction importante dans la réglementation 34de l’épissage. Nous croyons que cette alternative suppresseur de méthodes de dépistage subirait le même problème quand kuaytiaw a des rôles uniques, non remplaçables dans la croissance cellulaire.
Avant de réaliser le séquençage génomique, il est optimal à back-cross les colonies phénotypiquement récupérés, identifiés à partir du lecteur de plaque, avec les souches parentales afin de dégager le bagage génétique et obtenir des réplicats biologiques. De plus, le séquençage du génome entier profond identifie des centaines de modifications de nucléotides, dont la plupart n’est pas identique entre les réplicats biologiques qui sont de peu d’intérêt pour le dépistage. Par exemple, nous avons trouvé un total de 660 altérations génomiques à travers tous les trois chromosomes entre les deux elf1Δ P et les cinq souches différentes de S (Figure 3). Nous n’avons pas couramment observé des mutations identiques entre biologique séquencée réplique de chaque souche, suggérant que nouvelles mutations survenant au cours de la mise en culture des cellules de elf1Δ avant la construction de banque génomique ou des erreurs aléatoires peuvent être introduit lors de la construction de la bibliothèque et le séquençage. Isoler les mutations qui sont cohérentes entre les réplicats biologiques est donc un aspect important dans l’identification réussie de suppresseurs du séquençage du génome entier.
Nous avons identifié et confirmé deux suppresseurs dans les régions CDS dans cinq souches de S séquencées. Bien que les mutations dans SPBPJ4664.02 ont été détectées en S-A1 et A2-S souches, il est peu probable que le SPBPJ4664.02 est un suppresseur valide car S-A1 et A2-S ne contiennent pas un suppresseur sur le même gène, car ils ne sont pas complémentaires avec l’autre ( données non présentées). Nous avons également n’a pas confirmé rli1 en S-A3, qui ne pas séparer conjointement avec le phénotype S lorsque rétrocroisée avec elf1Δ. Sinon, nous avons trouvé des mutations spécifiques dans les régions non codantes dans S-A1, A2-S et S-A3. Il est possible que ces régions génomiques altérées non codantes alléger le phénotype elf1Δ , qui est traité dans nos futures études. Par rapport aux méthodes traditionnelles comme un test de liaison, qui peut prendre des années pour mapper une mutation génétique, nous avons identifié deux suppresseurs dans les deux mois après la confirmation qu’un élément monogénique a provoqué le phénotype S. Avec le développement rapide des technologies de séquençage de génomes complets, nous avons bon espoir que cette méthode sera plus efficace pour identifier les mutations génétiques cohérentes dans un avenir prévisible.
En résumé, ce protocole fournit des instructions étape par étape pour identifier avec succès des mutations pour n’importe quel gène suppresseur d’intérêt avec un défaut de croissance lente en milieu liquide. La simplicité de ce dosage permet le dépistage à grande échelle de multiples origines génétiques d’intérêt avec peu de formation pratique. Il n’y a place pour automatiser davantage le processus en utilisant un robot de manipulation de liquides pour effectuer des dilutions quotidiennes. Comme la manipulation de laboratoire de micro-organismes exige inévitablement croissance dans un milieu liquide, un processus qui est par nature sélectif pour le fitness, nous proposons que ce protocole peut être largement appliqué à d’autres organismes de modèle très peuplés comme les bactéries et autres espèces de levures.
Déclarations de divulgation
Les auteurs ne déclarent aucun endossement par les fabricants des instruments utilisés dans cette méthode et sans intérêts financiers concurrents.
Remerciements
Ce travail a été soutenu par le National Institute of General Medical Sciences, subventions 1R15GM119105-01 à K.Z. Nous remercions tous les relecteurs pour commentaires perspicaces. Nous remercions également James Tucker, Alicia Anderson, Elizabeth Black et Glen Marrs pour la discussion et les commentaires sur ce manuscrit.
matériels
| Name | Company | Catalog Number | Comments |
| Adenine, Powder | Acros Organics | 147441000 | Use at 75 mg/L to make liquid and solid rich media (YEA) |
| Bacteriology Petri Dish | Corning, Falcon | C351029 | 100×15mm, use to grow strains to single colonies on solid rich media |
| D-Glucose Anhydrous, Powder | Fisher Chemical | D16-1 | Use at 30 g/L to make liquid and solid rich media (YEA) |
| Difco Agar, Granuated | Becton, Dickinson and Co. | 214530 | Use at 20 g/L to make solid rich media (YEA) |
| DNA extraction buffer | 2% Triton X-100, 1% SDS, 100 mM NaCl, 10 mM Tris-Cl (pH 8.0), 1mM Na2-EDTA | ||
| Focused-ultrasonicator | Covaris Inc. | S220 | Alternatively, use QSonica Q800R sonicator/DNA and chromatin shearing system |
| Gen5 Data Collection and Analysis Software | Biotek, Inc. | GEN5SECURE | Or equivalent, must be compatible with the micro-plate reader, use to export data readings from the micro-plate reader |
| Hydrochloric Acid 1N, Liquid | Fisher Chemical | SA48-4 | Use to adjust pH to 5.5 in liquid and solid rich media |
| Liquid Rich Media (liquid YEA) | 30 g/L D-Glucose, 5 g/L Yeast Extract, 75 mg/L Adenine, pH adjusted to 5.5 with 1 M HCl | ||
| Microplate Reader, Synergy H1 Hybrid Multi-Mode Reader | Biotek, Inc. | BTH1MG | Or equivalent, must read visible light at 600nm wavelength range |
| Rich Media agar plates (YEA plates) | 30 g/L D-Glucose, 5 g/L Yeast Extract, 75 mg/L Adenine, 20 g/L Agar, pH adjusted to 5.5 with 1 M HCl. | ||
| RNase A/T1 mix | Thermo Fisher Scientific | EN0551 | Use according to manufacturer recommendation |
| Sterile Polystyrene Inoculating Loop | Corning, Inc. | OS101 | Or equivalent, use to transfer colonies from agar plates to 96-well plate |
| Sterile workspace and burners | |||
| Tissue Culture Plate, 96-well Optical Flat Bottom with Low Evaporation Lid | Corning, Falcon | C353072 | Or equivalent, must have optical flat bottom for micro-plate ready |
| TruSeq DNA PCR-Free LT/HT Library Prep Kit | Illumina, Inc. | 20015962 | Use to prepare the whole-genome sequencing library |
| Yeast Extract, Powder | Fisher Chemical | BP1422-500 | Use at 5 g/L to make liquid and solid rich media (YEA) |
Références
- McKay, J. K., Latta, R. G. Adaptive population divergence: Markers, QTL and traits. Trends in Ecology and Evolution. 17 (6), 285-291 (2002).
- Wood, V., Harris, M. A., et al. PomBase: A comprehensive online resource for fission yeast. Nucleic Acids Research. 40 (D1), (2012).
- de Visser, J. A. G. M., Cooper, T. F., Elena, S. F. The causes of epistasis. Proceedings of the Royal Society B: Biological Sciences. 278 (1725), 3617-3624 (2011).
- Sailer, Z. R., Harms, M. J. Detecting high-order epistasis in nonlinear genotype-phenotype maps. Genetics. 205 (3), 107911088 (2017).
- Kuzmin, E., Costanzo, M., Andrews, B., Boone, C. Synthetic genetic arrays: Automation of yeast genetics. Cold Spring Harbor Protocols. 2016 (4), 326-332 (2016).
- Tong, A. H. Y., Boone, C. Synthetic genetic array analysis in Saccharomyces cerevisiae. Methods in Molecular Biology. 313 (1), 171-192 (2006).
- Dixon, S. J., Costanzo, M., Baryshnikova, A., Andrews, B., Boone, C. Systematic Mapping of Genetic Interaction Networks. Annual Review of Genetics. 43 (1), 601-625 (2009).
- Boone, C., Bussey, H., Andrews, B. J. Exploring genetic interactions and networks with yeast. Nature Reviews Genetics. 8 (6), 437-449 (2007).
- Bai, X., Yang, Z., Jiang, H., Lin, S., Zon, L. I. Genetic suppressor screens in haploids. Methods in Cell Biology. , 129-136 (2011).
- Manson, M. D. Allele-specific suppression as a tool to study protein-protein interactions in bacteria. Methods. 20 (1), 18-34 (2000).
- Motter, A. E., Gulbahce, N., Almaas, E., Barabási, A. L. Predicting synthetic rescues in metabolic networks. Molecular Systems Biology. 4, 168 (2008).
- Peterson, R. T., Shaw, S. Y., et al. Chemical suppression of a genetic mutation in a zebrafish model of aortic coarctation. Nature Biotechnology. 22 (5), 595-599 (2004).
- Giorgini, F., Guidetti, P., Nguyen, Q., Bennett, S. C., Muchowski, P. J. A genomic screen in yeast implicates kynurenine 3-monooxygenase as a therapeutic target for Huntington disease. Nature Genetics. 37 (5), 526-531 (2005).
- Forsburg, S. L., Patton, E., et al. The art and design of genetic screens. Nature reviews. Genetics. 2 (9), 659-668 (2001).
- Johnston, D. S. The art and design of genetic screens. Genetics. 3 (March), 176-188 (2002).
- Jorgensen, E. M., Mango, S. E. The art and design of genetic screens: Caenorhabditis elegans. Nature Reviews Genetics. 3 (5), 356-369 (2002).
- Gocke, E., Müller, L. In vivo studies in the mouse to define a threshold for the genotoxicity of EMS and ENU. Mutation Research - Genetic Toxicology and Environmental Mutagenesis. 678 (2), 101-107 (2009).
- Suzuki, T., Hayashi, M., et al. A comparison of the genotoxicity of ethylnitrosourea and ethyl methanesulfonate in lacZ transgenic mice (Muta(TM)Mouse). Mutation Research - Genetic Toxicology and Environmental Mutagenesis. 395 (1), 75-82 (1997).
- Uttam, J., Alberico, C., De Stasio, E. ENU Mutagenesis. International C. elegans Meeting. , (1995).
- Putrament, A., Baranowska, H., Ejchart, A., Prazmo, W. Manganese Mutagenesis in Yeast. Methods in Cell Biology. 20, 25-34 (1978).
- Bose, J. L. Chemical and UV mutagenesis. Methods in Molecular Biology. 1373, 111-115 (2016).
- Ikehata, H., Ono, T. The Mechanisms of UV Mutagenesis. Journal of Radiation Research. 52 (2), 115-125 (2011).
- Shrivastav, N., Li, D., Essigmann, J. M. Chemical biology of mutagenesis and DNA repair: cellular responses to DNA alkylation. Carcinogenesis. 31 (1), 59-70 (2010).
- De Stasio, E. A., Dorman, S. Optimization of ENU mutagenesis of Caenorhabditis elegans. Mutation Research - Genetic Toxicology and Environmental Mutagenesis. 495 (1-2), 81-88 (2001).
- Probst, F. J., Justice, M. J. Mouse mutagenesis with the chemical supermutagen ENU. Methods in Enzymology. 477 (C), 297-312 (2010).
- Bähler, J., Wu, J. Q., et al. Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast. 14 (10), 943-951 (1998).
- Li, H., Durbin, R. Fast and accurate short read alignment with Burrows – Wheeler transform. Bioinformatics. 25 (14), 1754-1760 (2009).
- Van der Auwera, G. A., Carneiro, M. O., et al. From fastQ data to high-confidence variant calls: The genome analysis toolkit best practices pipeline. Current Protocols in Bioinformatics. 43, (2013).
- Mckenna, A., Hanna, M., et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research. 20, 1297-1303 (2010).
- Li, H., Handsaker, B., et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 25 (16), 2078-2079 (2009).
- Marayati, B. F., Drayton, A. L., et al. Loss of Elongation-Like Factor 1 Spontaneously Induces Diverse, RNase H-Related Suppressor Mutations in Schizosaccharomyces pombe. Genetics. 209 (4), 967-981 (2018).
- Harris, M. A., Lock, A., Bähler, J., Oliver, S. G., Wood, V. FYPO: The fission yeast phenotype ontology. Bioinformatics. 29 (13), 1671-1678 (2013).
- Xu, X., Wang, L., Yanagida, M. Whole-Genome Sequencing of Suppressor DNA Mixtures Identifies Pathways That Compensate for Chromosome Segregation Defects in Schizosaccharomyces pombe. G3: Genes|Genomes|Genetics. 8 (3), 1031-1038 (2018).
- Marayati, B. F., Hoskins, V., et al. The fission yeast MTREC and EJC orthologs ensure the maturation of meiotic transcripts during meiosis. RNA. 22 (9), 1349-1359 (2016).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.