
Method Article
Dépistage stratégique et caractérisation du complexe de signalisation des protéines VISUAL GPCR-mini-G pour une cristallisation réussie
Dans cet article
Résumé
Ce rapport décrit le dépistage de différents détergents pour la préparation du GPCR visuel, de la rhodopsine et de son complexe avec mini-Go. Des méthodes biochimiques caractérisant la qualité du complexe à différents stades de purification sont démontrées. Ce protocole peut être généralisé à d’autres complexes de protéines membranaires pour leurs futures études structurelles.
Résumé
La clé pour déterminer les structures cristallines des complexes de protéines membranaires est la qualité de l’échantillon avant la cristallisation. En particulier, le choix du détergent est essentiel, car il affecte à la fois la stabilité et la monodispersion du complexe. Nous avons récemment déterminé la structure cristalline d’un état actif de rhodopsine bovine couplée à une protéine G conçue, mini-Go, à 3,1 résolution. Ici, nous détaillons la procédure d’optimisation de la préparation du complexe rhodopsin-mini-Go. La rhodopsine à l’état foncé a été préparée dans des détergents classiques et neopentyl glycol (NPG), suivis d’une formation complexe avec mini-Go sous exposition légère. La stabilité de la rhodopsine a été évaluée par spectroscopie ultraviolette visible (UV-VIS), qui surveille la reconstitution en rhodopsine du ligand sensible à la lumière, rétinaire de 9 cis. La chromatographie automatisée de taille-exclusion (SEC) a été employée pour caractériser la monodispersité de la rhodopsine et du complexe de rhodopsin-mini-Go. L’électrophoresis SDS-polyacrylamide (SDS-PAGE) a confirmé la formation du complexe en identifiant un rapport molaire de 1:1 entre la rhodopsine et le mini-Go après avoir taché le gel avec le bleu Coomassie. Après avoir recoupé toutes ces données analytiques, nous avons éliminé les détergents inappropriés et continué avec le meilleur détergent candidat pour la préparation et la cristallisation à grande échelle. Un problème supplémentaire est né de l’hétérogénéité de la N-glycosylation. La rhodopsine héterologement-exprimée a été observée sur SDS-PAGE pour avoir deux populations différentes de N-glycosylated, qui auraient probablement entravé la cristallogenèse. Par conséquent, différentes enzymes de deglycosylation ont été testées, et endoglycosidase F1 (EndoF1) a produit de la rhodopsine avec une seule espèce de N-glycosylation. Avec ce pipeline stratégique pour caractériser la qualité des protéines, la préparation du complexe rhodopsin-mini-Go a été optimisée pour fournir la structure cristalline. Ce n’était que la troisième structure cristalline d’un complexe de signalisation de protéines GPCR-G. Cette approche peut également être généralisée pour d’autres protéines membranaires et leurs complexes afin de faciliter la préparation de l’échantillon et la détermination de la structure.
Introduction
La détermination des structures cristallines des protéines membranaires et de leurs complexes a toujours été difficile en raison des difficultés à obtenir des cristaux bien-diffracting. Contrairement aux protéines solubles, les protéines membranaires intégrales constituent un noyau hydrophobe qui s’étend sur la membrane cellulaire. Pour éliminer les protéines membranaires de la membrane cellulaire en tampon aqueux, les détergents doivent être utilisés pour former une micelle détergent-protéine, remplaçant ainsi les lipides autour du noyau hydrophobe des protéines membranaires. La stabilité, l’activité et l’intégrité des protéines membranaires dépendent directement des propriétés chimiques et structurelles du détergent1, et les propriétés du détergent déterminent également la taille de la micelle. Une grande micelle détergente peut occluder les surfaces hydrophiles d’une petite protéine membranaire, empêchant ainsi la cristallisation due au manque de contacts cristallins lors de l’utilisation de la méthode de diffusion de vapeur. Une petite micelle détergente est avantageuse pour la cristallographie, mais les détergents à chaîne courte sont généralement plus sévères et conduisent donc à la déstabilisation et à l’agrégation de la protéine membranaire. Par conséquent, avant la cristallisation, une procédure supplémentaire de dépistage des détergents est indispensable, ciblant généralement des détergents plus courts qui maintiennent encore la stabilité des protéines.
Les récepteurs couplés aux protéines G (GPCR) sont des protéines membranaires intégrales contenant sept transmembrane a-helices. Les RPGC existent dans deux États principaux, soit un état inactif stabilisé par des agonistes inverses ou des antagonistes, soit un état actif lié à un agoniste et stabilisé par une protéine G, bien qu’il soit probable qu’il existe une multitude de sous-états entre ces deux extrêmes. La détermination de la structure des GPCRs s’est d’abord concentrée sur les États inactifs liés aux agonistes et aux antagonistes inverses en raison de leur stabilité plus élevée que les États actifs2. Lorsque les GPCR sont activés sur la liaison agoniste, les récepteurs sont très dynamiques, et une fissure se forme transitoirement sur le visage cytoplasmique du récepteur pour l’accouplement de protéine G. On pense que ce dynamisme est la raison pour laquelle les GPCR à destination agoniste sont souvent plus instables que l’état inactif. Par conséquent, il devient essentiel de détergents appropriés à l’état conformationnel du récepteur à l’étude, car il est probable que des détergents plus doux seront nécessaires pour étudier un état actif par rapport à un état inactif.
Dans ce rapport, nous utilisons le GPCR visuel, rhodopsine bovine3, et son complexe avec mini-Go protéine4,5 pour les expériences de dépistage de détergent, représentant l’état inactif et l’état actif, respectivement. Le dépistage des détergents s’est concentré sur les détergents classiques de maltoside et de glucoside et les détergents de nopentyl glycol (NPG). Dans ce contexte, un détergent classique est construit à partir d’un groupe de tête de sucre et d’une chaîne d’alcyl, tandis que les détergents de type NPG contiennent deux détergents classiques identiques qui sont fusionnés par un carbone quaternaire à l’interface entre les sucres et les chaînes d’alcyl6,7,8.
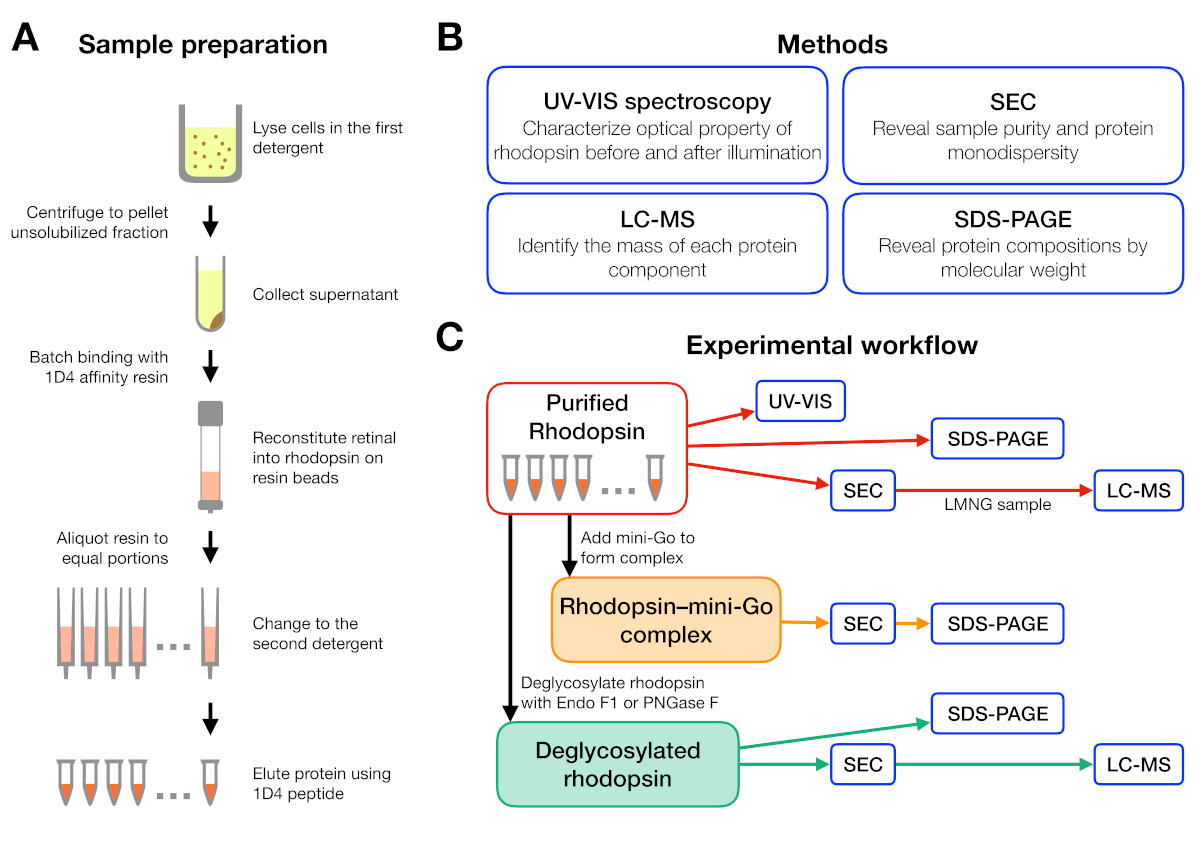
Un flux de travail expérimental a été conçu à partir de la purification de la rhodopsine dans différents détergents, suivi de la formation du complexe rhodopsin-mini-Go et se terminant par la caractérisation du complexe à l’aide de plusieurs méthodes(figure 1). Pour l’état inactif de rhodopsine, la reconstitution de la rétinale 9-cis sensible à la lumière a été surveillée par spectroscopie ultraviolette visible (UV-VIS). Le spectre révèle l’état physicochimique de la rétinienne et est indicatif de son environnement dans la poche rétinienne de liaison de rhodopsine. La chromatographie d’exclusion de taille (SEC) a été employée pour évaluer la monodispersité de la rhodopsine purifiée aussi bien que la formation du complexe de rhodopsin-mini-Go. Comme SEC différencie les molécules de protéines par leur taille et leur forme, la population de protéines agrégées peut être identifiée comme ils elute dans le volume vide. Pour confirmer la formation complexe, des fractions de SEC ont été évaluées par le gel de sulfate-polyacrylamide de sodium électrophoresis (SDS-PAGE) pour confirmer la présence de rhodopsine et de mini-Go.
Un autre facteur qui doit être pris en considération est les modifications post-traductionnelles (PTM) sur les protéines membranaires. PTM tels que N-glycosylation sont souvent observés sur les protéines de membrane eucaryote produites dans les systèmes d’expression des mammifères et des cellules d’insectes. Une souche limitée de N-glycosylation des cellules embryonnaires humaines de rein 293 (HEK293) a été développée par la suppression du gène codant N-acétylglucosaminyltransferase I (GnTI), ayant pour résultat la N-glycosylation homogène par GlcNAc2Man5 au site consensuel Asn-X-Ser/Thr. Bien que la N-glycosylation puisse être évitée en mutant un résidu d’acide aminé dans le site consensuel, cela peut également modifier la fonction de la protéine ou l’efficacité du pliage. Dans la rhodopsine bovine, la mutation des résidus N-glycosylated Asn15 conduit au pliage incorrect et à l’activation réduite de protéine de G9,10. Le rhodopsin utilisé dans ce rapport a été exprimé dans heK 293 GnTI-déficience lignée cellulaire. Cependant, SDS-PAGE a montré la présence de deux espèces de rhodopsine. Cette hétérogénéité pourrait empêcher la formation de cristaux et donc la deglycosylation à l’aide de peptide-N-glycosidase F (PNGase F) et endoglycosidase F1 (Endo F1) a été testée. Le produit déglycosylé a été caractérisé par SDS-PAGE et la spectrométrie de masse chromatographie liquide (LC-MS) pour identifier le niveau de glycosylation et son homogénéité.
Protocole
REMARQUE : Ce protocole de dépistage des détergents est détaillé pour 30 g de granules de cellules HEK293 comme matériau de départ.
1. Matériaux, produits chimiques et réactifs
REMARQUE : Toutes les solutions sont préparées à l’aide de réactifs de qualité analytique et d’eau ultrapure, qui est purifiée à partir d’eau déionisée pour atteindre une résistance de 18,2 M à 25 oC.
- Solutions de stock tampon
- Préparer le salin tampon 10x phosphate (10x PBS).
- Préparer HEPES tampon: 1 M, titré à pH 7.5 avec NaOH.
- Préparer 5 M NaCl.
- Préparer 2 M MgCl2.
REMARQUE : Toutes les solutions de stock sont passées par un filtre de 0,22 m pour maintenir leur stérilité.
- Solutions d’actions détergentes
- Préparer dodecyl maltoside (DDM), 10% (w/v).
- Préparer le décyl maltoside (DM), 10% (w/v).
- Préparer 6-cyclohexyl-hexyl maltoside (Cymal-6), 10% (w/v).
- Préparer 5-cyclohexyl-pentyl maltoside (Cymal-5), 10% (w/v).
- Préparer le glucoside nonyl (C9G), 10% (w/v).
- Préparer le neopentyl glycol de maltose de maltose de lauryl (GNL), 5 % (w/v).
- Préparer le neopentyl glycol de maltose décyl (DMNG), 10% (w/v).
- Préparer le cymal-6 neopentyl glycol (C6NG), 10% (w/v).
- Préparer le cymal-5 neopentyl glycol (C5NG), 10% (w/v).
- Préparer le neopentyl glycol de glucose octyle (OGNG), 10% (w/v).
REMARQUE : Pour une solution de stock de détergent de 10 %, dissoudre 1 g de poudre de détergent dans de l’eau ultrapure avec un basculement doux, puis ajuster le volume final à 10 ml. La solution de stock de détergent doit être maintenue à -20 oC pour le stockage à long terme et sur la glace pendant le travail.
CAUTION : Il est généralement recommandé de conserver les détergents en bouteille au congélateur de -20 oC. Les bouteilles contenant de la poudre de détergent doivent être réchauffées à température ambiante avant l’ouverture. La poudre de détergent est hygroscopique, ainsi l’équilibre de température empêchera la formation de condensation qui mouillera le détergent.
- Autres produits chimiques et réactifs
- Préparer la résine d’agarose d’immunoaffinité 1D4 : 10 ml de la boue de 50 %.
REMARQUE : L’agarose d’immunoaffinité 1D4 sont les perles d’agarose liées à l’anticorps rho1D4 monoclonal, qui lie les 9 derniers acides aminés de rhodopsin bovin TETSQVAPA comme épitope. L’agarose d’immunoaffinité 1D4 agit comme matériau de purification d’affinité pour capturer des protéines qui contiennent une séquence C-terminal 1D4. Ce matériau de purification peut être préparé9,11 ou acheté. - Préparer la solution rétinienne à 9 cis : 1 mM, dissous dans 100% d’éthanol.
REMARQUE : Prévenir l’exposition à la lumière à la rétinienne pendant la préparation et l’entreposage. - Préparer le peptide 1D4 (séquence TETSQVAPA) : 800 M, dissous dans l’eau.
- Préparer la résine d’agarose d’immunoaffinité 1D4 : 10 ml de la boue de 50 %.
- Tampons
REMARQUE : Tous les tampons sont mélangés des solutions de stock à la concentration désirée. Tous les tampons sont réfrigérés à 4 oC avant utilisation.- Préparer le tampon A: PBS, 0,04% DDM.
- Préparer buffer B: 20 mM HEPES pH 7.5, 150 mM NaCl, 0.04% DDM.
- Préparer le tampon C : 20 mM HEPES pH 7,5, 150 mM NaCl, et détergent à leur concentration de travail énumérées dans le tableau 1.
- Préparer buffer D: 20 mM HEPES pH 7.5, 150 mM NaCl.
- Préparer le tampon Elution : 20 mM HEPES pH 7,5, 150 mM NaCl, 80 M 1D4 peptide, et détergent à leur concentration de travail.
- Préparer le tampon SEC: 20 mM HEPES pH 7,5, 150 mM NaCl, 0,025% DDM; filtré à travers un filtre de 0,22 m.
- Solvant pour LC-MS
- Préparer le solvant A : acétonitrile contenant 0,1 % d’acide formic.
- Préparer le solvant B : eau ultrapure contenant 0,1 % d’acide formique.
- Préparer le solvant C : iso-propanol.
2. Solubilisation de membrane cellulaire et extraction de protéine
- Dégel 30 g de HEK293 GnTI- granule cellulaire exprimant le mutant bovin rhodopsin N2C/M257Y/D282C3,9 à température ambiante, ajouter 120 ml de tampon PBS 1x contenant un cocktail inhibiteur de la protéase et homogénéiser à l’aide d’un homogénéisant Dounce ou d’un homogénéisateur électrique (13 000 tr/min pour 30 s). Recueillir la suspension cellulaire homogénéisée dans un bécher et ajuster le volume à 150 ml.
REMARQUE : 30 g de granule cellulaire équivaut à 3 L de culture cellulaire à 2 x 106 cellules/mL densité. - Ajouter délicatement 10% de DDM aux cellules homogénéisées pour donner une concentration finale de 1,25%. Remuer sur la glace pendant 1 h.
- Centrifuge la cellule lysate à 4 oC et 150.000 x g pendant 45 min pour enlever les débris nonsolubilisés.
- Transférer le supernatant dans une bouteille de 500 ml et ajouter 10 ml de résine d’agarose d’immunoaffinité 1D4 (50% de boue). Mélanger délicatement le lysate solubilisé et la résine pendant 4 h ou toute la nuit à 4 oC.
- Charger le mélange de lysate/résine à une colonne ouverte pour recueillir la résine.
- Laver la résine avec 10 volumes de colonnes (CV) du tampon de lavage A.
REMARQUE : Le volume de la colonne est le volume des emballages (100 %) résine agarose utilisée. Dans ce cas, 1 CV est de 5 mL. - Réutilisez la résine avec 2 CV de Buffer A.
CAUTION: À partir de l’étape 2.8, les étapes qui doivent être effectuées dans un état de faible luminosité rouge sont étiquetés avec "[Dark]" au début de la description. - [Dark] Ajouter la rétinienne de 9 cis à la résine résuspendée à la concentration finale de 50 M. Mélanger délicatement à 4 oC pendant 4-16 h dans l’obscurité.
REMARQUE : Un temps d’incubation plus court peut mener à la reconstitution incomplète de la rétine. - [Dark] Retirez le flux à travers de la colonne. Laver la résine avec 20 CV Buffer A, suivi de 15 CV Buffer B.
- [Dark] Résuspendez la résine dans 2 CV Buffer B, puis divisez la suspension de résine également à 10 colonnes d’élimination de 10 ml.
- [Dark] Retirez le débit à travers de la colonne, puis réutilisez la résine dans 1 mL Buffer C. Incubate pendant 1 h à 4 oC.
- [Dark] Répéter l’étape 2.11.
- [Dark] Retirez le flux à travers de la colonne, puis réutilisez la résine dans 0,8 mL Elution Buffer pour chaque colonne. Mélanger délicatement pendant 2 h.
- [Dark] Recueillir l’elution de la colonne dans un tube de 2 ml.
- [Dark] Résuspendez la résine dans 0,7 ml d’Elution Buffer pour chaque colonne. Mélanger délicatement pendant 1 h.
- [Dark] Recueillir l’elution de la colonne dans le même tube.
3. Spectroscopie UV-VIS
- Préparer le spectrophotomètre pour couvrir la plage de mesure de 250-650 nm. Enregistrez la ligne de base à l’aide d’eau ou de tampon Elution.
- [Dark] Chargez la protéine elfée à la cuvette de quartz. Mesurer le spectre de l’échantillon de protéines.
- [Dark] Illuminez la protéine directement dans la cuvette pendant 2 min avec la lumière passée à travers un filtre de 495 nm de long-passage.
- Mesurer le spectre de l’échantillon illuminé.
- Effectuez la même mesure pour tous les échantillons de protéines purifiés dans les 9 autres détergents, les états sombres et illuminés.
- Tracer les courbes (absorption par rapport à longueur d’onde) dans le graphique de dispersion X-Y.
4. chromatographie automatisée de taille-exclusion de rhodopsine et rhodopsin-mini-Go complexe
- [Dark] Concentrez la protéine à 100 l par centrifugation à l’aide d’un concentrateur de spin avec un seuil de poids moléculaire (MWCO) de 30 kDa à 4 oC. Les échantillons surconcentrés peuvent être dilués à l’aide du débit à travers du concentrateur ou du tampon C. Pour déterminer la concentration de l’échantillon de protéines, mesurer l’absorption à 280 nm à l’aide d’un spectrophotomètre.
REMARQUE : À partir de l’étape 4.2, l’expérience ne nécessite pas un environnement sombre, et donc des échantillons peuvent être préparés sous une lumière normale. - Préparer la rhodopsine de 100 L à 0,7 mg/mL pour chaque état de détergent.
- Préparer 100 l de rhodopsine (0,7 mg/mL) et le mini-Go4,12 (0,2 mg/mL) pour chaque état de détergent. Complétez le mélange avec 1 mM MgCl2. Illuminer le mélange de lumière à partir d’un filtre à pas long de 495 nm et incuber pendant 30 min.
- Montez une colonne de filtration de gel de 24 ml avec une plage de fractionnement de 10-600 kDa d’une protéine globulaire sur un purificateur de chromatographie liquide. Équilibre la colonne avec tampon SEC.
REMARQUE : Le purificateur de chromatographie liquide est équipé d’un autosampler, d’un détecteur de longueur d’onde multiple et d’un collecteur fraction. - Transférer les échantillons sur les flacons d’autosampler et les placer dans le plateau de l’échantillon. Programmez un fichier de méthode pour automatiser les exécutions Séquentielles DE SEC pour chaque échantillon, avec l’autosampler chargeant 77 L de l’échantillon à la colonne, et le purificateur qui s’écoule de 24 ml de tampon SEC à un débit de 0,5 mL/min par course. Enregistrez l’absorption à 280 nm et 380 nm.
- Recueillir les fractions maximales de rhodopsine et de rhodopsine-mini-Go complexe au volume de rétention autour de 12,9 ml.
- Analyser les échantillons gauches de rhodopsine de l’étape 4.2 et les fractions maximales de rhodopsin-mini-Go complexe sur 4-12% SDS-dénaturing gels gradient avec colorant bleu Coomassie.
- Tracer l’chromatogramme d’elution (A280 ou A380 contre le volume de rétention).
5. Étude de la deglycosylation et de la LC-MS
- Pour l’étude LC-MS, n’utilisez que l’échantillon de rhodopsine purifié dans le détergent de GNL.
- Préparer un mélange de rhodopsine de 200 l à 1 mg/ml et PNGase F13 à 0,01 mg/mL. Bien mélanger et incuber à 4 oC pendant la nuit.
- Préparer un mélange de rhodopsine de 200 l à 1 mg/mL et Endo F113 à 0,01 mg/mL. Bien mélanger et incuber à 4 oC pendant la nuit.
- Analyser le résultat de digestion par SDS-PAGE et Coomassie colorant bleu.
- Concentrez les échantillons de rhodopsine non traités et endo F1 et faites l’objet d’une purification DE SEC dans Buffer D.
REMARQUE : Il s’agit de préparer l’échantillon avec une quantité minimale de détergent pour l’étude LC-MS. Buffer D ne contient aucun détergent, mais en raison du ralentissement du GNL provenant d’une protéine membranaire14,la rhodopsine ne s’agrége pas. - Recueillir la fraction maximale au volume de rétention autour de 12,9 ml. Concentrez-vous à 1 mg/mL à l’aide d’un concentrateur de spin (MWCO 30 kDa).
- Injecter 10 g de protéines dans une colonne Reprosil 200 C18-AQ et eluter la colonne à l’aide de la méthode de gradient linéaire avec la composition et les paramètres solvants énumérés dans le tableau 2. Le flux est divisé à 25 % pour le spectromètre de masse et à 75 % pour la détection UV.
Résultats
Le flux de travail expérimental pour la préparation et l’analyse de l’échantillon est résumé à la figure 1. L’utilisation de colonnes ouvertes pour la purification de l’affinité à petite échelle nous a permis de préparer des échantillons dans de nombreuses conditions de détergents différentes en parallèle(figure 1A). Une telle purification à petite échelle a donné suffisamment de protéines pour d’autres analyses à l’aide de la spectroscopie UV-VIS, de la SEC et du SDS-PAGE(figure 1B-C).
La spectroscopie UV-VIS a révélé la stabilité de la rhodopsine
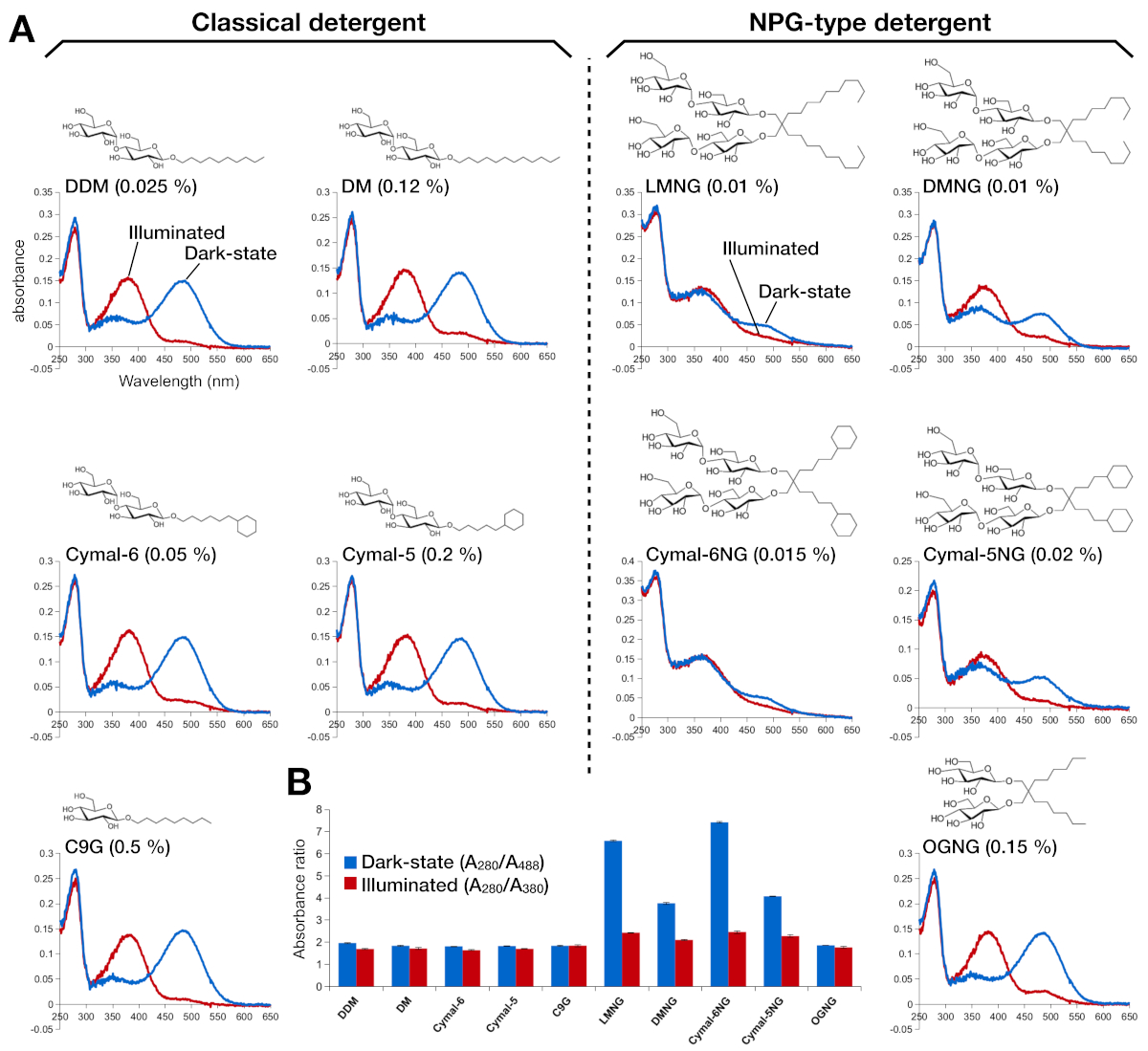
La stabilité de la rhodopsine reconstituée par la rétinienne a été évaluée par son absorption optique(figure 2). Dans l’état sombre, la rétine 9-cis est covalentement liée à Lys296 comme une base de Schiff protonated. Après l’illumination, le rétinien de 9 cis est isomerisé à l’isoforme tout-trans et le lien de base Schiff est déprotoné. La rétinienne protonnée de 9 cis donne un pic d’absorption à 488 nm, tandis que la rétinienne tout trans déprotonée a un pic à 380 nm. Les spectres UV-VIS de rhodopsine dans DDM ont montré l’absorption typique de la rhodopsine rétinienne et activée par la lumière de 9 cis, où un décalage bleu de 108 nm avec à peu près la même densité optique a été clairement observé(figure 2A, panneau supérieur gauche). Quand la rhodopsine est déstabilisée, puis la poche de liaison pour les changements rétiniens, qui a comme conséquence la déprotonation rétinienne et peut-être la dissociation. Si cela se produit, et puis le spectre montre la contribution de la déprotonation ainsi que la forme libre de rétinienne15. Par conséquent, nous avons déterminé l’efficacité de la reconstitution rétinienne en rhodopsine par le rapport d’absorption entre la protéine (280 nm) et la rétine (488 nm pour la rétinienne protonated 9-cis, 380 nm pour la rétination tout trans déprotonated) (figure 2B). Les échantillons de rhodopsine purifiés dans les détergents classiques (DDM, DM, Cymal-6, Cymal-5, C9G) présentent le même profil optique. Cependant, les échantillons purifiés dans les détergents NPG (LMNG, DMNG, Cymal-6NG, Cymal-5NG) montrent des profils optiques suggérant un environnement de liaison sous-optimal pour la rétinienne, à l’exception de l’échantillon OGNG, qui a donné le même profil optique que l’échantillon DDM.
La chromatographie de taille-exclusion a montré la pureté d’échantillon et la monodispersité de protéine.
SEC est un outil d’analyse efficace et robuste pour évaluer les échantillons de protéines pendant la préparation et le dépistage. Il valide la pureté de l’échantillon de l’étape de purification précédente ainsi que la monodispersion des molécules de protéines. Pour la rhodopsine et son mini-Go complexe, la qualité de l’échantillon a été interprétée à partir des courbes d’absorption à 280 nm et 380 nm(figure 3A). Les traces de 280 nm ont montré la présence de protéines, et la trace de 380 nm a montré la présence de rétinienne. Tous les signaux apparaissant dans le volume vide (environ 8 ml lors de l’utilisation de cette colonne) ont été attribués à des agrégats de protéines. Par conséquent, les résultats ont montré que les échantillons préparés dans les détergents classiques se trouvaient dans un état de monodisperse, à l’exception du C9G, où une partie de l’agrégat apparaissait. En revanche, les échantillons préparés à l’aide des détergents de type NPG contenaient beaucoup plus d’agrégats que l’échantillon C9G; Le GNL et le Cymal-6NG ont mené à la formation la plus globale, mais moins d’agrégats ont été observés dans DMNG et Cymal-5NG. L’exception était OGNG, qui a montré un profil similaire à DDM. Les agrégats protéiques qui s’échappaient au volume vide ont également eu une occupation rétinienne plus faible, comme le montre le rapport A280/A380 qui avait augmenté par rapport au pic au volume de rétention de 12,9 ml correspondant à 135 kDa. Une autre caractéristique que nous avons observée était que la rhodopsine et le rhodopsin-mini-Go s’est écoulé autour du même volume de rétention(figure 3B). Ce n’est pas surprenant, parce que le poids moléculaire apparent de la rhodopsine liée au détergent était de 120 kDa et celui de la rhodopsine-mini-Go 144 kDa. Nous n’avons donc pas pu déterminer la formation complexe simplement à partir des données de la SEC, de sorte que SDS-PAGE a été utilisé pour analyser davantage l’échantillon purifié par la SEC.
SDS-PAGE confirme la formation complexe
SDS-PAGE est une méthode standard pour identifier les composants protéiques d’un échantillon. La rhodopsine concentrée (avant la purification de LA SEC) a été analysée par SDS-PAGE pour confirmer sa pureté, et a montré deux bandes près de 37 kDa et une bande enduite au-dessus de 50 kDa (figure 4A). Les deux bandes inférieures ont été plus tard confirmées pour avoir différents états de N-glycosylation. La bande au-dessus de 50 kDa a été interprétée comme des oligomers agrégés de rhodopsine induits par le tampon d’échantillon SDS-PAGE parce que ces agrégats n’ont pas été observés dans SEC ou toute autre méthode de détection. Comme les données de la SEC n’ont pas pu confirmer la formation complexe, les fractions d’extraits de la SEC provenant d’échantillons de rhodopsin-mini-Go ont été analysées à l’aide de SDS-PAGE. Le SDS-PAGE a montré des bandes protéinées de rhodopsine et de mini-Go dans toutes les conditions de détergent, ce qui suggère que le complexe a été formé quel que soit le choix du détergent(figure 4B).
La spectrométrie LC-MS a identifié le modèle de N-glycosylation dans la rhodopsine
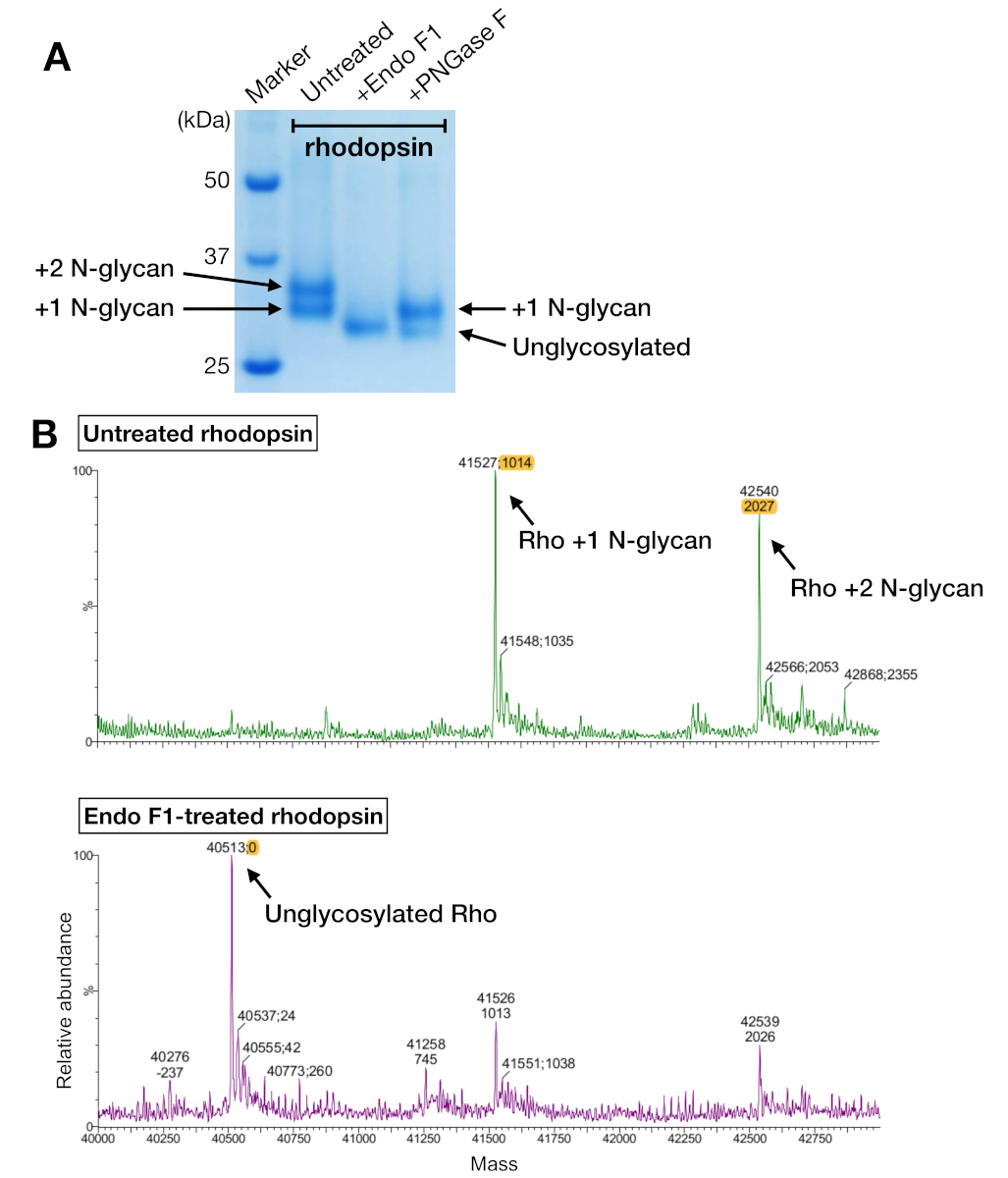
Des échantillons de rhodopsine provenant à la fois de la purification de l’affinité et de la SEC ont montré deux bandes protéiques qui ont migré avec un poids moléculaire apparent d’environ 37 kDa sur un gel SDS-PAGE, qui ne pouvait pas être séparé par SEC lors de l’utilisation d’une colonne de 24 ml. Différents modèles de N-glycosylation sur la rhodopsine hétérologue-exprimée de HEK 293 GnTI- cellules était l’explication la plus probable. Par conséquent, deux enzymes, PNGase F et Endo F1, ont été testées pour leur capacité à déglycosyler la rhodopsine. D’après les données de SDS-PAGE, Endo F1 a réduit le poids moléculaire des deux bandes protéiques en un seul produit, tandis que la digestion de PNGase F donnait encore deux populations(figure 5A). Les échantillons non digérés et traités par Endo F1 ont été analysés à l’aide de la spectrométrie LC-MS pour identifier les masses de différentes espèces. Les données ont montré que la rhodopsine produite dans HEK 293 GnTI- cellules contenait soit un ou deux N-glycans, avec une différence de masse de 1014-1 Da. Endo F1-traité rhodopsine ne contenait pas de N-glycans et avait une différence de masse de 2027-1 Da par rapport à la rhodopsine contenant deux N-glycans. Ces résultats sont compatibles avec l’absence de l’enzyme N-acétylglucosaminyltransferase I dans la lignée cellulaire utilisée pour exprimer la rhodopsine, ce qui entraîne tous les N-glycans ayant la structure GlcNAc2Homme5, (masse 1014 Da).

Figure 1 : Préparation et caractérisation de l’échantillon pour l’expérience de dépistage des détergents. (A) Préparation d’échantillons de rhodopsine dans différents détergents pendant la purification. (B) Méthodes utilisées dans le protocole : spectroscopie UV-VIS, chromatographie de taille-exclusion (SEC), SDS-PAGE et spectrométrie de masse de chromatographie liquide (LC-MS). (C) Flux de travail expérimental pour la caractérisation de la rhodopsine, de la rhodopsine-mini-Go,et du produit de deglycosylation de la rhodopsine. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 2 : spectroscopie UV-VIS de rhodopsine. (A) spectre UV-VIS de rhodopsine. Les spectres de la rhodopsine à l’état sombre, à 9 cis liés à la rétinienne, sont représentés dans des courbes bleues. Après l’illumination, la rétinienne de 9 cis est déprotonée et isomerise en rétinienne tout-trans, et les spectres de rhodopsine illuminée sont représentés comme des courbes rouges. La structure chimique de chaque détergent est indiquée comme un encart. (B) Les ratios deA 280/A488 (barre bleue) et A280/A380 (barre rouge) représentent la stabilité de la rhodopsine dans l’état sombre et l’état clair, respectivement. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 3 : Profils de chromatographie de taille-exclusion de rhodopsine et de rhodopsine-mini-Go complexe purifié dans 10 détergents différents. (A) Le panneau de gauche montre les profils SEC d’échantillons purifiés dans les détergents classiques. Le panneau de droite représente les profils SEC d’échantillons purifiés dans les détergents de type NPG. Le profil des protéines de marqueur standard est indiqué comme superposition avec l’échantillon DDM. L’interprétation des profils de pointe est montrée pour DMNG, avec le scénario idéal (pas d’agrégats) vu pour DDM, DM, Cymal-6, Cymal-5 et OGNG. (B) Le profil agrandi de l’échantillon OGNG dans le volume de rétention de 12-14 ml. Tous les échantillons ont été analysés à l’aide d’une colonne Superdex200 Increase 10/300 GL. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 4 : Analyse SDS-PAGE du complexe de rhodopsine et de rhodopsine/mini-Go. (A) Échantillons de Rhodopsine purifiés dans les détergents. La bande enduite au-dessus de 50 kDa est attribuée aux oligomers agrégés de rhodopsine induits par le tampon d’échantillon SDS-PAGE. (B) échantillons purifiés par sec du complexe de rhodopsine/mini-Go. Rhodopsin avec 1 et 2 N-glycan et mini-Go sont représentés. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}

Figure 5 : Identification de la glycosylation dans la rhodopsine. (A) Analyse SDS-PAGE de rhodopsine déglycosée à l’aide de PNGase F et Endo F1. (B) LC-MS spectra de rhodopsine sans (panneau supérieur) et avec deglycosylation par Endo F1 (panneau inférieur). Pour la préparation du complexe rhodopsin-mini-Go pour la cristallisation, nous avons choisi Endo F1 au-dessus de PNGase F parce qu’Endo F1 a livré une seule espèce homogène de rhodopsine. S’il vous plaît cliquez ici pour voir une version plus grande de ce chiffre.
{kind=link}
| Détergent | Concentration de travail (%) | Concentration critique de micelle (%) |
| Ddm | 0.025 | 0.0087 |
| Dm | 0.12 | 0.087 |
| Cymal-6 | 0.05 | 0.028 |
| Cymal-5 | 0.2 | 0.12 |
| C9G | 0.5 | 0.2 |
| GNL | 0.01 | 0.001 |
| DMNG DMNG | 0.01 | 0.0034 |
| Cymal-6NG | 0.015 | Non disponible; devrait être inférieur à 0,056 |
| Cymal-5NG | 0.02 | 0.0056 |
| OGNG (OGNG) | 0.15 | 0.058 |
Tableau 1 : Concentrations de détergent tampon C.
| Temps (min) | Solvant A (%) | Solvant B (%) | Solvant C (%) | Débit (ml/min) |
| 0 | 0 | 95 | 5 | 0.5 |
| 1 | 0 | 95 | 5 | 0.5 |
| 5 | 20 | 75 | 5 | 0.6 |
| 25 | 85 | 10 | 5 | 0.6 |
| 26 | 90 | 5 | 5 | 0.6 |
| 30 | 90 | 5 | 5 | 0.6 |
Tableau 2 : Paramètres d’elution de colonne.
Discussion
Le succès de la cristallisation des protéines repose fortement sur l’échantillon de protéines, en particulier les protéines membranaires et leurs complexes en raison de la complication causée par les détergents. Ce rapport démontre le dépistage et l’évaluation des détergents de la qualité de l’échantillon pour les complexes de signalisation des protéines GPCR-mini-G. Une variété de méthodes ont été largement utilisées pour étudier la propriété biochimique des protéines membranaires, par exemple, l’analyse de thermostabilité à l’aide de colorants fluorescents16,17, l’essai de liaison pour détecter la formation complexe en mesurant le changement dans le signal de fluorescence de tryptophane18 ou le transfert d’énergie de résonance avec des biocapteurs19. Cependant, les environnements chimiques utilisés dans ces méthodes sont très différents de ceux pour la préparation d’un échantillon de cristallisation, soit les protéines sont à une concentration mille fois plus faible pour la mesure à base de fluorescence, ou les protéines sont incorporées dans les bilayers lipidiques ou dans une condition de détergent fixe. Dans ce protocole, les méthodes utilisées sont également normalisées dans la préparation d’échantillons à grande échelle avant la cristallisation. Par conséquent, les paramètres optimisés peuvent être facilement transférés pour la préparation à l’échelle de cristallisation sans autre dépistage et optimisation majeurs.
L’objectif de ce protocole est d’optimiser la préparation d’un complexe de protéines GPCR-mini-G stable et homogène pour la cristallisation de diffusion de vapeur et la détermination de la structure par cristallographie aux rayons X. Le protocole intègre un ensemble de méthodes pour évaluer qualitativement l’impact du détergent et de la deglycosylation lors de la préparation du complexe rhodopsin-mini-Go. Rhodopsine à l’état inactif et l’état activé par la lumière lié avec et sans le peptide de transducine a été cristallisé lorsqu’il est purifié dans les détergents octyl glucoside (C8G)20,21,22 et C9G23,24. Comme le complexe rhodopsin-mini-Go purifié dans C8G et C9G n’a pas produit de cristaux (données non montrées), nous avons ensuite exploré un plus large éventail d’autres détergents à l’aide de la stratégie décrite(figure 1). En profitant de la sensibilité lumineuse de la rhodopsine, nous pourrions très bien suivre la reconstitution de la rétinienne à longueurs d’onde autres que 280 nm. Dans la spectroscopie UV-VIS et la SEC, nous avons détecté la rétine à 380 nm ou 488 nm. Cependant, la plupart des protéines membranaires n’ont pas un chromophore si pratique pour suivre la fonctionnalité pendant la purification. D’autres options seraient de rendre un ligand détectable en ajoutant un chromophore détectable de lumière ou en utilisant radioligand-liaison et les essais de décalage thermique25.
Rhodopsin a un poids moléculaire de 40 kDa. En raison de la masse de détergent qu’il lie, son poids moléculaire apparent sur SEC est d’environ 120 kDa. Il n’est donc pas surprenant que la liaison de mini-Go (24 kDa) n’ait pas été facilement détectée sur SEC, car cela nécessiterait une différenciation des protéines avec des masses apparentes de 120 kDa et 144 kDa. L’analyse des fractions sec par SDS-PAGE a donc été utilisée pour confirmer la pureté de l’échantillon et la formation complexe. Même si les profils de la SEC montrent un changement clair sur la formation complexe, il est toujours recommandé d’effectuer une analyse SDS-PAGE pour confirmer la formation complexe avec des partenaires contraignants corrects plutôt que d’autres contaminants protéiques co-purifiés.
Les rhodopsins et les mini-Go ont été purifiés en quantités de milligrammes, ce qui a permis l’utilisation de la détection de faible sensibilité des complexes, tels que l’absorption UV-VIS pendant SEC et Commassie Coloring bleu des gels SDS-PAGE. Lorsque les échantillons sont limités, une détection plus sensible devrait être utilisée, comme un purificateur LC équipé d’un détecteur de fluorescence pour retracer les signaux de tryptophane provenant de la protéine (280 nm excitation, émission de 350 nm) et la teinture d’argent pour les gels SDS-PAGE. Une autre approche consisterait à fusionner une protéine fluorescente, comme la protéine de fluorescence verte (GFP) à la protéine d’intérêt, ce qui permettrait également la détection même pendant l’expression protéique26, mais elle devrait être enlevée avant la cristallisation.
Il est essentiel de s’assurer que la protéine purifiée est également exempte d’hétérogénéité résultant de MPT variables. Dans le cas décrit ici, les deux populations de rhodopsine observées sur les gels SDS-PAGE ont été caractérisées comme ayant un ou deux N-glycans. La modification variable d’une protéine pourrait potentiellement empêcher la formation de cristaux bien-diffracting, ainsi nous avons donc deglycosylated rhodopsin. L’endoglycosidase Endo F1 a été le plus d’effet endoglycosidase testé et le traitement a conduit à une seule espèce de récepteur nonglycosylated, tandis que PNGase F seulement partiellement enlevé les glycans sur la rhodopsine et a eu comme conséquence un mélange de rhodopsine complètement unglycosylated ou avec un N-glycan est resté. Rhodopsine sans traitement de deglycosylase a été cristallisé avec succès3,27,28, et le N-glycan sur rhodopsin Asn15 est important pour former le contact cristallin dans ces cas. Dans le cas de rhodopsin-mini-Go, il est nécessaire d’enlever les N-glycans par Endo F1 pour obtenir des cristaux. Il n’existe pas de règle normalisée pour déglycosyler les protéines d’intérêt avant la cristallisation, mais l’élimination des SMT hétérogènes devrait être envisagée lorsque les protéines ne se cristallisent pas après de vastes essais de cristallisation.
Les données et la méthodologie décrites ici nous ont guidés à choisir OGNG comme le détergent le plus préféré pour la cristallisation du complexe rhodopsin-mini-Go en raison de sa petite taille de micelle et de sa capacité à stabiliser le complexe. Nous avons également utilisé Endo F1 pour nous assurer que la rhodopsine purifiée était une espèce homogène. Les cristaux ont ensuite été obtenus et nous avons déterminé la structure cristalline à 3,14, qui n’était que la troisième structure cristalline d’un complexe de signalisation de protéines GPCR-G14,29.
Pour les protéines membranaires liées avec et sans protéine partenaire, elles doivent être considérées comme deux protéines différentes. Une protéine à différents états fonctionnels a des conformations différentes et est à un niveau d’énergie différent. Par conséquent, il est recommandé d’optimiser le protocole de préparation pour chaque état fonctionnel car le paramètre de l’état inactif peut ne pas être entièrement transférable à l’état activé. Aussi, sans parler du changement de la propriété protéique compliquée par la liaison d’une protéine partenaire. Le protocole utilise des méthodes qui sont normalisées pour la préparation d’un échantillon de cristallisation pour préparer les protéines membranes inactives dans différents détergents, suivies par l’activation des protéines et la formation complexe, et pour caractériser la qualité des protéines. Ainsi, ce protocole peut facilement être généralisé à d’autres protéines membranaires et à leurs complexes pour des études structurelles avec modification mineure.
Déclarations de divulgation
La CGT est consultante et membre du Conseil consultatif scientifique de Sosei Heptares. Tous les autres auteurs n’ont rien à révéler.
Remerciements
Nous remercions le professeur Gebhard F. X. Schertler pour son soutien à long terme à ce projet, le Dr Roger J.P. Dawson et Hoffmann La Roche, pour son soutien à la culture cellulaire. Ce travail a été parrainé par la Fondation nationale suisse des sciences (subventions 210030-153145 et 310030B_173335 à GFXS), et le financement à la CGT du Conseil européen de recherches (EMPSI, 339995) et du Conseil de recherches médicales (MRC U105197215). FP reconnaît ETH Zurich par l’intermédiaire du National Center of Competence in Research Molecular Ultrafast Science and Technology (NCCR MUST) et des programmes ETH Femtosecond et Attosecond Science and Technology (ETH FAST). FP, JM, AB et CJT reconnaissent le soutien financier à long terme de l’Institut Paul Scherrer.
matériels
| Name | Company | Catalog Number | Comments |
| 1D4 peptide | Peptide2.0 | Under request | |
| 9-cis retinal | Sigma-Aldrich | R5754 | |
| Autosampler A-900 | GE Healthcare | Discontinued | |
| C9G | Anatrace | N324 | |
| cOmplete, EDTA-free protease inhibitor coctail | Roche | 5056489001 | |
| Cymal-5 | Anatrace | C325 | |
| Cymal-5NG | Anatrace | NG325 | |
| Cymal-6 | Anatrace | C326 | |
| Cymal-6NG | Anatrace | NG326 | |
| DDM | Anatrace | D310 | |
| DM | Anatrace | D322 | |
| DMNG | Anatrace | NG322 | |
| Econo column | Bio-Rad | 7372512 | |
| Ettan LC | GE Healthcare | Discontinued | |
| FRAC-950 | GE Healthcare | Discontinued | |
| HPLC Water 2795 Separation Module | Waters AG | 720000358EN | |
| InstantBlue Protein Stain | Expedeon | ISB1L | |
| LCT Premier mass spectrometer (ESI-TOF) | Waters AG | - | |
| LMNG | Anatrace | NG310 | |
| Monitor UV-900 | GE Healthcare | 18110835 | |
| Nanodrop 1000 | Witec AG/ThermoFisher | Discontinued | |
| NuPAGE 4-12% Bis-Tris gel 1.0 mm, 15 well | ThermoFisher | NP0323BOX | |
| NuPAGE MES SDS buffer (20x) | ThermoFisher | NP0002 | |
| OGNG | Anatrace | NG311 | |
| PAGEr Minigel Chamber | Lonza | 59905 | |
| Reprosil 200 C18-AQ column | Morvay Analytik GmbH | #s1503 | |
| Superdex 200 Increase GL column | GE Healthcare | 28990944 | |
| Tabletop centrifuge 5424R | Eppendorf | 5404000413 | |
| Ultracentrifuge Optima XE-100 | Beckmann Coulter | A94516 | |
| ULTRA-TURRAX T25 | IKA WERKE | 0003725003 | |
| UV-VIS spectrophotometer | Shimadzu | UV-2401PC | |
| Waters 2487 Dual λ Absorbance Detector | Waters AG | - |
Références
- Tate, C. G. Practical considerations of membrane protein instability during purification and crystallisation. Methods in Molecular Biology. 601, 187-203 (2010).
- Lebon, G., Bennett, K., Jazayeri, A., Tate, C. G. Thermostabilisation of an agonist-bound conformation of the human adenosine A(2A) receptor. Journal of Molecular Biology. 409 (3), 298-310 (2011).
- Deupi, X., et al. Stabilized G protein binding site in the structure of constitutively active metarhodopsin-II. Proceedings of the National Academy of Sciences. 109 (1), 119-124 (2012).
- Tsai, C. -. J., et al. Crystal structure of rhodopsin in complex with a mini-G o sheds light on the principles of G protein selectivity. Science Advances. 4 (9), (2018).
- Carpenter, B., Tate, C. G. Engineering a minimal G protein to facilitate crystallisation of G protein-coupled receptors in their active conformation. Protein Engineering Design and Selection. 29 (12), 583-594 (2016).
- Chae, P. S., et al. Maltose-neopentyl glycol (MNG) amphiphiles for solubilization, stabilization and crystallization of membrane proteins. Nature Methods. 7 (12), 1003-1008 (2010).
- Loll, P. J. Membrane proteins, detergents and crystals: what is the state of the art. Acta Crystallographica Section F Structural Biology Communications. 70 (12), 1576-1583 (2014).
- Chae, P. S., et al. Glucose-neopentyl glycol (GNG) amphiphiles for membrane protein study. Chemical communications. 49 (23), 2287-2289 (2013).
- Standfuss, J., Xie, G., Edwards, P. C., Burghammer, M., Oprian, D. D., Schertler, G. F. X. Crystal structure of a thermally stable rhodopsin mutant. Journal of Molecular Biology. 372 (5), 1179-1188 (2007).
- Kaushal, S., Ridge, K. D., Khorana, H. G. Structure and function in rhodopsin: the role of asparagine-linked glycosylation. Proceedings of the National Academy of Sciences of the United States of America. 91 (9), 4024-4028 (1994).
- Molday, L. L., Molday, R. S. 1D4: a versatile epitope tag for the purification and characterization of expressed membrane and soluble proteins. Methods in Molecular Biology. 1177 (604), 1-15 (2014).
- Carpenter, B., Tate, C. G. Expression and Purification of Mini G Proteins from Escherichia coli. Bio-Protocol. 7 (8), (2017).
- Grueninger-Leitch, F., D'Arcy, A., D'Arcy, B., Chène, C. Deglycosylation of proteins for crystallization using recombinant fusion protein glycosidases. Protein Science. 5 (12), 2617-2622 (1996).
- Rasmussen, S. G. F., et al. Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature. 477 (7366), 549-555 (2011).
- Loginova, M. Y., Rostovtseva, Y. V., Feldman, T. B., Ostrovsky, M. A. Light damaging action of all-trans-retinal and its derivatives on rhodopsin molecules in the photoreceptor membrane. Biochemistry (Moscow). 73 (2), 130-138 (2008).
- Alexandrov, A. I., Mileni, M., Chien, E. Y. T., Hanson, M. A., Stevens, R. C. Microscale Fluorescent Thermal Stability Assay for Membrane Proteins. Structure. 16 (3), 351-359 (2008).
- Sonoda, Y., et al. Benchmarking Membrane Protein Detergent Stability for Improving Throughput of High-Resolution X-ray Structures. Structure. 19 (1), 17-25 (2011).
- Maeda, S., et al. Crystallization scale preparation of a stable GPCR signaling complex between constitutively active rhodopsin and G-protein. PloS One. 9 (6), 98714 (2014).
- Boute, N., Jockers, R., Issad, T. The use of resonance energy transfer in high-throughput screening: BRET versus FRET. Trends in Pharmacological Sciences. 23 (8), 351-354 (2002).
- Singhal, A., Guo, Y., Matkovic, M., Schertler, G., Deupi, X., Yan, E. C. Y. Structural role of the T 94 I rhodopsin mutation in congenital stationary night blindness. EMBO Report. 17 (10), 1-10 (2016).
- Choe, H. -. W., et al. Crystal structure of metarhodopsin II. Nature. 471 (7340), 651-655 (2011).
- Mattle, D., et al. Ligand channel in pharmacologically stabilized rhodopsin. Proceedings of the National Academy of Sciences of the United States of America. 115 (14), 3640-3645 (2018).
- Okada, T., Fujiyoshi, Y., Silow, M., Navarro, J., Landau, E. M., Shichida, Y. Functional role of internal water molecules in rhodopsin revealed by X-ray crystallography. Proceedings of the National Academy of Sciences of the United States of America. 99 (9), 5982-5987 (2002).
- Blankenship, E., Vahedi-Faridi, A., Lodowski, D. T. The High-Resolution Structure of Activated Opsin Reveals a Conserved Solvent Network in the Transmembrane Region Essential for Activation. Structure. 23 (12), 2358-2364 (2015).
- Magnani, F., et al. A mutagenesis and screening strategy to generate optimally thermostabilized membrane proteins for structural studies. Nature Protocols. 11 (8), 1554-1571 (2016).
- Kawate, T., Gouaux, E. Fluorescence-detection size-exclusion chromatography for precrystallization screening of integral membrane proteins. Structure. 14 (4), 673-681 (2006).
- Standfuss, J., et al. The structural basis of agonist-induced activation in constitutively active rhodopsin. Nature. 471 (7340), 656-660 (2011).
- Singhal, A., et al. Insights into congenital stationary night blindness based on the structure of G90D rhodopsin. EMBO reports. 14 (6), 520-526 (2013).
- Carpenter, B., Nehmé, R., Warne, T., Leslie, A. G. W., Tate, C. G. Structure of the adenosine A(2A) receptor bound to an engineered G protein. Nature. 536 (7614), 104-107 (2016).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.