
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Imagerie de réflectance interférométrique à particule unique, caractérisation des vésicules extracellulaires individuelles et dynamique des populations
Dans cet article
Résumé
Ce protocole présente une imagerie de réflectance interférométrique à particule unique conçue pour les mesures complètes et à plusieurs niveaux de la taille des vésicules extracellulaires (VE), du nombre de VE, du phénotype de la VE, et de la colocalisation des biomarqueurs de VE.
Résumé
Les vésicules extracellulaires (VE) sont des vésicules de taille nanométrique avec une bicouche lipidique qui sont sécrétées par la plupart des cellules. Les VE transportent une multitude de molécules biologiques différentes, notamment des protéines, des lipides, de l’ADN et de l’ARN, et sont censés faciliter la communication de cellule à cellule dans divers tissus et organes. Récemment, les VE ont attiré beaucoup d’attention en tant que biomarqueurs pour le diagnostic et agents thérapeutiques pour diverses maladies. De nombreuses méthodes ont été développées pour la caractérisation des VE. Cependant, les méthodes actuelles d’analyse des VE ont toutes des limites différentes. Ainsi, le développement de méthodes efficaces et efficientes pour l’isolation et la caractérisation des VE reste l’une des étapes cruciales pour ce domaine de recherche de pointe à mesure qu’il mûrit. Nous présentons ici un protocole détaillé décrivant un capteur d’imagerie interférométrique par réflectance à particule unique (SP-IRIS), en tant que méthode capable de détecter et de caractériser les VE provenant de sources biologiques non purifiées et les VE purifiés par d’autres méthodologies. Cette technique avancée peut être utilisée pour des mesures multi-niveaux et complètes pour l’analyse de la taille des VE, du nombre de VE, du phénotype des VE et de la colocalisation des biomarqueurs.
Introduction
Les vésicules extracellulaires (VE) sont des vésicules membranaires d’origine cellulaire de taille nanométrique qui peuvent être isolées de nombreux fluides biologiques, notamment le sang, le lait maternel, la salive, l’urine, la bile, le suc pancréatique et les fluides céphalo-rachidien et péritonéal. La dérivation des VE se produit par trois mécanismes principaux : l’apoptose, la libération par fusion de corps multivésiculaires avec la membrane plasmique et le gonflement de la membrane plasmique1. Les preuves du transfert par EV de composants cellulaires du donneur vers des cellules et des tissus voisins ou éloignés suggèrent que ces paquets enfermés dans la membrane peuvent jouer un rôle important dans les cascades de signalisation paracrine ainsi que de longue distance ou endocrinienne 1,2,3. Parce que les VE peuvent fournir un instantané du phénotype d’une cellule, le potentiel de leur utilisation comme outils diagnostiques et thérapeutiques pour le traitement de diverses maladies est devenu un domaine de recherche actif 4,5,6,7,8.
De nombreuses méthodes visant à la caractérisation des VE ont été développées 9,10,11,12,13. La plupart de ces méthodes fournissent des informations uniques et précieuses sur les populations de VE, principalement en vrac. Bien qu’un sous-ensemble de ces techniques puisse fournir des détails sur les substances contenues dans ou sur des VE uniques, il peut y avoir des limites à la caractérisation des VE au niveau d’un seul VE. Par exemple, l’immuno-microscopie électronique peut être utilisée pour comprendre les VE uniques et leur composition, mais cette technique est à faible débit, très limitée dans sa capacité à être utilisée pour décrire la dynamique des populations et nécessite le développement de méthodes importantes14.
Récemment, le développement et la commercialisation de la technique du capteur d’imagerie de réflectance interférométrique à particule unique (SP-IRIS), via la plateforme ExoView, ont ouvert la caractérisation individuelle des VE à l’aide d’une méthode de collecte de données automatisée simple et routinière. Le cœur de cette technologie est la puce, une double couche Si/SiO2 de 1 cm x 1 cm, qui permet la mesure interférométrique de nanoparticules biologiques uniques. La puce est dotée d’un microréseau de points d’anticorps fonctionnalisés individuels, ce qui permet de détecter multiplexement jusqu’à six types de capture différents. Les puces standard incluent les marqueurs tétraspanines courants (CD81, CD63 et CD9) pour la capture pendant l’étape d’incubation, et l’utilisateur peut ajouter des points de capture personnalisés supplémentaires pour isoler des populations distinctes de VE distinctes des tétraspanines. Après l’étape d’incubation, chaque point de capture a lié à lui de nombreux EV qui expriment le marqueur correspondant. Ces EV capturés peuvent ensuite être simplement lavés, séchés et scannés dans le lecteur pour quantifier la taille des vésicules liées au point de capture entre 50 et 200 nm afin d’obtenir une distribution de taille pondérée en nombre via SP-IRIS15. Le système offre également trois canaux de détection fluorescente pour l’immunomarquage des VE capturés, et fournit à la fois l’intensité fluorescente moyenne, qui n’est pas limitée par la taille telle que les mesures SP-IRIS, et les aspects de colocalisation pour chaque coloration fluorescente. Cela permet à l’utilisateur de définir des populations de VE uniques sur la base de l’affichage de quatre biomarqueurs différents par VE (capture plus trois marqueurs immunofluorescents). Le système peut aller au-delà de la mesure des protéines de surface avec l’immunofluorescence, car un protocole de fret facultatif permet à l’utilisateur de sonder les protéines intérieures des VE capturés et les épitopes luminaux des marqueurs de surface couvrant la membrane, ainsi que de vérifier l’intégrité de la membrane EV. Dans cet article, nous fournissons un protocole détaillé décrivant les étapes nécessaires pour obtenir des données cohérentes concernant la taille et le nombre de VE, avec jusqu’à quatre biomarqueurs différents à un seul niveau de VE sur de grandes populations de VE. Cette technique peut être utilisée à la fois sur des fluides biologiques non traités et sur des VE isolés à l’aide d’un certain nombre de techniques, telles que l’ultracentrifugation, l’ultrafiltration, les agents précipitants, la capture d’immunoaffinité, la microfluidique et la chromatographie d’exclusion stérique.
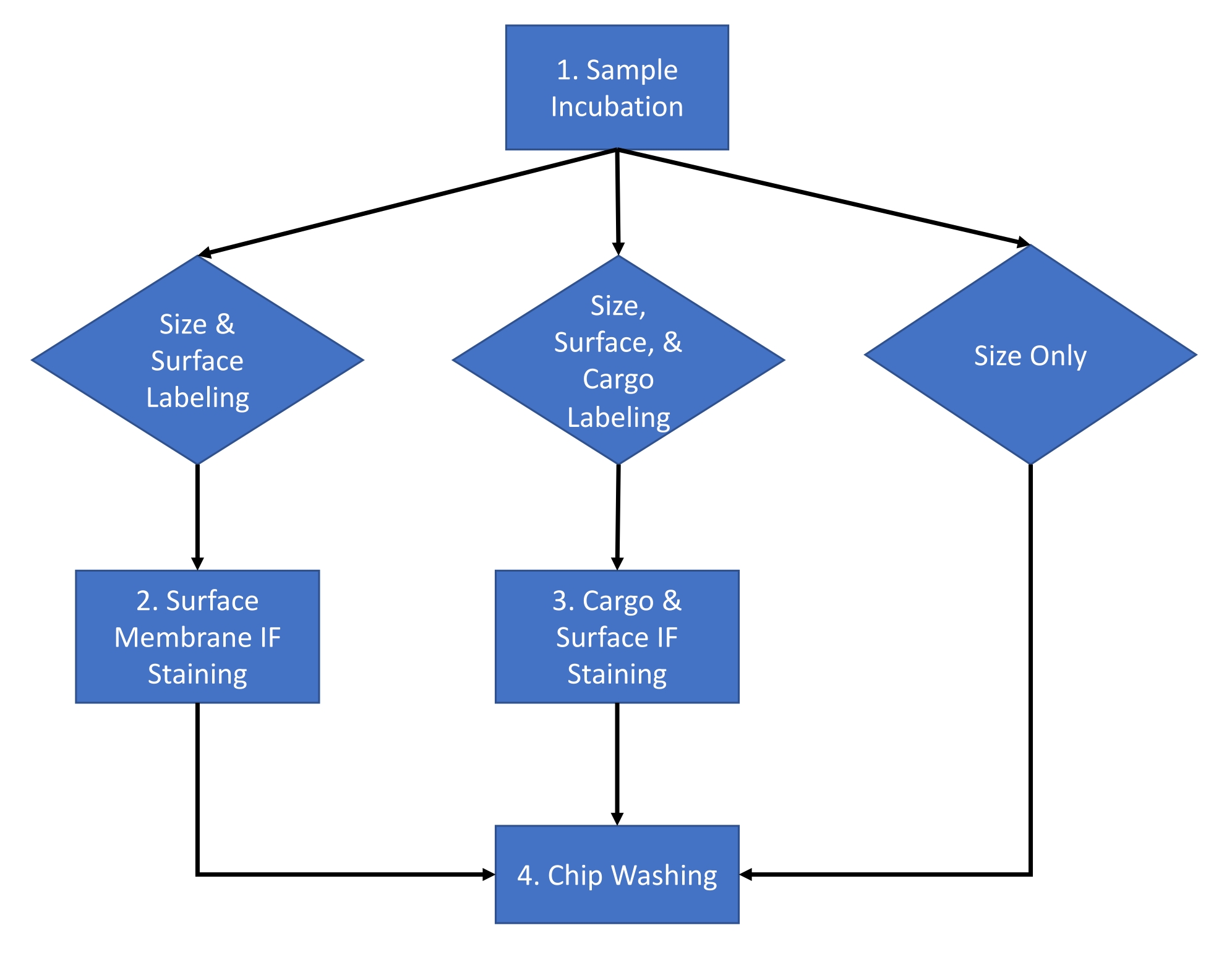
Le protocole décrit ci-dessous utilise des vésicules extracellulaires (VE) dérivées de milieux de culture cellulaire HEK 293 et du sérum de souris en utilisant une méthode d’isolement établie16. Le protocole a été appliqué à de nombreux autres fluides biologiques, milieux de culture cellulaire et vésicules extracellulaires purifiées isolées de fluides biologiques. Ce protocole est divisé en une procédure de deux jours avec le flux de travail d’une expérience typique illustrée à la figure 1.

Figure 1 : Flux de travail du dosage. Flux de travail d’analyse pour choisir le type d’analyse à effectuer pour l’échantillon entre la taille et le nombre, le nombre de tailles et la coloration de surface, et le nombre de tailles et la coloration de la cargaison. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
Access restricted. Please log in or start a trial to view this content.
Protocole
Des échantillons de sérum ont été prélevés sur des souris selon le protocole approuvé par l’Institutional Animal Care and Use Committee (IACUC) du centre médical de l’Université du Kansas (KUMC). L’utilisation de ces échantillons biologiques dans ces expériences a également été approuvée par le KUMC.
1. Préparation de l’échantillon (jour 1)
- Déterminez la concentration de VE à l’aide du suivi des nanoparticules ou d’une technique équivalente.
- Diluer l’échantillon avec la solution d’incubation à une concentration de 5 x 107-5 x 108 VE/mL ; un minimum de 50 μL est nécessaire.
REMARQUE : Pour les échantillons dont la concentration en EV est inconnue, on peut utiliser des protéines totales à 1 μg/mL comme mesure de substitution. Si l’on s’attend à ce que la concentration de l’échantillon soit faible, effectuer une dilution d’au moins 1:1 dans la solution d’incubation avant le chargement. - Placez une plaque à 24 puits sur une surface plane, exempte de vibrations et de mouvements brusques.
REMARQUE : Pour améliorer le contraste, une feuille de papier blanche peut être placée sous la plaque. - Ajoutez de l’eau aux abords des puits (figure 2).

Figure 2 : Disposition des plaques à 24 puits. L’emplacement de l’aliquote ddH2O (le colorant bleu a été ajouté à des fins de visualisation uniquement) et les puits dans lesquels les copeaux seront maintenus sont indiqués. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
2. Préparation et prénumérisation des puces
- Retirez la plaque scellée à 48 puits contenant les copeaux du réfrigérateur à 4-8 °C et laissez-les atteindre la température ambiante (~15 min) avant d’ouvrir le sceau.
REMARQUE : Ceci est essentiel pour éviter la condensation sur les copeaux qui peut endommager les taches. Si de la condensation est observée sur la surface lors du retrait de la puce du bloc, attendez plus longtemps avant d’en retirer d’autres. - Passez à l’étape 8 pour récupérer le mandrin afin de préparer le cycle de pré-balayage (Figure 3).
REMARQUE : Les données de pré-balayage seront utilisées pour identifier toutes les particules détectables sur les points de capture avant l’incubation avec l’échantillon afin qu’elles puissent être éliminées pendant l’étape d’analyse.

Figure 3 : Image du mandrin utilisé pour charger la puce dans la machine. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
- À l’aide d’une pince à épiler, retirez le nombre souhaité de copeaux (un copeau par échantillon) de la plaque à 48 puits et chargez les copeaux directement dans le mandrin pour le cycle de prébalayage. Lorsque toutes les puces destinées à être utilisées pour l’expérience ont été chargées et prébalayées comme décrit à l’étape 8, passez à l’étape 2.4.
REMARQUE : lorsque vous manipulez des puces, assurez-vous de ne pas toucher les carrés au centre, car les points de capture d’anticorps se trouvent dans cette région et seront endommagés s’ils sont touchés par la pince à épiler (Figure 4).

Figure 4 : Puce et manipulation correcte de la puce. (A) La ligne pointillée jaune indique l’emplacement des anticorps tachetés ou le côté fonctionnel de la puce. L’identifiant de la puce se trouve sous la ligne (« 58 »). La figure montre également une manipulation correcte. (B) Démontre une manipulation inappropriée de la puce. (C) Côté non fonctionnel de la puce. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
- Placez chaque puce pré-numérisée avec la surface fonctionnalisée face vers le haut dans la plaque à 24 puits pré-préparée. La surface fonctionnalisée est facilement reconnaissable à la numérotation distincte, aux grilles d’alignement et aux trois boîtes noires au milieu. Le côté non fonctionnalisé est une surface plate et uniforme en argent.
- À l’aide d’une pince à épiler à pointe fine, assurez-vous que chaque copeau est centré dans le puits (figure 5).
REMARQUE : Le centrage de la puce dans le puits est essentiel car si la puce touche la paroi du puits, l’échantillon peut être évacué après le chargement.

Figure 5 : Démonstration du bon placement des copeaux dans le puits. (A) Les copeaux doivent être placés au milieu du puits, sans qu’aucun coin ne touche les côtés du puits. (B) Représentation d’un mauvais placement de la puce, où les coins touchent les côtés du puits. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
3. Chargement et incubation de la puce et de l’échantillon
- Pipeter 35 μL de l’échantillon préparé (à partir de l’étape 1) sur la puce.
REMARQUE : Veillez à ne pas ajouter de bulles ou à ne pas toucher la puce avec la pointe de la pipette, car cela peut empêcher la répartition uniforme de l’échantillon sur la puce ou endommager les taches d’anticorps sur la puce. L’échantillon doit s’étendre sur toute la surface de la puce. Dans le cas où l’échantillon s’échappe de la puce pendant le chargement, ajoutez un échantillon supplémentaire dans le puits jusqu’à un volume de 250 μL ou ajoutez une solution d’incubation jusqu’à 250 μL et notez le facteur de dilution modifié pour cet échantillon. - Scellez la plaque à l’aide de la pellicule incluse dans le kit pour éviter l’évaporation de l’échantillon.
- Incuber l’échantillon/la puce pendant une nuit (~16 h) dans la plaque scellée à température ambiante dans un endroit exempt de vibrations ou de mouvements.
REMARQUE : Après l’incubation d’une nuit, l’utilisateur choisira l’option du jour 2 appropriée pour sa question de recherche. Si seules la taille et le nombre de véhicules électriques sont souhaités, passez à l’étape 4 (jour 2). Si vous souhaitez effectuer une analyse de plusieurs marqueurs de surface, passez à l’étape 5 (jour 2).
4. Déterminer la taille et le nombre de véhicules électriques (jour 2)
- Ajouter 1 mL de solution A sur le côté de chaque puits contenant une puce en prenant soin de ne pas ajouter directement la solution sur la puce ou de rayer la puce avec la pointe de la pipette.
- Placez la plaque sur un agitateur orbital tournant à ~500 tr/min pendant 3 min à température ambiante.
REMARQUE : Si les copeaux cliquettent sur la plaque, diminuez immédiatement la vitesse de sorte que le liquide tourbillonne mais qu’il n’y ait pas de cliquetis. - Retirer 750 μL de liquide. Évitez d’incliner la plaque pendant l’évacuation du liquide pour éviter le séchage accidentel de la puce.
- Ajouter 750 μL de la solution B en utilisant la technique décrite à l’étape 4.1 et agiter à ~500 tr/min pendant 3 min à température ambiante.
- Répétez les étapes 4.3 à 4.4 deux fois de plus pour un total de trois lavages avec la solution B.
- À la fin du dernier lavage, retirer 750 μL de la solution, en laissant 250 μL de solution B dans le puits.
- Ajouter 750 μL d’eau double distillée (ddH2O) et agiter à ~500 tr/min pendant 3 min à température ambiante.
- Remplissez une boîte de Pétri de 10 cm avec 50 ml de ddH2O et transférez une puce à la fois du puits dans la boîte à l’aide d’une pince à épiler.
REMARQUE : Prenez soin de transférer la puce horizontalement et assurez-vous qu’elle ne sèche pas. Jusqu’à huit copeaux peuvent être lavés avant de remplacer le ddH2O. - Dans le ddH2O, tenez la puce par les bords à l’aide d’une pince à épiler et faites tourner dans le plat pendant trois tours pour éliminer les débris.
- Retirez la puce à 45° hors de l’eau et placez-la sur du papier absorbant avec l’ID de la puce vers le haut (Figure 6).
REMARQUE : Les puces sont maintenant prêtes à être lues. Passez à l’étape 8 pour la numérisation sur le lecteur SP-IRIS.

Figure 6 : Méthode correcte pour retirer l’éclat de l’eau ddH2O à un angle de 45°. (A) Vue de dessus et (B) vue de côté montrant l’angle auquel retirer l’éclat. Veuillez cliquer ici pour voir une version agrandie de cette figure.
{kind=link}
5. Préparation de la solution d’anticorps (jour 2)
- Ajouter 300 μL de solution de blocage par puce dans un tube de taille appropriée (0,5 à 3 mL).
- Ajouter 0,6 μL d’anticorps par 300 μL de solution bloquante. Mélangez doucement en tapotant le tube et en essorant rapidement.
REMARQUE : Les anticorps contre les CD9, CD81 et CD63 humains ou les CD9, CD81 et CD63 murins sont inclus dans les kits appropriés. Si d’autres anticorps conjugués par fluorescence sont souhaités, déterminez la concentration optimale de coloration à l’aide d’un titrage typique sur une plage de 0,1 à 10 μg/mL avec une charge d’échantillon constante comme vous le feriez pour la cytométrie en flux.
6. Détermination de la taille, du nombre et du phénotypage des VE par coloration immunofluorescente
- Ajouter 1 mL de solution A sur le côté de chaque puits contenant une puce, en prenant soin de ne pas ajouter directement la solution sur la puce ou de rayer la puce avec la pointe de la pipette.
- Placez la plaque sur un agitateur orbital tournant à ~500 tr/min pendant 3 min à température ambiante.
REMARQUE : Si les copeaux cliquettent sur la plaque, diminuez immédiatement la vitesse de sorte que le liquide tourbillonne mais qu’il n’y ait pas de cliquetis. - Retirer 750 μL de liquide. Évitez d’incliner la plaque pendant l’évacuation du liquide pour éviter le séchage accidentel de la puce.
- Ajouter 750 μL de la solution A en utilisant la technique décrite à l’étape 6.1 et agiter à ~500 tr/min pendant 3 minutes à température ambiante.
- Répétez les étapes 6.3 et 6.4 deux fois pour un total de trois lavages avec la solution A.
- À la fin du dernier lavage, retirer 750 μL de la solution, en laissant 250 μL de solution A dans le puits.
REMARQUE : Si l’utilisateur souhaite une coloration de cargaison, passez à l’étape 7 à ce stade. Si ce n’est pas le cas, passez à l’étape 6.7 - Ajouter 250 μL de la solution d’anticorps (étape 5) dans chaque puce dans le puits. Couvrez la plaque d’une feuille d’aluminium pour la protéger de la lumière et agitez pendant 1 h sur l’agitateur orbital à température ambiante.
- Ajoutez 500 μL de solution A, de sorte que le volume total par puits soit de ~1 000 μL.
- Retirer immédiatement 750 μL et ajouter 750 μL de solution A et agiter à ~500 tr/min sur l’agitateur orbital pendant 3 min à température ambiante.
- Retirez 750 μL de la solution et ajoutez 750 μL de la solution B et agitez à ~500 tr/min pendant 3 minutes à température ambiante.
- Répétez l’étape 6.10 deux fois de plus pour un total de trois lavages.
- Ajouter 750 μL d’eau double distillée (ddH2O) et agiter à ~500 tr/min pendant 3 min à température ambiante.
- Remplissez une boîte de Pétri de 10 cm avec 50 ml de ddH2O et transférez une puce du puits dans la boîte à l’aide d’une pince à épiler.
REMARQUE : Prenez soin de transférer la puce horizontalement et assurez-vous qu’elle ne sèche pas. Jusqu’à huit copeaux peuvent être lavés avant d’être remplacés par du ddH2O frais. - Dans le ddH2O, tenez la puce par les bords à l’aide d’une pince à épiler et faites tourner dans le plat pendant trois tours pour éliminer les débris.
- Retirez la puce à 45° de l’eau et placez-la sur du papier absorbant avec l’ID de la puce vers le haut (Figure 6).
REMARQUE : Les puces sont maintenant prêtes à être lues. Passez à l’étape 8 (Collecte de données) pour obtenir des instructions sur la configuration des puces pour la numérisation sur le lecteur.
7. Coloration de la cargaison en option
REMARQUE : Ce protocole permet l’étiquetage simultané des marqueurs internes et de surface.
- Avec 250 μL de solution A restant dans chaque puits, ajoutez 250 μL de solution C dans chaque puits.
- Placez immédiatement sur l’agitateur orbital et réglez à ~200 tr/min pendant précisément 10 min.
REMARQUE : Dans cette étape et l’étape 7.8 ci-dessous, le timing et la vitesse d’agitation plus lente sont critiques ; Assurez-vous que la vitesse ne donne qu’un tourbillon lent dans les puits et utilisez une minuterie. - Après 10 min d’incubation avec la solution C, ajouter 500 μL de solution A dans chaque puits.
- Retirer immédiatement 750 μL et ajouter 750 μL de la solution A et agiter à ~500 tr/min sur l’agitateur orbital pendant 3 min à température ambiante.
- Répétez l’étape 7.4 deux fois de plus pour un total de trois lavages.
- À la fin du dernier lavage, retirez 750 μL de la solution, en laissant ~250 μL de la solution dans le puits avec la puce.
- Ajouter 250 μL de solution D dans chaque puits.
- Placez immédiatement sur l’agitateur orbital réglé à ~200 tr/min pendant précisément 10 min.
- Après 10 min d’incubation avec la solution C, ajouter 500 μL de solution A dans chaque puits.
- Retirer immédiatement 750 μL et ajouter 750 μL de solution A et agiter à ~500 tr/min sur l’agitateur orbital pendant 3 min à température ambiante.
- Répétez l’étape 7.10 deux fois pour un total de trois lavages.
- Retirer 750 μL de la solution après le dernier lavage et revenir à l’étape 6.7 ci-dessus pour la coloration et l’achèvement du protocole de dosage.
8. Collecte des données
REMARQUE : La procédure de collecte de données à partir des puces à l’aide de l’ExoView R100 est automatisée et ne nécessite aucune intervention de l’utilisateur. Vous trouverez des instructions détaillées dans le Guide de l’utilisateur et la vidéo correspondante pour le chargement du support de puce, ou « mandrin », et l’acquisition de données17.
- Allumez la plate-forme de caractérisation EV à l’aide des deux interrupteurs d’alimentation situés à l’arrière de l’instrument, puis démarrez le logiciel du scanner en double-cliquant sur l’icône du bureau.
REMARQUE : Lorsque le logiciel du scanner démarre, le lecteur se règle automatiquement sur l’écran d’accueil, puis invite l’utilisateur à « Ouvrir la porte pour charger les puces ». - Ouvrez la porte à l’avant du lecteur en soulevant la poignée argentée. Cela éjectera la platine, permettant à l’utilisateur d’accéder au mandrin et de configurer un nouveau balayage dans le logiciel.
- Identifiez l’emplacement où l’utilisateur souhaite enregistrer les données en cliquant sur le dossier Enregistrer et en sélectionnant l’emplacement souhaité pour les données à enregistrer.
REMARQUE : Il est souvent utile de nommer l’emplacement de sauvegarde de manière plus perspicace, comme le nom de l’expérience, puis de créer deux sous-dossiers, l’un pour les pré-analyses et l’autre pour les post-analyses. Cela facilite la recherche et la correspondance des données lors de l’analyse. - Pour analyser les puces, localisez les fichiers de puces sur l’ordinateur de contrôle.
REMARQUE : Les fichiers de puce sont des cartes de la disposition des taches d’anticorps sur la puce utilisée et permettent au scanner de savoir où scanner doit scanner sur chaque puce. Dans chaque kit, il y a une clé USB avec les chipfiles pour les puces à l’intérieur. Ceux-ci doivent être enregistrés à un emplacement sur l’ordinateur de contrôle dès réception du kit où les utilisateurs peuvent les trouver de manière fiable. - Chargez les copeaux sur le mandrin avec le numéro sur les copeaux face à la poignée du mandrin, puis dans le menu déroulant Puce de l’ordinateur, sélectionnez chaque copeau dans la liste des fichiers de puces et placez-le à l’emplacement correspondant approprié dans le mandrin virtuel dans le logiciel du scanner. Une fois terminé, une invite à l’écran demandant à l’utilisateur de placer le mandrin sur scène apparaîtra.
- Placez le mandrin chargé sur la scène ; l’alignement magnétique sur le mandrin le déplacera automatiquement au bon endroit sur la scène, puis cliquez sur OK à côté de Placer le mandrin sur la scène. Le scanner commencera alors la routine de collecte automatisée des données.
REMARQUE : Les données sont prêtes à être analysées une fois que le logiciel signale l’état de balayage de chaque puce comme réussi.
9. Analyse des données
- Double-cliquez sur ExoView Analyzer sur le bureau du PC de contrôle. Après le démarrage du logiciel, cliquez sur le bouton Données de pré-balayage et sélectionnez l’emplacement du dossier pour l’ensemble de données de pré-balayage dans la section 8.3.
- Cliquez sur le bouton Données Postscan et sélectionnez l’emplacement du dossier pour l’ensemble de données Postscan dans la section 8.3.
REMARQUE : Si les données sont correctement détectées dans les deux dossiers, pour au moins certaines des puces, le logiciel affichera le nombre « X » de puces détectées à côté du bouton d’emplacement du fichier Chipfile. Lorsqu’elles sont correctement enregistrées, les puces détectées doivent correspondre au nombre de puces analysées. - Cliquez sur Suivant en bas de la zone de chargement des données.
- Dans le menu déroulant de la table méta, chaque puce aura une cellule. Entrez les noms des échantillons, les facteurs de dilution et les marqueurs colorés dans chaque canal de détection en cliquant dans la case correspondante et en tapant les informations ; une fois terminé, cliquez à nouveau sur Suivant .
REMARQUE : Ces métadonnées seront enregistrées avec les données. - Préformez le contrôle de la qualité (CQ) en cliquant sur l’onglet Désactiver sous le bouton CQ en haut à gauche.
REMARQUE : Cette fonctionnalité permet de désactiver des sondes et des puces spécifiques pour l’analyse. Le logiciel fournira deux avertissements différents concernant les données, l’un pour les comptages élevés qui peuvent indiquer qu’une capture particulière est saturée, et l’autre pour un coefficient de variation (CV) élevé qui identifie quand l’une des réplications d’un type de capture est différente des autres.- Cliquez sur un point d’avertissement pour un CV élevé, examinez les points qui sont signalés par le logiciel et voyez s’il y a des dommages physiques évidents ; lorsque les dommages sont identifiés, cliquez sur le numéro de point pour désactiver ce point dans l’analyse.
- Répétez l’opération jusqu’à ce que tous les avertissements aient été évalués, puis cliquez sur Suivant.
- Effectuez l’analyse de coupure en cliquant sur l’onglet Coupure situé à côté de l’onglet Désactiver sous le bouton QC en haut à gauche.
REMARQUE : Cela présentera la réponse fluorescente du point de contrôle dans un tableau et offrira à l’utilisateur deux paramètres, un minimum et un maximum pour chaque canal de couleur (rouge/vert/bleu). Le réglage de la valeur de coupure pour chaque canal de détection fluorescente est le seul réglage des données qui doit être effectué et est relativement simple. Il est important de noter que la coupure finale choisie doit être cohérente dans l’ensemble d’une expérience et, comme indiqué, doit respecter les règles empiriques pour 300-400 UA en rouge et vert, et 400-700 UA pour le bleu dans les expériences typiques ; Le logiciel dispose de guides à code couleur pour l’utilisateur pour ces gammes également.- Cliquez sur l’onglet Couleur verte pour charger les données de ce canal et afficher les coupures actuelles. En examinant le point de contrôle négatif de l’isotype, on peut voir des événements de détection avec de très faibles intensités de fluorescence ; Dans ce cas, les utilisateurs doivent généralement s’attendre à mettre le vert entre 300 et 400 A.U.
- Augmentez le minimum pour chacun des canaux de détection jusqu’à ce que le « Moy % Inc » (le % moyen des particules détectées sur le point de contrôle inclus au-dessus de la limite) sous l’affichage des données n’ait pas d’avertissement, comme indiqué par une mise en surbrillance rouge ou jaune dans cette cellule.
REMARQUE : La valeur maximale n’a généralement pas besoin d’être ajustée par rapport à la valeur par défaut, mais peut être abaissée pour filtrer les particules brillantes ou limiter la détection fluorescente à une plage plus étroite. - Cliquez sur le bouton Suivant .
- Augmentez le minimum pour chacun des canaux de détection jusqu’à ce que le « Moy % Inc » (le % moyen des particules détectées sur le point de contrôle inclus au-dessus de la limite) sous l’affichage des données n’ait pas d’avertissement, comme indiqué par une mise en surbrillance rouge ou jaune dans cette cellule.
- Cliquez sur l’onglet Couleur rouge pour charger les données de ce canal et afficher les coupures actuelles. Encore une fois, en examinant la tache de contrôle négatif de l’isotype, on peut voir des événements de détection avec de très faibles intensités de fluorescence ; Dans ce cas, les utilisateurs doivent généralement s’attendre à régler le rouge entre 300 et 400 UA. Suivez les mêmes procédures que celles indiquées aux étapes 9.6.1.1 à 9.6.1.3.
- Cliquez sur l’onglet Couleur bleue pour charger les données de ce canal et afficher les coupures actuelles. Encore une fois, en examinant le point de contrôle négatif de l’isotype, suivez les mêmes procédures que celles indiquées aux étapes 9.6.1.1 à 9.6.1.3.
REMARQUE : Le canal bleu est unique en ce sens que l’anticorps dans le spot est auto-fluorescent à des niveaux suffisants pour nécessiter une coupure légèrement plus élevée de 400-700 u.a., même pour les puces à blanc.
- Cliquez sur l’onglet Couleur verte pour charger les données de ce canal et afficher les coupures actuelles. En examinant le point de contrôle négatif de l’isotype, on peut voir des événements de détection avec de très faibles intensités de fluorescence ; Dans ce cas, les utilisateurs doivent généralement s’attendre à mettre le vert entre 300 et 400 A.U.
- Une fois l’analyse Cutoff terminée, cliquez sur Suivant et le tracé de la carte thermique apparaîtra, qui donnera un aperçu de la liaison des particules à travers l’expérience. Il existe plusieurs outils de visualisation que l’utilisateur peut utiliser pour tracer les données. Ces tracés et les données brutes associées peuvent être ajoutés à un rapport en cliquant sur le bouton Ajouter un rapport de tracé .
REMARQUE : La carte thermique est l’affichage des données par défaut qui affiche une vue de tous les échantillons, de toutes les captures d’une détection particulière. L’affichage par défaut est le total qui quantifie les VE uniques liés à chaque point de capture. - Cliquez sur le bouton Exporter le rapport et sélectionnez un emplacement d’enregistrement pour le rapport.
REMARQUE : Le rapport s’ouvre dans un navigateur. Trouvez le fichier de tableur de la liste de particules filtrées joint au rapport. il s’agit des informations à soumettre à EVTRACK. - Continuez à ajouter des rapports à l’aide des différentes sélections d’échantillons et de types de capture dans les menus déroulants en haut de l’écran. Les données affichées sont contrôlées par la sélection des boutons de détection de canal en haut à gauche. Définissez un phénotype/sélection d’échantillon/canal de capture particulier en cliquant sur chaque bouton de couleur pour désactiver ce canal de détection (grisé) ou l’activer (coloré).
- Cliquez sur le bouton Ajouter un tracé au rapport pour agréger les tracés souhaités dans un rapport.
- Cliquez sur Exporter le rapport et définissez l’emplacement d’enregistrement du rapport final.
Access restricted. Please log in or start a trial to view this content.
Résultats
La figure 7 (panneau de gauche) montre une image composite tricolore de VE dérivées de supports conditionnés HEK293 liés au point CD63 sur la puce et colorés pour CD81, CD63 et CD9 dans les canaux suivants vert, rouge et bleu, respectivement. La figure 7 (panneau en haut à droite) est une image agrandie qui montre que chacun des véhicules électriques capturés peut afficher la co-localisation d’une ou plusieurs couleur...
Access restricted. Please log in or start a trial to view this content.
Discussion
Les méthodes actuelles de caractérisation des VE reposent en grande partie sur des VE purifiés, ce qui est limité par les limites expérimentales actuelles des méthodes de purification des VE 9,10,11,12,13. L’imagerie par réflectance interférométrique à particules uniques (SP-IRIS) est une technologie efficace qui p...
Access restricted. Please log in or start a trial to view this content.
Déclarations de divulgation
Clayton Deighan et George Daaboul sont des employés et des actionnaires de NanoView Biosciences Inc.
Remerciements
Ce travail a été parrainé en partie par le programme de prix d’approvisionnement en équipement et en ressources de recherche de la faculté de médecine de l’Université du Kansas. PCG, LKC, FD et AR ont été soutenus par des fonds de la NIA R21 AG066488-01.
Access restricted. Please log in or start a trial to view this content.
matériels
| Name | Company | Catalog Number | Comments |
| 10-cm sterile Petri dish | Fisher | FB0875712 | |
| 15mL sterile tube | n/a | various | |
| 24-well cell culture plate, flat bottom | Fisher | 08-772-1 | |
| Blocking Solution | NanoView Biosciences | EV-TETRA-C | Can be found in ExoView Human Tetraspanin Kit. |
| Chipfiles | NanoView Biosciences | EV-TETRA-C | Can be found in ExoView Human Tetraspanin Kit. |
| Chips | NanoView Biosciences | EV-TETRA-C | Can be found in ExoView Human Tetraspanin Kit. |
| Chuck | NanoView Biosciences | EV-TETRA-C | Can be found in ExoView Human Tetraspanin Kit. |
| Corning Easy Grip Disposable Polystyrene Sterile Bottles 250 ml | Fisher | 09-761-4 | |
| Corning Easy Grip Disposable Polystyrene Sterile Bottles 500 ml | Fisher | 09-761-10 | |
| Deionized (DI) water | Fisher | LC267404 | |
| EMS style tweezers with Carbon Fiber tips | Fisher | 50-193-0842 | |
| ExoView Human Tetraspanin Kit | NanoView Biosciences | EV-TETRA-C | Capture for hCD81, hCD9, hCD63, IgG Control + stains for hEV-A (hEV-CD63-647, hEV-CD81-555, hEV-CD9-488) 16 Chips per kit |
| ExoView R100 Imager | NanoView Biosciences | EV-R100 | Interferometric microscope including high specification camera including 3 color fluorescence and label free sizing and counting extracellular vesicles |
| Fluorescently labled huma CD9 IgG antibody | NanoView Biosciences | EV-TETRA-C | Can be found in ExoView Human Tetraspanin Kit. |
| Fluorescently labled human CD63 IgG antibody | NanoView Biosciences | EV-TETRA-C | Can be found in ExoView Human Tetraspanin Kit. |
| Fluorescently labled human CD81 IgG antibody | NanoView Biosciences | EV-TETRA-C | Can be found in ExoView Human Tetraspanin Kit. |
| Incubation Solution | NanoView Biosciences | EV-TETRA-C | Can be found in ExoView Human Tetraspanin Kit. |
| Orbital shaker or microplate shaker with digital settings capable of shaking at 500 rpm | n/a | various | |
| Plate Seal | NanoView Biosciences | EV-TETRA-C | Can be found in ExoView Human Tetraspanin Kit. |
| Solution A | NanoView Biosciences | EV-TETRA-C | Can be found in ExoView Human Tetraspanin Kit. |
| Solution B | NanoView Biosciences | EV-TETRA-C | Can be found in ExoView Human Tetraspanin Kit. |
| Solution C | NanoView Biosciences | EV-TETRA-C | Can be found in ExoView Human Tetraspanin Kit. |
| Solution D | NanoView Biosciences | EV-TETRA-C | Can be found in ExoView Human Tetraspanin Kit. |
| Square/flat tip tweezer | Fisher | 50-239-62 | |
| Straight strong point Boley style tweezers | Fisher | 16-100-124 | |
| Thermo Scientific Adhesive PCR Plate Seals | Fisher | AB-0558 |
Références
- Maas, S. L. N., Breakefield, X. O., Weaver, A. M. Extracellular vesicles: Unique intercellular delivery vehicles. Trends in Cell Biology. 27 (3), 172-188 (2017).
- Shah, R., Patel, T., Freedman, J. E. Circulating extracellular vesicles in human disease. The New England Journal of Medicine. 379 (10), 958-966 (2018).
- Deng, F., Miller, J. A review on protein markers of exosome from different bio-resources and the antibodies used for characterization. Journal of Histotechnology. 42 (4), 226-239 (2019).
- Cohen, O., et al. ’Golden’ exosomes as delivery vehicles to target tumors and overcome intratumoral barriers: in vivo tracking in a model for head and neck cancer. Biomaterials Science. 9 (6), 2103-2114 (2021).
- Han, Y., et al. Overview and update on methods for cargo loading into extracellular vesicles. Processes (Basel). 9 (2), 356(2021).
- Lee, M., Im, W., Kim, M. Exosomes as a potential messenger unit during heterochronic parabiosis for amelioration of Huntington's disease. Neurobiology of Disease. 155, 105374(2021).
- Sun, B., et al. Characterization and biomarker analyses of post-COVID-19 complications and neurological manifestations. Cells. 10 (2), 386(2021).
- Jiang, L., Gu, Y., Du, Y., Liu, J. Exosomes: Diagnostic biomarkers and therapeutic delivery vehicles for cancer. Molecular Pharmaceutics. 16 (8), 3333-3349 (2019).
- Bachurski, D., et al. Extracellular vesicle measurements with nanoparticle tracking analysis - An accuracy and repeatability comparison between NanoSight NS300 and ZetaView. Journal of Extracellular Vesicles. 8 (1), 1596016(2019).
- Carmicheal, J., et al. Label-free characterization of exosome via surface enhanced Raman spectroscopy for the early detection of pancreatic cancer. Nanomedicine. 16, 88-96 (2019).
- Greening, D. W., Xu, R., Ji, H., Tauro, B. J., Simpson, R. J. A protocol for exosome isolation and characterization: evaluation of ultracentrifugation, density-gradient separation, and immunoaffinity capture methods. Methods in Molecular Biology. 1295, 179-209 (2015).
- Wu, Y., Deng, W., Klinke, D. J. Exosomes: improved methods to characterize their morphology, RNA content, and surface protein biomarkers. The Analyst. 140 (19), 6631-6642 (2015).
- van de Vlekkert, D., Qiu, X., Annunziata, I., d'Azzo, A. Isolation and characterization of exosomes from skeletal muscle fibroblasts. Journal of Visualized Experiments: JoVE. (159), (2020).
- Ayala-Mar, S., Donoso-Quezada, J., Gallo-Villanueva, R. C., Perez-Gonzalez, V. H., Gonzalez-Valdez, J. Recent advances and challenges in the recovery and purification of cellular exosomes. Electrophoresis. 40 (23-24), 3036-3049 (2019).
- Daaboul, G. G., et al. Digital detection of exosomes by interferometric imaging. Scientific Reports. 6, 37246(2016).
- Pohler, K. G., et al. Circulating microRNA as candidates for early embryonic viability in cattle. NMolecular Reproduction and Development. 84 (8), 731-743 (2017).
- NanoView Biosciences. ExoView R100 User Guide. v240.4. NanoView Biosciences. , 202(2021).
- Daaboul, G. G., et al. Enhanced light microscopy visualization of virus particles from Zika virus to filamentous ebolaviruses. PLoS One. 12, 0179728(2017).
- ET Consortium. EV-TRACK: transparent reporting and centralizing knowledge in extracellular vesicle research. Nature Methods. 14, 228-232 (2017).
Access restricted. Please log in or start a trial to view this content.
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationExplorer plus d’articles
This article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.