Method Article
Sondare la cinetica dell'mRNA nello spazio e nel tempo in Escherichia coli utilizzando la fluorescenza a singola molecola a due colori in ibridazione situ
In questo articolo
Riepilogo
Questo protocollo descrive un'applicazione di fluorescenza a singola molecola in ibridazione situ (smFISH) per misurare la cinetica in vivo della sintesi e della degradazione dell'mRNA.
Abstract
La fluorescenza a singola molecola nell'ibridazione in situ (smFISH) consente di contare il numero assoluto di mRNA nelle singole cellule. Qui, descriviamo un'applicazione di smFISH per misurare i tassi di trascrizione e degradazione dell'mRNA in Escherichia coli. Poiché smFISH si basa su cellule fisse, eseguiamo smFISH in più punti di tempo durante un esperimento di corso nel tempo, cioè quando le cellule sono sottoposte a cambiamenti sincronizzati in caso di induzione o repressione dell'espressione genica. In ogni momento, le sotto-regioni di un mRNA sono spettralmente distinte per l'allungamento della trascrizione della sonda e la terminazione prematura. Il risultato di questo protocollo consente anche di analizzare la localizzazione intracellulare degli mRNA e l'eterogeneità nei numeri di copia dell'mRNA tra le cellule. Utilizzando questo protocollo è possibile elaborare molti campioni (50 USD) entro 8 h, come la quantità di tempo necessaria per pochi campioni. Discutiamo come applicare questo protocollo per studiare la trascrizione e la cinetica di degradazione di diversi mRNA nelle cellule batteriche.
Introduzione
Il flusso di informazioni genetiche dal DNA all'mRNA e alle proteine è uno dei processi cellulari più fondamentali, la cui regolazione è importante per la forma fisicacellulare 1. Il numero di mRNA in una cella è determinato da due processi dinamici, la trascrizione e la degradazione dell'mRNA. Tuttavia, il modo in cui la trascrizione e la degradazione dell'mRNA sono regolate nel tempo e nello spazio di una singola cellula non rimane completamente compreso, in gran parte a causa della carenza di metodi sperimentali per misurare quantitativamente la loro cinetica in vivo.
I metodi basati su mRNA totali estratti da una popolazione di cellule, come Northern blot, RT-PCR, RNA sequencing e microarray di espressione genica, possono misurare la differenza relativa nei livelli di mRNA e sono stati ampiamente utilizzati per analizzare il tasso di allungamento della trascrizione2,3,4,5 o il tasso di degradazione dell'mRNA6,7. Tuttavia, non forniscono il numero assoluto di mRNA per cellula e, di conseguenza, non sono adatti per sondare il tasso di iniziazione della trascrizione8. Inoltre, poiché gli mRNA vengono estratti da una popolazione di cellule, la distribuzione spaziale degli mRNA all'interno di una singola cellula e la variabilità dei numeri di copia dell'mRNA tra le cellule non possono essere misurati.
Il sequenziamento dell'RNA di nuova generazione su singole cellule (scRNAseq) può quantificare il numero di mRNA per cellula in una scala genomica9. Tuttavia, rimane difficile utilizzare questa tecnica per misurare la cinetica della trascrizione, a causa di sfide con la preparazione del campione e il costo elevato. In particolare, l'applicazione di scRNAseq ai batteri è stata tecnicamente difficile a causa della bassa abbondanza di mRNA10,11.
La fluorescenza a singola molecola in situ ibridazione (smFISH) si basa sull'ibridazione di sonde a singolo filamento etichettate fluorescente le cui sequenze sono complementari all'mRNA di destinazionedi interesse 12,13. Il concetto di ibridazione specifica della sequenza è simile a quello utilizzato in Northern blot o RT-PCR, ma l'ibridazione viene eseguita in situ all'interno di celle fisse, per preservare la localizzazione nativa degli mRNA. Il segnale di un singolo mRNA viene amplificato utilizzando molte sonde, 20 nucleotidi (nt) di lunghezza, ibridando in diverse parti di un mRNA (Figura 1A)13. In questo approccio di sonda "piastrella", il numero di sonde necessarie per rilevare un singolo mRNA imposta un limite inferiore alla lunghezza dell'mRNA che può essere analizzato. In alternativa, l'mRNA di interesse può essere scritratto in una matrice non codificante di sequenze di operatori lac tandem, in modo che più copie di una sonda lacO fluorescente si ibride in un singolo mRNA (Figura 1B)14.
smFISH è stato utilizzato per quantificare il numero di mRNA per cellula a stato costante (cioè, quando la sintesi e il decadimento sono in equilibrio) e per analizzare la media e la variabilità degli mRNA tra le cellule batteriche15,16,17. Recentemente, smFISH è stato applicato per quantificare i numeri di mRNA allo stato non stabile, subito dopo l'induzione o la repressione dell'espressione genica in E. coli18,19,20. Le variazioni temporali nei numeri di copia dell'mRNA assoluto sono state quindi utilizzate per calcolare il tasso di iniziazione della trascrizione, allungamento e terminazione, nonché il tasso di degradazione dell'mRNA. Per questa applicazione, le procedure smFISH convenzionali possono essere ingombranti perché ci sono molti campioni, ognuno dei quali rappresenta un punto di tempo, che devono passare attraverso più passaggi di scambio di buffer (ad esempio, centrifugazione e lavaggio). Qui, descriviamo un protocollo smFISH, in cui le fasi di movimentazione del campione sono notevolmente semplificate avendo cellule aderenti alla superficie di un coverslip e aspirando liquidi con un sistema di filtrazione del vuoto14,19. Utilizzando come esempio l'espressione di lac in E. coli, viene dimostrato il flusso di lavoro completo (Figura 2), inclusa l'analisi dell'immagine (Figura 3) che produce la cinetica in vivo della trascrizione (iniziazione, allungamento e terminazione) e la degradazione dell'mRNA, la variabilità da cellula a cellula nell'espressione mRNA e la localizzazione dell'mRNA. Prevediamo che il protocollo è ampiamente applicabile per sondare cinetica in vivo e localizzazione di altri mRNA in varie specie di batteri.
Protocollo
1. Preparazione delle sonde smFISH
NOTA: per etichettare le sonde smFISH con un singolo fluorforo, seguire un protocollo standard per l'etichettatura degli oligonucleotidi dell'acido nucleico basato sulla chimica dell'ester NHS21.
- Progettare sonde smFISH. Decidere se utilizzare sonde "piastrellate" o sonde "array" (Figura 1) per il gene di interesse. Vedere la sezione Discussione su come prendere la decisione.
- Per sonde di "affiancamento" (Figura 1A), utilizzare uno strumento di progettazione di probe online (ad esempio, vedere Tabella dei materiali).
- Per i probe "array" (Figura 1B), eseguire una ricerca di sequenza BLAST per assicurarsi che la sequenza probe non sia complementare ad altre sequenze mRNA.
- Per studiare la trascrizione e la cinetica di degradazione dell'mRNA, utilizzare due serie di 24 sonde, ciascuna delle quali copre la prima e l'ultima regione di 1 kb di lacz (3.072 bp)19.
NOTA: Questi set di sonde sono, in seguito, indicati rispettivamente come "sonda mRNA da 5'" e "sonda mRNA da 3'" . Le sequenze di queste sonde sono elencate nella Tabella dei Materiali.
- Ordinare sequenze di sonda come oligonucleotidi di DNA con un amino linker C6 alla fine 5 '. Sciogliere le singole sonde in acqua a 1 mM.
- Combinare quantità equimolari di sonde per i set "sonda mRNA da 5'" e "sonda mRNA da 3'". Ad esempio, per la sonda mRNA da 5' impostata per lac, combinare 20 L di ogni sonda (totale 24 tipi di sonde nel set).
- Eseguire la precipitazionedell'etanolo 22 delle sonde combinate per rimuovere eventuali contaminazioni di ammine primarie e secondarie (come Tris, glicina e sali di ammonio) che possono inibire la reazione di coniugazione. Alla fine, sciogliere il pellet di DNA in 100 L di acqua (producendo 4,5 mM di DNA in un set di sonda).
NOTA: Questo passaggio è consigliato anche se le sonde sono state sottoposte a una purificazione di desalt standard da parte del produttore. Una purificazione standard basata su filtri può funzionare al posto e in aggiunta alla precipitazione dell'etanolo. - Scegli due fluorofori spettralmente distinti con una moietà monofunzionale dell'ester NHS, in modo che i set di sonde mRNA da 5' e 3' possano essere etichettati in modo differenziale. Ad esempio, preparare l'ester Cy5 NHS per le sonde a mRNA da 5' e l'ester Cy3B NHS per le sonde mRNA da 3'. Sciogliere ogni tipo di fluoroforo in UN'anhydrous DMSO fino a 20 mg/mL finali (25 mM).
- Preparare 0,1 M di bicarbonato di sodio (pH 8.5) subito prima di ogni reazione di etichettatura. L'esposizione all'aria per lungo tempo ridurrà il suo pH e ridurrà l'efficienza di etichettatura.
- Per la reazione di coniugazione, unire quanto segue: 15 L del broroforo di fluoroforo Cy5 (dal punto 1.5), 4 L del set di sornate da 5' mRNA (dal punto 1.4), 75 L di bicarbonato di sodio (dal punto 1.6) e 7 L di acqua. Avvolgere il tubo con un foglio di alluminio e agitare a temperatura ambiente per 3-6 h.
NOTA: L'incubazione più lunga non comporta necessariamente una maggiore efficienza di etichettatura. Inoltre, la reazione può essere scalata verso l'alto o verso il basso se le concentrazioni dei componenti sono mantenute. - Ripetere il passo precedente per il set di sonda mRNA da 3' e il fluoroforo corrispondente (ad esempio, Cy3B NHS-ester).
- Eseguire la precipitazionedell'etanolo 22 per rimuovere le molecole di colorante non reagite. Sciogliere il pellet in 50 dollari di buffer TE (10mM Tris-HCl pH 8.0 con 1mM EDTA).
- Stimare le concentrazioni di DNA e fluoroforo utilizzando uno spettrometro UV-Vis.
- Misurare l'assorbimento a 260 nm e 559 nm (Cy3B) o 649 nm (Cy5). Se il campione è troppo concentrato per produrre una misurazione accurata, diluire 1 L del campione a 10 L.
- Convertire l'assorbimento nella concentrazione:



εDNA - 0,2 M-1 (per il DNA a 20-nt a filamento singolo), εCy5 - 0,25M -1e εCy3B - 0,13-1
NOTA: [DNA] è la concentrazione di sonde totali all'interno della soluzione. La concentrazione delle singole sonde è di circa 24 volte inferiore. La concentrazione delle sonde totali sarà utilizzata come "concentrazioni di sonda" da questo punto. Se il rapporto tra [DNA] e [dye] è 1, il seguente passaggio HPLC può essere saltato23e il campione deve essere diluito al 4-5 M finale nel buffer TE.
- (Scelta consigliata) Purificare le sonde etichettate da sonde senza etichetta e coloranti liberi utilizzando HPLC.
NOTA: Anche se questo passaggio di purificazione aggiuntivo porterà alla perdita di campione, è vantaggioso per le applicazioni a valle. La rimozione di sonde di DNA senza etichetta aumenterà il segnale di fluorescenza dai bersagli mRNA e la rimozione di coloranti nonactd ridurrà la fluorescenza di fondo.- Preparare HPLC con una colonna C18 analitica standard, 0,1 M di acetato di trietilammonium (TEAA) come buffer A e acetonitrile come buffer B.
- Aggiungere 1 M TEAA al campione (dal punto 1.9) per fare 0,1 M TEAA.
- Impostare il programma di gradiente come segue: 0-5 min con 0% B, 5-35 min con un gradiente lineare 0-30% di B, 35-37 min con un gradiente lineare del 30-100% di B e 37-40 min con 0% B. Mantenere la velocità di flusso a 0,1 mL/min e registrare i cromatogrammi a 260 e 649 nm (per i campioni con etichetta Cy5) o a 260 e 559 nm (per i campioni con etichetta Cy3B).
- Raccogliere il campione elato quando l'assorbimento aumenta sia nei canali del DNA che nel fluoroforo.
- Concentrare il campione elmo utilizzando un concentratore di vuoto e sospendere nuovamente il pellet in un buffer TE da 50 a 100 dollari.
- Controllare la concentrazione di DNA e fluoroforo utilizzando uno spettrometro UV-Vis (c'è punto 1.10). Diluire, se necessario, per effettuare la concentrazione finale intorno a 4-5 M. Conservare le sonde a -20 gradi centigradi
2. Preparazione delle soluzioni
- Preparare un grande volume di acqua e cuscinetti trattati con DEPC (Tabella 1). Queste soluzioni possono durare più di un anno a temperatura ambiente.
- Preparare la soluzione di fissaggio 4x e la soluzione di lavaggio (Tabella 1).
- Preparare la soluzione di pre-ibridazione e la soluzione di ibridazione del probe (Tabella 1). Preparare la soluzione di ibridazione della sonda durante l'incubazione al punto 5.1 o 6.1, quindi conservare la soluzione in uno shaker da banco da 37 gradi centigradi per 20-40 min con un coperchio per ridurre al minimo l'esposizione alla luce.
NOTA: le concentrazioni di formamide, SSC e sonda sono state ottimizzate per i set di sonde lac per ridurre al minimo la fluorescenza dello sfondo massimizzando al contempo il segnale reale. Vedere la sezione Discussione per informazioni dettagliate su come modificare queste concentrazioni per le diverse applicazioni.
3. Preparazione di coperture e vetrini
- Pulire coperture e scivoli di vetro.
- Posizionare le singole copertine e le diapositive in un barattolo Coplin utilizzando le forcetti. Assicurarsi che le coperture e le diapositive siano separate e non si tocchino a vicenda.
- Riempire il barattolo con etanolo al 100% e chiudere il coperchio. Mettere il barattolo in un detergente ad ultrasuoni bagno d'acqua e sonicare per 15-20 min.
NOTA: Per il sonicatore water-bath, si consiglia di disattivare la funzione di riscaldamento. - Versare l'etanolo e lavare con acqua ultrapura 3-4x. Utilizzare l'acqua che scorre direttamente dalla macchina per la purificazione dell'acqua.
- Versare l'acqua dal barattolo e riempirla con il 70% di etanolo. Chiudere il coperchio ed eseguire la sonicazione per 15-20 min e lavare con acqua ultrapura.
- Riempire il barattolo con acqua ultrapura e sonicare per 15-20 min.
NOTA: Le coperture e i vetrini possono essere conservati durante la notte nel barattolo Coplin pieno di acqua ultrapura. - Prendere uno scivolo o un coperture dal barattolo Coplin utilizzando le forcetti pulite e asciugarlo con gas N2. Ripetere questa operazione per le diapositive rimanenti e coverslips.
- Mettere i vetrini essiccati in una scatola di stoccaggio pulita fino all'uso al punto 7.5. Collocare i coperture essiccate in una scatola vuota di punta di pipetta da 1.000 lL, che servirà da "camera" nella procedura rimanente.
- Utilizzando un marcatore idrofobico, disegnare cerchi sulle coperture seguendo fori circolari nella casella della punta della pipetta. Questi cerchi (0,5 cm di diametro) fungeranno da "pozzi". Attendere almeno 5-10 min per il marcatore di essere completamente essiccato.
NOTA: Tenere sempre chiuso il coperchio della casella della punta. - Applicare un calo di 20-L dello 0,1% di poli-lsensilina ad ogni pozzo. Incubare per 10-50 min a temperatura ambiente.
NOTA: regolare questo volume in base alle dimensioni del pozzo. Assicurarsi che la soluzione copra completamente l'area del pozzo. Per un'incubazione più lunga, fare attenzione a evitare l'evaporazione. - Dopo l'incubazione aspirare poli-L-lisina senza toccare la superficie in quanto questo raschierà la poli-L-lisina off. Quindi applicare una goccia di acqua DEPC (20 usd) ai pozzi trattati con poli-L-lisina. Chiudere il coperchio della "camera" per evitare l'evaporazione fino al punto 5.1.
4. Esperimento di tempo e fissazione del campione
- Coltivare le cellule E. coli in coltura liquida da 20 mL in un pallone da 250 mL. Tenere il flacone in uno shaker per il bagno d'acqua (30 gradi centigradi) e continuare a tremare. Fermare lo shaker solo quando si prendono campioni.
NOTA: I risultati presentati in questo documento sono ottenuti da cellule MG1655 coltivate in media minima M9 integrato con 0,2% glicerolo, 0,1% di casaminoici e 1 mg/L diamina ad una fase di crescita esponenziale (OD600s.0.2). - Aggiungere 250 L della soluzione di fissaggio 4x in un tubo vuoto da 1,5 mL. Ripetere e preparare più tubi, quanti sono i punti di tempo da prendere. Etichettare i tubi con i numeri di punto di tempo e tenerli a temperatura ambiente.
- Prendi 750 L di coltura cellulare (OD600s.0.2) prima di iniziare un esperimento di tempo-corso. Aggiungere le impostazioni cultura a un tubo contrassegnato per "time zero" (dal passaggio 4.2). Invertire delicatamente il tubo per mescolare le cellule con la soluzione di fissaggio.
NOTA: Non pipetta su e giù per mescolare, vortice, o "essere ruvido" sulle celle. Questo esempio rappresenta lo stato represso e verrà utilizzato come controllo per calcolare l'intensità della fluorescenza di un singolo mRNA (vedere il passaggio 9.4). - Aggiungere 0,02-1 mM di isopropile β-D-1-thiogalactopyranoside (IPTG) alla coltura liquida per indurre l'espressione lac. Avviare un timer a questo punto (t e 0 min) e campionare a un determinato intervallo di tempo (ad esempio, ogni 1 min) da quel momento in poi. Per il campionamento, ripetere il passaggio 4.3.
- Aggiungere 5 mM orthonitropheynl-β-D-fucopyranoside (ONPF) o 500 mMglucosio 24 in un determinato momento durante l'esperimento di tempo-corso (ad es. a t - 1,5 min) per reprimere l'espressione lac. Dopo una ri-repressione, continuare a campionare le colture (passaggio 4.3) per tenere traccia della degradazione dell'mRNA.
NOTA: La repressione può essere fatta anche con la rifampicina di 400 g/mL, un inibitore dell'iniziazione dellatrascrizione 25. - Per la fissazione, incubare i tubi contenenti celle campiove a temperatura ambiente per 15 min, seguiti da incubazione nel ghiaccio per 30 min.
- Per rimuovere i fissativi, centrifugare i tubi a 4.500 x g per 4 min a temperatura ambiente. Rimuovere il supernatant con una pipetta.
NOTA: Assicurarsi di scartare la formaldeide in un contenitore di rifiuti separato seguendo il protocollo di sicurezza. - Aggiungere 1 mL DEPC-PBS e sospendere nuovamente le celle. Ripetere la centrifugazione e la sospensione 2 volte.
NOTA: Le cellule fisse sono fragili e necessitano di un trattamento delicato. Sospendere con attenzione il pellet ed evitare le bolle. - Dopo la fase di lavaggio finale, sospendere nuovamente le cellule in DEPC-PBS da 30 USD.
5. Permeabilizzazione delle membrane cellulari
- Applicare ogni campione di punto di tempo a diversi pozzetti sulla coperture (30 dollari l per pozzo). Attendere 10-30 min a temperatura ambiente per le cellule di aderire sulla superficie. Evitare la fusione delle gocce liquide tra i pozzetti.
- Per risciacquare le cellule non associate, aspirare il liquido e applicare il PBS DEPC DEPC da 20 dollari a ciascun pozzo. Aspirare DEPC PBS in pochi minuti.
- Permeabili le membrane cellulari applicando 15 L del 70% di etanolo ad ogni pozzo per 4 min. Aspirare l'etanolo dopo i 4 minuti e assicurarsi che i pozzi siano completamente asciutti.
NOTA: È fondamentale limitare il trattamento dell'etanolo per 4 minuti. Un trattamento più lungo si tradurrà in un'over-permeabiliization. - Applicare 30 L della soluzione di lavaggio ad ogni pozzo.
6. Ibridazione della sonda
- Aspirare la soluzione di lavaggio da ogni pozzo. Applicare 30 L della soluzione di pre-ibridazione per ogni pozzo. Incubare la camera nel forno da 37 gradi per 30 minuti.
NOTA: Aggiungere 50 mL di acqua sul fondo della camera per fornire umidità. - Aspirare la soluzione di pre-ibridazione da ogni pozzo. Applicare 30 dollari della soluzione di ibridazione della sonda per ogni pozzo. Coprire la camera con un foglio di alluminio e incubare nel forno da 37 gradi per 2 h.
NOTA: prima di questo passaggio, assicurarsi che la soluzione di ibridazione della sonda si trova nello shaker del piano di lavoro a 37 gradi centigradi. Evitare la fusione di liquidi tra pozzetti. Applicare un volume più piccolo della soluzione a ciascun pozzo, se necessario.
7. Lavaggio post-ibridazione e preparazione per l'imaging
- Utilizzando una pipetta multicanale, applicare 30 dollari della soluzione di lavaggio a ciascuno bene tutto in una volta. Aspirare e ripetere 3-5 volte di lavaggio. Incubare la camera nel forno da 37 gradi per 15-30 min.
- Ripetere il passaggio 7.1 altre due volte.
- Lavare bene ogni pozzo con DEPC-PBS 5 volte. Seguire il metodo utilizzato nel passaggio 7.1 ma ignorare il processo di incubazione.
- Aspirare il liquido dal coverslip. Applicare 4 L di DEPC-PBS a ciascun pozzo.
- Utilizzando le forcelle, sollevare e capovolgere il cosepe e posizionarlo delicatamente su uno scivolo di vetro (dal punto 3.2). Evitare le bolle.
- Sigillare i bordi del coperture con gomma dentale in silicone.
- Attendere che la gomma si solidifica. Si può mettere in pausa qui e memorizzare lo scivolo durante la notte a 4 gradi centigradi.
NOTA: Altri protocolli smFISH suggeriscono l'aggiunta di reagenti di scavenging dell'ossigeno (ad esempio, ossidasi/catalasi del glucosio) o l'utilizzo di un mezzo di montaggio anti-dissolvenzacommerciale 14,26 per aumentare la fototuarmizzabilità dei fluorofori.
8. Imaging
- Per trovare un'area di interesse, utilizzare la modalità live di imaging a contrasto di fase. Cambiare il campo di vista all'interno di un pozzo manovrando il joystick del palco. Scegliere un'area in cui la densità delle celle è ottimale (ad esempio, ci sono molte celle che sono per lo più separate). Regolare lo stato attivo z in modo che le immagini delle celle a contrasto di fase siano a fuoco.
- Scattare istantanee nell'ordine di Cy5 (esposizione 4-s), Cy3 (esposizione 2-s) e contrasto di fase (0,2-s esposizione).
NOTA: le molecole di colorante Cy3B sono state immagini nel canale Cy3 e le immagini sono denominate immagini Cy3. - Ripetere i passaggi 8.1-8.2 per acquisire immagini di 10 aree diverse all'interno di un pozzo.
- Spostare l'obiettivo in un altro pozzo e ripetere i passaggi 8.1-8.3.
- Esportare le immagini come file TIFF.
- (Facoltativo) Perline multi colore per le immagini adsorbite sulla superficie coverslip nei canali Cy5 e Cy3 per determinare lo spostamento spaziale tra i canali Cy5 e Cy3 ai fini della registrazione delle immagini.
- Applicare 10 dollari di perline fluorescenti multi colore (diametro 0,2 m) su una superficie pulita di coverslip e attendere 10-30 min. Dopo il lavaggio con 50 dollari di PBS, applicare 5 L di PBS e sandwich il coperture con uno scivolo di vetro. Sigillare e montare al microscopio.
- Perline di immagine in entrambi i canali Cy5 e Cy3.
9. Analisi delle immagini
NOTA: il codice Matlab utilizzato in questo passaggio è disponibile nel seguente sito Web GitHub: https://github.com/sjkimlab/Code_Publication/tree/master/JoVE_2020. La cartella GitHub contiene tutto il necessario per l'analisi dell'immagine, inclusi i valori dei parametri per la segmentazione delle celle e l'identificazione dei macchia. La procedura descritta in questo passaggio è ulteriormente illustrata nello script principale, denominato "FISHworkflow.m".
- Aprire uno strumento di segmentazione delle celle, ad esempio microbeTracker27 o Oufti28,e caricare le immagini di contrasto della fase. Scegliere "Cornici indipendenti" e premere un pulsante denominato "Tutti i fotogrammi" per avviare il processo di segmentazione, da cui vengono identificate le celle e i relativi contorni (Figura 3B,C).
NOTA: i protocolli dettagliati per l'utilizzo di questi pacchetti software sono disponibili online (ad esempio, oufti.org). - Caricare le immagini di fluorescenza Cy5 nella funzione spotFinder di microbeTracker o Oufti e premere il pulsante "Esegui" per iniziare l'identificazione dello spot e la quantificazione in base al raccordo gaussiano 2D (Figura 3B,C). Ripetere questo passaggio per le immagini di fluorescenza Cy3 per analizzare le macchie nel canale Cy3. Questo passaggio produce un elenco di punti in ogni cella, incluse le loro intensità e coordinate.
- (Facoltativo) Filtrare i punti dim (falsi positivi) utilizzando una soglia, come spiegato nel file FISHworkflow.m.
NOTA: esaminare le macchie fluorescenti nel controllo negativo (ad esempio MG1655- lac) e determinare la soglia per filtrare i falsi positivi. - Per ottenere l'intensità spot di un singolo mRNA, utilizzare un elenco di intensità spot misurate al tempo zero (prima di aggiungere IPTG) e adattare la distribuzione delle intensità spot con un modello di miscela gaussiana con due componenti della miscela. Prendere la posizione di picco della prima popolazione gaussiana (linea nera in Figura 3D,E) come l'intensità spot di un singolo mRNA. Eseguire questo per le macchie Cy5 e Cy3 separatamente per ottenere l'intensità spot di un singolo mRNA lac 5' e 3'3'.
NOTA: Ripetere questa operazione in ogni esperimento di tempo-corso perché l'intensità spot di un singolo mRNA può variare leggermente in diversi esperimenti. - Dividere l'intensità della fluorescenza di un punto con l'intensità di un singolo mRNA (dal punto 9.4) per ottenere il numero di mRNA all'interno di un punto. Sommare le intensità dei punti normalizzati all'interno di una cella per calcolare il numero totale di mRNA in una cella (Figura 3F). Eseguire questi calcoli per 5' e 3' mRNA separatamente.
- Calcolare e tracciare i numeri di mRNA medio per cella in ogni punto temporale (ad esempio, Figura 4B) e analizzare la cinetica in vivo della trascrizione e della degradazione dell'mRNA dalla variazione temporale dei livelli di mRNA medio (Figura 4B).
- Per ottenere il tasso di allungamento della trascrizione, eseguire un montaggio minimo quadrati di una linea all'aumento iniziale nei segnali mRNA 5' e 3' e identificare le intercettazioni ai livelli basali (Figura 4B). La differenza tra queste intercettazioni indica il tempo medio per gli RNAP di viaggiare dalla regione della sonda 5' alla regione della sonda 3'. Dividere la distanza tra due set di probe (2 kb) con questo tempo per ottenere la velocità media di allungamento della trascrizione.
- Per ottenere la velocità di degradazione dell'mRNA, adattare una funzione di decadimento esponenziale, y , a - A - exp (-t/) alla regione di decadimento finale dei segnali mRNA 5' e 3' (ad esempio, Figura 4B). Il parametro di adattamento, , è la durata media dell'mRNA.
- (Facoltativo) Analizzare la variazione da cellula a cellula nell'espressione genica (ad esempio, la risposta a livello di cellula all'induzione mostrata nella figura 4C), in base alla distribuzione dei numeri di mRNA in ogni cellula (calcolata al punto 9.5).
- (Facoltativo) Utilizzando le informazioni sulla posizione spot lungo gli assi principali e minori di una cella (ottenute dal passaggio 9.2), analizzare la localizzazione di mRNA (Figura 4D,E).
- (Facoltativo) Analizzare la co-localizzazione di 5' e 3' mRNA (Figura 5) confrontando la localizzazione dei punti rilevati nei canali Cy5 e Cy3.
- Caricare immagini di perline multi colore (passaggio 8.6) nella funzione spotFinderF in microbeTracker e ottenere le coordinate dei centriidi di perline nei canali Cy5 e Cy3. Utilizzare l'elenco delle coordinate centroidi per calcolare la matrice di trasformazione affine, che informa come i canali Cy5 e Cy3 vengono spostati e ruotati l'uno rispettoall'altro 29.
- Applicare la matrice di trasformazione affine alle immagini Cy5 e Cy3 FISH per convertire le immagini Cy3 nella coordinata Cy5. Classifica se un punto è co-localizzato con un altro punto in un canale diverso. Ad esempio, un punto nel canale Cy5 è considerato co-localizzato con un altro punto nel canale Cy3 se la distanza tra i relativi centroidi è inferiore a 150 nm (Figura 5).
- Analizza quanti punti Cy5 sono classificati come "co-localizzati" con punti Cy3 in ogni punto di tempo. Analizzare inoltre l'intensità delle macchie co-localizzate (Figura 5).
Risultati
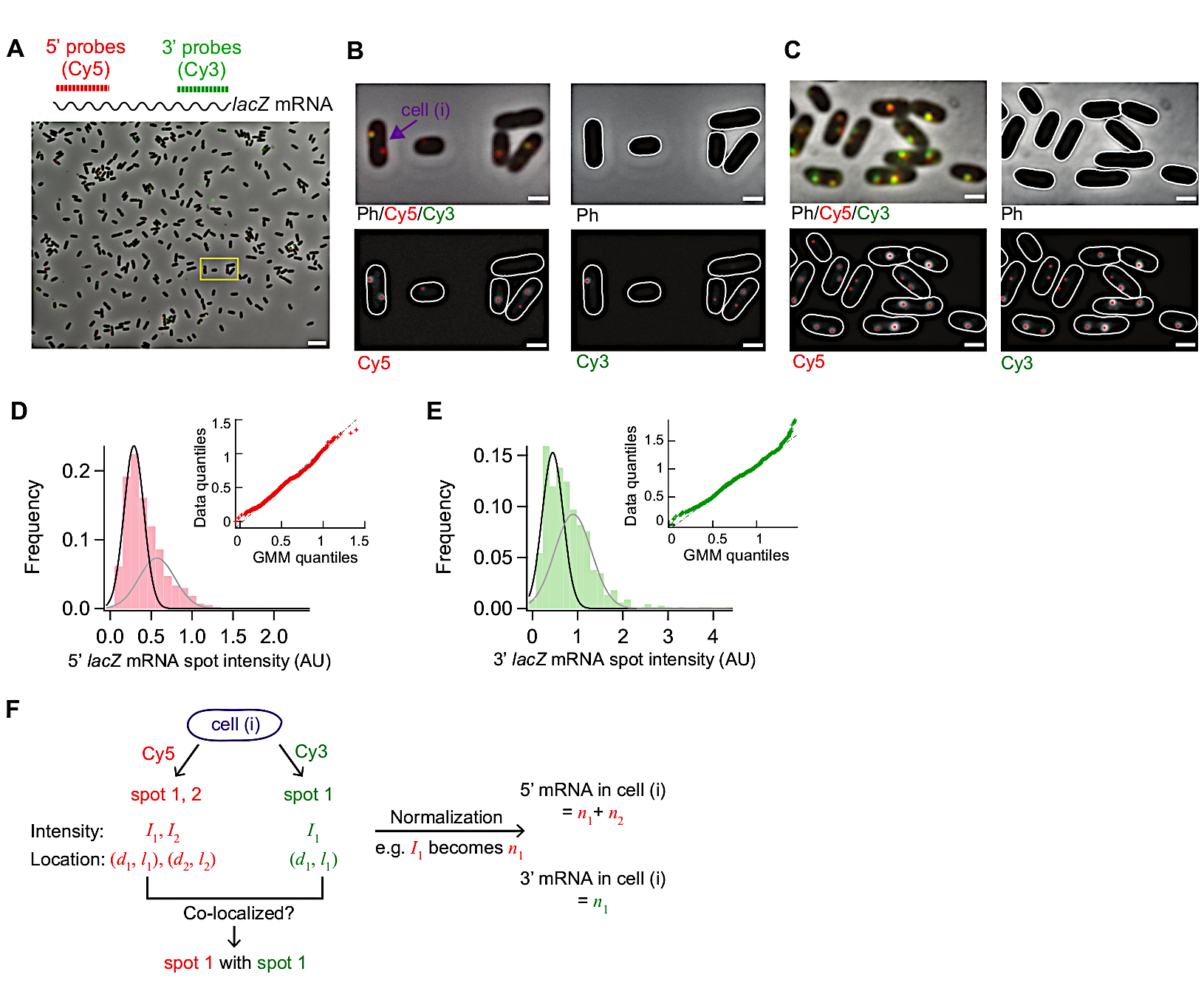
La figura 3 mostra le immagini rappresentative di questo protocollo smFISH. Un campo di vista completo (86,7 m x 66,0 m utilizzando l'impostazione della microscopia descritta nella Tabella dei materiali) mostra le celle E. coli dislocate in tutto il campo (Figura 3A). Se la densità delle celle è molto più alta di quella mostrata in questa immagine, la segmentazione automatica delle cellule diventa difficile poiché gli algoritmi di segmentazione non identificano in modo affidabile le singole celle quando le celle si toccano. È necessario regolare la concentrazione delle cellule e il tempo di incubazione utilizzato per l'aderenza alla superficie (punto 5.1) per ottenere la densità ottimale delle cellule nel campo di vista.
La morfologia delle cellule nella fase delle immagini di contrasto dovrebbe rimanere paragonabile a quella delle cellule vive ai fini della segmentazione(Figura 3A-C). Se le cellule sono sovra permeabili, la morfologia cellulare cambia (come "fantasmi"; (Figura 1)). In tal caso, si può ridurre la durata del 70% del trattamento con etanolo al punto 5.3.
Prima dell'induzione, il livello medio di espressione lac è stato di 0,03 mRNA per cella, in linea con i rapporti precedenti15,30. Inoltre, la distribuzione delle intensità spot di mRNA lac prima dell'induzione non si adattava bene con una distribuzione normale o una distribuzione di Poisson a causa della presenza di macchie con intensità elevate ( Figura3D,E). Ciò suggerisce che la maggior parte delle macchie rilevate sotto lo stato represso rappresentano un singolo mRNA lac, ma una piccola popolazione di macchie contiene più di un mRNA lac. Per isolare la popolazione con un singolo mRNA lac, è stato utilizzato un modello di miscela gaussiana con due componenti di miscela (insets in Figura 3D,E). Quindi, la media del primo gaussiano è stata presa come intensità media di un singolo punto di mRNA (ad esempio, il picco della curva nera nella figura 3D)e utilizzata per convertire l'intensità spot al numero di mRNA, per tutti i punti rilevati nell'esperimento del corso di tempo. Per calcolare il numero totale di MRNA all'interno di una cella, le intensità di punti normalizzate sono state sommate in ogni cella (Figura 3F)19.
Quando il livello di espressione dell'mRNA lac è basso, ci sono una o due macchie di mRNA lac, limitate dalla diffrazione, separate spazialmente all'interno di una cellula. Quindi, le immagini di questi punti possono essere analizzate da adattamento gaussiano 2D per la loro intensità e localizzazione.
Quando il livello di espressione è alto, in modo che le macchie si sovrappongano tra loro all'interno di una cella, il raccordo gaussiano 2D non esegue una quantificazione affidabile. In tal caso, il livello di mRNA deve essere calcolato dividendo il segnale di fluorescenza totale sottratto allo sfondo all'interno di una cella con l'intensità media di un singolo mRNA19.
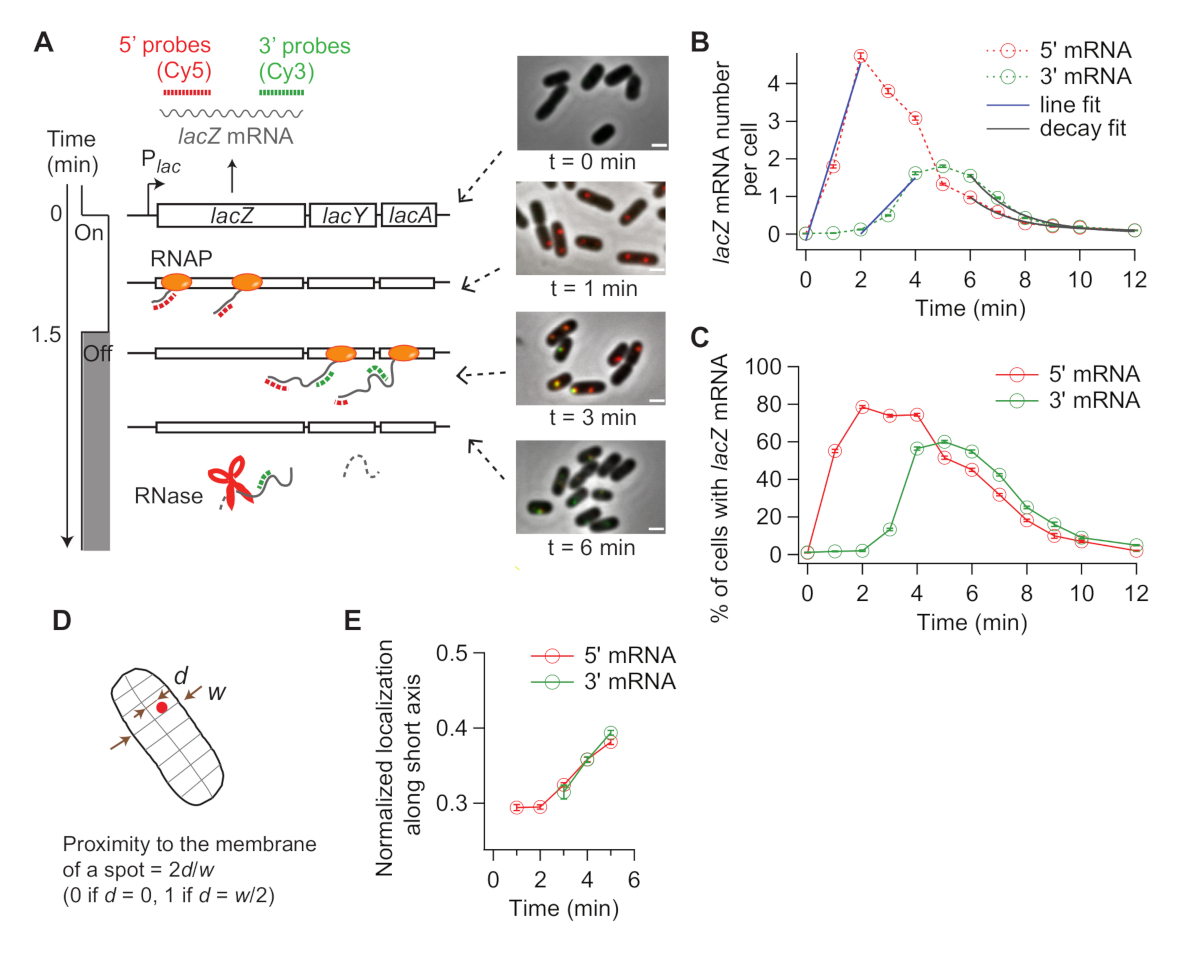
Quando viene indotta l'espressione di lac, il segnale di mRNA 5' lac' aumenta per primo e quello di 3' lac'mRNA aumenta più tardi (Figura 4B). Se l'espressione di lac è repressa, entrambi i segnali mRNA 5' e 3' lac diminuiscono con un certo ritardo tra (Figura 4B). Per ottenere il tasso di allungamento della trascrizione, l'aumento dei segnali di 5' e 3' è il primo adattamento con le linee (Figura 4B), e la differenza nelle intercettazioni x viene presa come il tempo per gli RNAP per percorrere la distanza tra due regioni della sonda (2.000 nt). Il tasso di allungamento della trascrizione può essere misurato da ogni esperimento del corso di tempo e le deviazioni standard possono essere calcolate da duplicati sperimentali. Il tasso medio di allungamento della trascrizione è stato di 15-30 nt/s nelle nostre condizioni sperimentali19.
Inoltre, il tasso di degradazione dell'mRNA (inverso della durata media dell'mRNA) è stato ottenuto adattando la regione di decadimento con una funzione esponenziale (Figura 4B). I nostri dati di tempo-corso contiene la degradazione dell'mRNA durante e dopo latrascrizione 31. Ci adattiamo ai punti di tempo dopo che 3' mRNA ha iniziato a decadere (t > 6 min) per sondare la degradazione degli mRNA rilasciati. Abbiamo ottenuto 90 s come una durata media di 5' o 3' lac' mRNA19.
Il tasso di inizio della trascrizione può essere calcolato dalla pendenza dell'aumento del segnale di 5' dopo l'induzione(Figura 4B, blu), o dal numero medio di mRNA allo stato costante (che è il tasso di iniziazione diviso per il tasso di degradazione). Inoltre, si può stimare la probabilità di terminazione prematura della trascrizione, sia prendendo il rapporto tra la pendenza di 3' aumento del segnale rispetto a quello di 5' aumento delsegnale 32 o tra i livelli di stato costante di 3' e 5' mRNA regioni19.
Poiché smFISH è una tecnica a cella singola, possiamo analizzare la variabilità da cellula a cellula nella trascrizione. Ad esempio, è possibile analizzare la percentuale di celle che esprimono mRNA lac dopo l'aggiunta di IPTG ( Figura4C). Si può anche affrontare se la localizzazione dell'mRNA cambia dopo l'induzione. Abbiamo osservato che le macchie di mRNA 5' e 3' lac si muovono leggermente verso l'esterno, lontano dal centro della cella (Figura 4D,E), coerente con un precedente rapporto33.
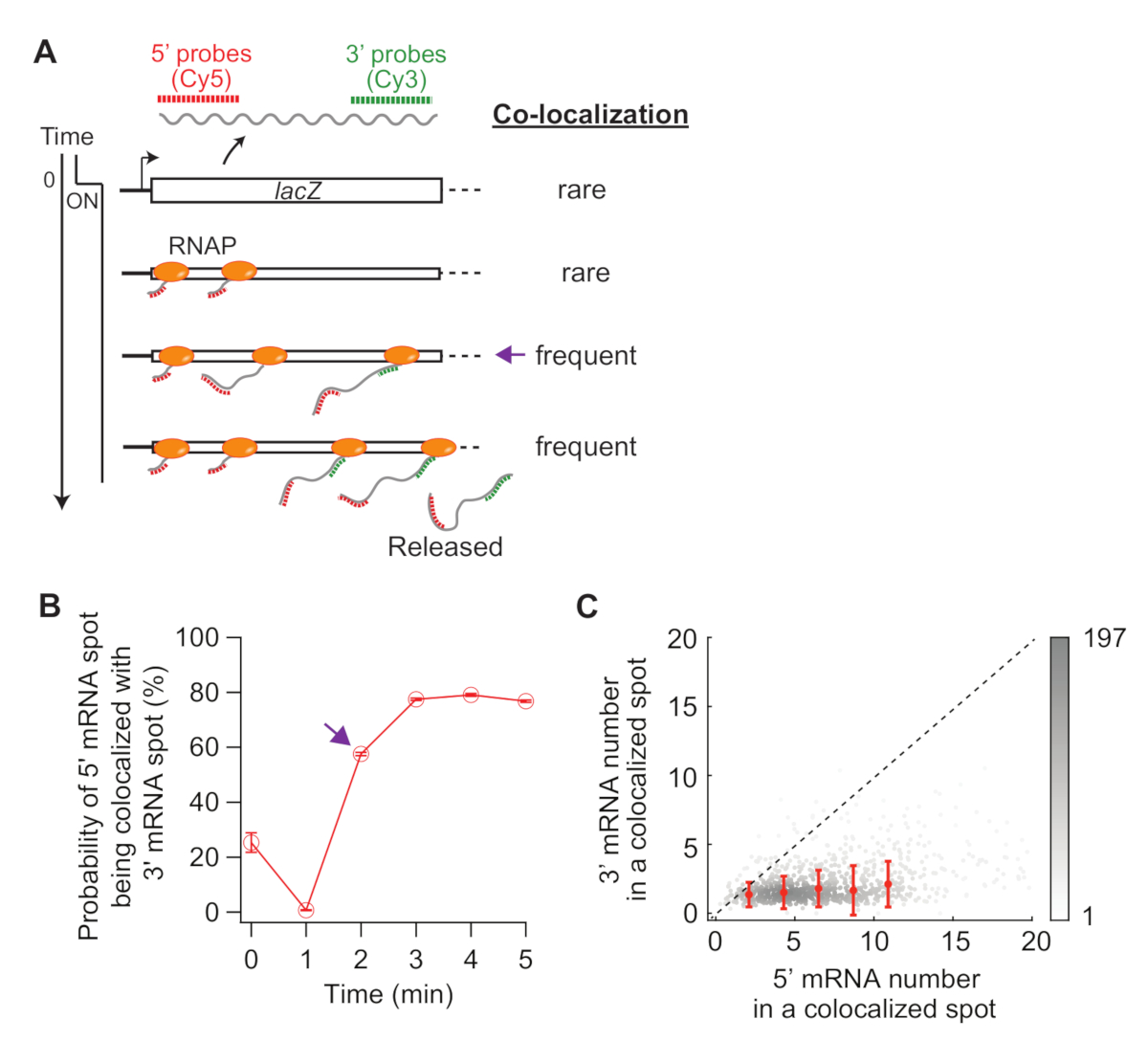
Infine, l'analisi della co-localizzazione tra i punti di mRNA da 5' e 3' può essere informativa(Figura 5A). Ad esempio, nello stato represso (tempo zero), circa il 25% delle macchie di mRNA da 5' sono co-localizzate con una macchia di mRNA di 3'. A 1 min, poiché molti loci genici hanno 5' sintesi di mRNA, ma non ancora 3' sintesi mRNA, la maggior parte delle macchie di mRNA 5' sono da sole senza segnale mRNA 3' (cioè, bassa probabilità di co-localizzazione). Tuttavia, quando viene visualizzato l'mRNA 3' (ad esempio, t e 2 min), aumenta la probabilità di co-localizzazione (freccia viola nella figura 5A,B). Questo punto di tempo, quando la co-localizzazione diventa frequente, dipende dal tasso di allungamento della trascrizione. Il grafico a densità 2D di numeri di mRNA 5' e 3' lac' all'interno di ogni punto di co-localizzazione rilevato in questo momento può essere utilizzato per dedurre la densità degli RNA sul gene lac . Come riportato inprecedenza 19, i numeri di mRNA 5' in questo grafico indicano che la maggior parte dei loci lac ha meno di 10 RNAps sul DNA quando l'espressione lac è indotta da 1 mM IPTG. Inoltre, i numeri di mRNA 3' in questo grafico sono correlati al clustering di RNA34. Il fatto che il numero di mRNA 3' sia vicino a uno significa che circa un RNAP entra nella regione della sonda 3'. Ciò suggerisce che gli RNAP sul gene lac è separati spazialmente, invece di formare un ammasso (o "convoglio").

Figura 1: Progettazione di sonde smFISH per un mRNA di interesse. (A) Un metodo di affiancamento. Vengono scelte sequenze di oligonucleotidi di DNA corti (20 bp di lunghezza) in modo che possano coprire l'mRNA di interesse. Le sonde oligonucleotide sono etichettate con una molecola colorante fluorescente. (B) Un metodo di matrice. Una matrice non codificante di sequenze tandem (ad esempio, "lacO array ") è scrizionalmente fusa all'mRNA di interesse. La sonda con etichetta fluorescente complementare all'unità di ripetizione (ad esempio, sonda lacO di 17 bp di lunghezza) viene utilizzata per amplificare il segnale di un mRNA. Si prega di fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

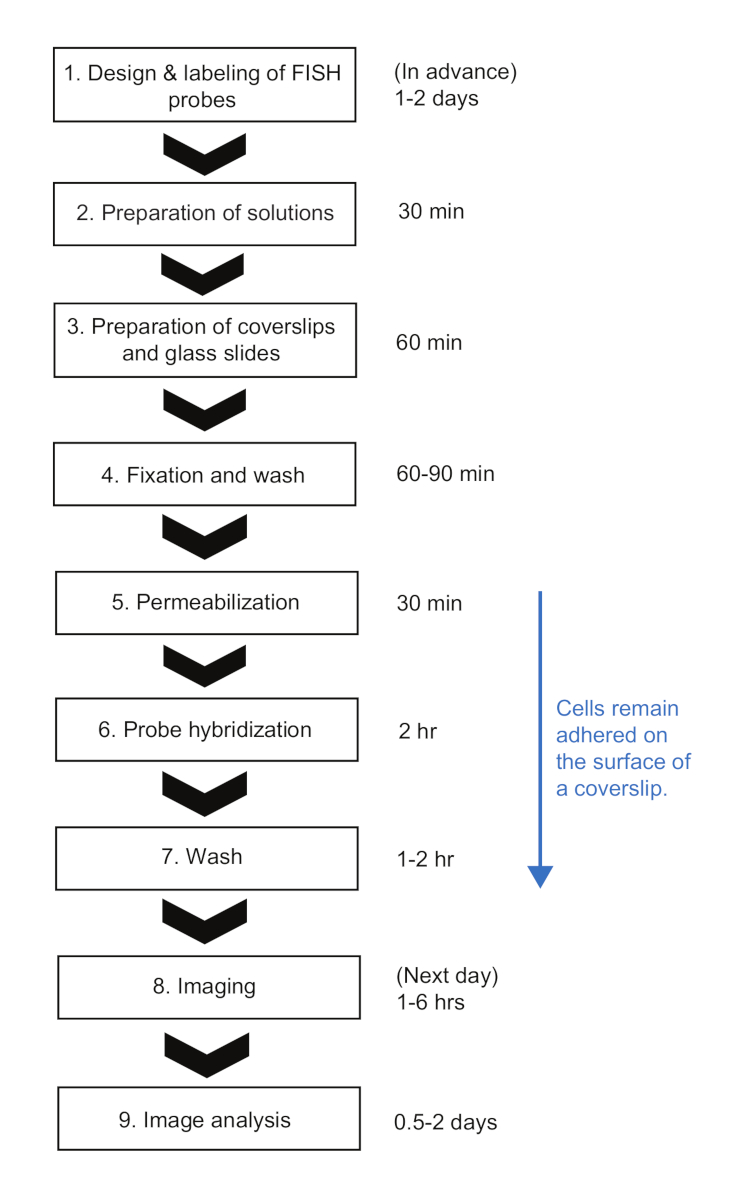
Figura 2: Schema della procedura sperimentale smFISH e durata del tempo di ogni passaggio. Si prega di fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 3: analisi delle immagini smFISH. (A-C) immagine di microscopia smFISH di mRNA (rosso) e 3' mRNA (verde) in tipo selvatico E. coli (MG1655) coltivata in M9 media minima integrata con glicerolo 0,2%, 0,1% di casamino acidi, e 1 mg/L tiamina a 30 gradi centigradi (A) Un'immagine rappresentativa di un campione da t - 3 min dopo l'induzione con 0,05 mM IPTG a t - 0 min e repressione con glucosio 500 mM a t - 1,5 min. Il contrasto di fase e due immagini di fluorescenza di Cy5 (per 5' lac, rosso) e Cy3 (per 3' mRNA lac, verde) sono stati sovrapposti con pseudo-colorazione. L'immagine mostra un intero campo di 86,7 m x 66,0 m. Barra di scala, 5 m. (B) Versione ingrandita di una piccola area (casella gialla) in (A). I contorni delle celle sono visualizzati in bianco e le macchie di fluorescenza identificate dall'analisi dell'immagine sono mostrate con punti rossi. Barra di scala, 1 m. (C) Rilevamento dei contorni delle cellule e delle macchie fluorescenti in condizioni di espressione elevata (t - 4 minuti dopo l'induzione con 1 mM IPTG). Barra di scala, 1 m. (D-E) Distribuzioni di 5' e 3' mRNA spot intensità misurate prima di aggiungere IPTG (lo stato represso). Gli istogrammi sono mostrati con due funzioni gaussiani (nero e grigio) i cui valori medio sono del modello di miscela gaussiana. L'inset mostra un grafico quantile-quantile di numeri casuali generati dai modelli di miscela gaussiana e intensità spot di mRNA misurati sperimentalmente (n - 1040 per mRNA 5' e 680 per mRNA 3'). (F) Informazioni ottenute per una singola cella puntata nel pannello (B). Per una determinata cella (i), le macchie sono state identificate nei canali Cy5 e Cy3, e la loro intensità (I) e la loro coordinata lungo l'asse breve e lungo di una cella(d, l ) sono state quantificate dal raccordo gaussiano 2D. Dopo che le intensità del punto di normalizzazione sono state sommate per produrre il numero totale di 5' o 3' mRNA in questa cella. Inoltre, è possibile analizzare la co-localizzazione tra punti di canali diversi, come nell'esempio illustrato nella Figura 5. Si prega di fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 4: Analisi della cinetica in vivo della trascrizione e della degradazione dell'mRNA. (A) Immagini schematiche e rappresentative di esperimenti smFISH a due colori che misurano i cambiamenti nei livelli di mRNA lac nel tempo. Le linee tratteggiate rosse e verdi indicano sonde oligonucleotide etichettate Cy5 o Cy3B che si ibridano rispettivamente nelle regioni mRNA lunghe 1 kb e 3' dell'mRNA lac in E. coli. Vengono inoltre mostrate sovrapposizioni di due immagini di fluorescenza con un'immagine di contrasto di fase nei punti di tempo indicati dopo l'induzione con 0,2 mM IPTG a t e 0 min. La trascrizione è stata repressa con glucosio di 500 mM a t - 1,5 min. Barra di scala, 1 m. La cifra è stata modificata da Kim et al19. (B) 5' e 3' numeri di mRNA lac per cellula nel corso dell'esperimento descritto nel pannello (A). Le barre di errore sono SEM bootstrap. Sono state analizzate almeno 1.200 cellule per punto di tempo. L'aumento iniziale dei segnali mRNA da 5' e 3' era in forma con una linea (blu). La differenza nelle intercettazioni x è stata di 1,93 min, producendo il tasso medio di allungamento della trascrizione di 17,3 nt/s. Il decadimento finale dei segnali mRNA da 5' e 3' era in forma con una funzione di decadimento esponenziale (grigio). I parametri di adattamento indicano che la durata media dell'mRNA è di 1,52 min per 5' mRNA e 1,66 min per 3' mRNA. (C) Percentuale di cellule con una o più macchie di mRNA lac durante l'esperimento descritto in (A). Le barre di errore sono SEM bootstrap. (D) Localizzazione di un punto lungo l'asse corto di una cella. È possibile quantificare la vicinanza di un punto alla membrana dividendo la posizione lungo l'asse corto (d) con la metà della cella (w). (E) Cambiamento nella localizzazione di macchie di mRNA 5' e 3' lac ' lungo l'asse corto delle cellule durante l'esperimento descritto in (A). Si prega di fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 5: Analisi della co-localizzazione di macchie di mRNA da 5' e 3'. (A) Schematico che mostra la co-localizzazione prevista tra 5' e 3' macchie di mRNA dopo l'induzione. Quando si fa 3' mRNA, aumenta la probabilità che una macchia di mRNA da 5' venga co-localizzata con una macchia di mRNA di 3' (freccia viola). (B) La probabilità di co-localizzazione dopo l'induzione con 1 mM IPTG. La freccia viola indica il punto di tempo in cui la probabilità di co-localizzazione diventa prima frequente in base allo schema nel pannello (A). (C) Il numero di mRNA da 5' e 3' lac' all'interno di un punto di co-localizzazione rilevato a t - 2 min dopo l'induzione con 1 mM IPTG (totale 841 punti). I punti grigi rappresentano singole macchie co-localizzate, mentre i punti rossi rappresentano la media dei dati collocati. Le barre di errore sono SEM. L'ombreggiatura di grigio indica la densità dei punti in una determinata area del grafico. La linea tratteggiata indica una pendenza di 1. Si prega di fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}

Figura 6: Ottimizzazione della condizione di ibridazione della sonda. Sono stati utilizzati due tipi di campioni: cellule MG1655 coltivate come descritto nella figura 3 e rimangono non indotto (blu) o trattate con 0,5 mM IPTG per 20 min (rosso). La soluzione di ibridazione della sonda è stata realizzata con diverse concentrazioni di sonde (totale 72 sonde coniugate cy5 che piastrellano l'intera regione lac) e di formamide. Le concentrazioni di formamide sono state regolate anche nella soluzione di pre-ibridazione e la soluzione di lavaggio, di conseguenza. "Nessuna sonda" (linea grigia) indica il livello di fluorescenza delle cellule aggiunte da IPTG trattate senza sonde durante la fase di ibridazione. L'intensità media della fluorescenza normalizzata dall'area cellulare (AU) è stata calcolata da 300-800 cellule. Le barre di errore sono SEM bootstrap. Si prega di fare clic qui per visualizzare una versione più grande di questa figura.
{kind=link}
Figura supplementare 1: Morfologie cellulari distorte dovute alla permeabilizzazione. Sovrapposizione del contrasto di fase (scala di grigi), 5' lac mRNA (Cy5, rosso) e 3' lac' mRNA (Cy3, verde) immagini di mg1655 celle 5 min dopo l'induzione con 1 mM IPTG. (A) Un esempio che mostra la miscela di cellule normali e cellule eccessivamente permeabili prive di morfologia normale (indicata con frecce rosa). (B) Un esempio che mostra le celle "fantasma" raggruppate insieme. Barra della scala : 1 m. Fare clic qui per scaricare questo file.
{kind=link}
| ACQUA DEPC | ||||
| Aggiungere lo 0,1% di DEPC all'acqua ultrapura e incubare la bottiglia (coperta) nel forno a 37 gradi durante la notte e l'autoclave il giorno successivo. | ||||
| DEPC PBS (10X) | ||||
| Mescolare quanto segue: | ||||
| 80 g | NaCl (finale 1,37 M) | |||
| 2 g | KCl (finale 27 mM) | |||
| 14,2 g | Na2HPO4 (finale 100 mM) | |||
| 2,7 g | KH2PO4 (finale 20 mM) | |||
| Acqua ultrapura a 1L | ||||
| Filtrare (0,22 m) in una bottiglia di vetro. | ||||
| Aggiungere 0,1% DEPC e seguire le istruzioni per l'acqua DEPC. | ||||
| Per fare la soluzione 1X, diluire 10 volte con acqua DEPC. | ||||
| 1M DEPC tampone di fosfato di sodio, pH 7.4 | ||||
| Mescolare quanto segue: | ||||
| 115 g | Na2HPO4 | |||
| 22,8 g | NaH2PO4 | |||
| Acqua ultrapura a 1 L | ||||
| Filtrare (0,22 m) in una bottiglia di vetro. | ||||
| Aggiungere 0,1% DEPC e seguire le istruzioni per l'acqua DEPC. | ||||
| Soluzione di fissaggio 4X (16% di formaldeide) | ||||
| 5 mL | 20% di formaldeide | |||
| 500 L | Acqua DEPC | |||
| 750 L | 1M DEPC tampone di fosfato di sodio, pH 7.4 | |||
| Conservare a 4 gradi centigradi per un massimo di 2-4 settimane. | ||||
| CAUTION: La formaldeide è tossica. Indossare guanti e utilizzare un cappuccio di fumi quando si effettua questa soluzione. | ||||
| Soluzione di lavaggio | ||||
| Mescolare quanto segue: | ||||
| 10 mL | Formamide (finale 25%) | |||
| 4 mL | 20X SSC (finale 2X) | |||
| Riempire l'acqua DEPC a 40 mL | ||||
| Filtro (0,22 m) e conservare a 4 | ||||
| CAUTION: La formamide è tossica. Indossare guanti e utilizzare un cappuccio di fumi quando si effettua questa soluzione. | ||||
| Soluzione di pre-ibridazione | ||||
| 200 L | Formamide (20%) finale | |||
| 100 L | 20X SSC (finale 2X) | |||
| 10 L | 100X VRC (finale 1X) | |||
| 25 L | 4% (w/v) BSA (finale 0,1%) | |||
| 685 L | Acqua DEPC | |||
| NOTA: Vortex lo stock VRC prima di prendere 10 L fuori. | ||||
| CAUTION: Formamide è tossico e un noto teratogeno. Indossare guanti e maneggiarlo sotto un cappuccio di fumi. | ||||
| Soluzione di ibridazione delle sonde | ||||
| 200 L | Formamide (20%) finale | |||
| 100 L | 20X SSC (finale 2X) | |||
| 10 L | 100X VRC (finale 1X) | |||
| 25 L | 4% (w/v) BSA (finale 0,1%) | |||
| 10 L | 40 mg/mL E. coli tRNA (finale 0,4 mg/mL) | |||
| 200 L | 50% solfato dextran (10%) finale | |||
| x LL | 5' set di sonda mRNA (dal punto 1.12) alla finale 4 nM. | |||
| y L | 3' set di sonda mRNA (dal punto 1.12) alla finale 4 nM. | |||
| - | ACQUA DEPC per rendere il volume totale 1 mL | |||
| NOTA: Aggiungere per ultimo il solfato dextran. Poiché è molto viscoso, tagliare l'estremità di una punta di pipette prima di prendere 200 L fuori dal 50% stock. Dopo aver aggiunto solfato di dextran, pipetta su e giù per omogeneizzare la soluzione. | ||||
Tabella 1: Ricette delle soluzioni utilizzate.
Discussione
Qui, abbiamo presentato un protocollo smFISH per misurare la cinetica dell'mRNA in E. coli. Nei protocolli smFISH pubblicati in precedenza per i batteri23, le cellule sono state tenute nei tubi fino alla fine del protocollo, vale a di qui fino a quando non sono pronte per l'imaging. Anche se ha molti vantaggi, come l'associazione minima non specifica di sonde fluorescenti sulla superficie coverslip23, è difficile seguire questi protocolli quando ci sono molti campioni da un esperimento di corso di tempo. In primo luogo, un volume relativamente grande di cellule (>1 mL) deve essere campionato e persino raccolto prima della fissazione. In secondo luogo, i campioni di cellule devono essere centrifugati più volte per scambiare soluzioni e lavarsi dopo la fase di ibridazione. Nel nostro protocollo, un piccolo volume (<1 mL) di coltura viene direttamente miscelato con una soluzione di fissaggio in un tubo da 1,5 mL, contribuendo a "congelare" rapidamente lo stato della cella al momento del campionamento. Inoltre, le cellule rimangono attaccate alla superficie per tutta la procedura e diverse soluzioni possono essere scambiate rapidamente aspirando liquidi con un sistema di filtrazione del vuoto e applicando gocce di soluzione contemporaneamente con una pipetta multicanale. Questa differenza rende il nostro protocollo altamente vantaggioso quando un gran numero di campioni devono essere elaborati contemporaneamente. Utilizzando il nostro protocollo, 12-48 campioni possono essere gestiti contemporaneamente e l'intera procedura FISH può essere completata entro 8 ore, circa una quantità di tempo simile necessaria per alcuni campioni (Figura 2). Anche se abbiamo usato come esempio l'espressione di lac in E. coli, il protocollo è ampiamente applicabile a diversi geni e specie batteriche con considerazioni discusse di seguito.
Per i diversi geni, la prima cosa da considerare sono le sonde smFISH. Si possono progettare sonde oligonucleotide che affiancare l'mRNA di interesse (Figura 1A)13. In questo approccio di sonda "piastrellante", ogni sonda è lunga 20 dollari ed etichettata con un fluoroforo al capolinea 5' o 3'. Questa strategia è conveniente in quanto non è necessaria alcuna manipolazione genetica. In alternativa, una ripetizione tandem della sequenza di 20 bp, estranea alla sequenza genomica (ad esempio, una matrice di sequenza lacO in Caulobacter crescentus14), può essere inserita nella regione non tradotta di un gene di interesse e una singola sonda complementare all'unità di ripetizione viene utilizzata per etichettare l'mRNA (approccio "array"; Figura 1B). In entrambi i casi, più fluorofori decorano un mRNA, dando segnale di fluorescenza amplificato che può essere facilmente differenziato da una singola sonda legata in modo non specifico all'interno di una cellula.
La scelta di approcci "affiancamento" o "array" dipende dal controllo negativo, un campione in cui viene testata l'associazione non specifica delle sonde perché manca dell'mRNA di destinazione. Per le sonde di affiancamento (Figura 1A), un ceppo mutante senza il gene di interesse o una condizione, in cui il gene non è trascritto (ad esempio, la repressione del lac) può servire come controllo negativo per testare il legame non specifico delle sonde. Per l'oggetto basato su matrice ( ), un ceppo di tipo selvatico privo della matrice può fungere da controllo negativo perché non contiene siti di associazione per i probe.
Le condizioni di ibridazione ottimali possono dipendere dalle sequenze di sonda e persino dalla scelta dei coloranti fluoroforo. Abbiamo ottimizzato la condizione di ibridazione per i set di sonde lac, mantenendo la temperatura di ibridazione a 37 gradi centigradi e testando diverse concentrazioni di set di sonde e formamide nella soluzione di ibridazione. Concentrazioni più elevate di formamide tendono a ridurre sia l'associazione non specifica che la specificaassociazione 26,35. Si consiglia di cambiare sistematicamente l'ibridazione e le sue condizioni di lavaggio mantenendo lo stesso tempo di ibridazione e temperatura. Man mano che la condizione diventa più rigorosa, diminuiscono sia l'associazione non specifica che la diminuzione dell'associazione (Figura 6). È importante trovare un punto in cui l'associazione non specifica inizia a raggiungere al di sotto di una soglia accettabile senza compromettere ulteriormente l'associazione specifica. Ad esempio, è stato utilizzato il livello di segnale ottenuto senza sonde ("nessuna sonda") come soglia (Figura 6).
Il metodo smFISH a due colori che etichetta due regioni separate di un mRNA è limitato a geni lunghi. Per misurare il tasso di allungamento della trascrizione, abbiamo approfittato del fatto che la lac è lunga (3075 bp) e la sua espressione può essere indotta da IPTG. Quando un gene è breve, è difficile progettare due set di sonde di affiancamento (vicino a 5' e 3' estremità) e risolvere il ritardo di tempo tra le apparizioni di regioni di mRNA da 5' e 3'. In questo caso, si possono contare gli mRNA nascenti allo stato costante di smFISH e analizzarne la distribuzione con un modello analitico con il tasso di allungamento della trascrizione come parametrodi adattamento 20. Inoltre, quando un gene di interesse non è inducibile, si possono trattare le cellule con rifampicina al momento zero e misurare il cambiamento temporale nelle sottosocieti di mRNA 5' e 3'. Il ritardo dalla diminuzione del segnale mRNA di 5' a quello del segnale mRNA 3' può quindi essere utilizzato per calcolare il tasso di allungamento della trascrizione come fatto inprecedenza 31.
Infine, il protocollo smFISH è versatile e può essere combinato con altri schemi di etichettatura. In precedenza, il locus del DNA è stato visualizzato insieme agli mRNA combinando mRNA FISH con DNA FISH14 o sistema fluorescente reporter-operatore20. I prodotti proteici possono essere visualizzati eseguendo l'immunofluorescenza insieme all'mRNA FISH14,36. Inoltre, può essere combinato con la microscopia tridimensionale super-risoluzione37 per visualizzare gli mRNA in tutte e tre ledimensioni 38,39.
Divulgazioni
Gli autori dichiarano di non avere interessi finanziari concorrenti.
Riconoscimenti
Questo protocollo è stato sviluppato da S.K. durante la sua ricerca post-dottorato nel laboratorio della dott.ssa Christine Jacobs-Wagner presso l'Howard Hughes Medical Institute e il Microbial Sciences Institute dell'Università di Yale. Ringraziamo il Dr. Jacobs-Wagner e i suoi membri del laboratorio per vari input durante lo sviluppo del metodo e Laura Troyer per la lettura critica del manoscritto. S.K. riconosce il sostegno del Searle Scholars Program; K.V. riconosce il sostegno del James Scholar Preble Research Award dell'Università dell'Illinois.
Materiali
| Name | Company | Catalog Number | Comments |
| Bacterial strain | |||
| Escherichia coli MG1655 | |||
| Chemicals, peptides, and others | |||
| Acetonitrile | Sigma-Aldrich | 34851 | UPLC buffer B |
| Ammonium chloride | Fisher Chemical | A661-500 | To make M9 medium |
| Bovine serum albumin (BSA) | Sigma-Aldrich | B2518 | Probe hybridization |
| Calcium chloride | Acros Organics | 349610250 | To make M9 medium |
| Casamino acid | BD Difco | 223050 | To make M9 medium |
| Cy3B NHS ester | GE Healthcare Life Sciences | PA63101 | Fluorophore for FISH probes |
| Cy5 NHS ester | GE Healthcare Life Sciences | PA15101 | Fluorophore for FISH probes |
| DEPC | Sigma-Aldrich | D5758 | |
| Dextran sulfate | Millipore | S4030 | Probe hybridization |
| Dextrose | Fisher Chemical | D16 | To repress the expression of lacZ |
| Dimethyl sulfoxide (DMSO) | Sigma-Aldrich | D8418 | To dissolve fluorophores |
| E. coli tRNA | Sigma-Aldrich | R1753 | Probe hybridization |
| Ethanol | Decon Laboratories | 2701 | Used in DNA purification, lysis, and cleaning coverslips |
| FISH probes | Biosearch Technologies | Sequences are published in ref#16 | |
| Formaldehyde | Ladd Research Industries | 20295 | Fixation |
| Formamide | American Bio | AB00600 | Probe hybridization, pre-hybridization, and wash |
| Glycerol | Americanbio | AB00751-01000 | To make M9 medium |
| Isopropylthio-β-galactoside (IPTG) | Invitrogen | 15529019 | lacZ induction |
| Magnesium sulfate | Fisher Chemical | M65-500 | To make M9 medium |
| 2-Nitrophenyl β-D-fucopyranoside (ONPF) | Santa Cruz Biotechnology | sc-216258 | lacZ repression |
| Picodent twinsil 22 | Picodent | 1300 1000 | Sealant |
| Poly-L-lysine | Sigma-Aldrich | P8920 | To treat the coverslip surface |
| Potassium chloride | Fisher BioReagents | BP366-500 | To make PBS |
| Potassium phosphate monobasic | Fisher BioReagents | BP362-500 | To make PBS and M9 medium |
| Rifampicin | Sigma-Aldrich | R3501 | To stop transcription initiation |
| Saline-sodium citrate buffer (SSC) | Invitrogen | AM9763 | Probe hybridization, pre-hybridization, and wash |
| sodium bicarbonate | Fisher BioReagents | BP328-500 | Fluorophore-probe conjugation |
| Sodium chloride | Fisher BioReagents | BP358-1 | For DNA purification, PBS and M9 medium |
| Sodium phosphate dibasic | Fisher BioReagents | BP332-500 | To make PBS and M9 medium |
| Sodium phosphate monobasic | Fisher BioReagents | BP329-500 | To make a sodium phosphate buffer |
| Super PAP Pen | Invitrogen | 8899 | Hydrophobic marker for coverslips |
| TetraSpek microspheres | Invitrogen | T7280 | Controls for multi-channel registration |
| Thiamine | Sigma-Aldrich | T1270 | To make M9 medium |
| Triethylammonium acetate | Sigma-Aldrich | 90358 | UPLC buffer A |
| Vanadyl ribonucleoside complex (VRC) | Sigma-Aldrich | 94742 | Probe hybridization and pre-hybridization |
| Equipment | |||
| C18 column | Waters | Acquity BEH C18 column | |
| Countertop centrifuge | Eppendorf | 5425 | |
| Countertop incubator | Eppendorf | Thermomixer F1.5 | |
| Incubator (Oven) | Thermo Scientific | 51030514 | Gravity convection |
| Water purification system | Millipore | Milli-Q Reference | |
| Nanodrop | Thermo Scientific | 2000C | |
| Nitrogen gas | Building | For blow-drying coverslips and glass slides | |
| UPLC | Waters | Acquity UPLC system | |
| Vacuum and aspirator | Building | Aspirator is made of a filtration flask with a side arm. | |
| Vacuum concentrator | Labconco | 7810010 | Centrivap; to dry samples collected from UPLC. |
| Vortexer | Scientific Industries | Genie-2 SI-0236 | |
| Water bath shaker | New Brunswick | Innova 3100 | Critical for time-course experiments |
| Water bath sonicator | VWR | 97043-960 | To clean coverslips and glass slides |
| Tools | |||
| 1.5-mL tubes | Eppendorf | 22431021 | DNA lobind tubes |
| 1000-uL pipette tip box | Denville Scientific | P1126 | An empty box after using all the tips |
| Coplin jar | SPI | 01240-AB | To clean coverslips and glass slides |
| Coverslip | Fisher Scientific | 22-050-230 | 24x60 No1 |
| Filtered pipette tips | Denville Scientific | P1121,P1122,P1126 | SHARP® Precision Barrier Tips |
| Forceps | SPI | K35a | To handle clean coverslips and glass slides |
| Glass slide | Fisher Scientific | 12-544-1 | |
| Gloves | Microflex | MK-296-M | |
| Multichannel pipetter | Eppendorf | 2231300045 | To use in the washing step (#7) |
| Pipette | Gilson | P1000, P200, P20 | |
| Reagent reservoir | MTC Bio | P8025-1S | To use in the washing step (#7) |
| Syringe filter (0.22 um) | Millipore | SLGS033SS | |
| Timer | VWR | 62344-641 | |
| Software and algorithms | |||
| MATLAB | Mathworks | R2013 and up | https://www.mathworks.com |
| MicrobeTracker or Oufti | https://www.github.com/JacobsWagnerLab/MicrobeTracker | ||
| https://oufti.org/ | |||
| Stellaris Probe Designer | Biosearch Technologies | https://www.biosearchtech.com/support/tools/design-software/stellaris-probe-designer | |
| Microscope | |||
| CCD camera | Hamamatsu Photonics | Orca-II-ER | |
| Cy3 filter set | Chroma | 49004 | |
| Cy5 filter set | Chroma | 49006 | |
| Epi-fluorescence microscope | Nikon | Eclipse Ti | For phase-contrast and epi fluorescence |
| Fluorescence excitation source | Lumencor | SOLA-E | |
| Nikon Elements software | Nikon | software that controls the microscope setup | |
| Phase-contrast 100x objective | Nikon | Plan Apochromat (NA 1.45) | |
| Probe sequence | |||
| DNA oligos with C6 amino modification at the 5' end | Biosearch Technologies Inc | ||
| lacZ1 | GTGAATCCGTAATCATGGTC | 5' mRNA | |
| lacZ2 | TCACGACGTTGTAAAACGAC | 5' mRNA | |
| lacZ3 | ATTAAGTTGGGTAACGCCAG | 5' mRNA | |
| lacZ4 | TATTACGCCAGCTGGCGAAA | 5' mRNA | |
| lacZ5 | ATTCAGGCTGCGCAACTGTT | 5' mRNA | |
| lacZ6 | AAACCAGGCAAAGCGCCATT | 5' mRNA | |
| lacZ7 | AGTATCGGCCTCAGGAAGAT | 5' mRNA | |
| lacZ8 | AACCGTGCATCTGCCAGTTT | 5' mRNA | |
| lacZ9 | TAGGTCACGTTGGTGTAGAT | 5' mRNA | |
| lacZ10 | AATGTGAGCGAGTAACAACC | 5' mRNA | |
| lacZ11 | GTAGCCAGCTTTCATCAACA | 5' mRNA | |
| lacZ12 | AATAATTCGCGTCTGGCCTT | 5' mRNA | |
| lacZ13 | AGATGAAACGCCGAGTTAAC | 5' mRNA | |
| lacZ14 | AATTCAGACGGCAAACGACT | 5' mRNA | |
| lacZ15 | TTTCTCCGGCGCGTAAAAAT | 5' mRNA | |
| lacZ16 | ATCTTCCAGATAACTGCCGT | 5' mRNA | |
| lacZ17 | AACGAGACGTCACGGAAAAT | 5' mRNA | |
| lacZ18 | GCTGATTTGTGTAGTCGGTT | 5' mRNA | |
| lacZ19 | TTAAAGCGAGTGGCAACATG | 5' mRNA | |
| lacZ20 | AACTGTTACCCGTAGGTAGT | 5' mRNA | |
| lacZ21 | ATAATTTCACCGCCGAAAGG | 5' mRNA | |
| lacZ22 | TTTCGACGTTCAGACGTAGT | 5' mRNA | |
| lacZ23 | ATAGAGATTCGGGATTTCGG | 5' mRNA | |
| lacZ24 | TTCTGCTTCAATCAGCGTGC | 5' mRNA | |
| lacZ25 | ACCATTTTCAATCCGCACCT | ||
| lacZ26 | TTAACGCCTCGAATCAGCAA | ||
| lacZ27 | ATGCAGAGGATGATGCTCGT | ||
| lacZ28 | TCTGCTCATCCATGACCTGA | ||
| lacZ29 | TTCATCAGCAGGATATCCTG | ||
| lacZ30 | CACGGCGTTAAAGTTGTTCT | ||
| lacZ31 | TGGTTCGGATAATGCGAACA | ||
| lacZ32 | TTCATCCACCACATACAGGC | ||
| lacZ33 | TGCCGTGGGTTTCAATATTG | ||
| lacZ34 | ATCGGTCAGACGATTCATTG | ||
| lacZ35 | TGATCACACTCGGGTGATTA | ||
| lacZ36 | ATACAGCGCGTCGTGATTAG | ||
| lacZ37 | GATCGACAGATTTGATCCAG | ||
| lacZ38 | AAATAATATCGGTGGCCGTG | ||
| lacZ39 | TTTGATGGACCATTTCGGCA | ||
| lacZ40 | TATTCGCAAAGGATCAGCGG | ||
| lacZ41 | AAGACTGTTACCCATCGCGT | ||
| lacZ42 | TGCCAGTATTTAGCGAAACC | ||
| lacZ43 | AAACGGGGATACTGACGAAA | ||
| lacZ44 | TAATCAGCGACTGATCCACC | ||
| lacZ45 | GGGTTGCCGTTTTCATCATA | ||
| lacZ46 | TCGGCGTATCGCCAAAATCA | ||
| lacZ47 | TTCATACAGAACTGGCGATC | ||
| lacZ48 | TGGTGTTTTGCTTCCGTCAG | ||
| lacZ49 | ACGGAACTGGAAAAACTGCT | 3' mRNA | |
| lacZ50 | TATTCGCTGGTCACTTCGAT | 3' mRNA | |
| lacZ51 | GTTATCGCTATGACGGAACA | 3' mRNA | |
| lacZ52 | TTTACCTTGTGGAGCGACAT | 3' mRNA | |
| lacZ53 | GTTCAGGCAGTTCAATCAAC | 3' mRNA | |
| lacZ54 | TTGCACTACGCGTACTGTGA | 3' mRNA | |
| lacZ55 | AGCGTCACACTGAGGTTTTC | 3' mRNA | |
| lacZ56 | ATTTCGCTGGTGGTCAGATG | 3' mRNA | |
| lacZ57 | ACCCAGCTCGATGCAAAAAT | 3' mRNA | |
| lacZ58 | CGGTTAAATTGCCAACGCTT | 3' mRNA | |
| lacZ59 | CTGTGAAAGAAAGCCTGACT | 3' mRNA | |
| lacZ60 | GGCGTCAGCAGTTGTTTTTT | 3' mRNA | |
| lacZ61 | TACGCCAATGTCGTTATCCA | 3' mRNA | |
| lacZ62 | TAAGGTTTTCCCCTGATGCT | 3' mRNA | |
| lacZ63 | ATCAATCCGGTAGGTTTTCC | 3' mRNA | |
| lacZ64 | GTAATCGCCATTTGACCACT | 3' mRNA | |
| lacZ65 | AGTTTTCTTGCGGCCCTAAT | 3' mRNA | |
| lacZ66 | ATGTCTGACAATGGCAGATC | 3' mRNA | |
| lacZ67 | ATAATTCAATTCGCGCGTCC | 3' mRNA | |
| lacZ68 | TGATGTTGAACTGGAAGTCG | 3' mRNA | |
| lacZ69 | TCAGTTGCTGTTGACTGTAG | 3' mRNA | |
| lacZ70 | ATTCAGCCATGTGCCTTCTT | 3' mRNA | |
| lacZ71 | AATCCCCATATGGAAACCGT | 3' mRNA | |
| lacZ72 | AGACCAACTGGTAATGGTAG | 3' mRNA | |
Riferimenti
- Bervoets, I., Charlier, D. Diversity, versatility and complexity of bacterial gene regulation mechanisms: opportunities and drawbacks for applications in synthetic biology. FEMS Microbiology Reviews. 43 (3), 304-339 (2019).
- Epshtein, V., Nudler, E. Cooperation between RNA polymerase molecules in transcription elongation. Science. 300 (5620), 801-805 (2003).
- Vogel, U., Jensen, K. F. The RNA chain elongation rate in Escherichia coli depends on the growth rate. Journal of Bacteriology. 176 (10), 2807-2813 (1994).
- Tennyson, C. N., Klamut, H. J., Worton, R. G. The human dystrophin gene requires 16 hours to be transcribed and is cotranscriptionally spliced. Nature Genetics. 9, 184 (1995).
- Singh, J., Padgett, R. A. Rates of in situ transcription and splicing in large human genes. Nature Structural & Molecular Biology. 16, 1128 (2009).
- Selinger, D. W., Saxena, R. M., Cheung, K. J., Church, G. M., Rosenow, C. Global RNA Half-Life Analysis in Escherichia coli Reveals Positional Patterns of Transcript Degradation. Genome Research. 13 (2), 216-223 (2003).
- Bernstein, J. A., Khodursky, A. B., Lin, P. -. H., Lin-Chao, S., Cohen, S. N. Global analysis of mRNA decay and abundance in Escherichia coli at single-gene resolution using two-color fluorescent DNA microarrays. Proceedings of the National Academy of Sciences. 99 (15), 9697-9702 (2002).
- Pérez-Ortín, J. E., Medina, D. A., Chávez, S., Moreno, J. What do you mean by transcription rate. BioEssays. 35 (12), 1056-1062 (2013).
- Tang, F., et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nature Methods. 6 (5), 377-382 (2009).
- Kuchina, A., et al. Microbial single-cell RNA sequencing by split-pool barcoding. BioRxiv. , 869248 (2019).
- Blattman, S. B., Jiang, W., Oikonomou, P., Tavazoie, S. Prokaryotic single-cell RNA sequencing by in situ combinatorial indexing. Nature Microbiology. , (2020).
- Femino, A., Fay, F., Fogarty, K., Singer, R. Visualization of single RNA transcripts in situ. Science. 280 (5363), 585-590 (1998).
- Raj, A., vanden Bogaard, P., Rifkin, S. A., van Oudenaarden, A., Tyagi, S. Imaging individual mRNA molecules using multiple singly labeled probes. Nature Methods. 5 (10), 877-879 (2008).
- Montero Llopis, P., et al. Spatial organization of the flow of genetic information in bacteria. Nature. 466 (7302), 77-81 (2010).
- So, L. -. h., et al. General properties of transcriptional time series in Escherichia coli. Nature Genetics. 43 (6), 554-560 (2011).
- Taniguchi, Y., et al. Quantifying E. coli proteome and transcriptome with single-molecule sensitivity in single cells. Science. 329 (5991), 533-538 (2010).
- Jones, D. L., Brewster, R. C., Phillips, R. Promoter architecture dictates cell-to-cell variability in gene expression. Science. 346 (6216), 1533-1536 (2014).
- Iyer, S., Park, B. R., Kim, M. Absolute quantitative measurement of transcriptional kinetic parameters in vivo. Nucleic Acids Research. 44 (18), 142 (2016).
- Kim, S., Beltran, B., Irnov, I., Jacobs-Wagner, C. Long-Distance Cooperative and Antagonistic RNA Polymerase Dynamics via DNA Supercoiling. Cell. 179 (1), 106-119 (2019).
- Wang, M., Zhang, J., Xu, H., Golding, I. Measuring transcription at a single gene copy reveals hidden drivers of bacterial individuality. Nature Microbiology. 4 (12), 2118-2127 (2019).
- Joo, C., Ha, T. . Labeling DNA (or RNA) for single-molecule FRET. 2012 (9), 1005-1008 (2012).
- Sambrook, J., Russell, D. W. . Standard Ethanol Precipitation of DNA in Microcentrifuge Tubes. 2006 (1), 4456 (2006).
- Skinner, S. O., Sepúlveda, L. A., Xu, H., Golding, I. Measuring mRNA copy number in individual Escherichia coli cells using single-molecule fluorescent in situ hybridization. Nature Protocols. 8 (6), 1100-1113 (2013).
- Adesnik, M., Levinthal, C. The synthesis and degradation of lactose operon messenger RNA in E. coli. Cold Spring Harbor Symposia on Quantitative Biology. 35, 451-459 (1970).
- Campbell, E. A., et al. Structural mechanism for rifampicin inhibition of bacterial RNA polymerase. Cell. 104 (6), 901-912 (2001).
- Raj, A., Tyagi, S., Walter, N. G. . Methods in Enzymology. 472, 365-386 (2010).
- Sliusarenko, O., Heinritz, J., Emonet, T., Jacobs-Wagner, C. High-throughput, subpixel precision analysis of bacterial morphogenesis and intracellular spatio-temporal dynamics. Molecular Microbiology. 80 (3), 612-627 (2011).
- Paintdakhi, A., et al. Oufti: an integrated software package for high-accuracy, high-throughput quantitative microscopy analysis. Molecular Microbiology. 99 (4), 767-777 (2016).
- Moffitt, J. R., Zhuang, X., Filonov, G. S., Jaffrey, S. R. . Methods in Enzymology. 572, 1-49 (2016).
- Yu, J., Xiao, J., Ren, X., Lao, K., Xie, X. S. Probing gene expression in live cells, one protein molecule at a time. Science. 311 (5767), 1600-1603 (2006).
- Chen, H., Shiroguchi, K., Ge, H., Xie, X. S. Genome-wide study of mRNA degradation and transcript elongation in Escherichia coli. Molecular Systems Biology. 11 (1), 781 (2015).
- Vogel, U., Sørensen, M., Pedersen, S., Jensen, K. F., Kilstrup, M. Decreasing transcription elongation rate in Escherichia Coli exposed to amino acid starvation. Molecular Microbiology. 6 (15), 2191-2200 (1992).
- Yang, S., et al. Transcription and translation contribute to gene locus relocation to the nucleoid periphery in E. coli. Nature Communications. 10 (1), 5131 (2019).
- Zenklusen, D., Larson, D. R., Singer, R. H. Single-RNA counting reveals alternative modes of gene expression in yeast. Nature Structural & Molecular Biology. 15 (12), 1263-1271 (2008).
- Fontenete, S., Guimarães, N., Wengel, J., Azevedo, N. F. Prediction of melting temperatures in fluorescence in situ hybridization (FISH) procedures using thermodynamic models. Critical Reviews in Biotechnology. 36 (3), 566-577 (2016).
- Sepúlveda, L. A., Xu, H., Zhang, J., Wang, M., Golding, I. Measurement of gene regulation in individual cells reveals rapid switching between promoter states. Science. 351 (6278), 1218-1222 (2016).
- Huang, B., Wang, W., Bates, M., Zhuang, X. Three-Dimensional Super-Resolution Imaging by Stochastic Optical Reconstruction Microscopy. Science. 319 (5864), 810-813 (2008).
- Moffitt, J. R., Pandey, S., Boettiger, A. N., Wang, S., Zhuang, X. Spatial organization shapes the turnover of a bacterial transcriptome. eLife. 5, 13065 (2016).
- Fei, J., et al. Determination of in vivo target search kinetics of regulatory noncoding RNA. Science. 347 (6228), 1371-1374 (2015).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati