
このコンテンツを視聴するには、JoVE 購読が必要です。 サインイン又は無料トライアルを申し込む。
Method Article
動的細胞突起におけるソフトウェア支援トラッキングのためのグラフィカルユーザインタフェース
要約
我々は、動的細胞突起の長さに沿った相対的なタンパク質濃度の半自動追跡のためのソフトウェアソリューションを提示する。
要約
フィロポディアは、移動および細胞 - 細胞通信に関連する動的で指様の細胞突起である。糸球体の開始、伸長、その後の安定化または収縮の基礎となる複雑なシグナル伝達機構をよりよく理解するためには、これらの動的構造における時空間タンパク質活性を決定することが重要である。フィロポディアのタンパク質機能を解析するために、我々は最近、フィロポードの形状変化に適応する半自動追跡アルゴリズムを開発した。ここでは、最適化された細胞操作、画像取得およびソフトウェア分析のための詳細な段階的プロトコールを提示する。また、画像解析やデータ表示の際にオプション機能を使用するための手順や、重要なステップすべてに関するトラブルシューティングのガイドラインを提供します。最後に、dの比較フィロポディア定量化のために利用可能な他のプログラムと共に、エスケープされた画像分析ソフトウェア。一緒に、提示されたプロトコルは、画像分析ソフトウェアを用いて糸状突起におけるタンパク質動態の正確な分析のためのフレームワークを提供する。
概要
アクチン調節タンパク質の時空間制御は、フィロポジウムダイナミクス1,2に関連している。したがって、これらの動的構造の開始、伸長、安定化または崩壊の根底にある機構の理解を促進するためには、時間の経過とともに糸状体の長さに沿って空間的に分解されたタンパク質濃度を追跡することが重要である 3,4 。線維柱は、絶えず5つ折りになって屈曲する動的な微細構造であり、したがって、ラインスキャンのような単純なアプローチを用いた分析が不可能である細胞質ゾルにおけるタンパク質分析とは異なり、
人工肛門形状を追跡するためのさまざまなソフトウェアソリューションが利用可能です(6,7,8,9)。類似細胞体内のタンパク質動態をレシオメトリックに追跡するソフトウェアが開発されました10,11 。我々は、有形形状の自動追跡と時空間タンパク質解析を組み合わせるために、最近、凸包アルゴリズム12に基づく画像解析ソフトウェアを開発した。この新しい分析方法は、グラフィカルユーザーインターフェース(GUI) を介して操作され、初めて、繊維口径および成長速度に沿った相対的なタンパク質濃度を組み合わせることにより、これらの動きに関係なく時空間タンパク質分布の正確な測定を可能にする動的構造12 。
ソフトウェアの背後にある考え方(ソースコードは自由に利用可能です、以下を参照)は、凸包の頂点の1つがフィリピンの先端と一致することです( 図1A )。次のフレームfoを見ることによってrは凸包の最も近い頂点であり、ムービングチップは映画全体を通して追跡することができる。先端が各フレームで検出されると、フィリピンの基底の基準点( 図1B )と先端を結合することによって、その位置を軸の描画に使用します。最後に、軸に直交する線に沿った最大強度を有する中央値画素によって位置が決定される等距離節点を使用して、骨棘形状に従う骨格を決定する。この適応性のあるバックボーンを利用して、キモグラフを作成して、糸状体の長さに沿って最大3チャンネルの糸球体の成長およびタンパク質濃度を追跡する( 図1C )。

図1:イメージ解析ソフトウェアの動作原理 ( A )後ろのアルゴリズムソフトウェア。ステップ1では、ユーザは、フィロポジウムの基準(ベース)および頂点(先端)を指定する。ステップ2-1では、最大輝度値を有する中央値ピクセルを用いてフィリピンのバックボーンが得られる。ステップ3では、バックボーンを空間タンパク質強度プロファイルに使用する。ステップ2-2では、ソフトウェアは次のフレームの先端を自動的に追跡する。全体の手順が繰り返されます。 ( B )追跡に使用されている凸包のような重要な要素を導入している実在極を持つアルゴリズムのスナップショット。 ( C )アルゴリズムで測定可能なパラメータの概要。この数字は参考文献12から変更されています。 この図の拡大版を見るには、ここをクリックしてください。
{kind=link}
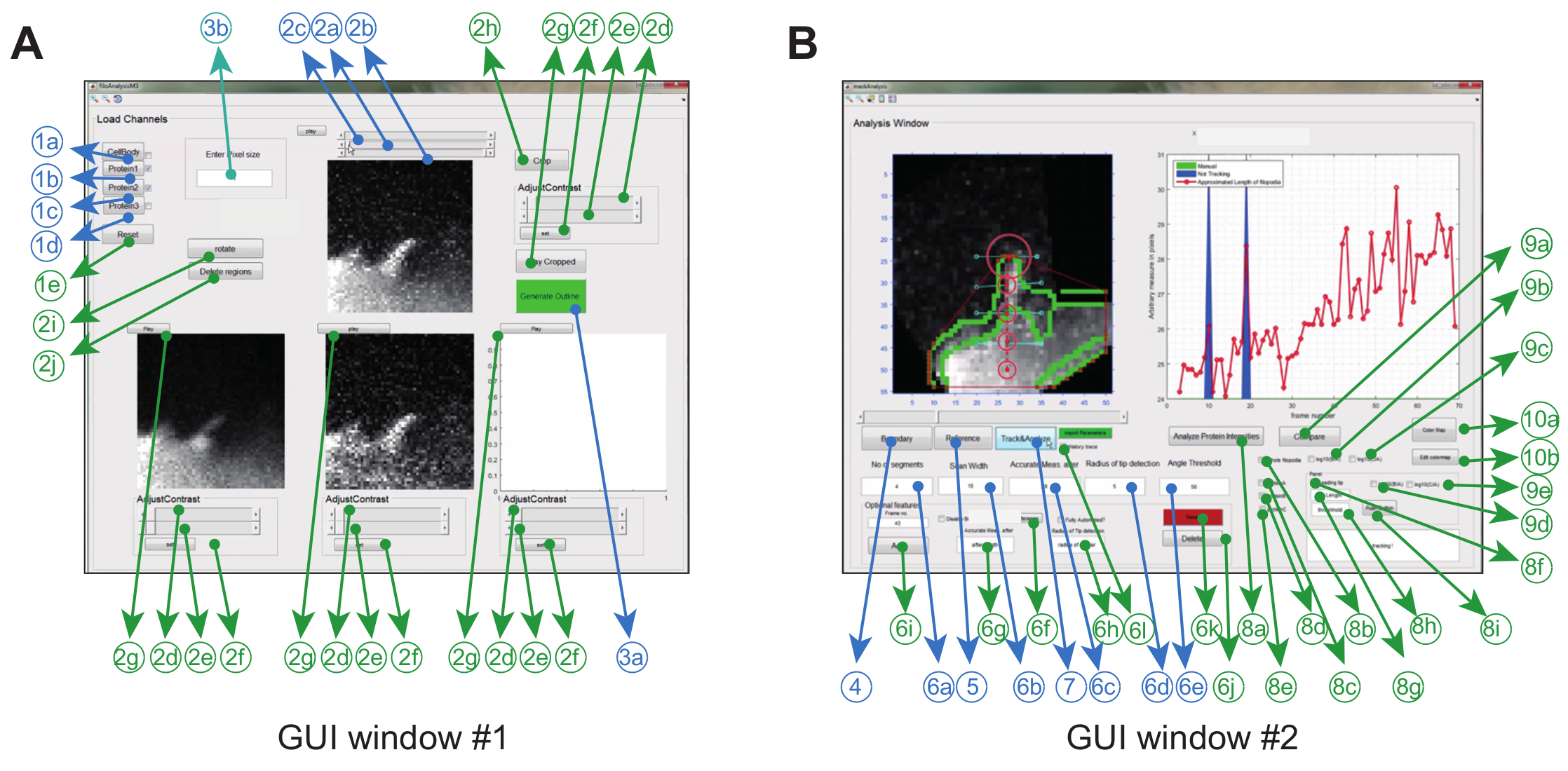
画像解析ソフトウェアは、グラフィカルな使用によってMatlab(プログラミングソフトウェアと呼ばれる)で操作されますrインターフェース。特定の実験設定の柔軟性と堅牢性を最大限にするために、ユーザーは一連のトラッキングパラメータ(許容される曲げ角度やフレーム間移動など)を調整したり、ムービーの一部の修正(クロッピング、回転、 ( 図2Aおよび表1) 。
| GUI | いいえ。 | 必須 | 説明 | 名前(GUI内) | ||||
| #1 | 1a | Y | (ボックスをチェックインして)セル本体を表す積み上げられた.tiffファイルをロードするか、チャンネルからスーパーインポーズされたセル本体を作成する | CellBody | ||||
| #1 | 1b | Y | タンパク質1に対応する積み上げ画像ファイルを読み込む | タンパク質1 | ||||
| #1 | 1c | Y | タンパク質2に対応する積み重ね画像ファイルを読み込む | プロテイン2 | ||||
| #1 | 1d | Y | タンパク質3に対応する積み重ねられた画像ファイルを読み込む | プロテイン3 | ||||
| #1 | 1e | N | プリロードされた積み重ねられた画像ファイルにすべてをリセットします。 | リセット | ||||
| #1 | 2a | Y | スクロールバーを使用して、GUIウィンドウ#2での分析の初期フレームを決定します。 | NA | ||||
| #1 | 2b | Y | スクロールバーを使用して、GUIウィンドウ#2で解析するための最終フレームを決定します | NA | ||||
| #1 | 2c | Y | curreを表すスクロールバーNTフレーム | NA | ||||
| #1 | 2d | N | すべてのピクセルがゼロに設定されるピクセルのグレー値 | NA | ||||
| #1 | 2e | N | すべてのピクセルが最大値に設定されるピクセルのグレー値 | NA | ||||
| #1 | 2f | N | <2e>&<2f>で指定されたピクセルの輝度値を設定します。 | セット | ||||
| #1 | 2g | N | 強度調整されたムービーを再生する | 遊びます | ||||
| #1 | 2時間 | N | クロップ画像 | 作物 | ||||
| #1 | 2i | N | イメージを回転する | 回転する | ||||
| #1 | 2j | N | スタック全体の領域を削除する | リージョンを削除する | ||||
| #1 | 3a | Y | クリックすると「解析ウィンドウ」(GUIウィンドウ#2)が開きます。 | 追跡ウィンドウ | ||||
| #1 | 3b | Y | ピクセルのサイズをミクロン単位で入力します | ピクセルサイズを入力してください | ||||
| #2 | 4 | Y | クリックすると、重ね合わされたセル本体の境界/エッジ画像が生成されます | 境界 | ||||
| #2 | 5 | Y | クリックして、糸状虫の基部と先端を選択します | Referernce | ||||
| #2 | 6a | Y | セグメントまたはノードの数を入力してください | セグメント数 | ||||
| #2 | 6b | Y | スキャンの長さを入力します(軸に垂直) | スキャン幅 | ||||
| #2 | 6c | Y | フィロポディアが曲がり始める長さを入力してください | 後の正確な測定 | ||||
| #2 | 6d | Y | 先端検出円の半径(すなわち、次のフレームで頂点が局在化できる領域)を入力します。 | 先端検出の半径 | ||||
| #2 | 6e | Y | フィリピンが垂直軸から曲げることができる最大角度を入力します | 角度の閾値 | ||||
| #2 | 6f | N | その特定のフレームのベースとチップの参照点を追加する | リファレンスを選択 | ||||
| #2 | 6g | N | 特定のフレームの曲がり始める長さを入力します | 後の正確な測定 | ||||
| #2 | 6h | N | 特定のフレームの検出円の半径を入力します | 先端検出の半径 | ||||
| #2 | 6i | N | 特定のフレームのすべてのパラメータを入力した後、その値をメモリおよびファイルに保存して参照する | 追加 | ||||
| #2 | 6j | N | クリックすると、そのフレームの手動パラメータが削除されます。 | 削除 | ||||
| #2 | 6k | N | クリックすると、すべてのフレームで 'オプション機能パネル'を使用して手動で保存されたすべてのパラメータが削除されます | リセット | ||||
| #2 | 6l | N | 追跡する前にチェックインして、すべての追跡結果を将来の参照のためにメモリに格納する | 履歴トレース | ||||
| #2 | 7 | Y | トラッキングを開始するにはクリックしてください | トラックと分析 | ||||
| #2 | 8a | N | クリックしてタンパク質チャネル強度の追跡を開始する | タンパク質の強度を分析する | ||||
| #2 | 8b | N | チェックインして、糸状体の長さに沿ったタンパク質チャネルの強度を追跡する | 完全な糸状仮足 | ||||
| #2 | 8c | N | リファレンスタンパク質またはプロテインAを追跡するためにチェックイン | プロテインA | ||||
| #2 | 8d | N | タンパク質Bを追跡するためにチェックイン | ProteinB | ||||
| #2 | 8e | N | タンパク質Cを追跡するためにチェックイン | ProteinC | ||||
| #2 | 8f | N | タンパク質の平均強度を追跡するためにチェックインする電子チップ | リーディングチップ | ||||
| #2 | 8g | N | 先端の長さを入力してください | 先端の長さ | ||||
| #2 | 8時間 | N | チップが形成を開始するベースからの最小距離を入力します | 閾値 | ||||
| #2 | 8i | N | 先頭のチップ解析結果をファイルに保存する場合にクリックします | ボタンを押す | ||||
| #2 | 9a | N | レシオメトリックなタンパク質分析を開始するにはクリックしてください | 比較 | ||||
| #2 | 9b | N | タンパク質BをAと比較するためにチェックインする | log10(B / A) | ||||
| #2 | 9c | N | チェックインして、プロテインCとAを比較する | log10(C / A) | ||||
| #2 | 9d | N | 先端のAに関してプロテインBを比較するためにチェックインする | log10(B / A) | ||||
| #2 | 9e | N | 先端のAに関してプロテインCを比較するためにチェックインする | log10(C / A) | ||||
| #2 | 10a | N | 他のカラーマップを選択してください(デフォルト:Jetplot) | カラーマップ | ||||
| #2 | 10b | N | カラーマップを編集する | カラーマップを編集 | ||||
表1:GUIに存在するすべての機能の概要Windows#1および#2。
これが完了すると、プログラムは凸包を作成し、ムービー全体で自動的にヒントを追跡します。レシオメトリックなキモグラフ、成長速度、およびフィリピンの長さなど、映画から抽出されたパラメータa表示され、画像として、またデータファイルとして作業フォルダに保存されます。その後、生存期間、成長速度および収縮速度などの他のパラメータを抽出し、格納されたデータファイルからさらに分析することができる( 図2B )。

図2:イメージ解析ソフトウェアを使用するためのグラフィカルユーザーインターフェイス ( A )GUIウィンドウ#1は画像の読み込みと処理に使用されます。プログラムは最大3つのタンパク質チャネルをロードすることができ、それによって2つのチャネルが対に比較される。 ( B )GUIウィンドウ#2は、フィロポジウムと時空間および比率メトリックタンパク質分析を追跡するために使用されます。ここでも、オプション機能は緑色でマークされています。この数字は変更されています参考文献12から編集されている。 この図の拡大版を見るには、ここをクリックしてください。
{kind=link}
ここでは、サンプルの準備とソフトウェア処理のための詳細なプロトコールを示します。細胞の培養と画像分析に最適化されたムービーの取得に関する詳細な手順から始めます。このデータ取得セクションに続いて、画像分析ソフトウェアを操作するための詳細な説明が続きます。プロトコル全体を通して、データの収集と処理の際に考慮する必要がある重要なステップとオプション機能を紹介します。最後に、我々は画像解析ソフトウエアを用いて様々なモデルシステムからの糸状仮説を解析し、記述された画像解析ソフトウェアと、有棘線量定量化のための他のプログラムとの比較および制限および将来の方向性に関する議論を終了する。
プロトコル
1.細胞培養
- 4.5g / LのD-グルコース、L-アラニン-L-グルタミンジペプチド、ピルビン酸塩、10%ウシ胎児血清、および10単位/ mlのペニシリン/ストレプトマイシンを含むダルベッコ変法イーグル培地(DMEM)中の培養HeLaまたはCOS細胞。 L-グルタミン、グルタミン酸またはアスパラギン酸を含まない培養培地中の培養ニューロンで、0.5mMのL-アラニン-L-グルタミンジペプチド、無血清ニューロン補充物および10単位/ mlのペニシリン/ストレプトマイシンを補充した。
- 40%のコンフルエントに達したら、製造業者の指示に従ってトランスフェクション試薬を用いて細胞を選択したコンストラクトでトランスフェクションする。 37℃、5%CO 2のインキュベーターにトランスフェクションした細胞を15〜18時間保管してください。
- 画像取得中のpHおよび浸透圧の変化を減少させるために、20 mM 4-(2-ヒドロキシエチル)-1を含む37℃の予熱した培地で90%まで細胞培養チャンバーを満たす(イメージング前) - ピペラジンエタンスルホン酸(HEPES)。封印する蓋の内側に真空グリースの薄層を塗布し、トランスフェクトされた細胞を含む培養チャンバー上に軽く押し付ける。
2.画像取得
注:糸状仮足の長さは2〜10μmです。フィロポディアは平均速度0.05-0.1μm/ sで増殖する13,14 。
- 60Xまたは100Xの対物レンズを使用し、ピクセルビニングなしの顕微鏡(ここでは、回転円板共焦点顕微鏡)を使用して画像を取得します。フィリピンのダイナミクスを追跡するには、1ヘルツ(Hz)以上の取得速度を使用します。焦点外れアーチファクトを最小限に抑えるために、基底膜( すなわち、基材表面)に近い画像糸状仮足(filopodia)。
- スムーズなトラッキングを保証するには、信号対雑音比(SNR)が4を超えるようにカメラの露光時間とレーザー強度を調整します。個々のチャネルの飽和を避ける8ビット画像では255、16ビット画像では65,535)、以後の画像解析はできません。
- ブリードスルーを避けるために、顕微鏡のレーザーラインとフィルターに適合する蛍光タグのみを使用してください(詳細は参考文献15を参照)。
3.画像前処理
注:ImageJまたは他の利用可能なソフトウェアを使用して画像16,17を前処理します。
- サンプルが動いている場合は、分析前に利用可能なソフトウェア( 例: https : //github.com/NMSchneider/fixTranslation-Macro_for_ImageJ)を使用して横方向ドリフトを補正します。アキシャルドリフト(z方向の動き)でムービーを除外します。
- バックグラウンド( すなわち 、細胞外領域のグレー値)、漂白( すなわち 、蛍光タンパク質の損傷による蛍光強度の場合の連続的な損失)および可能なブリードトラフ( すなわち 、1つのインフルエンザからのシグナル( 例えば、 http://imagej.net/Category:Pluginsおよび16,17)を使用して、2つのチャンネルの蛍光プローブ(orescence probe)
注:個々のチャンネルの蛍光強度はソフトウェアによって変更されません。 - 後続の画像解析を確実にするために、特定のタンパク質チャンネルに対応するムービーを、グレースケールで、ワークフォルダ内の「.tiff」スタックファイル形式で保存します。
注:スタックの寸法(サイズ、長さ)は、すべてのチャネルで同じでなければなりません。
4.画像解析 - ステップ1:画像を読み込む
注記:ここに記載されているソフトウェアはMatlab(プログラミングソフトウェアと呼ばれます)で書かれており、このプログラムでのみ実行されます。
- 次のサイトから、画像解析に必要なファイルがすべて入っているzipフォルダをダウンロードしてください。https://campus.uni-muenster.de/en/einrichtungen/impb0/nanoscale-forces-in-cells/softw/である。ファイルを解凍してワークフォルダにコピーします。
- インストールが完了したら、プログラミングソフトウェアを開き、 'filopodiaAnalysisM3.fig'を実行します。 GUIウィンドウ#1が図2Aに示すように開く。
- プロテインAの場合は<1b>、プロテインBの場合は<1c>、プロテインCの場合は<1d>を使用して、保存したスタックされた '.tiff'ファイルをGUI Window#1にロードします。 図2および表1を参照してください。
注記:GUIウィンドウ#1に表示されているプロテインAは、最終的なレシオメトリック解析の参照チャネルとして機能します。 - <1a>をクリックして、タンパク質チャンネルから細胞のスーパーインポーズ画像を作成します。
- <2a>をクリックして最初のフレームを、<2b>を解析に使用した最後のフレームに割り当てます。
- 必要に応じて、ボタン<2h>を使用して目的のフィリピンを含む関心領域(ROI)をクロッピングし、イメージを回転させます歌ボタン<2i>を押すか、フリーハンド描画ツール<2j>を使用して不要な領域を削除します。
注:イメージ解析を合理化するため、ImageJを使用して、ROI(クロップ、回転、削除)と他の前処理ステップ(バックグラウンド減算、漂白補正など)を分離することをお勧めします。 - 各フレームのスライダ<2c>を品質管理のために動かし、フィリピンが映画全体を通して明確に見えるかどうかをチェックします。
5.画像解析 - ステップ2:トレースを生成する
- GUIウィンドウ#1のボタン<3a>をクリックしてGUIウィンドウ#2を開きます ( 図2Bを参照)。
- GUIウィンドウ#2のボタン<4>をクリックして、(<1a>をクリックした後にGUIウィンドウ#1で生成された)重ね合わされたセル本体のマスクを生成します。また、マスクの境界線が生成され、頂点を得るために凸包が実装されます。
- クリckボタン<5>;カーソルが表示されます。カーソルを使用して、ベースを選択します(ここからフィリポダイアルの先端の距離が測定されます)。最初に現れたフレーム内のフィリポディアの先端が続きます。これを行うには、ウィンドウ#2のスライダーを動かします)。
注記:データ出力のエラーを最小限に抑えるには、ベースポイントを軸に沿ってフィリピン先端の下に垂直に配置します。基点を他の場所に配置する( 例えば横方向にシフトさせる)ことにより、細胞体内の蛍光値に指向性バイアスを導入することができる。 - <6c>を使用して閾値の長さを選択します(これを超えると、フィロポディアは曲がります)。
注:この長さは、(<5>を使用して選択された)基点と細胞体の境界( すなわち 、糸状仮足が成長し始める領域)との間の距離として定義される。測定距離はピクセル単位です。 - <6a>のフィロポディアの形状を近似するために使用するセグメントの数を指定します。
注:m最も多くのセグメントの数は、フィロポジウムによって到達される最大長に依存するが、フィロポジウムがどのように曲がるかにも依存する。ベースとチップの間のピクセル数( つまり 、スレッショルド長)よりも大きなセグメント数を選択しないでください。手順5.4で定義したしきい値の長さよりも多くのセグメントを選択する。 ( すなわち 、基部と先端との間のピクセル数)は、フィロポジウムの長さを過大評価する結果となる。 - <6b>にノードを配置する水平スキャナとして機能するスキャン幅を指定します。
注記:これらのノードは、基盤と先端をフィロソームの本体に結合する線( すなわち 、バックボーンを作成する線)に適合させるためにプログラムによって使用されます。開始点として、最大長に係数を掛けた値に等しいピクセル値を入力します 。
。 - ボックス<6d>にスキャン半径(ピクセル単位)を指定します。 obより約50%大きい値を使用するインターフレーム先端変位を提供した。
注:非常に大きなスキャン半径を使用すると、実際の糸状裂先端が存在しないフレーム( たとえば 、平面外の動きまたは低いSNR)で、不要な凸包点を取得する可能性があります。 - ボックス<6e>に曲げ角度を指定します。
注:角度閾値は、全解析中にフィリピンが曲がる最大角度によって決定されます。角の閾値を指定することにより、フィロポディアが細胞体に向かって曲がるときに、フィロポディアの側面から成長する望ましくない構造の追跡をソフトウェアが排除するのに役立つ。このプログラムは、垂直軸から45度未満の傾斜角を有する糸門のために確実に機能する。 - トラッキングを開始するには、GUIウィンドウ#2のボタンをクリックします。 GUIウインドウ#2の「履歴トレース」ボックスをクリックして、将来の参照のために追跡プロトコル全体を保存します。
注記:追跡手順が完了した後、各フレームのフィロポジウムの長さが将来の参考のために 'dynamics.xlsx'の 'length_vel'という名前のシート。同様に、他のすべてのトラッキングパラメータは、 'dynamics.xlsx'の 'parameters'という名前のシートに保存されます。 - 必要に応じて、フィロポディアがすべてのフレームで自動的に検出されない場合は、次の手順を実行して手動で修正します。
- ウィンドウ#2のスライダを使用してそれぞれのフレームにアクセスし、フィリピンの先端を手動で選択します。
注:凸包点が検出されないフレームは、GUIウィンドウ#2のトラッキングウィンドウで青色の領域で表されます。そのフレームの節座標にアクセスするために追跡プログラムが開始される前に、「履歴トレース」ボタンをチェックすることが必須です。 - GUIウィンドウ#2で<6f>を使用して基準点(ベースに続いて先端)を選択します。 「スキャン長さ」、「正確な測定後」、「最大曲げ角度」などの他のパラメータを指定します。;ステップ5.4-5.8で説明したようにそのフレームに対して
- ボタン<6i>をクリックして、新しいパラメータ(その1つのフレームに固有)を保存します。このステップは、青色領域によって示されるすべてのフレームについて繰り返される。
- 終了したら、<7>を使用してトラッキング手順を再初期化します。
注記:この修正は、映画の中で突然1つの留学生から別の留学生に切り替える場合にも使用できます。代わりに、複数の糸状仮足を含むムービーのクロッピングを検討してください。
- ウィンドウ#2のスライダを使用してそれぞれのフレームにアクセスし、フィリピンの先端を手動で選択します。
画像解析 - ステップ3:時空間タンパク質分析
- 時空間解析のために、<8b>で表されるボックスを選択し、続いて目的のタンパク質チャネル(<8c>および/または<8d>および/または<8e>)を選択する。
注:プロテインAを参照として使用するので、レシオメトリック分析(<9a>、<9b>および/または<9c>)の前にプロテインAを選択することが必須です。 - <8a>をクリックします。ステップ5.9で生成されたトレースを使用して、糸道長に沿ってタンパク質の追跡を開始する。
7.画像解析 - ステップ4:比率測定タンパク質分析
- チェックボックス<9b>または<9c>を選択し、ボタン<9a>をクリックして時空間レシオメトリックプロットを取得します。
注:相対タンパク質濃度の誤った表現を避けるために、レシオメトリック画像はX / Yとしてプロットされず、log(X / Y)としてプロットされます。将来の使用のために、レシオメトリックプロットは '.png'および '.fig'形式のファイルでエクスポートされ、生のプロットデータは 'dynamics.xlsx'ファイル内に保存されます。
8.画像解析 - ステップ5:口腔先端解析
- チェックボックス<8f>を選択し、<8g>と<8h>を使用してベースからチップの長さとしきい値の長さを指定します。 <押しボタン>をクリックして、データを「dynamics.xlsx」ファイルに保存します。 をクリックすると、線維先端チップのタンパク質強度の痕跡を測定し、レシオメトリック分析のために保存する。
注:チップの長さは、分析に使用されるチップのピクセル数を決定します。ソフトウェアは、各フレームの先端のピクセルの平均強度値を返します。閾値長さは、基底部から先端までの最短距離を決定します(フィリポダイアル先端部が成長を開始する(そして、プログラムが先端部の強度値を追跡するとき)。したがって、チップの長さはしきい値の長さよりも小さくなければなりません。 - <9d>または<9e>を使用して分析する希望の比率をクリックします。
- 比較ボタン<9a>をクリックしてレシオメトリックデータを生成します。
結果
線維性アクチン(f-トラクチン18 、赤色)および細胞質ゾル基準(緑色)のマーカーでトランスフェクトしたCOS細胞を用いて、アクチンに富む糸状突起を見出した( 図3A 、上部パネル)。時系列は、糸状仮足が急速に伸び縮みすることを示した( 図3A 、中央パネル)。画像解析ソフトウェアを使用して、...
ディスカッション
ここでは、凸包アルゴリズムを用いて、これらの動的構造における糸球体成長ダイナミクスおよび相対タンパク質濃度の分析のための詳細なプロトコルを提示する。このソフトウェアを使用すると、1回の操作で最大3つのチャネルをペアワイズで比較することができます。これにより、2つのチャネル(タンパク質)の相対濃度が伸長/収縮サイクルを通じて決定され、画像ファイルとデータフ...
開示事項
著者は何も開示することはない。
謝辞
著者らは、DFG(EXC-1003からMG)への資金提供を認めている。
資料
| Name | Company | Catalog Number | Comments |
| DMEM | Life Technologies | 31966-021 | |
| 10% Fetal bovine serum | Biochrom AG | L11-044 | |
| Lipofectamine 2000 | Life Technologies | 11668-027 | |
| 1% penicillin/streptomycin | Biochrom AG | 12212 | |
| Neurobasal Medium | Life Technologies | 21103-049 | |
| B27 | Life Technologies | 17504-044 | |
| HEPES (1M stock solution) | Life Technologies | 15630 | |
| Citrine-N1 | Addgene | 54593 | |
| Labtech | Thermo | 155411 | |
| Glutamax-I | Thermo | 35050-061 | |
| Hela | Leibniz Institute DSMZ | ACC-57 | |
| COS 7 | Leibniz Institute DSMZ | ACC-60 | |
| 3T3 cells | Leibniz Institute DSMZ | ACC-59 | |
| Microscope | Nicon Eclipse | ||
| Camera | Andor | DU888 Ultra | |
| Confocal Unit | Yokagawa | CSU-X1 | |
| Pyruvate | Gibco | 31966-021 |
参考文献
- Dunaevsky, A., Tashiro, A., Majewska, A., Mason, C., Yuste, R. Developmental regulation of spine motility in the mammalian central nervous system. Proc Natl Acad Sci U S A. 96 (23), 13438-13443 (1999).
- Matus, A., Brinkhaus, H., Wagner, U. Actin dynamics in dendritic spines: a form of regulated plasticity at excitatory synapses. Hippocampus. 10 (5), 555-560 (2000).
- Galic, M., et al. Dynamic recruitment of the curvature-sensitive protein ArhGAP44 to nanoscale membrane deformations limits exploratory filopodia initiation in neurons. Elife. 3, e03116 (2014).
- Hotulainen, P., et al. Defining mechanisms of actin polymerization and depolymerization during dendritic spine morphogenesis. J Cell Biol. 185 (2), 323-339 (2009).
- Leijnse, N., Oddershede, L. B., Bendix, P. M. Helical buckling of actin inside filopodia generates traction. Proc Natl Acad Sci U S A. 112 (1), 136-141 (2015).
- Tsygankov, D., et al. CellGeo: a computational platform for the analysis of shape changes in cells with complex geometries. J Cell Biol. 204 (3), 443-460 (2014).
- Xiong, Y., et al. Automated characterization of cell shape changes during amoeboid motility by skeletonization. BMC Syst Biol. 4, 33 (2010).
- Styner, M., Gerig, G., Lieberman, J., Jones, D., Weinberger, D. Statistical shape analysis of neuroanatomical structures based on medial models. Med Image Anal. 7 (3), 207-220 (2003).
- Blum, H., Wathen-Dunn, W. A transformation for extracting new descriptors of shape. Models for the Perception of Speech and Visual Form: Proceedings of a Symposium. , 362-380 (1967).
- Barry, D. J., Durkin, C. H., Abella, J. V., Way, M. Open source software for quantification of cell migration, protrusions, and fluorescence intensities. J Cell Biol. 209 (1), 163-180 (2015).
- Machacek, M., et al. Coordination of Rho GTPase activities during cell protrusion. Nature. 461 (7260), 99-103 (2009).
- Saha, T., et al. Automated analysis of filopodial length and spatially resolved protein concentration via adaptive shape tracking. Mol Biol Cell. 27 (22), 3616-3626 (2016).
- Argiro, V., Bunge, M. B., Johnson, M. I. A quantitative study of growth cone filopodial extension. J Neurosci Res. 13 (1-2), 149-162 (1985).
- Mogilner, A., Rubinstein, B. The physics of filopodial protrusion. Biophys J. 89 (2), 782-795 (2005).
- Shaner, N. C., Steinbach, P. A., Tsien, R. Y. A guide to choosing fluorescent proteins. Nat Methods. 2 (12), 905-909 (2005).
- Abramoff, M. D., Magalhaes, P. J., Ram, S. J. Image Processing with ImageJ. Biophotonics International. 11, 36-42 (2004).
- Courtney, J., Woods, E., Scholz, D., Hall, W. W., Gautier, V. W. MATtrack: A MATLAB-Based Quantitative Image Analysis Platform for Investigating Real-Time Photo-Converted Fluorescent Signals in Live Cells. PLoS One. 10 (10), e0140209 (2015).
- Schell, M. J., Erneux, C., Irvine, R. F. Inositol 1,4,5-trisphosphate 3-kinase A associates with F-actin and dendritic spines via its N terminus. J Biol Chem. 276 (40), 37537-37546 (2001).
- Korobova, F., Svitkina, T. Molecular architecture of synaptic actin cytoskeleton in hippocampal neurons reveals a mechanism of dendritic spine morphogenesis. Mol Biol Cell. 21 (1), 165-176 (2010).
- Cheadle, L., Biederer, T. The novel synaptogenic protein Farp1 links postsynaptic cytoskeletal dynamics and transsynaptic organization. J Cell Biol. 199 (6), 985-1001 (2012).
- Tarnok, K., et al. A new tool for the quantitative analysis of dendritic filopodial motility. Cytometry A. 87 (1), 89-96 (2015).
- Hendricusdottir, R., Bergmann, J. H. F-dynamics: automated quantification of dendrite filopodia dynamics in living neurons. J Neurosci Methods. 236, 148-156 (2014).
- Fanti, Z., Martinez-Perez, M. E., De-Miguel, F. F. NeuronGrowth, a software for automatic quantification of neurite and filopodial dynamics from time-lapse sequences of digital images. Dev Neurobiol. 71 (10), 870-881 (2011).
- Costantino, S., et al. Semi-automated quantification of filopodial dynamics. J Neurosci Methods. 171 (1), 165-173 (2008).
- Nilufar, S., Morrow, A. A., Lee, J. M., Perkins, T. J. FiloDetect: automatic detection of filopodia from fluorescence microscopy images. BMC Syst Biol. 7, 66 (2013).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved