
このコンテンツを視聴するには、JoVE 購読が必要です。 サインイン又は無料トライアルを申し込む。
Method Article
文化、ゼブラフィッシュ初代培養細胞のトランスフェクション
要約
トランスフェクション ライブセル イメージングと大人ゼブラフィッシュ脳から一次電池を準備するためのプロトコルのゼブラフィッシュ胚の一次電池文化を準備するための効率的で簡単に使用できるプロトコルを提案します。
要約
ゼブラフィッシュ胚は透過的であり、母は、そのままで、発展途上の脊椎動物の生物学的プロセスの動的優れた生体内イメージングすることができます外に急速に開発。ただし、細胞および細胞レベル下の構造の形態の詳細な画像は、全体のマウントで制限されます。したがって、私たちはゼブラフィッシュ胚および成体組織から文化ライブ初代培養細胞に効率的で簡単に使用できるプロトコルを設立しました。
簡単に言うと、2 の dpf zebrafish の胚は、dechorionated、deyolked、殺菌し、コラゲナーゼを持つ単一のセルに分離したが。ろ過ステップ後初代培養細胞はガラス底培養皿上にメッキ、数日のために栽培されます。限り長い期間同もの、新鮮な文化は、高分解能共焦点イメージング研究に使用できます。文化には、異なる種類細胞、横紋筋細胞と神経細胞がポリ L リジン コーティングに顕著にはが含まれています。蛍光マーカー蛋白質による、細胞内構造を具体的にラベルする方法にまたニューロンを含む異なったセルタイプにプラスミド DNA トランスフェクションができるエレクトロポレーション プロトコルを確立しました。したがって、演算子の存在下で定義された刺激、複雑型細胞の動作、およびプライマリ ゼブラフィッシュ細胞の細胞内動態評価できます高時空間分解能を持つ。さらに、大人ゼブラフィッシュ脳を使用して、基本的な培養条件と同様に説明した解離手法も大人ゼブラフィッシュ組織の仕事を紹介します。
概要
ゼブラフィッシュ (動脈分布, D. 学) は、様々 な分野の基礎は、医学研究1脊椎動物人気モデルです。ゼブラフィッシュ胚子宮 ex急速に開発、透明なとこのように脊椎動物における生物を勉強するための優秀な前提条件を提供する、顕微鏡下でフィット。ゼブラフィッシュ2の遺伝の少ない原因は、特定の細胞集団の観察を可能にする様々 な蛍光マーカーの細胞型特異的発現と安定した遺伝子改変レポーター行数を確立されています。ゼブラフィッシュのコミュニティを提供していますさまざまな合成 Kal4TA4 (または KalTA3 相当 GalFF) を表現する遺伝子を運ぶいわゆる Gal4 ドライバー行ウイルスの転写活性化する酵母の Gal4 DNA 結合ドメインを持つ遺伝子が融合しました。細胞型特異エンハンサーの制御下にあるドメイン。これらのドライバーの行系列から成る定義済み上流活性化 (UAS) レポーター遺伝子と融合遺伝子を運ぶエフェクター ラインに交差しています。Kal4TA4 タンパク質はレポーター遺伝子3,4の細胞型選択的発現の活性化、UAS 要素にバインドします。このアプローチは、ダブル トランスジェニック動物のほぼすべての利用可能なエンハンサーと記者要素の非常に多様な組み合わせ研究できます。
ただし、個々 の細胞または細胞レベル下の内容の焦点と詳細なライブ イメージング全体と絶えず変化する胚は限られます。最高の解像度を持つ生物学的質問を特定のセルに対処するためは、細胞培養の使用はしばしば望ましいです。ゼブラフィッシュのいくつかのセルのラインが存在するが、大きくと見なされます選択した5,6,7との伝播時間がかかり。さらに、すべての使用可能なセルの行、線維芽細胞派生、1 種類の細胞に細胞培養を用いた実験を制限することです。したがって、私たちはゼブラフィッシュ胚およびアプローチ文化の寿命を増加し、栽培の多様性を広げると共に、大人のゼブラフィッシュ脳から直接細胞を準備する両方効率的で簡単に使用できるプロトコルを設立細胞の種類。さらに、蛍光細胞小器官のマーカーの表現構造と胚初代培養細胞を使って手順を提案する.したがって、細胞の形態と細胞内の構造は、彼らの主要な機能を保持する種類の異なる細胞で高時空分解能で分析できます。
プロトコル
ここで説明したすべての動物の仕事は、法律に基づき (EU の指令 2010年/63) です。ブラウンシュヴァイク工科大学と消費者保護の下ザクセン州事務所食品安全 (ラーベス、オルデンブルク、ドイツの代表的な動物の福祉、地方自治体によって承認されているメンテナンスと魚の取り扱いアリゾナ州 § 4 (02.05) TSchB TU BS)。
1. ゼブラフィッシュ胚初代培養細胞の準備
- 2 日間の準備投稿受精 (dpf) ゼブラフィッシュ胚
- 1 日目は、ゼブラフィッシュのひずみ、ゼブラフィッシュ施設マネージャー8の仕様によると選択のいくつかの交差を設定します。
- 2 日目: チームメイトの魚 10 cm シャーレ (プラスチック) の産卵直後後の卵8を収集しています。パスツール ピペット (プラスチック) 死んだまたは汚染された卵を削除します。洗浄は卵への 0.0001% (w/v) メチレン ブルー Danieau 30% (5.8 mM 塩化ナトリウム、塩化カリウム 0.07 mM、0.04 mM 硫酸マグネシウム、硝酸カルシウム 0.06 mM、5 mM 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic 酸、pH 7.2)8 1 x です。Danieau に培地交換メチレン ブルー、メチレン ブルーが、自発を引き起こす、28 ° C で一晩卵を孵化なし 30%
注: は、細胞の十分な量を得るために魚のラインあたり 100 個の卵の最小値と開始します。1 つのシャーレに 150 以上の胚は発生しません。 - 3 日目: 死んだまたは汚染された胚を取り外して 30 %danieau に培地を交換します。胚の数をカウントすることによって決定します。28 ° C で 1 泊以上の胚を孵化します。
注: 蛍光レポーターを表現する形質転換線を使用している場合、1 dpf や 2 dpf でスクリーニングが必要かもしれません。
注: 大量の胚は、勧めしますそれぞれシャーレ (図 1 a) の黒と白の画像を撮影し、イメージング ソフトウェアと胚の数を定量化します。 - 4 日目: 胚 2 dpf されます。絨毛を削除し、次に Danieau 308の 10 mL に 1 mg/mL の濃度 1 mL プロナーゼを追加すべての絨毛になるまで室温でシェーカーで胚を孵化させなさい (周囲温度によって 20-40 分) を分離しました。Danieau で洗ってプロナーゼと絨毛の両方を削除し、さらに使用するまで室温で胚を保持 30%。
- 再利用可能なポリ L リジン コーティング ガラス底培養皿の準備
注: 市販ガラス底培養皿シングル専用に設計された、高価です。次の手順では、標準的な実験教材から再利用可能な独立独行ガラス底培養皿を準備する方法について説明します。- 標準的な細胞培養皿 (直径 6 cm、プラスチック製) (図 1 b) の底に直径 10 mm の穴を開けます。皿の底掘削ほこりを削除する水道水で十分に洗ってください。
- 皿の底の下面に穴の周りのシリコン グリスを広める、接着剤としてグリースを使用して観察を取り付けます。グリース シール皿の底、coverslip の間のギャップであることを確認します。
注: 使用される coverslips の厚さが後でイメージング アプリケーションに適していることを確認します。 - 自作ガラス底培養皿を徹底的に、しかし慎重に、冷たい水道水と石鹸で洗います。ガラス底培養皿 3 回石鹸を削除する脱イオン水ですすいでください。料理の蓋を風乾し、底の皿クリーン ボックスにさらに使用するまで保管します。
- 文化準備の日 (4 日は、1.1.4 を参照): 両方の内側を湿らせて蓋を皿、皿の底の 70% (v/v) エタノール。皿蓋を置き、底層流と紫外線滅菌作業台の内側を上向きに料理します。エタノールが蒸発するまで空気乾燥し、20 分間紫外線を適用します。この処理の後料理は組み立て、滅菌と見なされます。
- コーティング、ポリ-L-リジン (0.1 mg/mL) 各ガラス底皿の真ん中に 200 μ L をピペット、ピペット チップの表面張力を壊すことによって、coverslip の液体が 。60 分間乾燥させ、滅菌リン酸緩衝生理食塩水 (PBS) x と 1 x を洗います。液体を削除します。さらに使用するまでベンチの下で料理をしてください。
注: その他のコーティングは、実験の目的に応じてテスト可能性があります。任意付加的なコーティングが線維芽細胞様細胞 (図 1E, F) の成長のために好ましいように見えたなしプラスチックを扱うに対し、我々 はポリ-L-リジン ニューロンの成長をサポートするのに十分なを発見します。
注: 自分で作ったガラス底培養皿を何度も使用できます。Coverslip の交換、ぬるま湯で洗って、慎重に、coverslip のデタッチ、70% (v/v) エタノールと石鹸で残りのグリースを削除します。
- 準備及び一次電池のめっき
- 文化準備の日 (4 日は、1.1.4 を参照): 無菌細胞培養皿に胚を転送 (直径 6 cm) 新鮮なプラスチック パスツール ピペットを使用しています。直径約 2 cm 以下の大幅な落ち込みに集まっているすべての胚まで液体をステップごと削除します。
- 無菌作業台内の胚と皿を置き、CO2を追加-独立した媒体 (10% を添加した (v/v) 濾過されるウシ血清、グルタミンと 1.2% (v/v) 10,000 x 1 U ペニシリン-ストレプトマイシン; すべてのサプリメントと媒体は、次「細胞培養培地」という料理が半分になるまで。
注: 代替 CO2-独立した媒体は、たとえば、neurobasal 培地、DMEM 培地やリーボビッツの L-15 媒体として実験の目的に応じてテスト可能性があります。CO2-独立した中小企業リーボビッツの L-15 媒体 CO2インキュベーターを必要としないという利点があります。 - 200 μ L ピペット チップを使用して上下の胚のピペット、卵黄を削除します。成功した deyolking は、培地の濁りが認識できます。
- エタノール 70% (v/v) と細胞培養ディッシュや新鮮な細胞培養培地と別の細胞培養皿を埋めます。ハンドル (40 μ m と無菌細胞ストレーナーに胚を転送するのにカットオフ 1,000 μ L ピペット チップを使用します。図 1)。ハンドルでストレーナを取り出して、すべての胚が 5 の水中に沈むので、エタノールが付いている皿にそれをつける s のすぐにその後、新鮮な細胞培養液を皿に胚とストレーナーが水没します。
注: セル ストレーナー再利用できる数回。水道水、ストア 70% エタノールで、乾燥、紫外線治療を実行しているの下で柔らかいブラシできれいな滅菌作業の下でそれらは使用する前に直接ベンチ (1.2.4 を参照してください)。 - 滅菌 1.5 mL 反応管 (1 つの管で約 100 胚) に胚を転送します。コラゲナーゼ (タイプ 2) セル培地 1 mL の容量で 4 mg/mL の最終濃度に希釈を追加します。常温で 45 分の毎分 30 回転と垂直管回転胚とチューブを孵化させなさい。
- 1,000 μ L ピペット先端上下胚コラゲナーゼ混合物をピペットで残りの細胞塊を切り離して考えます。ガス抜きスロット (40 μ m と無菌細胞ストレーナーを介して細胞懸濁液をフィルターします。図 1)50 mL コニカル チューブ。約 10 ml の新鮮な細胞培養液中のストレーナーをすすいでください。
- 180 x gで 3 分間遠心分離によって細胞をペレットします。ペレットは、ほぼ表示ことがあります。慎重に上澄みを除去し、30 もともと使用されていた胚あたり 200 μ L 新鮮な細胞培養の培地で細胞を再懸濁します。
注: 表示されているペレットを得るためには、お勧め 100 胚の最小値と開始します。 - 1.3.7 自作ポリ L リジン コーティング ガラス底皿のガラス面積の直接の手順で得られた細胞懸濁液 200 μ L をピペット (1.2 を参照してください)。無菌作業台の下で室温で 60 分間インキュベートします。新鮮な細胞培養液 6 mL を加えるし、28 ° C で一次電池を孵化させなさい
- 28 ° C で、倒立顕微鏡を使用して目的のイメージング アプリケーションを実行します。細胞培養液を毎日交換します。文化は (dap) めっき後数日間イメージングに使用できます。

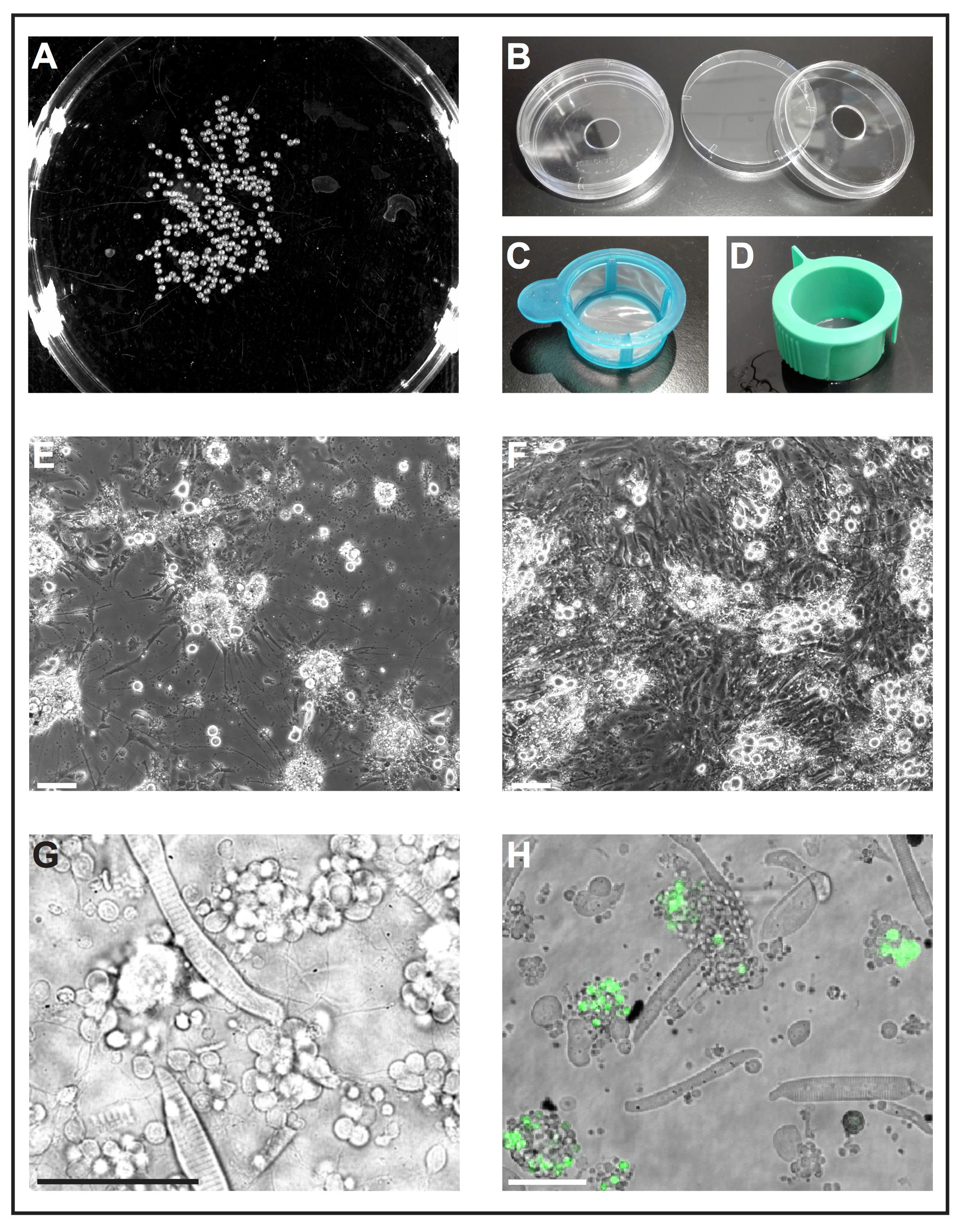
図 1: ゼブラフィッシュ胚の一次電池文化。(A) 1 の白黒画像 dap 胚は、胚の数を分析するためのソフトウェア ツールによって処理することができます。(B) 細胞培養皿 (直径 6 cm) とドリル穴 (直径 10 mm) を使用する再利用可能な独立独行ガラス底培養皿。(C) deyolked 胚をエタノールに浸すと新鮮な細胞培養液中にそれらをすばやく転送する単純なハンドルを持つセル ストレーナー (40 μ m)、「ランディング ネット」として使用されます。(D) セルのスロットを抜きにストレーナー (40 μ m)、コラゲナーゼを介した解離後セルをフィルター処理に使用します。(E) 5 dap 後、一次電池シード主にポリ L-リシンでコーティングされたガラスに顕著な拡張子を持つニューロンを形成します。スケールバー = 100 μ m. (F) 後 5 dap 処理プラスチック塗装, 線維芽細胞のような細胞にはびこる文化なし。スケールバー = 100 μ m. (E) 及び (F) epifluorescent 顕微鏡によって買収されました。(G) 1 で野生型ゼブラフィッシュに由来初代培養細胞の感染光イメージ dap。横紋筋細胞と薄いプロセスを拡張するニューロンのクラスターは容易に観察できます。スケールバー = 50 μ m. (H) 培養細胞形質転換線 Tg (ptf1a: eGFP) 中 1、菱脳でほとんど gaba 作動性ニューロンの神経前駆細胞と網膜細胞集団29,のサブセットの eGFP を表現します。30,31。 スケールバー = 50 μ m. (G) と (H) 共焦点レーザー顕微鏡 (B) に示すように作ったガラス底培養皿を使用してによって買収されました。この図の拡大版を表示するのにはここをクリックしてください。
{kind=link}
2. プライマリのトランスフェクション プラスミッド DNA が付いている細胞します。
- 再懸濁します濾過され、播種細胞細胞培養液ではなく 1 × PBS で 1.3.7 のステップします。細胞懸濁液カウント部屋9の細胞数を決定するためトリパン ブルーの因数を染色します。
- 50 万セルの反作用の管を 1.5 mL の 10 μ g の超純粋なプラスミッド DNA を混ぜて、1 × PBS 100 μ L に容量を調整します。
注: 実験主に使用しましたプラスミド pCS2 +10に基づく表現の構文。開いたリーディング ・ フレームの pCS2 + 多重クローニング サイトに複製の表現は、人間のサイトメガロ ウイルス (CMV プロモーター) のユビキタスのプロモーターによって駆動されます。(代表結果と図 2 と図 3を参照) その他の表現の構文とプロモーターをテストできます。 - エレクトロポレーション キュベット (0.4 cm) に直ちに転送細胞の DNA ミックス キュヴェットをエレクトロポレーションのデバイスの次の設定で electroporate: ワンタイム パルス、指数関数的減衰、280 V、950 μ F。
- エレクトロポレーション後に、直接新鮮な細胞培養培地の 300 μ L で 1.5 mL 反応チューブに細胞 DNA ミックスを転送します。
- 1.3.8 で説明したように細胞懸濁液 200 μ L をプレートし、進む数時間後または翌日に使用される表現の構成要素によって 1.3.9 で解説した、蛍光タンパク質の発現が検出かもしれない。
3. 固定細胞の染色
注: 細胞内構造物は、蛍光融合タンパク質レポーターを使用する代わりに古典的な染色によっても視覚化できます。ゼブラフィッシュの一次電池、蛍光マーカーで模範的な染色の核、F-アクチン、チューブリンのアセチル化に次の標準的なプロトコルを使用します。
- ポリ L リジン コーティング カバー スリップ上のプレートのセルは、(1.3.8 を参照) 上記のように、細胞培養皿または汚損プレートに配置されます。
- 修正して、メディアを取り出してし、1x PBS で 4% パラホルムアルデヒドとセルをカバーします。シェーカーで 4 ° C で 10 分間のセルを孵化させなさい。洗浄のセル室温で 1 × PBS の各 5 分の 3 倍します。1 × PBS がセルを完全にカバーすることを確認してくださいし、シェーカーで洗濯の手順に従います。
- ブロック、固定セルを permeabilize 5% 脱脂粉乳および 0.3% を含む 1 × PBS のセルをカバーするトリトン X-100。シェーカーで室温で 10 分間セルを配置します。3.2 で説明されているように、細胞を洗ってください。
- チューブリンのアセチル化をラベル、軸索11、マーカー希釈 1% 脱脂粉乳を含む 1x PBS で一次抗体 1:2,000 します。このソリューションでは細胞をカバーし、シェーカーで 4 ° c 夜の間それらを孵化させなさい。次の日に 3.2 で説明されているように細胞を洗ってください。
- 1% 脱脂粉乳を含む 1x PBS で緑 fluorochrom フルオレセインのイソチオ シアン酸 (FITC) 1: 100 と共役二次抗体を希釈し、暗闇の中で部屋の温度で 1 時間このソリューションでセルを孵化させなさい (、皿などボックス カバーやアルミ箔) シェーカーで。3.2 で説明されているように、細胞を洗ってください。
- 同時にアクチン細胞骨格と核を染色、ファロイジン12赤色螢光色素 (1:50) と 4', 6-diamidino-2-phenylindole (DAPI)13 (100 ng/mL) の部屋で 10 分の共役を添加した 1 × PBS のセルを孵化させなさい。シェーカーで暗闇の中温。3.2 で説明されているように、細胞を洗ってください。
- イメージ用細胞を準備、きれいな表面にガラス オブジェクト キャリア (顕微鏡スライド) を入れて、それでメディアをマウントのドロップに。ピンセットを使用して皿の固定と染色の細胞とカバー スリップを取るし、オブジェクトのキャリアに直面するセルとドロップの上に置きます。そのメディアをマウント カバー スリップの領域全体に広がっていることを確認します。暗闇の中で乾燥させます。
- 店は固定され、目的のイメージング アプリケーションが実行されるまで、4 ° C で暗闇の中で細胞をマウントします。

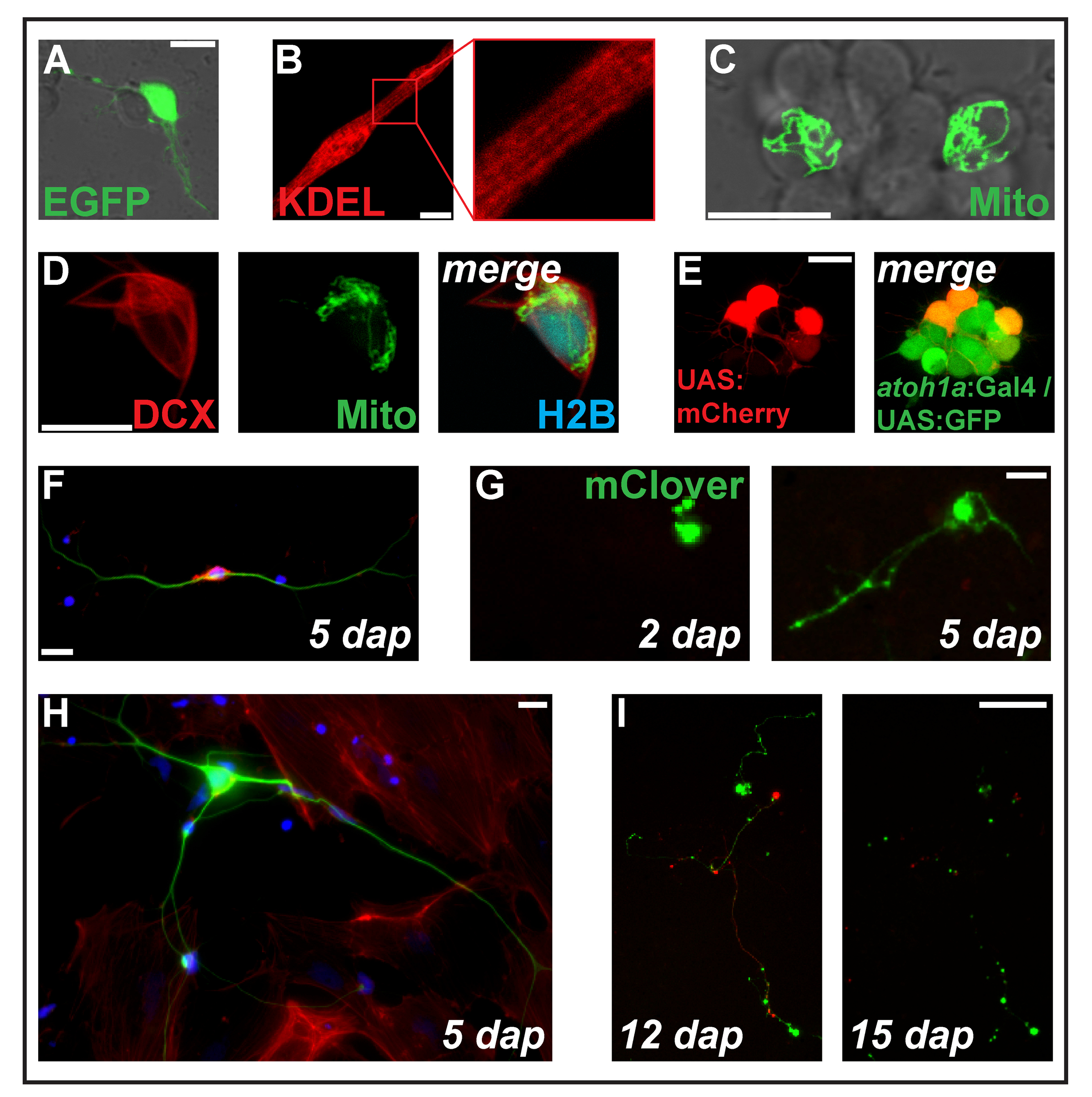
図 2: 式のトランスフェクションをエレクトロポレーションにより構築します。(A) 1 Pc eGFP をトランスフェクトした延髄ニューロン dap。(B) 横紋筋細胞 (2 dap) タンパク質の小胞体をターゲットとした ss RFP KDEL を表現します。(C) 2 つのニューロンで 2 Pc MitoTag YFP をトランスフェクトした神経クラスター内で dap します。(D) セル (2 dap) トリプル-トランスフェクトした Pc-DCX-tdTomato、Pc MitoTag YFP と Pc H2B mseCFP。(E) pSK-transgenes Tg を運ぶ二重遺伝子胚由来初代培養細胞 (1 dap) に UAS:mCherry electroporated (atoh1a: Gal4TA4) hzm222と GFP 発現結果 Tg (4xUAS:KGFPGI) hzm332菱脳の神経前駆細胞。スケール バー = 10 μ m. (A ~ E) 共焦点レーザー顕微鏡図 1 bに示すように作ったガラス底培養皿を使用してによって買収されました。Dap は 5 で固定ゼブラフィッシュ一次ニューロンの蛍光 (F) 染色します。青: DAPI (核);赤: ファロイジン (アクチン);緑: チューブリン (ニューロン) をアセチル化しました。スケール バー = 10 μ m (G) 神経細胞のような細胞の Pc mClover をトランスフェクトしました。2 で dap は、拡張子が表示されていません。5 で dap は、突起状の構造が形成されました。スケール バー = 25 μ m. (H) (F) のセルとして同じ準備からニューロン線維芽細胞様細胞に囲まれています。スケール バー = 10 μ m. (私) ニューロン transgene Tg を運ぶ遺伝子胚由来 (XITubb: した DsRed) Pc mClover zf14828をトランスフェクトしました。12 と 15 の dap は、神経突起は、大規模な変性を受けること。スケールバー = 100 μ m (F・私) で示したセル播種 (F, H) ポリ L リジン コーティングされたガラスまたはプラスチック (G、私)、10% 存在 L-15 媒体で培った濾過されるウシ血清と神経サプリメント B-27 (希釈 1:50) epifluorescent 顕微鏡とイメージングと。この図の拡大版を表示するのにはここをクリックしてください。
{kind=link}
4. 大人のゼブラフィッシュ脳から一次電池の準備
-
脳領域抽出
- 少なくとも 90 日齢の成魚を選択します。特定のセル型での作業が必要な場合細胞特異蛍光レポーター式が、目的の細胞を可視化する遺伝子組換えの行を選択します。
- Danieau 30% で麻酔 Tricaine8 (0.2%) 200 ml ビーカーの中の魚を配置します。魚の移動が停止するまで待ちます。それを安楽死させるのに 15 分間、200 mL の氷の水が入ったビーカーに麻酔下の魚を浸します。
- シャーレを埋める (直径 6 cm) 70% (v/v) エタノール。ピンセットのペアで尻尾を魚を保持し、エタノールに浸し。魚は 5 のためのエタノールで完全に浸漬を確保する s。
- 次の adaptions とグプタとマリンズの14のプロトコルに従って脳を抽出: のみまたはオートクレーブ滅菌パックのツールを使用し、滅菌 1x PBS で頭を解剖。
- 直接抽出後シャーレに脳を配置 (直径 3 cm) 滅菌 1x PBS で満たされ、細胞培養のための無菌作業台の下皿に移動します。
-
脳解離と一次電池へのめっき
- 場所 2 セット (オートクレーブ) 滅菌ピンセット、滅菌シャーレ (直径 6 cm) 満ちている 70% (v/v) エタノール、滅菌シャーレ (直径 10 cm) 満ちているリーボビッツの L-15 培 10% (v/v) 濾過されるウシ血清、B-27 (1:50)、1.2% (v/v) 10,000 U ペニシリン-ストレプトマイシンとハンドル (40 μ m と 1 つの無菌細胞ストレーナー図 1)下クリーン ベンチ。
- 場所ペトリ皿にセル ストレーナー エタノールと液体のレベルが少なくとも 5 mm のストレーナーの底より高いことを確認します。
- ピンセットの最初のセットを使用すると、エタノールに配置済みストレーナーに脳を転送し、それは完全に液体で覆われていることを確認します。1 後 s、転送上記リーボビッツの L-15 媒体を含んでいるペトリ皿に脳とストレーナーがサプリメントを説明しました。
- ピンセットの 2 番目のセットを使用して、転送 500 μ L ・ リーボヴィッツ L-15 媒体上満ちている滅菌 1.5 mL の反応管に脳は、サプリメントをについて説明します。1 mL の容量で 4 mg/mL の最終的な集中にコラゲナーゼ (2 種類) を追加します。
- 部屋の温度で 35 分の毎分 30 回転垂直管内回転子の管、インキュベートします。機械的解離過程を支援するために 1,000 μ L ピペット チップを使用して上下ピペッティングによる残りの組織塊を切り離して考えます。
- 目に見える粒子が溶液中に残ってないときに解離を停止します。ガス抜きスロット (40 μ m と無菌細胞ストレーナーを介して細胞懸濁液をフィルタ リングします。図 1)50 mL コニカル チューブ。約 10 ml の新鮮な細胞培養液中のストレーナーをすすいでください。
注: 単一細胞懸濁液を達成したとき、濾過手順は胚解離の場合と同様に、必要ありません。脳は軟部組織とその単一細胞懸濁液に解離が均一になりやすいです。 - 180 x gで 5 分間遠心分離によって細胞をペレットし、上記の説明補足を新鮮なリーボビッツの L-15 媒体の 1 mL に細胞ペレットを再懸濁します。
- 1 つ自作ポリ L リジン コーティング ガラス底皿の上の細胞懸濁液 (得られる細胞の 50%) 500 μ L をピペット (1.2 を参照してください) または 24 ウェル プレートの井戸。小さい表面 (96 ウェル プレートのウェルのすなわち125 μ L 溶液) の場合ダウンスケー リングします。無菌作業台の下で室温で 60 分間インキュベートします。特定のコンテナーを記入し、28 ° C で一次電池をインキュベートする新鮮な培地に必要な量を追加します。
- 28 ° C で、倒立顕微鏡を使用して目的のイメージング アプリケーションを実行します。文化は、めっき後数日間イメージングに使用できます。日常的にメディアの 50% を交換してください。
結果
図 1は、典型的な文化に由来する横紋筋細胞と神経細胞のような細胞の最も豊富な房を持つ野生型胚の透過光像を示しています。特定の細胞のタイプを識別しやすく、蛍光タンパク質の細胞型特異的発現と形質転換線をすることができます (図 1 H) を使用します。
PCS2 + のトラン...
ディスカッション
ここで、2 の dpf ゼブラフィッシュ胚または大人ゼブラフィッシュ脳から培養細胞を 2 つの異なるプロトコルを紹介します。
2 dpf ゼブラフィッシュからの一次電池文化の準備は基本的な細胞培養の技術の経験とのだれでもの実行する比較的簡単です。しかし、良いと再現性のある結果を得るに十分な開始材料として胚数は重要な (最小値は 100)。胚の発生、中には、細菌や...
開示事項
著者が明らかに何もありません。
謝辞
T. フリッチュ、A. 狼 Asseburg、I. リンデと s ・ M に感謝します優秀なアニマル ・ ケアおよびテクニカル サポートのための Tokarski。強烈な役に立つ議論のコスター ラボのすべてのメンバーに感謝しております。ドイツ研究振興協会 (KO 1949/5-1) と Niedersächsisches Vorab (VWZN2889) 低いザクセンの連邦国家によって資金を感謝します。
資料
| Name | Company | Catalog Number | Comments |
| Fish lines | |||
| AB (wild-type) | established by Streisinger and colleagues, available from the Zebrafish International Resource Center (ZIRC) | ||
| Tg(ptf1a:eGFP)jh1 | stable transgenic line in which the enhancer of the zebrafish gene ptf1a drives expression of the fluorescent protein EGFP (Parsons et al., 2007) | ||
| Tg(XITubb:DsRed)zf148 | stable transgenic line in which the Xenopus neural-specific beta tubulin promoter drives expression of the fluorescent protein DsRed (Peri and Nüsslein-Volhard, 2008) | ||
| Name | Company | Catalog Number | Comments |
| Equipment | |||
| centrifuge | Eppendorf | model 5804 R | |
| ChemiDoc MP imaging system | BioRad | model XRS+, used to acquire black-and-white images of Petri dishes containing 1 da embryos | |
| confocal laser scanning microscope | Leica microsystems | model SP8, equipped with 28 °C temperature box and a 63X objective | |
| epifluorescent microscope | Leica microsystems | model DM5500B, equipped with 28 °C temperature box and a 40X objective | |
| Gene Pulser Xcell with capacitance extender | BioRad | 1652661 | electroporation device |
| Horizontal shaker | GFL | model 3011 | |
| incubator for cell culture (28 °C) | Memmert | model incubator I | |
| incubator for embryos (28 °C) | Heraeus | type B6120 | |
| light microscope | Zeiss | model TELAVAL 31 | |
| micro pipettes | Gilson | ||
| sterile work bench | Bio Base | with laminar flow and UV light | |
| tweezers | Dumont | Style 5, Inox | |
| vertical tube rotator | Labinco B.V. | model LD-79 | |
| Name | Company | Catalog Number | Comments |
| Software | |||
| Image Lab Software | BioRad | for the ChemiDoc MP imaging system from BioRad | |
| ImageJ | National Institutes of Health | used for counting 1 dpf embryos by applying the Count particles-tool to the respective black-and-white images; Rasband, W.S., ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA, https://imagej.nih.gov/ij/. (1997-2016). | |
| LAS X | Leica Microsystems | for both confocal and epifluorescent microscopes from Leica Microsystems | |
| Name | Company | Catalog Number | Comments |
| Plasmids | |||
| pCS-DCX-tdTomato | Köster Lab | # 1599 | based on the backbone pCS2+ (Rupp et al., 1994) |
| pCS-eGFP | Köster Lab | # 7 | based on the backbone pCS2+ (Rupp et al., 1994) |
| pCS-H2B-mseCFP | Köster Lab | # 2379 | based on the backbone pCS2+ (Rupp et al., 1994) |
| pCS-mClover | Köster Lab | # 3865 | based on the backbone pCS2+ (Rupp et al., 1994) |
| pCS-MitoTag-YFP | Köster Lab | # 2199 | based on the backbone pCS2+ (Rupp et al., 1994) |
| pCS-ss-RFP-KDEL | Köster Lab | # 4330 | based on the backbone pCS2+ (Rupp et al., 1994) |
| pCS-VAMP1-mCitrine | Köster Lab | # 2291 | based on the backbone pCS2+ (Rupp et al., 1994) |
| pSK-UAS:mCherry | Köster Lab | # 1062 | based on the pBluescript-backbone of Stratagene |
| Plasmid numbers refer to the database entries of the Köster lab. Plasmids are available upon request. | |||
| Name | Company | Catalog Number | Comments |
| Plastic and glass ware | |||
| BD Falcon Cell Strainer (40 µm) | FALCON | REF 352340 | distributed by BD Bioscience, used as “landing net” to dip deyolked embryos into ethanol and to transfer them quickly to fresh cell culture medium |
| 1.5 mL reaction tubes | Sarstedt | 72690550 | |
| 24-well plate | Sarstedt | 83.3922 | |
| 50 mL falconic tube | Sarstedt | 62.547.004 | |
| 96-well plate | Sarstedt | 83.3924.005 | |
| EasyStrainer (40 µm) | Greiner Bio-One | 542 040 | with venting slots; used to filter cells after collagenase-mediated dissociation |
| electroporation cuvette (0.4 cm) | Kisker | 4905022 | |
| glass coverslips | Heinz Herenz Medizinalbedarf GmbH | 1051201 | |
| Microscope slides | Thermo Fisher Scientific (Menzel Gläser) | 631-0845 | |
| Neubauer chamber | Henneberg-Sander GmbH | 9020-01 | |
| Pasteur pipettes (plastic; 3 mL) | A. Hartenstein | PP05 | |
| Petri dishes (plastic; diameter 10 cm) | Sarstedt | 821473 | for zebrafish embryos |
| pipette tips | Sarstedt | Blue (1000 µl): 70762; Yellow (200 µl): 70760002; White (10 µL): 701116 | |
| sterile cell culture dishes (plastic; diameter 3 cm) | TPP Techno Plastic Products AG | 93040 | |
| sterile cell culture dishes (plastic; diameter 6 cm) | Sarstedt | 72690550 | |
| sterile Petri dishes (plastic; diameter 10 cm) | Sarstedt | 83.3902 | for brain dissection |
| Name | Company | Catalog Number | Comments |
| Chemicals and Reagents | |||
| sodium chloride | Roth | 0601.1 | |
| 4 % paraformaldehyde in 1x PBS | Sigma-Aldrich | 16005 | |
| 4',6-diamidino-2-phenylindole (DAPI) | Thermo Fisher Scientific | D1306 | |
| calcium nitrate tetrahydrate | Sigma-Aldrich | C1396 | |
| ethanol p.a. 100% | Sigma-Aldrich | 46139 | |
| goat α-mouse IgG (Fc specific) FITC conjugated | Thermo Fisher Scientific | 31547 | |
| HEPES | Roth | 9105.4 | |
| high vacuum grease | DOW CORNING | 3826-50 | silicon grease used for self-made glass bottom dishes |
| magnesium sulfate heptahydrate | Merck | 105886 | |
| methylene blue | Serva | 29198.01 | |
| Monoclonal Anti-Tubulin, Acetylated antibody | Sigma-Aldrich | T6793 | |
| Aqua-Poly/Mount (mounting medium) | Polyscience | 18606 | |
| poly-L-lysine | Biochrom | L 7240 | |
| potasssion chloride | Merck | 104938 | |
| Skim milk | Roth | 68514-61-4 | |
| Texas Red-X Phalloidin | Thermo Fisher Scientific | T7471 | |
| Tricaine | Sigma-Aldrich | E10521 | Synonym: Ethyl 3-aminobenzoate methanesulfonate |
| Triton X-100 | BioRad | 1610407 | |
| Trypan Blue | Gibco by Life Technologies | 15250061 | |
| Name | Company | Catalog Number | Comments |
| Enzymes | |||
| collagenase (Type 2) | Thermo Fisher Scientific | 17101015 | dissolve powder in cell culture medium (8 mg/mL) and sterile-filter the solution, store aliquots at -20 °C |
| pronase (from Streptomyces griseus) | Roche | 11459643001 | distributed by Sigma-Aldrich, dissolve in 30% Danieau (10 mg/mL) and store aliquots at -20 °C |
| Name | Company | Catalog Number | Comments |
| Medium and solutions for cell culture | |||
| 1x PBS (Dulbecco's Phosphate Buffered Saline) | Gibco by Life Technologies | 14190-169 | distributed by Thermo Fisher Scientific |
| CO2-independent medium | Gibco by Life Technologies | 18045054 | distributed by Thermo Fisher Scientific |
| filtrated bovine serum (FBS) | PAN-Biotech | individual batch | |
| glutamine 100x | Gibco by Life Technologies | 25030081 | distributed by Thermo Fisher Scientific |
| Leibovitz's L-15 medium | Gibco by Life Technologies | 11415049 | distributed by Thermo Fisher Scientific |
| PenStrep (10,000 U/mL) | Gibco by Life Technologies | 15140148 | distributed by Thermo Fisher Scientific |
参考文献
- Ablain, J., Zon, L. I. Of fish and men: using zebrafish to fight human diseases. Trends in Cell Biology. 23, 584-586 (2013).
- Sassen, W. A., Köster, R. A molecular toolbox for genetic manipulation of zebrafish. Advances in Genomics and Genetics. , 151 (2015).
- Scheer, N., Campos-Ortega, J. A. Use of the Gal4-UAS technique for targeted gene expression in the zebrafish. Mechanisms of Development. 80, 153-158 (1999).
- Köster, R. W., Fraser, S. E. Tracing transgene expression in living zebrafish embryos. Developmental Biology. 233, 329-346 (2001).
- Driever, W., Rangini, Z. Characterization of a cell line derived from zebrafish (Brachydanio rerio) embryos. In Vitro Cellular & Developmental Biology - Animal. 29A, 749-754 (1993).
- Badakov, R., Jaźwińska, A. Efficient transfection of primary zebrafish fibroblasts by nucleofection. Cytotechnology. 51, 105-110 (2006).
- Senghaas, N., Köster, R. W. Culturing and transfecting zebrafish PAC2 fibroblast cells. Cold Spring Harbor Protocols. , (2009).
- Westerfield, M. . The zebrafish book. A guide for the laboratory use of zebrafish (Danio rerio). , (2007).
- Basic methods in cellular and molecular biology. Using a hemacytometer to count cells. Journal of Visualized Experiments Available from: https://www.jove.com/science-education/5048/using-a-hemacytometer-to-count-cells (2017)
- Rupp, R. A., Snider, L., Weintraub, H. Xenopus embryos regulate the nuclear localization of XMyoD. Genes & Development. 8, 1311-1323 (1994).
- Piperno, G., Fuller, M. T. Monoclonal antibodies specific for an acetylated form of alpha-tubulin recognize the antigen in cilia and flagella from a variety of organisms. Journal of Cell Biology. 101 (6), 2085-2094 (1985).
- Barden, J. A., Miki, M., Hambly, B. D., Dos Remedios, C. G. Localization of the phalloidin and nucleotide-binding sites on actin. European Journal of Biochemistry. 162 (3), 583-588 (1987).
- Kapuscinski, J. DAPI: a DNA-specific fluorescent probe. Biotechnic & Histochemistry. 70 (5), 220-233 (1995).
- Gupta, T., Mullins, M. C. Dissection of organs from the adult zebrafish. Journal of Visualized Experiments. 37, E1717 (2010).
- Chalfie, M., Tu, Y., Euskirchen, G., Ward, W. W., Prasher, D. C. Green fluorescent protein as a marker for gene expression. Science. 263, 802-805 (1994).
- Stornaiuolo, M. KDEL and KKXX retrieval signals appended to the same reporter protein determine different trafficking between endoplasmic reticulum, intermediate compartment, and Golgi complex. Molecular Biology of the Cell. 14, 889-902 (2003).
- Lithgow, T. Targeting of proteins to mitochondria. FEBS Letters. 476, 22-26 (2000).
- Nagai, T., Ibata, K., Park, E. S., Kubota, M., Mikoshiba, K., Miyawaki, A. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nature Biotechnology. 20, 87-90 (2002).
- Sassen, W. A., Lehne, F., Russo, G., Wargenau, S., Dübel, S., Köster, R. W. Embryonic zebrafish primary cell culture for transfection and live cellular and subcellular imaging. Developmental Biology. 430, 18-31 (2017).
- Horesh, D., et al. Doublecortin, a stabilizer of microtubules. Human Molecular Genetics. 8, 1599-1610 (1999).
- Shaner, N. C., Campbell, R. E., Steinbach, P. A., Giepmans, B. N. G., Palmer, A. E., Tsien, R. Y. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nature Biotechnology. 22, 1567-1572 (2004).
- Distel, M., Hocking, J. C., Volkmann, K., Köster, R. W. The centrosome neither persistently leads migration nor determines the site of axonogenesis in migrating neurons in vivo. Journal of Cell Biology. 191, 875-890 (2010).
- Matsuda, T., Miyawaki, A., Nagai, T. Direct measurement of protein dynamics inside cells using a rationally designed photoconvertible protein. Nature Methods. 5, 339-345 (2008).
- Archer, B. T., Ozçelik, T., Jahn, R., Francke, U., Südhof, T. C. Structures and chromosomal localizations of two human genes encoding synaptobrevins 1 and 2. Journal of Biological Chemistry. 265, 17267-17273 (1990).
- Griesbeck, O., Baird, G. S., Campbell, R. E., Zacharias, D. A., Tsien, R. Y. Reducing the environmental sensitivity of yellow fluorescent protein. Mechanism and applications. Journal of Biological Chemistry. 276, 29188-29194 (2001).
- Shaner, N. C., et al. A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nature Methods. 10, 407-409 (2013).
- Campbell, R. E., et al. A monomeric red fluorescent protein. Procedings of the National Academy of Sciences of the United States of America. 99, 7877-7882 (2002).
- Peri, F., Nüsslein-Volhard, C. Live imaging of neuronal degradation by microglia reveals a role for v0-ATPase a1 in phagosomal fusion in vivo. Cell. 133, 916-927 (2008).
- Godinho, L., et al. Targeting of amacrine cell neurites to appropriate synaptic laminae in the developing zebrafish retina. Development. 132, 5069-5079 (2005).
- Jusuf, P. R., Harris, W. A. Ptf1a is expressed transiently in all types of amacrine cells in the embryonic zebrafish retina. Neural Development. 4, 34 (2009).
- Kani, S., et al. Proneural gene-linked neurogenesis in zebrafish cerebellum. Developmental Biology. 343, 1-17 (2010).
- Distel, M., Wullimann, M. F., Köster, R. W. Optimized Gal4 genetics for permanent gene expression mapping in zebrafish. Procedings of the National Academy of Sciences of the United States of America. 106, 13365-13370 (2009).
- Choorapoikayil, S., Overvoorde, J., den Hertog, J. Deriving cell lines from zebrafish embryos and tumors. Zebrafish. 10, 316-332 (2013).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved