
A subscription to JoVE is required to view this content. Sign in or start your free trial.
Method Article
Culture and Transfection of Zebrafish Primary Cells
In This Article
Summary
We present an efficient and easy-to-use protocol for preparing primary cell cultures of zebrafish embryos for transfection and live cell imaging as well as a protocol to prepare primary cells from adult zebrafish brain.
Abstract
Zebrafish embryos are transparent and develop rapidly outside the mother, thus allowing for excellent in vivo imaging of dynamic biological processes in an intact and developing vertebrate. However, the detailed imaging of the morphologies of distinct cell types and subcellular structures is limited in whole mounts. Therefore, we established an efficient and easy-to-use protocol to culture live primary cells from zebrafish embryos and adult tissue.
In brief, 2 dpf zebrafish embryos are dechorionated, deyolked, sterilized, and dissociated to single cells with collagenase. After a filtration step, primary cells are plated onto glass bottom dishes and cultivated for several days. Fresh cultures, as much as long term differenciated ones, can be used for high resolution confocal imaging studies. The culture contains different cell types, with striated myocytes and neurons being prominent on poly-L-lysine coating. To specifically label subcellular structures by fluorescent marker proteins, we also established an electroporation protocol which allows the transfection of plasmid DNA into different cell types, including neurons. Thus, in the presence of operator defined stimuli, complex cell behavior, and intracellular dynamics of primary zebrafish cells can be assessed with high spatial and temporal resolution. In addition, by using adult zebrafish brain, we demonstrate that the described dissociation technique, as well as the basic culturing conditions, also work for adult zebrafish tissue.
Introduction
The zebrafish (Danio rerio, D. rerio) is a popular model vertebrate for numerous fields of basic and biomedical research1. Zebrafish embryos develop rapidly ex utero, are transparent, and fit under a microscope, thus providing excellent prerequisites for studying vertebrate development in a living organism. Due to the genetic tractability of zebrafish2, many stable transgenic reporter lines with cell type-specific expression of various fluorescent markers have been established allowing for the observation of specific cell populations. The zebrafish community offers a broad variety of so-called Gal4-driver lines which carry a transgene expressing the synthetic Kal4TA4 (or the KalTA3-equivalent GalFF) gene with the Gal4-DNA-binding domain of yeast fused to viral transcriptional activation domains under the control of cell type-specific enhancers. These driver lines are crossed to effector lines which carry transgenes consisting of a defined upstream activating sequence (UAS) fused to a reporter gene. The Kal4TA4 protein binds to the UAS element, thus activating the cell type-selective expression of the reporter gene3,4. This approach allows for highly diverse combinatorial studies of almost all available enhancer and reporter elements in double-transgenic animals.
However, in-depth live imaging with focus on individual cells or their subcellular contents is limited in a whole and constantly changing embryo. To address specific cell biological questions with highest resolution, the use of cell cultures is often preferable. Some cell lines of zebrafish exist, but they are considered as heavily selected5,6,7 and their propagation is often time-consuming. Furthermore, all the available cell lines are fibroblast derived, limiting experiments using cell culture to one type of cells. Therefore, we established both an efficient and easy-to-use protocol to prepare primary cells directly from zebrafish embryos and adult zebrafish brain, together with approaches to increase the longevity of the culture and to broaden the diversity of cultivated cell types. In addition, we present a procedure to transfect embryonic primary cells with expression constructs for fluorescent organelle markers. Thus, cellular morphologies and subcellular structures can be analyzed with high spatial and temporal resolution in distinct cell types which retain their key features.
Protocol
All animal work described here is in accordance with legal regulations (EU-Directive 2010/63). Maintenance and handling of fish has been approved by local authorities and by the animal welfare representative of the Braunschweig University of Technology and the Lower Saxony State Office of Consumer Protection and Food Safety (LAVES, Oldenburg, Germany; Az. §4 (02.05) TSchB TU BS).
1. Preparation of Primary Cells from Zebrafish Embryos
- Preparation of 2 days post fertilization (dpf) zebrafish embryos
- Day 1: Set up several crossings of the zebrafish strain of choice and according to the specifications of your zebrafish facility manager8.
- Day 2: Mate fish and collect eggs8 directly after spawning in a 10 cm Petri dish (plastic). Remove dead or contaminated eggs with a Pasteur pipette (plastic). Wash eggs 1x with Danieau 30% (5.8 mM sodium chloride, 0.07 mM potassium chloride, 0.04 mM magnesium sulfate, 0.06 mM calcium nitrate, 5 mM 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid, pH 7.2)8 with 0.0001% (w/v) methylene blue. Exchange the medium to Danieau 30% without methylene blue, since methylene blue may cause autofluorescence, and incubate eggs over night at 28 °C.
NOTE: Start with a minimum of 100 eggs per fish line in order to obtain a sufficient amount of cells. Do not raise more than 150 embryos in one Petri dish. - Day 3: Remove dead or contaminated embryos and exchange the medium to Danieau 30%. Determine the number of embryos by counting. Incubate embryos over night at 28 °C.
NOTE: When using a transgenic line expressing a fluorescent reporter, screening at 1 dpf or 2 dpf may be necessary.
NOTE: For larger amounts of embryos, it is recommended to take a black-and-white image of the respective Petri dish (Figure 1A) and to quantify the number of embryos with an imaging software. - Day 4: Embryos are now 2 dpf. To remove chorions, add 1 mL pronase with a concentration of 1 mg/mL to 10 mL of Danieau 30%8 and incubate embryos on a shaker at room temperature until all chorions are detached (20–40 min depending on ambient temperature). Wash with Danieau 30% to remove both pronase and chorions and keep embryos at room temperature until further use.
- Preparation of reusable poly-L-lysine coated glass bottom dishes
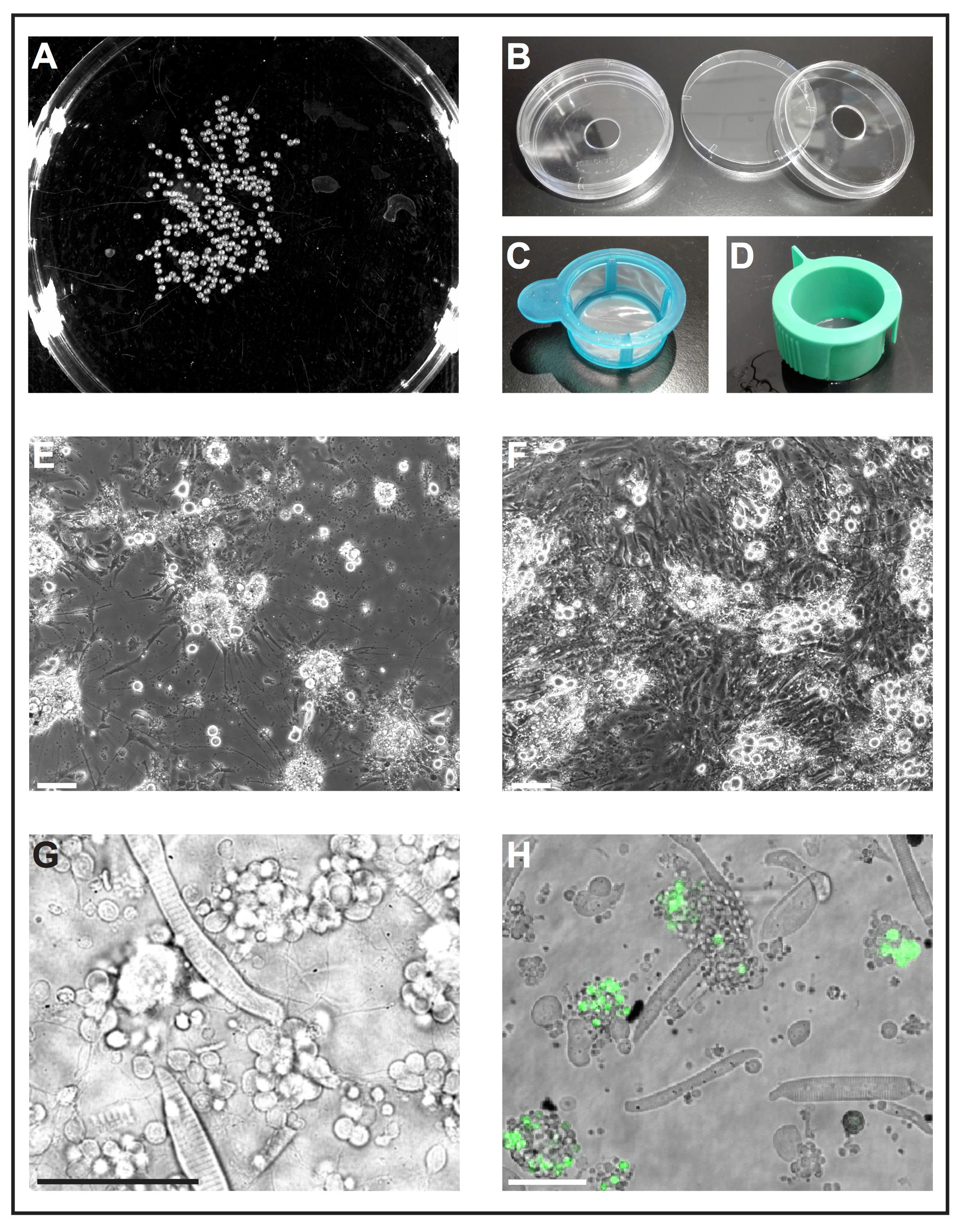
NOTE: Commercially available glass bottom dishes are designed for single-use only and are expensive. The following procedure describes how to prepare reusable self-made glass bottom dishes from standard lab materials.- Drill a hole with a diameter of 10 mm into the bottom of standard cell culture dishes (diameter 6 cm, plastic) (Figure 1B). Wash dish bottoms thoroughly with tap water to remove drilling dust.
- Spread silicon grease around the hole at the underside of the dish bottoms and attach a coverslip using the grease as glue. Make sure that the grease seals the gap between dish bottom and coverslip.
NOTE: Make sure that the thickness of the used coverslips is appropriate for the later imaging application. - Wash self-made glass bottom dishes thoroughly, but carefully, with cold tap water and soap. Rinse glass bottom dishes three times with deionized water to remove soap. Air-dry dish lids and dish bottoms and store them in a clean box until further use.
- At the day of culture preparation (Day 4, see 1.1.4): Moisten the inside of both dish lids and dish bottoms with 70% (v/v) ethanol. Place dish lids and dish bottoms facing the inner side up in a sterile work bench with laminar flow and UV light. Air-dry until the ethanol is evaporated, then apply UV light for 20 min. After this treatment, dishes are assembled and considered as sterile.
- For coating, pipette 200 µL of poly-L-lysine (0.1 mg/mL) in the middle of each glass bottom dish and spread the liquid on the coverslip by breaking surface tension with a pipette tip. Let dry for 60 min, then wash 1x with sterile 1x phosphate-buffered saline (PBS). Remove the liquid. Keep dishes under the bench until further use.
NOTE: Other coatings may be tested depending on the aim of the experiment. We found poly-L-lysine sufficient to support the growth of neurons, whereas treated plastic without any additional coating appeared to be favorable for the growth of fibroblast-like cells (Figure 1E, F).
NOTE: Self-made glass bottom dishes can be used many times. To exchange the coverslip, wash with warm tap water, carefully detach the coverslip and remove remaining grease with 70% (v/v) ethanol and soap.
- Preparation and plating of primary cells
- At the day of culture preparation (Day 4, see 1.1.4): Transfer embryos into a sterile cell culture dish (diameter 6 cm) by using a fresh plastic Pasteur pipette. Remove the liquid step-wise until all embryos are gathered in a big drop with a diameter of about 2 cm or less.
- Place the dish with embryos in the sterile work bench and add CO2-independent medium (supplemented with 10% (v/v) filtrated bovine serum, 1 x glutamine and 1.2% (v/v) 10,000 U Penicillin-Streptomycin; medium with all supplements is in the following referred to as “cell culture medium”) until the dish is half-filled.
NOTE: Alternatives to CO2-independent medium may be tested depending on the aim of the experiment, as for example neurobasal medium, DMEM medium or Leibovitz’s L-15 medium. CO2-independent medium and Leibovitz’s L-15 medium have the advantage of not requiring a CO2 incubator. - To remove the yolk, pipette embryos up and down using a 200 µL-pipette tip. Successful deyolking can be recognized by the clouding of the medium.
- Fill a cell culture dish with 70% (v/v) ethanol and another cell culture dish with fresh cell culture medium. Use a cut-off 1,000 µL-pipette tip to transfer embryos into a sterile cell strainer with handle (40 µm; Figure 1C). Take the strainer by the handle and dip it into the dish with ethanol so that all embryos are submerged for 5 s. Immediately afterwards, submerge the strainer with embryos in the dish with fresh cell culture medium.
NOTE: Cell strainers can be re-used several times. Clean with a soft brush under running tap water, store them in 70% ethanol, and dry and UV-treat them under the sterile work bench directly before use (see also 1.2.4). - Transfer embryos into sterile 1.5 mL reaction tubes (approximately 100 embryos in one tube). Add collagenase (Type 2) diluted in cell culture medium to a final concentration of 4 mg/mL in a total volume of 1 mL. Incubate tubes with embryos on a vertical tube rotator with 30 revolutions per min for 45 min at room temperature.
- Dissociate remaining cell clumps by pipetting the embryo-collagenase mixture up and down with a 1,000 µL-pipette tip. Then filter the cell suspension through a sterile cell strainer with venting slot (40 µm; Figure 1D) into a 50 mL conical tube. Rinse the strainer with approximately 10 mL of fresh cell culture medium.
- Pellet cells by centrifugation for 3 min at 180 x g. The pellet may be nearly invisible. Carefully remove the supernatant and resuspend the cells in 200 µL fresh cell culture medium per 30 originally used embryos.
NOTE: To obtain a visible pellet, it is recommended to start with a minimum of 100 embryos. - Pipette 200 µL of the cell suspension obtained in step 1.3.7 directly on the glass area of a self-made poly-L-lysine-coated glass bottom dish (see 1.2). Incubate for 60 min at room temperature under the sterile work bench. Add 6 mL of fresh cell culture medium and incubate primary cells at 28 °C.
- Perform the desired imaging application using an inverted microscope at 28 °C. Exchange the cell culture medium daily. Cultures can be used for imaging for several days after plating (dap).

Figure 1: Primary cell culture of zebrafish embryos. (A) Black-and-white image of 1 dap embryos, which can be processed by a software tool to analyze the number of embryos. (B) Cell culture dishes (diameter 6 cm) with a drilled hole (diameter 10 mm) are used to prepare reusable self-made glass bottom dishes. (C) Cell strainers (40 µm) with a simple handle are used as "landing nets" to dip deyolked embryos into ethanol and to transfer them quickly to fresh cell culture medium. (D) Cell strainers (40 µm) with venting slots are used to filter cells after collagenase-mediated dissociation. (E) After 5 dap, primary cells seeded on glass coated with poly-L-lysine primarily form neurons with pronounced extensions. Scale bar = 100 µm. (F) After 5 dap on treated plastic without coating, fibroblast-like cells overgrow the culture. Scale bar = 100 µm. (E) and (F) were acquired by an epifluorescent microscope. (G) Transmitted light image of primary cells derived from wild type zebrafish at 1 dap. Striated myocytes and clusters of neurons extending thin processes can be easily observed. Scale bar = 50 µm. (H) Cultured cells of the transgenic line Tg(ptf1a:eGFP)jh1, which expresses eGFP in neuronal progenitors of mostly GABAergic neurons in the hindbrain and a subset of retinal cell populations29,30,31. Scale bar = 50 µm. (G) and (H) were acquired by a confocal laser scanning microscope using the glass bottom dishes made as illustrated in (B). Please click here to view a larger version of this figure.
{kind=link}
2. Transfection of Primary Cells with Plasmid DNA
- Resuspend filtrated and pelleted cells obtained in step 1.3.7 in 1x PBS instead of cell culture medium. Stain an aliquot of the cell suspension with Trypan Blue to determine the cell number in a counting chamber9.
- Mix 0.5 million cells with 10 µg ultra-pure plasmid DNA in a 1.5 mL reaction tube and adjust the total volume to 100 µL with 1x PBS.
Note: For our experiments, we mainly used expression constructs based on the plasmid pCS2+10. Expression of open reading frames cloned into the multiple cloning site of pCS2+ is driven by the ubiquitous promoter of the human cytomegalovirus (CMV promoter). Other expression constructs and promoters may be tested (see also Representative Results and Figure 2 and Figure 3). - Transfer the cell-DNA mix immediately to an electroporation cuvette (0.4 cm), place the cuvette in an electroporation device and electroporate with the following settings: one-time pulse, exponential decay, 280 V, 950 µF.
- Directly after the electroporation, transfer the cell-DNA mix into a 1.5 mL reaction tube with 300 µL of fresh cell culture medium.
- Plate 200 µL of the cell suspension as described in 1.3.8 and proceed as described in 1.3.9 Depending on the used expression construct, expression of fluorescent proteins may be detectable after a few hours or at the next day.
3. Staining of Fixed Primary Cells
NOTE: Subcellular structures can also be visualized by classic immunostaining instead of using fluorescent fusion protein reporters. For zebrafish primary cells, we use the following standard protocol to exemplary stain nucleus, F-actin and acetylated tubulin with fluorescent markers.
- Plate cells on poly-L-lysine coated cover slips placed in a cell culture dish or multiwell plate as described above (see 1.3.8).
- For fixing, remove the medium and cover the cells with 4% paraformaldehyde in 1x PBS. Incubate the cells for 10 min at 4 °C on a shaker. Wash cells 3x for 5 min each with 1x PBS at room temperature. Make sure that the 1x PBS covers the cells completely and perform the washing steps on a shaker.
- To block and to permeabilize the fixed cells, cover the cells with 1x PBS containing 5% skim milk and 0.3% Triton X-100. Place the cells for 10 min at room temperature on a shaker. Wash cells as described in 3.2.
- To label acetylated tubulin, a marker for axons11, dilute the primary antibody 1:2,000 in 1x PBS containing 1% skim milk. Cover the cells with this solution and incubate them over night at 4 °C on a shaker. At the next day, wash cells as described in 3.2.
- Dilute the secondary antibody conjugated with the green fluorochrom fluorescein isothiocyanate (FITC) 1:100 in 1x PBS containing 1% skim milk and incubate the cells with this solution for 1 h at room temperature in the dark (cover the dish e.g. with a box or aluminium foil) on a shaker. Wash cells as described in 3.2.
- To simultaneously stain the actin cytoskeleton and nuclei, incubate the cells in 1x PBS supplemented with Phalloidin12 conjugated with a red fluorochrome (1:50) and 4',6-diamidino-2-phenylindole (DAPI)13 (100 ng/mL) for 10 min at room temperature in the dark on a shaker. Wash cells as described in 3.2.
- To prepare cells for imaging, put a glass object carrier (microscope slide) on a clean surface and place on drop of mounting medium on it. Take a cover slip with fixed and stained cells out of a dish using tweezers and place it on the drop with the cells facing the object carrier. Make sure that mounting medium spreads over the whole area of the cover slip. Let dry in the dark.
- Store fixed and mounted cells in the dark at 4 °C until the desired imaging application is performed.

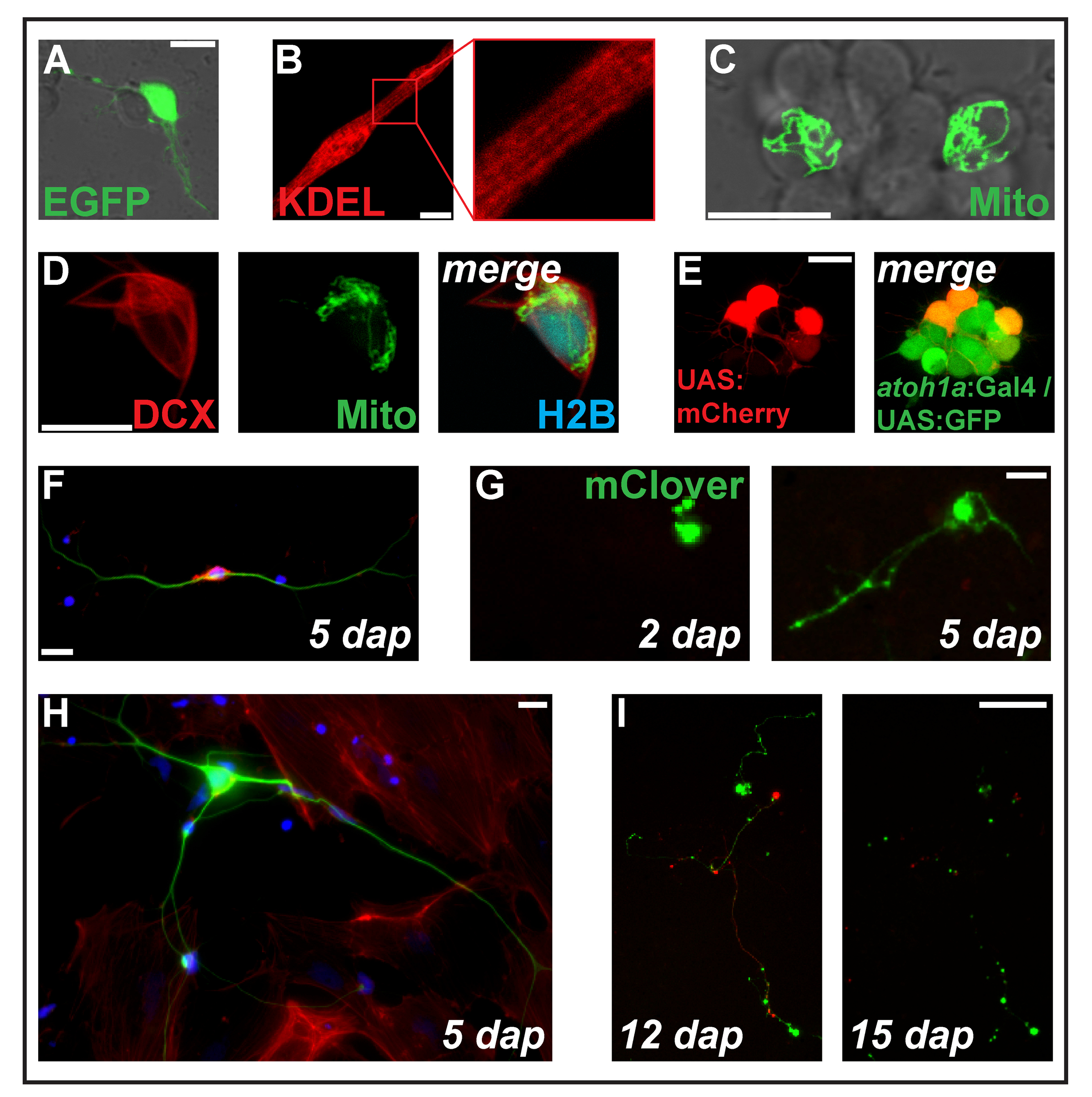
Figure 2: Transfection of expression constructs by electroporation. (A) Putative neuron transfected with pCS-eGFP at 1 dap. (B) Striated myocyte (2 dap) expressing the endoplasmic reticulum-targeted protein ss-RFP-KDEL. (C) Two neurons within a neuronal cluster transfected with pCS-MitoTag-YFP at 2 dap. (D) Cell (2 dap) triple-transfected with pCS-DCX-tdTomato, pCS-MitoTag-YFP and pCS-H2B-mseCFP. (E) pSK-UAS:mCherry electroporated into primary cells (1 dap) derived from double-transgenic embryos carrying the transgenes Tg(atoh1a:Gal4TA4)hzm222 and Tg(4xUAS:KGFPGI)hzm332 resulting in GFP expression in neuronal progenitors of the hindbrain. Scale bars = 10 µm. (A-E) were acquired by a confocal laser scanning microscopy using the glass bottom dishes made as illustrated in Figure 1B. (F) Fluorescent staining of fixed zebrafish primary neurons at 5 dap. Blue: DAPI (nucleus); Red: Phalloidin (F-actin); Green: Acetylated tubulin (neurons). Scale bar = 10 µm. (G) Neuron-like cell transfected with pCS-mClover. At 2 dap, no extension is visible. At 5 dap, a neurite-like structure has formed. Scale bar = 25 µm. (H) Neuron from the same preparation as the cell in (F), surrounded by fibroblasts-like cells. Scale bar = 10 µm. (I) Neuron derived from a transgenic embryo carrying the transgene Tg(XITubb:DsRed)zf14828 transfected with pCS-mClover. Between 12 and 15 dap, the neurites undergo massive degeneration. Scale bar = 100 µm. Cells shown in (F-I) were seeded on poly-L-lysine coated glass (F, H) or plastic (G, I), cultivated in L-15 medium in the presence of 10% filtrated bovine serum and the neuronal supplement B-27 (diluted 1:50)and imaged with an epifluorescent microscope. Please click here to view a larger version of this figure.
{kind=link}
4. Preparation of Primary Cells from Adult Zebrafish Brain
- Brain extraction
- Select an adult fish of at least 90 days of age. If working with a specific cell type is required, choose a transgenic line in which cell-specific fluorescent reporter expression will allow to visualize the desired cells.
- Place the fish in a beaker filled with 200 mL of the anesthetic Tricaine8 (0.2%) in Danieau 30%. Wait until the fish stops moving. Immerse the anesthetized fish in a beaker filled with 200 mL ice water for 15 min to euthanize it.
- Fill a Petri dish (diameter 6 cm) with 70% (v/v) ethanol. Hold the fish by the tail with a pair of tweezers and dip it into the ethanol. Ensure the fish is completely submerged in ethanol for 5 s.
- Extract the brain according to the protocol of Gupta and Mullins14 with the following adaptions: use only autoclaved or sterile-packed tools and dissect the head in sterile 1x PBS.
- Directly after extraction, place the brain in a Petri dish (diameter 3 cm) filled with sterile 1x PBS and move the dish under a sterile work bench for cell culture.
- Brain dissociation and plating of primary cells
- Place two sets of sterile tweezers (autoclaved), a sterile Petri dish (diameter 6 cm) filled with 70% (v/v) ethanol, a sterile Petri dish (diameter 10 cm) filled with Leibovitz’s L-15 medium supplemented with 10% (v/v) filtrated bovine serum, B-27 (1:50) and 1.2% (v/v) 10,000 U Penicillin-Streptomycin and one sterile cell strainer with handle (40 µm; Figure 1C) under the clean bench.
- Place the cell strainer into the Petri dish filled with ethanol and ensure the liquid level is at least 5 mm higher than the bottom of the strainer.
- Using the first set of tweezers, transfer the brain into the strainer already placed in the ethanol and ensure it is fully covered by the liquid. After 1 s, transfer the strainer with the brain into the Petri dish containing Leibovitz’s L-15 medium with the above described supplements.
- Using the second set of tweezers, transfer the brain into a sterile 1.5 mL reaction tube filled with 500 µL Leibovitz’s L-15 medium with the above described supplements. Add collagenase (Type 2) to a final concentration of 4 mg/mL in a total volume of 1 mL.
- Incubate the tube on a vertical tube rotator with 30 revolutions per min for 35 min at room temperature. Mechanically dissociate the remaining tissue clumps by pipetting up and down using a 1,000 µL pipette tip to aid the dissociation process.
- Stop the dissociation when no visible particles remain in solution. Filter the cell suspension through a sterile cell strainer with venting slot (40 µm; Figure 1D) into a 50 mL conical tube. Rinse the strainer with approximately 10 mL of fresh cell culture medium.
NOTE: When single cell suspension is achieved, the filtration step is not as necessary as in case of embryos dissociation. The brain is a soft tissue and as such it is more prone to be homogeneously dissociated into single cell suspension. - Pellet cells by centrifugation for 5 min at 180 x g and resuspend the cell pellet into 1 mL of fresh Leibovitz’s L-15 medium with the above described supplements.
- Pipette 500 µL of cell suspension (50% of the obtained cells) on one self-made poly-L-lysine coated glass bottom dish (see 1.2) or in a well of a 24-well plate. Downscale in case of smaller surfaces (i.e., 125 µL solution for a well of a 96-well plate). Incubate for 60 min at room temperature under the sterile work bench. Then add the necessary volume of fresh medium to fill the specific container and incubate primary cells at 28 °C.
- Perform the desired imaging application using an inverted microscope at 28 °C. Cultures can be used for imaging for several days after plating. Replace 50% of the medium on a daily basis.
Results
Figure 1G shows a transmitted light image of a typical culture derived from wild type embryos with striated myocytes and clusters of neuron-like cells being most abundant. To identify certain cell types more easily, a transgenic line with cell type-specific expression of a fluorescent protein can be used (Figure 1H).
Transfection of a pCS2+-based plasmid
Discussion
Here, we present two different protocols to culture primary cells from either 2 dpf zebrafish embryos or adult zebrafish brain.
The preparation of primary cell cultures from 2 dpf zebrafish is relatively easy to perform for anyone with experience in basic cell culture techniques. However, to obtain good and reproducible results, a sufficient number of embryos as starting material is crucial (100 is the minimum). During the raising of the embryos, all possible sources of contamination must be a...
Disclosures
The authors have nothing to disclose.
Acknowledgements
We thank T. Fritsch, A. Wolf-Asseburg, I. Linde and S.-M. Tokarski for excellent animal care and technical support. We are grateful to all members of the Köster lab for intense and helpful discussions. We gratefully acknowledge funding by the Deutsche Forschungsgemeinschaft (KO 1949/5-1) and the Federal State of Lower Saxony, Niedersächsisches Vorab (VWZN2889).
Materials
| Name | Company | Catalog Number | Comments |
| Fish lines | |||
| AB (wild-type) | established by Streisinger and colleagues, available from the Zebrafish International Resource Center (ZIRC) | ||
| Tg(ptf1a:eGFP)jh1 | stable transgenic line in which the enhancer of the zebrafish gene ptf1a drives expression of the fluorescent protein EGFP (Parsons et al., 2007) | ||
| Tg(XITubb:DsRed)zf148 | stable transgenic line in which the Xenopus neural-specific beta tubulin promoter drives expression of the fluorescent protein DsRed (Peri and Nüsslein-Volhard, 2008) | ||
| Name | Company | Catalog Number | Comments |
| Equipment | |||
| centrifuge | Eppendorf | model 5804 R | |
| ChemiDoc MP imaging system | BioRad | model XRS+, used to acquire black-and-white images of Petri dishes containing 1 da embryos | |
| confocal laser scanning microscope | Leica microsystems | model SP8, equipped with 28 °C temperature box and a 63X objective | |
| epifluorescent microscope | Leica microsystems | model DM5500B, equipped with 28 °C temperature box and a 40X objective | |
| Gene Pulser Xcell with capacitance extender | BioRad | 1652661 | electroporation device |
| Horizontal shaker | GFL | model 3011 | |
| incubator for cell culture (28 °C) | Memmert | model incubator I | |
| incubator for embryos (28 °C) | Heraeus | type B6120 | |
| light microscope | Zeiss | model TELAVAL 31 | |
| micro pipettes | Gilson | ||
| sterile work bench | Bio Base | with laminar flow and UV light | |
| tweezers | Dumont | Style 5, Inox | |
| vertical tube rotator | Labinco B.V. | model LD-79 | |
| Name | Company | Catalog Number | Comments |
| Software | |||
| Image Lab Software | BioRad | for the ChemiDoc MP imaging system from BioRad | |
| ImageJ | National Institutes of Health | used for counting 1 dpf embryos by applying the Count particles-tool to the respective black-and-white images; Rasband, W.S., ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA, https://imagej.nih.gov/ij/. (1997-2016). | |
| LAS X | Leica Microsystems | for both confocal and epifluorescent microscopes from Leica Microsystems | |
| Name | Company | Catalog Number | Comments |
| Plasmids | |||
| pCS-DCX-tdTomato | Köster Lab | # 1599 | based on the backbone pCS2+ (Rupp et al., 1994) |
| pCS-eGFP | Köster Lab | # 7 | based on the backbone pCS2+ (Rupp et al., 1994) |
| pCS-H2B-mseCFP | Köster Lab | # 2379 | based on the backbone pCS2+ (Rupp et al., 1994) |
| pCS-mClover | Köster Lab | # 3865 | based on the backbone pCS2+ (Rupp et al., 1994) |
| pCS-MitoTag-YFP | Köster Lab | # 2199 | based on the backbone pCS2+ (Rupp et al., 1994) |
| pCS-ss-RFP-KDEL | Köster Lab | # 4330 | based on the backbone pCS2+ (Rupp et al., 1994) |
| pCS-VAMP1-mCitrine | Köster Lab | # 2291 | based on the backbone pCS2+ (Rupp et al., 1994) |
| pSK-UAS:mCherry | Köster Lab | # 1062 | based on the pBluescript-backbone of Stratagene |
| Plasmid numbers refer to the database entries of the Köster lab. Plasmids are available upon request. | |||
| Name | Company | Catalog Number | Comments |
| Plastic and glass ware | |||
| BD Falcon Cell Strainer (40 µm) | FALCON | REF 352340 | distributed by BD Bioscience, used as “landing net” to dip deyolked embryos into ethanol and to transfer them quickly to fresh cell culture medium |
| 1.5 mL reaction tubes | Sarstedt | 72690550 | |
| 24-well plate | Sarstedt | 83.3922 | |
| 50 mL falconic tube | Sarstedt | 62.547.004 | |
| 96-well plate | Sarstedt | 83.3924.005 | |
| EasyStrainer (40 µm) | Greiner Bio-One | 542 040 | with venting slots; used to filter cells after collagenase-mediated dissociation |
| electroporation cuvette (0.4 cm) | Kisker | 4905022 | |
| glass coverslips | Heinz Herenz Medizinalbedarf GmbH | 1051201 | |
| Microscope slides | Thermo Fisher Scientific (Menzel Gläser) | 631-0845 | |
| Neubauer chamber | Henneberg-Sander GmbH | 9020-01 | |
| Pasteur pipettes (plastic; 3 mL) | A. Hartenstein | PP05 | |
| Petri dishes (plastic; diameter 10 cm) | Sarstedt | 821473 | for zebrafish embryos |
| pipette tips | Sarstedt | Blue (1000 µl): 70762; Yellow (200 µl): 70760002; White (10 µL): 701116 | |
| sterile cell culture dishes (plastic; diameter 3 cm) | TPP Techno Plastic Products AG | 93040 | |
| sterile cell culture dishes (plastic; diameter 6 cm) | Sarstedt | 72690550 | |
| sterile Petri dishes (plastic; diameter 10 cm) | Sarstedt | 83.3902 | for brain dissection |
| Name | Company | Catalog Number | Comments |
| Chemicals and Reagents | |||
| sodium chloride | Roth | 0601.1 | |
| 4 % paraformaldehyde in 1x PBS | Sigma-Aldrich | 16005 | |
| 4',6-diamidino-2-phenylindole (DAPI) | Thermo Fisher Scientific | D1306 | |
| calcium nitrate tetrahydrate | Sigma-Aldrich | C1396 | |
| ethanol p.a. 100% | Sigma-Aldrich | 46139 | |
| goat α-mouse IgG (Fc specific) FITC conjugated | Thermo Fisher Scientific | 31547 | |
| HEPES | Roth | 9105.4 | |
| high vacuum grease | DOW CORNING | 3826-50 | silicon grease used for self-made glass bottom dishes |
| magnesium sulfate heptahydrate | Merck | 105886 | |
| methylene blue | Serva | 29198.01 | |
| Monoclonal Anti-Tubulin, Acetylated antibody | Sigma-Aldrich | T6793 | |
| Aqua-Poly/Mount (mounting medium) | Polyscience | 18606 | |
| poly-L-lysine | Biochrom | L 7240 | |
| potasssion chloride | Merck | 104938 | |
| Skim milk | Roth | 68514-61-4 | |
| Texas Red-X Phalloidin | Thermo Fisher Scientific | T7471 | |
| Tricaine | Sigma-Aldrich | E10521 | Synonym: Ethyl 3-aminobenzoate methanesulfonate |
| Triton X-100 | BioRad | 1610407 | |
| Trypan Blue | Gibco by Life Technologies | 15250061 | |
| Name | Company | Catalog Number | Comments |
| Enzymes | |||
| collagenase (Type 2) | Thermo Fisher Scientific | 17101015 | dissolve powder in cell culture medium (8 mg/mL) and sterile-filter the solution, store aliquots at -20 °C |
| pronase (from Streptomyces griseus) | Roche | 11459643001 | distributed by Sigma-Aldrich, dissolve in 30% Danieau (10 mg/mL) and store aliquots at -20 °C |
| Name | Company | Catalog Number | Comments |
| Medium and solutions for cell culture | |||
| 1x PBS (Dulbecco's Phosphate Buffered Saline) | Gibco by Life Technologies | 14190-169 | distributed by Thermo Fisher Scientific |
| CO2-independent medium | Gibco by Life Technologies | 18045054 | distributed by Thermo Fisher Scientific |
| filtrated bovine serum (FBS) | PAN-Biotech | individual batch | |
| glutamine 100x | Gibco by Life Technologies | 25030081 | distributed by Thermo Fisher Scientific |
| Leibovitz's L-15 medium | Gibco by Life Technologies | 11415049 | distributed by Thermo Fisher Scientific |
| PenStrep (10,000 U/mL) | Gibco by Life Technologies | 15140148 | distributed by Thermo Fisher Scientific |
References
- Ablain, J., Zon, L. I. Of fish and men: using zebrafish to fight human diseases. Trends in Cell Biology. 23, 584-586 (2013).
- Sassen, W. A., Köster, R. A molecular toolbox for genetic manipulation of zebrafish. Advances in Genomics and Genetics. , 151 (2015).
- Scheer, N., Campos-Ortega, J. A. Use of the Gal4-UAS technique for targeted gene expression in the zebrafish. Mechanisms of Development. 80, 153-158 (1999).
- Köster, R. W., Fraser, S. E. Tracing transgene expression in living zebrafish embryos. Developmental Biology. 233, 329-346 (2001).
- Driever, W., Rangini, Z. Characterization of a cell line derived from zebrafish (Brachydanio rerio) embryos. In Vitro Cellular & Developmental Biology - Animal. 29A, 749-754 (1993).
- Badakov, R., Jaźwińska, A. Efficient transfection of primary zebrafish fibroblasts by nucleofection. Cytotechnology. 51, 105-110 (2006).
- Senghaas, N., Köster, R. W. Culturing and transfecting zebrafish PAC2 fibroblast cells. Cold Spring Harbor Protocols. , (2009).
- Westerfield, M. . The zebrafish book. A guide for the laboratory use of zebrafish (Danio rerio). , (2007).
- Basic methods in cellular and molecular biology. Using a hemacytometer to count cells. Journal of Visualized Experiments Available from: https://www.jove.com/science-education/5048/using-a-hemacytometer-to-count-cells (2017)
- Rupp, R. A., Snider, L., Weintraub, H. Xenopus embryos regulate the nuclear localization of XMyoD. Genes & Development. 8, 1311-1323 (1994).
- Piperno, G., Fuller, M. T. Monoclonal antibodies specific for an acetylated form of alpha-tubulin recognize the antigen in cilia and flagella from a variety of organisms. Journal of Cell Biology. 101 (6), 2085-2094 (1985).
- Barden, J. A., Miki, M., Hambly, B. D., Dos Remedios, C. G. Localization of the phalloidin and nucleotide-binding sites on actin. European Journal of Biochemistry. 162 (3), 583-588 (1987).
- Kapuscinski, J. DAPI: a DNA-specific fluorescent probe. Biotechnic & Histochemistry. 70 (5), 220-233 (1995).
- Gupta, T., Mullins, M. C. Dissection of organs from the adult zebrafish. Journal of Visualized Experiments. 37, E1717 (2010).
- Chalfie, M., Tu, Y., Euskirchen, G., Ward, W. W., Prasher, D. C. Green fluorescent protein as a marker for gene expression. Science. 263, 802-805 (1994).
- Stornaiuolo, M. KDEL and KKXX retrieval signals appended to the same reporter protein determine different trafficking between endoplasmic reticulum, intermediate compartment, and Golgi complex. Molecular Biology of the Cell. 14, 889-902 (2003).
- Lithgow, T. Targeting of proteins to mitochondria. FEBS Letters. 476, 22-26 (2000).
- Nagai, T., Ibata, K., Park, E. S., Kubota, M., Mikoshiba, K., Miyawaki, A. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nature Biotechnology. 20, 87-90 (2002).
- Sassen, W. A., Lehne, F., Russo, G., Wargenau, S., Dübel, S., Köster, R. W. Embryonic zebrafish primary cell culture for transfection and live cellular and subcellular imaging. Developmental Biology. 430, 18-31 (2017).
- Horesh, D., et al. Doublecortin, a stabilizer of microtubules. Human Molecular Genetics. 8, 1599-1610 (1999).
- Shaner, N. C., Campbell, R. E., Steinbach, P. A., Giepmans, B. N. G., Palmer, A. E., Tsien, R. Y. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nature Biotechnology. 22, 1567-1572 (2004).
- Distel, M., Hocking, J. C., Volkmann, K., Köster, R. W. The centrosome neither persistently leads migration nor determines the site of axonogenesis in migrating neurons in vivo. Journal of Cell Biology. 191, 875-890 (2010).
- Matsuda, T., Miyawaki, A., Nagai, T. Direct measurement of protein dynamics inside cells using a rationally designed photoconvertible protein. Nature Methods. 5, 339-345 (2008).
- Archer, B. T., Ozçelik, T., Jahn, R., Francke, U., Südhof, T. C. Structures and chromosomal localizations of two human genes encoding synaptobrevins 1 and 2. Journal of Biological Chemistry. 265, 17267-17273 (1990).
- Griesbeck, O., Baird, G. S., Campbell, R. E., Zacharias, D. A., Tsien, R. Y. Reducing the environmental sensitivity of yellow fluorescent protein. Mechanism and applications. Journal of Biological Chemistry. 276, 29188-29194 (2001).
- Shaner, N. C., et al. A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nature Methods. 10, 407-409 (2013).
- Campbell, R. E., et al. A monomeric red fluorescent protein. Procedings of the National Academy of Sciences of the United States of America. 99, 7877-7882 (2002).
- Peri, F., Nüsslein-Volhard, C. Live imaging of neuronal degradation by microglia reveals a role for v0-ATPase a1 in phagosomal fusion in vivo. Cell. 133, 916-927 (2008).
- Godinho, L., et al. Targeting of amacrine cell neurites to appropriate synaptic laminae in the developing zebrafish retina. Development. 132, 5069-5079 (2005).
- Jusuf, P. R., Harris, W. A. Ptf1a is expressed transiently in all types of amacrine cells in the embryonic zebrafish retina. Neural Development. 4, 34 (2009).
- Kani, S., et al. Proneural gene-linked neurogenesis in zebrafish cerebellum. Developmental Biology. 343, 1-17 (2010).
- Distel, M., Wullimann, M. F., Köster, R. W. Optimized Gal4 genetics for permanent gene expression mapping in zebrafish. Procedings of the National Academy of Sciences of the United States of America. 106, 13365-13370 (2009).
- Choorapoikayil, S., Overvoorde, J., den Hertog, J. Deriving cell lines from zebrafish embryos and tumors. Zebrafish. 10, 316-332 (2013).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved