
Un abonnement à JoVE est nécessaire pour voir ce contenu. Connectez-vous ou commencez votre essai gratuit.
Method Article
Culture et la Transfection des cellules primaires de poisson-zèbre
Dans cet article
Résumé
Nous présentons un protocole efficace et facile à utiliser pour la préparation de cultures de cellules primaires d’embryons de poisson-zèbre pour la transfection et l’imagerie de cellules vivantes, mais aussi un protocole pour préparer les cellules primaires du cerveau adulte poisson-zèbre.
Résumé
Des embryons de poisson-zèbre sont transparentes et se développeront rapidement en dehors de la mère, permettant ainsi d’excellents en vivo imagerie des processus biologiques dynamiques chez un vertébré intacte et en développement. Cependant, l’imagerie détaillée de la morphologie de types cellulaires distincts et des structures subcellulaires est limité dans toute montures. Donc, nous avons établi un protocole efficace et facile à utiliser pour la culture des cellules vivantes primaire par des embryons de poisson-zèbre et de tissus adultes.
En bref, 2 embryons de poisson-zèbre dpf sont déchorionés, deyolked, stérilisé et se dissocie des cellules individuelles avec la collagénase. Après une étape de filtration, les cellules primaires sont ensemencés sur des plats en verre bas et cultivées pendant plusieurs jours. Des cultures fraîches, au long terme différencié ceux, peuvent être utilisés pour les études d’imagerie confocale haute résolution. La culture contient différents types de cellules, avec des myocytes striés et les neurones étant visible sur revêtement poly-L-lysine. À spécifiquement étiquette structures subcellulaires de protéines marqueur fluorescent, nous avons aussi établi un protocole d’électroporation qui permet la transfection d’ADN plasmidique dans différents types de cellules, y compris les neurones. Ainsi, en présence de l’opérateur défini des stimuli, comportement cellulaire complexe et dynamique intracellulaire des cellules primaires de poisson-zèbre peut être évaluée avec une haute résolution spatiale et temporelle. En outre, en utilisant le cerveau adulte poisson-zèbre, nous démontrons que la technique de dissociation décrites, ainsi que les conditions de culture base, également travaillent pour des tissus de poisson-zèbre adulte.
Introduction
Le poisson zèbre (Danio rerio, d. rerio) est un modèle populaire vertébré pour nombreux domaines de la recherche biomédicale et de base1. Des embryons de poisson-zèbre se développent rapidement ex utero, sont transparents et s’adaptent au microscope, offrant ainsi des conditions excellentes pour l’étude du développement des vertébrés dans un organisme vivant. En raison de la traçabilité génétique du poisson-zèbre2, nombreuses lignes de journaliste transgéniques stable avec l’expression spécifique de type cellule de différents marqueurs fluorescents ont été établies permettant l’observation des populations de cellules spécifiques. La communauté de poissons zèbres vous propose un large éventail de ce que l'on appelle lignes de Gal4-pilote muni d’un transgène exprimant les Kal4TA4 synthétiques (ou l’équivalent de KalTA3-GalFF) gène avec le domaine de Gal4-DNA-binding de levure fondue à l’activation de la transcription virale domaines relevant d’exhausteurs de spécifiques au type de cellule. Ces lignes de pilote sont croisent aux lignes effectrices qui transportent les transgènes consistant en une séquence activatrice en amont (UAS) déterminée fusionnée à un gène rapporteur. La protéine Kal4TA4 se lie à l’élément de SAMU, activant ainsi l’expression de type sélectif de cellule du journaliste gene3,4. Cette approche permet de très diverses études combinatoires de presque tous les éléments disponibles d’enhancer et reporter chez les animaux double-transgéniques.
Cependant, profondeur imagerie live mettant l’accent sur des cellules individuelles ou leur contenu subcellulaire est limité dans un embryon entier et en constante évolution. Pour répondre à des questions biologiques spécifiques de cellules avec la résolution la plus élevée, l’utilisation de cultures cellulaires est souvent préférable. Certaines lignées cellulaires de poisson-zèbre existent, mais elles sont considérées comme fortement sélectionné5,6,7 et leur propagation est souvent fastidieux. En outre, toutes les lignées de cellules disponibles sont des fibroblastes dérivés, limitant les expériences de culture cellulaire à un seul type de cellules. Par conséquent, nous avons établi un protocole à la fois efficace et facile à utiliser pour préparer les cellules primaires directement à partir des embryons de poisson-zèbre et le cerveau de poisson zèbre adulte, ainsi que des approches pour augmenter la longévité de la culture et d’élargir la diversité de la culture types de cellules. En outre, nous présentons une procédure pour transfecter les cellules embryonnaires primaires avec les constructions de l’expression des marqueurs fluorescents organite. Ainsi, les morphologies cellulaires et subcellulaires structures peuvent être analysées avec haute résolution spatiale et temporelle de types cellulaires distincts qui conservent leurs caractéristiques principales.
Protocole
Tous les animaux décrits ici on travaille conformément aux prescriptions légales (EU-Directive 2010/63). Entretien et maniement de poissons a été approuvé par les autorités locales et par le bien-être des animaux représentatif de l’Université technique de Braunschweig et la Basse Saxe État Office de la Protection des consommateurs et la sécurité alimentaire (Lavesarchitecte construction, Oldenburg, Allemagne ; § 4 AZ (02.05) TU TSchB BS).
1. préparation des cellules primaires d’embryons de poisson-zèbre
- Préparation de 2 jours après la fécondation (dpf) des embryons de poisson-zèbre
- Jour 1 : Mettre en place plusieurs passages de la souche de poisson-zèbre de choix et selon les spécifications de votre poisson-zèbre installation manager8.
- Jour 2 : Mate poisson et recueillir des œufs8 directement après le frai dans une boîte de Pétri (plastique) de 10 cm. Retirer les œufs morts ou contaminées avec une pipette Pasteur (plastique). Lavez les oeufs 1 x avec Danieau 30 % (5,8 mM chlorure de sodium, chlorure de potassium 0,07 mM, sulfate de magnésium de 0,04 mM, 0. 06 mM nitrate de calcium, acide 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic 5 mM, pH 7,2)8 avec le bleu de méthylène 0,0001 % (p/v). Échanger le milieu à Danieau 30 % sans le bleu de méthylène, étant donné que le bleu de méthylène peut causer autofluorescence et incuber des oeufs pendant la nuit à 28 ° C.
NOTE : Démarrer avec un minimum de 100 oeufs par ligne de poissons afin d’obtenir une quantité suffisante de cellules. Ne levez pas plus de 150 embryons dans une boîte de Pétri. - Jour 3 : Supprimer des embryons morts ou contaminés et d’échanger le milieu à Danieau 30 %. Déterminer le nombre d’embryons en comptant. Incuber les embryons pendant la nuit à 28 ° C.
Remarque : Lorsque vous utilisez une lignée transgénique exprimant un journaliste fluorescent, projection au 1 dpf ou 2 dpf peut être nécessaire.
Remarque : Pour les quantités plus grandes d’embryons, il est recommandé de prendre une image en noir et blanc de la boîte de Pétri respectifs (Figure 1 a) et de quantifier le nombre d’embryons avec un logiciel d’imagerie. - Jour 4 : Les embryons sont maintenant 2 dpf. Pour supprimer les chorions, ajouter 1 mL la pronase avec une concentration de 1 mg/mL à 10 mL de Danieau 30 %8 et incuber des embryons sur un agitateur à température ambiante jusqu'à ce que tous les chorions sont détachés (20 à 40 min selon la température ambiante). Laver avec Danieau 30 % pour supprimer la pronase et chorions et garder les embryons à température ambiante jusqu'à une utilisation ultérieure.
- Préparation de plats de fond verre réutilisable poly-L-lysine
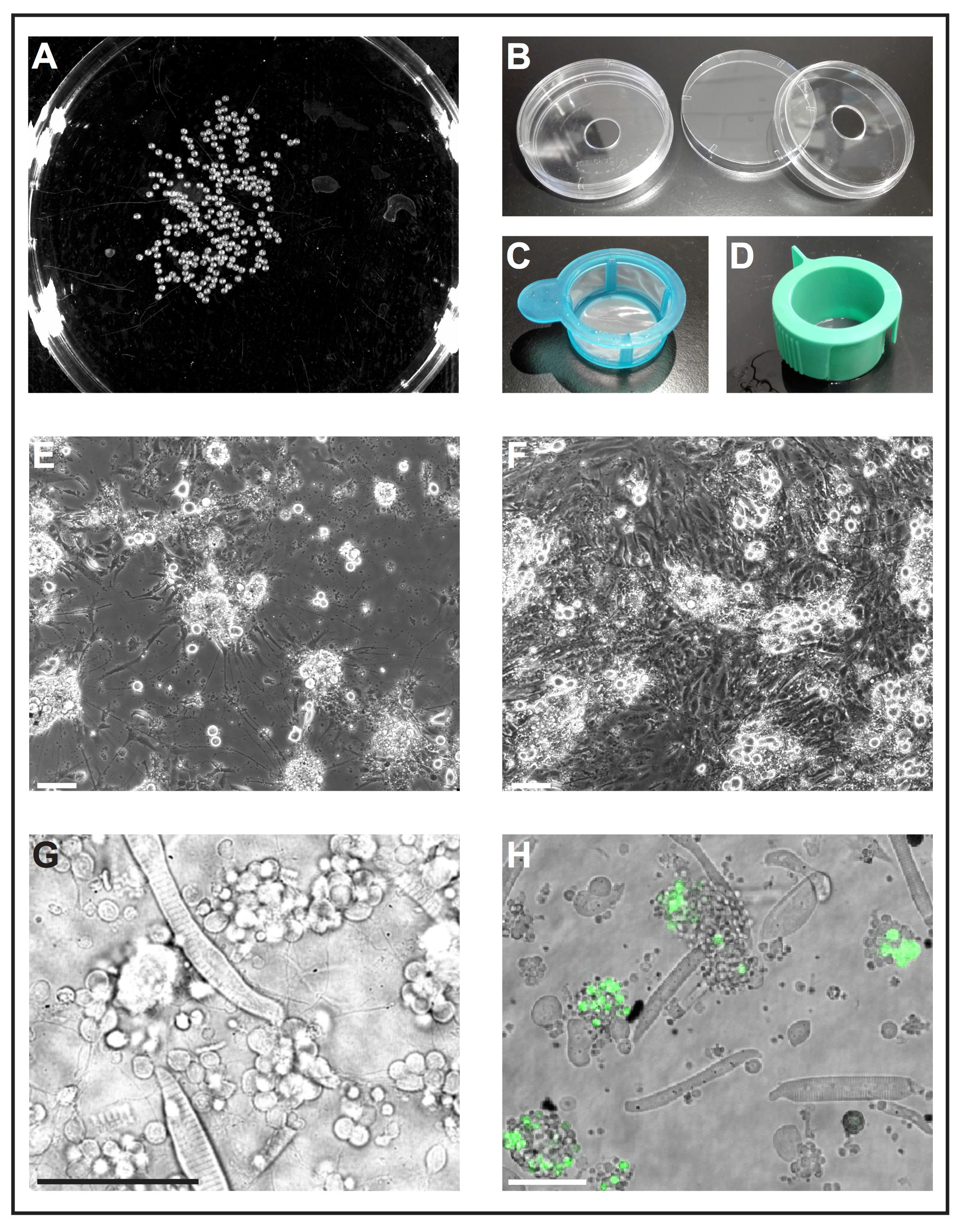
NOTE : Plats bas en verre disponibles dans le commerce sont conçus pour usage unique et sont chers. La procédure suivante décrit comment préparer des plats fond verre réutilisables de self-made à partir de matériaux standard de laboratoire.- Percer un trou d’un diamètre de 10 mm dans le fond des récipients de culture cellulaire standard (diamètre 6 cm, plastique) (Figure 1 b). Lavez à fond plat avec de l’eau du robinet pour enlever la poussière de perçage.
- Répandre la graisse de silicone autour du trou trouve sous le fond plat et fixez un lamelle couvre-objet à l’aide de la graisse comme colle. Veillez à ce que la graisse scelle l’écart entre le fond du plat et lamelle.
Remarque : Assurez-vous que l’épaisseur des lamelles utilisé est approprié pour l’application d’imagerie plus tard. - Vaisselle verre self-made bas complètement, mais avec précaution, avec eau froide du robinet et du savon. Rincer la vaisselle fond verre trois fois avec de l’eau déionisée pour enlever le savon. Sécher à l’air couvercles plat et plat fond et stockez-les dans une boîte propre avant réutilisation.
- Lors de la journée de préparation de la culture (jour 4, voir 1.1.4) : mouiller l’intérieur des deux couvercles à vaisselle et plat à fond avec l’éthanol à 70 % (v/v). Placer les couvercles plat et plat à fond le côté intérieur vers le haut dans un banc de travail stérile à flux laminaire et la lumière UV. Sécher à l’air jusqu'à ce que l’éthanol s’évapore, puis appliquez les ultraviolets pendant 20 min. Après ce traitement, les plats sont assemblés et considérés comme stériles.
- Pour le couchage, distribuer 200 µL de la poly-L-lysine (0,1 mg/mL) au milieu de chaque plat de dessous de verre et répandre le liquide sur la lamelle couvre-objet en brisant la tension superficielle avec un embout de la pipette. Laissez sécher pendant 60 minutes, puis laver 1 fois avec stérile 1 x solution saline tamponnée au phosphate (PBS). Retirer le liquide. Garder la vaisselle sous le banc jusqu'à une utilisation ultérieure.
Remarque : Les autres revêtements peuvent être testés selon le but de l’expérience. Nous avons trouvé poly-L-lysine suffisante pour soutenir la croissance des neurones, tandis que les traités de plastique sans tout revêtement supplémentaire semble être favorable à la croissance des cellules fibroblastes (Figure 1E, F).
NOTE : Self-made verre fond plats peuvent être utilisés plusieurs fois. Pour échanger la lamelle, lavez-les à l’eau du robinet, soigneusement détacher la lamelle et dégraisser restants avec l’éthanol à 70 % (v/v) et du savon.
- Préparation et placage des cellules primaires
- Lors de la journée de préparation de la culture (jour 4, voir 1.1.4) : transfert d’embryons dans une boîte de Petri stérile cellulaire (diamètre 6 cm) en utilisant une nouvelle pipette Pasteur en plastique. Retirer le liquide de lavage jusqu'à ce que tous les embryons sont rassemblés dans une grosse chute avec un diamètre d’environ 2 cm ou moins.
- Placer le plat avec des embryons dans le banc de travail stérile et ajouter CO2-média indépendant (additionné de 10 % (v/v) filtrée sérum, 1 x glutamine et 1,2 % (v/v) 10 000 U pénicilline-streptomycine ; moyenne avec tous les suppléments est par la suite dénommé « milieu de culture cellulaire ») jusqu'à ce que le plat est à moitié plein.
Remarque : Des solutions de rechange au CO2-support autonome peut être testé selon le but de l’expérience, comme par exemple neurobasal milieu, milieu DMEM ou moyen L-15 de Leibovitz. CO2-support autonome et support L-15 de Leibovitz ont l’avantage de ne pas nécessiter un incubateur à CO2 . - Pour enlever le jaune d’oeuf, Pipetter embryons et descendre à l’aide d’un embout de 200 µL-pipette. Vitellus réussie se distingue par l’opacification du milieu.
- Remplir une boîte de Petri de cellule avec l’éthanol à 70 % (v/v) et un autre plat de culture cellulaire avec milieu de culture de cellules fraîches. Une coupure de 1 000 µL-de pipette permet de transférer les embryons dans une passoire de cellule stérile avec poignée (40 µm ; La figure 1). Prendre la crépine par la poignée et trempez-le dans le plat avec de l’éthanol afin que tous les embryons sont submergées pendant 5 s. immédiatement après, plonger la crépine avec des embryons dans le plat avec un milieu de culture cellulaire fraîches.
Remarque : Les crépines de cellule peuvent être réutilisées plusieurs fois. Nettoyer avec une brosse douce sous l’eau du robinet, magasin dans l’éthanol à 70 % et sec et UV-régal sous le travail stérile banc directement avant utilisation (voir aussi 1.2.4). - Transfert des embryons dans des tubes de réaction stérile de 1,5 mL (environ 100 embryons dans un tube). Ajouter la collagénase (Type 2) dilué dans un milieu de culture cellulaire à une concentration finale de 4 mg/mL dans un volume total de 1 mL. Incuber les tubes avec des embryons sur un rotateur de tube vertical avec 30 tours / min pendant 45 min à température ambiante.
- Dissocier des touffes de cellules restantes en pipettant également, le mélange de l’embryon-collagénase haut et en bas avec une pointe de 1 000 µL-pipette. Puis filtrez la suspension cellulaire à travers un tamis de cellule stérile avec fente de ventilation (40 µm ; La figure 1) dans un tube conique de 50 mL. Rincer la crépine avec environ 10 mL de milieu de culture de cellules fraîches.
- Cellules de granule par centrifugation pendant 3 min à 180 g. Les granulés peuvent être presque invisibles. Avec précaution, retirez le surnageant et remettre en suspension les cellules dans le milieu de culture de 200 µL cellulaires fraîches pour 30 embryons initialement utilisés.
Remarque : Pour obtenir une boulette de visible, il est recommandé de commencer avec un minimum de 100 embryons. - Pipeter 200 µL de la suspension cellulaire obtenue à l’étape 1.3.7 directement sur la surface de verre d’un plat de fond de verre de poly-L-lysine-enduit self-made (voir 1.2). Incuber pendant 60 min à température ambiante sous le banc de travail stérile. Ajouter 6 mL de milieu de culture de cellules fraîches et incuber les cellules primaires à 28 ° C.
- Effectuer l’application d’imagerie souhaitée à l’aide d’un microscope inversé à 28 ° C. Change tous les jours le milieu de culture cellulaire. Cultures peuvent être utilisés pour l’imagerie pendant plusieurs jours après l’ensemencement (dap).

Figure 1 : culture de cellules primaires des embryons de poissons zèbres. (A) une image en noir et blanc de 1 dap embryons qui peuvent être traitées par un logiciel pour analyser le nombre d’embryons. (B) récipients de culture de cellules (diamètre 6 cm) avec un trou (diamètre 10 mm) sont utilisées pour préparer des plats de fond verre réutilisables de self-made. (C) tamis cellulaire (40 µm) avec une poignée simple sont utilisés comme « épuisettes » deyolked embryons de plonger dans l’éthanol et de les transférer rapidement sur milieu de culture de cellules fraîches. (D) Cell crépines (40 µm) avec évacuation des machines à sous sont utilisés pour filtrer les cellules après dissociation induite par la collagénase. (E) après 5 dap, les cellules primaires ensemencées sur verre recouvert de poly-L-lysine principalement forment des neurones avec des extensions prononcées. Echelle = 100 µm. (F) après 5 dap sur plastique traitée sans enduit, des fibroblastes-comme des cellules proliférer la culture. Echelle = 100 µm. (E) et (F) ont été acquises par un microscope à épifluorescence. (G) image lumineuse transmise des cellules primaires dérivés de poisson zèbre type sauvage à 1 dap. Myocytes striés et groupes de neurones qui s’étend de processus minces peuvent être facilement observées. Echelle = 50 µm. (H) des cellules de la lignée transgénique Tg (ptf1a: eGFP) jh1, exprimant eGFP progéniteurs neurones des surtout GABAergique neurones dans le cerveau postérieur et un sous-ensemble de cellules rétiniennes populations29, 30 , 31. Echelle = 50 µm. (G) et (H) ont été acquises par un confocal laser scanning microscope à l’aide de la vaisselle en verre bas faite conformément à (B). S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}
2. la transfection du primaire les cellules avec l’ADN de plasmide
- Remettre filtrée et granulés cellules obtenues à l’étape 1.3.7 dans du PBS 1 x au lieu de milieu de culture cellulaire. Colorer une partie aliquote de la suspension cellulaire avec le bleu Trypan pour déterminer le nombre de cellules dans un comptage de la chambre9.
- 0,5 millions de cellules se mêlent 10 µg ultra pure l’ADN de plasmide dans un tube de réaction de 1,5 mL et ajuster le volume total à 100 µL avec du PBS 1 x.
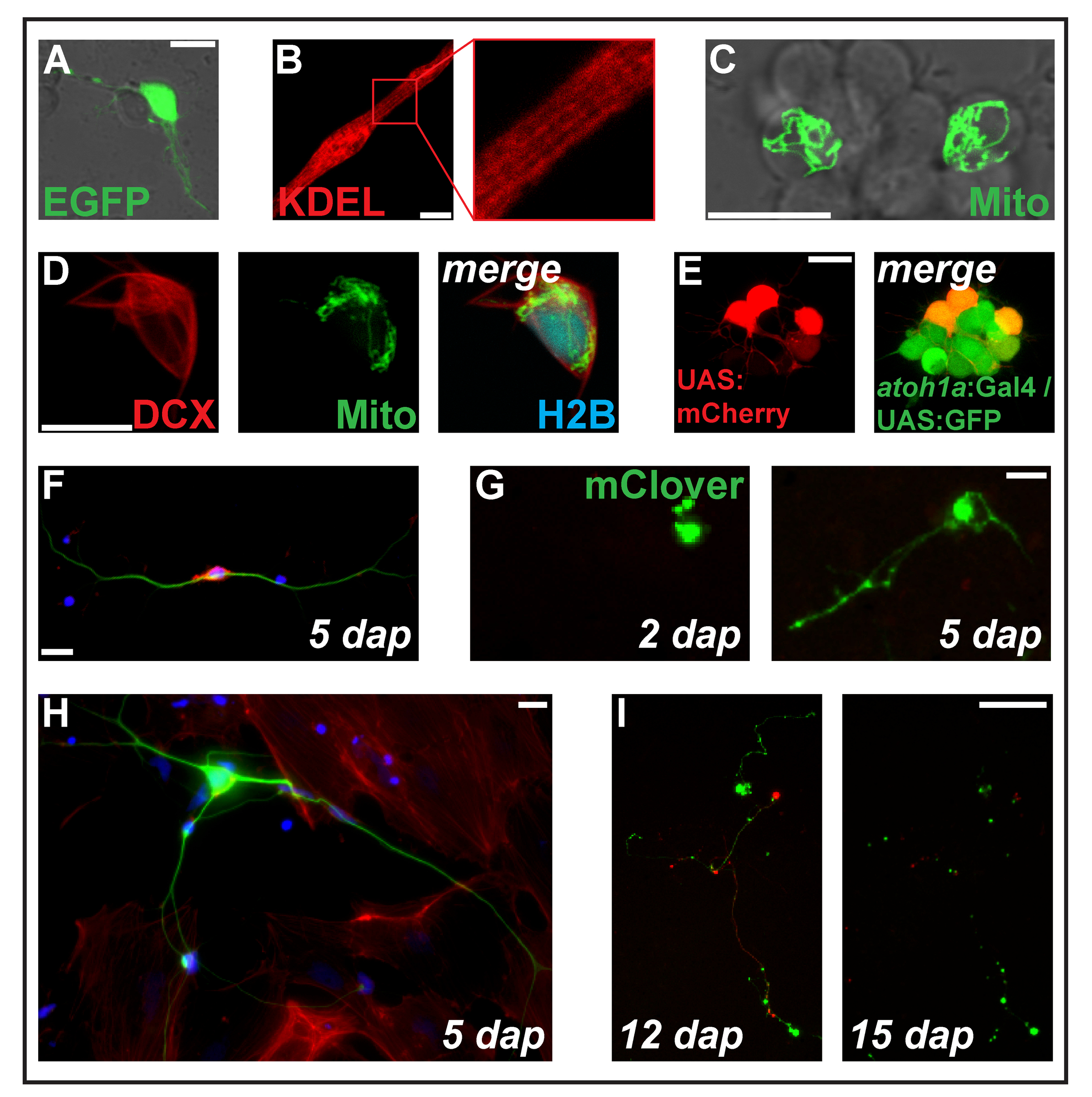
Remarque : Pour nos expériences, nous avons utilisé principalement des constructions d’expression fondées sur le plasmide Bureau2 +10. Expression des cadres ouverts de lecture clonés dans le site multiple de clonage de Bureau2 + est entraînée par le promoteur omniprésent de la cytomégalovirus humain (promoteur de CMV). Autres constructions de l’expression et les promoteurs peuvent être testés (voir aussi les résultats de représentant et de la Figure 2 et Figure 3). - Transfert de l’ADN cellulaire mix immédiatement à une cuvette d’électroporation (0,4 cm), placer la cuve dans un dispositif d’électroporation et oligonucléotides avec les paramètres suivants : impulsion unique, décroissance exponentielle, 280 V, 950 µF.
- Directement après l’électroporation, transférer le mélange de l’ADN cellulaire dans un tube de réaction de 1,5 mL avec 300 µL de milieu de culture de cellules fraîches.
- Plaque de 200 µL de la suspension cellulaire comme décrit dans 1.3.8 et procéder comme décrit dans 1.3.9 selon la construction de l’expression utilisée, l’expression de protéines fluorescentes peut-être être détectable après quelques heures ou lors de la prochaine journée.
3. la coloration des cellules primaires fixes
NOTE : Structures subcellulaires peuvent également être visualisées par immunomarquage classique au lieu d’utiliser des reporters de protéine de fusion fluorescent. Pour les cellules primaires de poisson-zèbre, nous utilisons le protocole standard suivant à noyau exemplaire tache, F-actine et acétylée tubuline avec marqueurs fluorescents.
- Cellules de la plaque sur les lamelles revêtus de poly-L-lysine placé dans une plaque de plat ou multiwell de culture cellulaire comme décrit plus haut (voir 1.3.8).
- Pour la fixation, enlever le milieu et couvrir les cellules avec du paraformaldéhyde de 4 % en solution 1 PBS x. Incuber les cellules pendant 10 min à 4 ° C dans un agitateur. Laver les cellules 3 x de 5 min chacun avec du PBS 1 x à température ambiante. S’assurer que la solution de PBS 1 x couvre les cellules complètement et effectuez les opérations de lavage sur un agitateur.
- Pour bloquer et pour permeabilize les cellules fixes, couvrir les cellules avec du PBS 1 x contenant 5 % de lait écrémé et 0,3 % Triton X-100. Placer les cellules pendant 10 min à température ambiante sur un agitateur. Laver les cellules comme décrit au point 3.2.
- Pour étiqueter la tubuline acétylée, un marqueur des axones11, diluer l’anticorps primaire de 1:2,000 en 1 x PBS contenant 1 % de lait écrémé. Couvrir les cellules grâce à cette solution et les incuber pendant la nuit à 4 ° C dans un agitateur. Lors de la prochaine journée, laver les cellules comme décrit au point 3.2.
- Diluer l’anticorps secondaire conjugué avec le fluorochrom vert fluorescéine isothiocyanate (FITC) au 1/100 dans du PBS 1 x contenant 1 % de lait écrémé et incuber les cellules avec cette solution pendant 1 h à température ambiante dans l’obscurité (couvrir le plat par exemple avec une boîte ou feuille d’aluminium) sur un agitateur. Laver les cellules comme décrit au point 3.2.
- Pour simultanément souiller le cytosquelette d’actine et les noyaux, incuber les cellules dans du PBS 1 x additionné de phalloïdine12 conjugué avec un fluorochrome rouge (01:50) et 4', 6-diamidino-2-phénylindole (DAPI)13 (100 ng/mL) pendant 10 min à la chambre température dans l’obscurité sur un agitateur. Laver les cellules comme décrit au point 3.2.
- Pour préparer des cellules pour l’imagerie, mettre un support d’objet de verre (lame de microscope) sur une surface propre et placez sur la goutte de milieu de montage là-dessus. Prendre une lamelle couvre-objet avec cellules fixes et colorées sur un plat à l’aide de la pince à épiler et placez-le sur la chute avec les cellules vers le transporteur de l’objet. Veillez à ce que ce support de fixation s’étend sur toute la surface de la lamelle couvre-objet. Laisser sécher dans l’obscurité.
- Store fixe et monté des cellules dans l’obscurité à 4 ° C jusqu'à ce que l’application d’imagerie désirée est effectué.

Figure 2 : Transfection d’expression construit par électroporation. (A) neurone Putative transfecté avec pCS-eGFP 1 dap. Myocyte strié (B) (2 dap) exprimant la protéine ciblée réticulum endoplasmique ss-DP-KDEL. (C) deux neurones dans un cluster neuronal transfectées avec PC-MitoTag-YFP à 2 dap. (D) Cell (2 dap) triple transfectées avec pCS-DCX-tdTomato, PC-MitoTag-YFP et pCS-H2B-mseCFP. (E) pSK-électroporation de UAS:mCherry dans les cellules primaires (1 dap) provenant d’embryons transgéniques double transportant les transgènes Tg (atoh1a: Gal4TA4) hzm222 et Tg (4xUAS:KGFPGI) hzm332 aboutissant à l’expression de la GFP dans progéniteurs neurones du rhombencéphale. Barreaux de l’échelle = 10 µm. (A-E) ont été acquises par un laser confocal, microscopie en utilisant les verre fond plats comme illustré dans la Figure 1 b. (F) coloration fluorescente de neurones primaires zebrafish fixe à 5 dap. Bleu : DAPI (noyau) ; Rouge : Phalloïdine (actine-F) ; Vert : Acétylé tubuline (neurones). Echelle = 10 µm. (G) neurone cellules transfectées avec pCS-mClover. 2 dap, aucune extension n’est visible. 5 dap, a formé une structure en forme de neurites. Echelle = 25 µm. (H) neurone de la même préparation que la cellule en (F), entourée de cellules semblables à des fibroblastes. Echelle = 10 µm. (j’ai) neurone dérivées d’un embryon transgénique porteurs du transgène Tg (XITubb: DsRed) zf14828 transfectées avec pCS-mClover. Entre 12 et 15 dap, les neurites subissent une dégénérescence massive. Echelle = 100 µm. les cellules montrés (F-I) ont été ensemencés sur verre revêtu de poly-L-lysine (F, H) ou filtrée de plastique (G, I), cultivé en L-15 en présence de 10 % de sérum de bovin et le neuronale Supplément B-27 (dilué 01:50) et photographié avec un microscope à épifluorescence. S’il vous plaît cliquez ici pour visionner une version agrandie de cette figure.
{kind=link}
4. préparation des cellules primaires de cerveau de poisson-zèbre adulte
-
Extraction du cerveau
- Sélectionnez un poisson adult d’au moins 90 jours d’âge. Si vous travaillez avec un type de cellule spécifique est nécessaire, choisissez une lignée transgénique dans laquelle expression spécifique des cellules Rapporteur fluorescent permettra de visualiser les cellules souhaitées.
- Placer le poisson dans un bécher contenant 200 mL de l’anesthésique tricaïne8 (0,2 %) à Danieau 30 %. Attendez que le poisson ne bouge. Immerger les poissons anesthésiés dans un bécher rempli avec 200 mL d’eau glacée pendant 15 min à euthanasier il.
- Remplir une boîte de Pétri (diamètre 6 cm) avec l’éthanol à 70 % (v/v). Tenir le poisson par la queue avec une paire de pincettes et trempez-le dans l’éthanol. S’assurer que le poisson soit totalement immergé dans l’éthanol pendant 5 s.
- Extraire le cerveau selon le protocole du Gupta et Mullins14 , avec les adaptations suivantes : utiliser uniquement des outils stérilisés à l’autoclave ou stérile emballé et disséquer la tête dans du PBS stérile 1 x.
- Directement après l’extraction, placer le cerveau dans une boîte de Pétri (diamètre 3 cm) remplie avec du PBS stérile 1 x et passer le plat sous un banc de travail stérile pour la culture cellulaire.
-
Dissociation du cerveau et le placage des cellules primaires
- Placer deux ensembles de pinces stériles (autoclave), une boîte de Petri stérile (diamètre 6 cm) rempli d’éthanol à 70 % (v/v), une boîte de Petri stérile (diamètre 10 cm) rempli L-15 additionné de Leibovitz de 10 % (v/v) filtrée de sérum de bovin, de B-27 (01:50) et de 1,2 % (v / v) 10 000 U pénicilline-streptomycine et une crépine d’aspiration cellule stérile avec poignée (40 µm ; La figure 1) sous le banc propre.
- Place la crépine de la cellule dans la boîte de Pétri remplie avec de l’éthanol et s’assurer que le niveau de liquide soit au moins 5 mm de plus que le fond de la passoire.
- À l’aide de la première série de pincettes, transférer le cerveau dans la passoire déjà placée dans l’éthanol et s’assurer qu'il est entièrement recouverte par le liquide. Après 1 s, transfert la crépine avec le cerveau dans la boîte de Pétri contenant un milieu L-15 de Leibovitz avec ce qui précède décrit des suppléments.
- En utilisant le deuxième ensemble de pinces à épiler, transfert le cerveau dans un tube à essais stérile de 1,5 mL rempli de milieu L-15 de 500 µL Leibovitz avec ce qui précède décrit des suppléments. Ajouter collagénase (Type 2) à une concentration finale de 4 mg/mL dans un volume total de 1 mL.
- Incuber le tube sur un rotateur de tube vertical avec 30 tours / min pour 35 min à température ambiante. Mécaniquement, se dissocient des touffes de tissus restants de pipetage et descendre à l’aide d’une pointe de pipette 1 000 µL pour faciliter le processus de dissociation.
- Arrêter la dissociation quand aucuns particules visibles ne restent en solution. Filtrer la suspension cellulaire à travers un tamis de cellule stérile avec fente de ventilation (40 µm ; La figure 1) dans un tube conique de 50 mL. Rincer la crépine avec environ 10 mL de milieu de culture de cellules fraîches.
Remarque : Lorsque la suspension monocellulaire est obtenue, l’étape de filtration n’est pas aussi nécessaire qu’en cas de dissociation des embryons. Le cerveau est un tissu mou et comme tel, il est plus enclin à être homogène dissociés en suspension cellulaire unique. - Cellules de granule par centrifugation pendant 5 min à 180 x g et Resuspendre le culot dans 1 mL de milieu L-15 de Leibovitz frais avec les suppléments décrits ci-dessus.
- Pipeter 500 µL de suspension cellulaire (50 % des cellules obtenues) sur un plat de bas de self-made verre revêtu de poly-L-lysine (voir 1.2) ou dans un puits d’une plaque 24 puits. Réduire en cas de petites surfaces (c.-à-d. 125 µL de solution pour un puits d’une plaque de 96 puits). Incuber pendant 60 min à température ambiante sous le banc de travail stérile. Puis ajouter la quantité nécessaire de milieu frais pour remplir le conteneur spécifique et incuber les cellules primaires à 28 ° C.
- Effectuer l’application d’imagerie souhaitée à l’aide d’un microscope inversé à 28 ° C. Cultures peuvent être utilisés pour l’imagerie pendant plusieurs jours après l’ensemencement. Remplacez 50 % de la moyenne sur une base quotidienne.
Résultats
Figure 1 montre une image de lumière transmise d’une culture typique provenant d’embryons de type sauvage avec des myocytes striés et amas de cellules de neurone comme étant plus abondant. Pour identifier plus facilement certains types de cellules, une lignée transgénique avec expression spécifiques à un type de cellule d’une protéine fluorescente peut être utilisée (Figure 1 H).
Discussion
Nous présentons ici deux protocoles différents de cellules en culture primaire de 2 embryons de poisson-zèbre dpf ou cerveau de poisson-zèbre adulte.
La préparation des cultures de cellules primaires de 2 dpf poisson zèbre est relativement facile à réaliser pour quelqu'un ayant une expérience dans les techniques de culture cellulaire de base. Toutefois, pour obtenir des résultats reproductibles satisfaisants, un nombre suffisant d’embryons comme matériel de départ est crucial (10...
Déclarations de divulgation
Les auteurs n’ont rien à divulguer.
Remerciements
Nous remercions T. Fritsch, A. Wolf-Asseburg, I. Linde et S.-M. Tokarski pour animaux excellent et un support technique. Nous sommes reconnaissants à tous les membres du laboratoire Köster de discussions intenses et utiles. Nous reconnaissons avec financement par la Deutsche Forschungsgemeinschaft (KO 1949/5-1) et le Land de Basse-Saxe, Niedersächsisches Vorab (VWZN2889).
matériels
| Name | Company | Catalog Number | Comments |
| Fish lines | |||
| AB (wild-type) | established by Streisinger and colleagues, available from the Zebrafish International Resource Center (ZIRC) | ||
| Tg(ptf1a:eGFP)jh1 | stable transgenic line in which the enhancer of the zebrafish gene ptf1a drives expression of the fluorescent protein EGFP (Parsons et al., 2007) | ||
| Tg(XITubb:DsRed)zf148 | stable transgenic line in which the Xenopus neural-specific beta tubulin promoter drives expression of the fluorescent protein DsRed (Peri and Nüsslein-Volhard, 2008) | ||
| Name | Company | Catalog Number | Comments |
| Equipment | |||
| centrifuge | Eppendorf | model 5804 R | |
| ChemiDoc MP imaging system | BioRad | model XRS+, used to acquire black-and-white images of Petri dishes containing 1 da embryos | |
| confocal laser scanning microscope | Leica microsystems | model SP8, equipped with 28 °C temperature box and a 63X objective | |
| epifluorescent microscope | Leica microsystems | model DM5500B, equipped with 28 °C temperature box and a 40X objective | |
| Gene Pulser Xcell with capacitance extender | BioRad | 1652661 | electroporation device |
| Horizontal shaker | GFL | model 3011 | |
| incubator for cell culture (28 °C) | Memmert | model incubator I | |
| incubator for embryos (28 °C) | Heraeus | type B6120 | |
| light microscope | Zeiss | model TELAVAL 31 | |
| micro pipettes | Gilson | ||
| sterile work bench | Bio Base | with laminar flow and UV light | |
| tweezers | Dumont | Style 5, Inox | |
| vertical tube rotator | Labinco B.V. | model LD-79 | |
| Name | Company | Catalog Number | Comments |
| Software | |||
| Image Lab Software | BioRad | for the ChemiDoc MP imaging system from BioRad | |
| ImageJ | National Institutes of Health | used for counting 1 dpf embryos by applying the Count particles-tool to the respective black-and-white images; Rasband, W.S., ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA, https://imagej.nih.gov/ij/. (1997-2016). | |
| LAS X | Leica Microsystems | for both confocal and epifluorescent microscopes from Leica Microsystems | |
| Name | Company | Catalog Number | Comments |
| Plasmids | |||
| pCS-DCX-tdTomato | Köster Lab | # 1599 | based on the backbone pCS2+ (Rupp et al., 1994) |
| pCS-eGFP | Köster Lab | # 7 | based on the backbone pCS2+ (Rupp et al., 1994) |
| pCS-H2B-mseCFP | Köster Lab | # 2379 | based on the backbone pCS2+ (Rupp et al., 1994) |
| pCS-mClover | Köster Lab | # 3865 | based on the backbone pCS2+ (Rupp et al., 1994) |
| pCS-MitoTag-YFP | Köster Lab | # 2199 | based on the backbone pCS2+ (Rupp et al., 1994) |
| pCS-ss-RFP-KDEL | Köster Lab | # 4330 | based on the backbone pCS2+ (Rupp et al., 1994) |
| pCS-VAMP1-mCitrine | Köster Lab | # 2291 | based on the backbone pCS2+ (Rupp et al., 1994) |
| pSK-UAS:mCherry | Köster Lab | # 1062 | based on the pBluescript-backbone of Stratagene |
| Plasmid numbers refer to the database entries of the Köster lab. Plasmids are available upon request. | |||
| Name | Company | Catalog Number | Comments |
| Plastic and glass ware | |||
| BD Falcon Cell Strainer (40 µm) | FALCON | REF 352340 | distributed by BD Bioscience, used as “landing net” to dip deyolked embryos into ethanol and to transfer them quickly to fresh cell culture medium |
| 1.5 mL reaction tubes | Sarstedt | 72690550 | |
| 24-well plate | Sarstedt | 83.3922 | |
| 50 mL falconic tube | Sarstedt | 62.547.004 | |
| 96-well plate | Sarstedt | 83.3924.005 | |
| EasyStrainer (40 µm) | Greiner Bio-One | 542 040 | with venting slots; used to filter cells after collagenase-mediated dissociation |
| electroporation cuvette (0.4 cm) | Kisker | 4905022 | |
| glass coverslips | Heinz Herenz Medizinalbedarf GmbH | 1051201 | |
| Microscope slides | Thermo Fisher Scientific (Menzel Gläser) | 631-0845 | |
| Neubauer chamber | Henneberg-Sander GmbH | 9020-01 | |
| Pasteur pipettes (plastic; 3 mL) | A. Hartenstein | PP05 | |
| Petri dishes (plastic; diameter 10 cm) | Sarstedt | 821473 | for zebrafish embryos |
| pipette tips | Sarstedt | Blue (1000 µl): 70762; Yellow (200 µl): 70760002; White (10 µL): 701116 | |
| sterile cell culture dishes (plastic; diameter 3 cm) | TPP Techno Plastic Products AG | 93040 | |
| sterile cell culture dishes (plastic; diameter 6 cm) | Sarstedt | 72690550 | |
| sterile Petri dishes (plastic; diameter 10 cm) | Sarstedt | 83.3902 | for brain dissection |
| Name | Company | Catalog Number | Comments |
| Chemicals and Reagents | |||
| sodium chloride | Roth | 0601.1 | |
| 4 % paraformaldehyde in 1x PBS | Sigma-Aldrich | 16005 | |
| 4',6-diamidino-2-phenylindole (DAPI) | Thermo Fisher Scientific | D1306 | |
| calcium nitrate tetrahydrate | Sigma-Aldrich | C1396 | |
| ethanol p.a. 100% | Sigma-Aldrich | 46139 | |
| goat α-mouse IgG (Fc specific) FITC conjugated | Thermo Fisher Scientific | 31547 | |
| HEPES | Roth | 9105.4 | |
| high vacuum grease | DOW CORNING | 3826-50 | silicon grease used for self-made glass bottom dishes |
| magnesium sulfate heptahydrate | Merck | 105886 | |
| methylene blue | Serva | 29198.01 | |
| Monoclonal Anti-Tubulin, Acetylated antibody | Sigma-Aldrich | T6793 | |
| Aqua-Poly/Mount (mounting medium) | Polyscience | 18606 | |
| poly-L-lysine | Biochrom | L 7240 | |
| potasssion chloride | Merck | 104938 | |
| Skim milk | Roth | 68514-61-4 | |
| Texas Red-X Phalloidin | Thermo Fisher Scientific | T7471 | |
| Tricaine | Sigma-Aldrich | E10521 | Synonym: Ethyl 3-aminobenzoate methanesulfonate |
| Triton X-100 | BioRad | 1610407 | |
| Trypan Blue | Gibco by Life Technologies | 15250061 | |
| Name | Company | Catalog Number | Comments |
| Enzymes | |||
| collagenase (Type 2) | Thermo Fisher Scientific | 17101015 | dissolve powder in cell culture medium (8 mg/mL) and sterile-filter the solution, store aliquots at -20 °C |
| pronase (from Streptomyces griseus) | Roche | 11459643001 | distributed by Sigma-Aldrich, dissolve in 30% Danieau (10 mg/mL) and store aliquots at -20 °C |
| Name | Company | Catalog Number | Comments |
| Medium and solutions for cell culture | |||
| 1x PBS (Dulbecco's Phosphate Buffered Saline) | Gibco by Life Technologies | 14190-169 | distributed by Thermo Fisher Scientific |
| CO2-independent medium | Gibco by Life Technologies | 18045054 | distributed by Thermo Fisher Scientific |
| filtrated bovine serum (FBS) | PAN-Biotech | individual batch | |
| glutamine 100x | Gibco by Life Technologies | 25030081 | distributed by Thermo Fisher Scientific |
| Leibovitz's L-15 medium | Gibco by Life Technologies | 11415049 | distributed by Thermo Fisher Scientific |
| PenStrep (10,000 U/mL) | Gibco by Life Technologies | 15140148 | distributed by Thermo Fisher Scientific |
Références
- Ablain, J., Zon, L. I. Of fish and men: using zebrafish to fight human diseases. Trends in Cell Biology. 23, 584-586 (2013).
- Sassen, W. A., Köster, R. A molecular toolbox for genetic manipulation of zebrafish. Advances in Genomics and Genetics. , 151 (2015).
- Scheer, N., Campos-Ortega, J. A. Use of the Gal4-UAS technique for targeted gene expression in the zebrafish. Mechanisms of Development. 80, 153-158 (1999).
- Köster, R. W., Fraser, S. E. Tracing transgene expression in living zebrafish embryos. Developmental Biology. 233, 329-346 (2001).
- Driever, W., Rangini, Z. Characterization of a cell line derived from zebrafish (Brachydanio rerio) embryos. In Vitro Cellular & Developmental Biology - Animal. 29A, 749-754 (1993).
- Badakov, R., Jaźwińska, A. Efficient transfection of primary zebrafish fibroblasts by nucleofection. Cytotechnology. 51, 105-110 (2006).
- Senghaas, N., Köster, R. W. Culturing and transfecting zebrafish PAC2 fibroblast cells. Cold Spring Harbor Protocols. , (2009).
- Westerfield, M. . The zebrafish book. A guide for the laboratory use of zebrafish (Danio rerio). , (2007).
- Basic methods in cellular and molecular biology. Using a hemacytometer to count cells. Journal of Visualized Experiments Available from: https://www.jove.com/science-education/5048/using-a-hemacytometer-to-count-cells (2017)
- Rupp, R. A., Snider, L., Weintraub, H. Xenopus embryos regulate the nuclear localization of XMyoD. Genes & Development. 8, 1311-1323 (1994).
- Piperno, G., Fuller, M. T. Monoclonal antibodies specific for an acetylated form of alpha-tubulin recognize the antigen in cilia and flagella from a variety of organisms. Journal of Cell Biology. 101 (6), 2085-2094 (1985).
- Barden, J. A., Miki, M., Hambly, B. D., Dos Remedios, C. G. Localization of the phalloidin and nucleotide-binding sites on actin. European Journal of Biochemistry. 162 (3), 583-588 (1987).
- Kapuscinski, J. DAPI: a DNA-specific fluorescent probe. Biotechnic & Histochemistry. 70 (5), 220-233 (1995).
- Gupta, T., Mullins, M. C. Dissection of organs from the adult zebrafish. Journal of Visualized Experiments. 37, E1717 (2010).
- Chalfie, M., Tu, Y., Euskirchen, G., Ward, W. W., Prasher, D. C. Green fluorescent protein as a marker for gene expression. Science. 263, 802-805 (1994).
- Stornaiuolo, M. KDEL and KKXX retrieval signals appended to the same reporter protein determine different trafficking between endoplasmic reticulum, intermediate compartment, and Golgi complex. Molecular Biology of the Cell. 14, 889-902 (2003).
- Lithgow, T. Targeting of proteins to mitochondria. FEBS Letters. 476, 22-26 (2000).
- Nagai, T., Ibata, K., Park, E. S., Kubota, M., Mikoshiba, K., Miyawaki, A. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nature Biotechnology. 20, 87-90 (2002).
- Sassen, W. A., Lehne, F., Russo, G., Wargenau, S., Dübel, S., Köster, R. W. Embryonic zebrafish primary cell culture for transfection and live cellular and subcellular imaging. Developmental Biology. 430, 18-31 (2017).
- Horesh, D., et al. Doublecortin, a stabilizer of microtubules. Human Molecular Genetics. 8, 1599-1610 (1999).
- Shaner, N. C., Campbell, R. E., Steinbach, P. A., Giepmans, B. N. G., Palmer, A. E., Tsien, R. Y. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nature Biotechnology. 22, 1567-1572 (2004).
- Distel, M., Hocking, J. C., Volkmann, K., Köster, R. W. The centrosome neither persistently leads migration nor determines the site of axonogenesis in migrating neurons in vivo. Journal of Cell Biology. 191, 875-890 (2010).
- Matsuda, T., Miyawaki, A., Nagai, T. Direct measurement of protein dynamics inside cells using a rationally designed photoconvertible protein. Nature Methods. 5, 339-345 (2008).
- Archer, B. T., Ozçelik, T., Jahn, R., Francke, U., Südhof, T. C. Structures and chromosomal localizations of two human genes encoding synaptobrevins 1 and 2. Journal of Biological Chemistry. 265, 17267-17273 (1990).
- Griesbeck, O., Baird, G. S., Campbell, R. E., Zacharias, D. A., Tsien, R. Y. Reducing the environmental sensitivity of yellow fluorescent protein. Mechanism and applications. Journal of Biological Chemistry. 276, 29188-29194 (2001).
- Shaner, N. C., et al. A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nature Methods. 10, 407-409 (2013).
- Campbell, R. E., et al. A monomeric red fluorescent protein. Procedings of the National Academy of Sciences of the United States of America. 99, 7877-7882 (2002).
- Peri, F., Nüsslein-Volhard, C. Live imaging of neuronal degradation by microglia reveals a role for v0-ATPase a1 in phagosomal fusion in vivo. Cell. 133, 916-927 (2008).
- Godinho, L., et al. Targeting of amacrine cell neurites to appropriate synaptic laminae in the developing zebrafish retina. Development. 132, 5069-5079 (2005).
- Jusuf, P. R., Harris, W. A. Ptf1a is expressed transiently in all types of amacrine cells in the embryonic zebrafish retina. Neural Development. 4, 34 (2009).
- Kani, S., et al. Proneural gene-linked neurogenesis in zebrafish cerebellum. Developmental Biology. 343, 1-17 (2010).
- Distel, M., Wullimann, M. F., Köster, R. W. Optimized Gal4 genetics for permanent gene expression mapping in zebrafish. Procedings of the National Academy of Sciences of the United States of America. 106, 13365-13370 (2009).
- Choorapoikayil, S., Overvoorde, J., den Hertog, J. Deriving cell lines from zebrafish embryos and tumors. Zebrafish. 10, 316-332 (2013).
Réimpressions et Autorisations
Demande d’autorisation pour utiliser le texte ou les figures de cet article JoVE
Demande d’autorisationThis article has been published
Video Coming Soon
À PROPOS DE JoVE
Copyright © 2025 MyJoVE Corporation. Tous droits réservés.