
È necessario avere un abbonamento a JoVE per visualizzare questo. Accedi o inizia la tua prova gratuita.
Method Article
Cultura e transfezione di cellule primarie di Zebrafish
In questo articolo
Riepilogo
Vi presentiamo un protocollo efficiente e facile da usare per la preparazione di colture cellulari primarie di embrioni di zebrafish per la transfezione e live cell imaging nonché un protocollo per preparare cellule primarie dal cervello di zebrafish adulto.
Abstract
Embrioni di zebrafish sono trasparenti e sviluppano rapidamente di fuori della madre, consentendo in tal modo eccellente in vivo imaging dei processi biologici dinamici in un vertebrato intatto e in via di sviluppo. Tuttavia, le immagini dettagliate delle morfologie di tipi distinti delle cellule e strutture subcellulari sono limitato in tutto monta. Quindi, abbiamo stabilito un protocollo efficiente e facile da usare per coltura primaria di cellule vive da embrioni di zebrafish e tessuto adulto.
In breve, di 2 embrioni di zebrafish dpf sono dechorionated, deyolked, sterilizzato e dissociato di singole cellule con collagenasi. Dopo una fase di filtrazione, cellule primarie sono piastrate su piatti di vetro inferiore e coltivate per diversi giorni. Colture fresche, per quanto a lungo termine differenziata quelli, possono essere utilizzati per studi di imaging confocale ad alta risoluzione. La cultura contiene diversi tipi di cellule, con miociti striati e neuroni essendo prominente sul rivestimento di poli-L-lisina. A label specificamente le strutture subcellulari di proteine marker fluorescente, abbiamo anche stabilito un protocollo di elettroporazione che permette la transfezione del DNA del plasmide in tipi differenti delle cellule, compresi i neuroni. Così, in presenza di operatore stimoli definiti, comportamento delle cellule complesse e dinamiche intracellulare delle cellule zebrafish primario può essere valutata con elevata risoluzione spaziale e temporale. Inoltre, utilizzando il cervello adulto zebrafish, dimostriamo che la tecnica descritta dissociazione, come pure le condizioni di coltura di base, funzionano anche per il tessuto di zebrafish adulto.
Introduzione
Il pesce zebra (Danio rerio, d. rerio) è un modello popolare vertebrato per numerosi campi della ricerca biomedica di base1. Embrioni di zebrafish ex uterosi sviluppano rapidamente, sono trasparenti e si adattano al microscopio, fornendo così eccellenti prerequisiti per lo studio di sviluppo vertebrato in un organismo vivente. A causa della genetica trattabilità di zebrafish2, molte linee di reporter transgenici stabile con espressione di tipo specifico delle cellule di vari marcatori fluorescenti sono state stabilite consentendo l'osservazione di popolazioni cellulari specifici. La comunità di zebrafish offre un'ampia varietà di cosiddette linee Gal4-driver che trasportano un transgene esprimendo il Kal4TA4 sintetico (o il GalFF di KalTA3-equivalente) gene con il dominio di legame del DNA, Gal4 di lievito fusa per attivazione trascrizionale virale domini sotto il controllo di rinforzatori di tipo specifico di cellule. Queste linee di driver sono incrociate alle linee dell'effettore che trasportano transgeni che consiste di una sequenza d'attivazione a Monte definita (UAS) fusa a un gene reporter. La proteina Kal4TA4 si lega all'elemento UAS, attivando così l'espressione di tipo selettivo delle cellule del reporter gene3,4. Questo approccio consente studi altamente diversificati combinatoriali di quasi tutti gli elementi disponibili enhancer e reporter in animali transgenici doppio.
Tuttavia, approfondita formazione immagine dal vivo con focus su singole celle o del loro contenuto subcellulare è limitata in un embrione intero e in costante evoluzione. Per rispondere a domande biologiche cellulari specifici con risoluzione più alta, l'uso di colture cellulari è spesso preferibile. Alcune linee di cellule di zebrafish esistono, ma sono considerati come fortemente selezionato5,6,7 e la loro propagazione è spesso richiede molto tempo. Inoltre, tutte le linee cellulari disponibili sono dei fibroblasti derivati, limitando esperimenti utilizzando colture cellulari per un tipo di cellule. Quindi, abbiamo stabilito un protocollo sia efficiente e facile da usare per preparare cellule primarie direttamente da embrioni di zebrafish e cervello di zebrafish adulto, insieme ad approcci per aumentare la longevità della cultura e di ampliare la diversità di coltivato tipi di cellule. Inoltre, presentiamo una procedura per transfect cellule embrionali primari con costrutti di espressione per i marcatori fluorescenti organello. Così, cellulari morfologie e strutture subcellulari possono essere analizzati con elevata risoluzione spaziale e temporale in tipi distinti delle cellule che mantengono le loro caratteristiche chiave.
Protocollo
Tutto il lavoro animale descritto qui è secondo disposizioni di legge (EU-Direttiva 2010/63). Manutenzione e gestione di pesce è stato approvato dalle autorità locali e dal benessere animale rappresentanza l'Università tecnica di Braunschweig, Bassa Sassonia stato ufficio della protezione dei consumatori e sicurezza alimentare (LAVES, Oldenburg, Germania; AZ. § 4 (02.05) TSchB TU BS).
1. preparazione di cellule primarie da embrioni di Zebrafish
- Preparazione di 2 giorni dopo fertilizzazione (dpf) embrioni di zebrafish
- Giorno 1: Impostare diversi attraversamenti del ceppo zebrafish di scelta e secondo le specifiche del vostro zebrafish facility manager8.
- Giorno 2: Mate pesce e raccogliere le uova8 direttamente dopo la deposizione in un 10cm di Petri (plastica). Rimuovere le uova contaminate o morte con una pipetta di Pasteur (plastica). Lavare le uova 1 x con Danieau 30% (5,8 mM di cloruro di sodio, 0,07 mM di cloruro di potassio, solfato di magnesio di 0,04 mM, 0,06 mM nitrato di calcio, acido 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic 5 mM, pH 7,2)8 con blu di metilene 0,0001% (p/v). Cambio medio-Danieau 30% senza il blu di metilene, poiché il blu di metilene può causare autofluorescenza e incubare uova tutta la notte a 28 ° C.
Nota: Iniziare con un minimo di 100 uova per ogni riga di pesce al fine di ottenere una quantità sufficiente di cellule. Non sollevare più di 150 embrioni in una capsula di Petri. - Giorno 3: Rimuovere gli embrioni morti o contaminati e scambiare il mezzo a Danieau 30%. Determinare il numero di embrioni contando. Incubare gli embrioni tutta la notte a 28 ° C.
Nota: Quando si utilizza una linea transgenica che esprimono un reporter fluorescente, proiezione al 1 dpf o 2 dpf può essere necessario.
Nota: Per grandi quantità di embrioni, si consiglia di prendere un immagine in bianco e nero dei rispettivi di Petri (Figura 1A) e di quantificare il numero di embrioni con un software di imaging. - Giorno 4: Gli embrioni sono ora 2 dpf. Per rimuovere chorions, aggiungere 1 mL pronase con una concentrazione di 1 mg/mL a 10 mL di Danieau 30%8 e incubare embrioni su un agitatore a temperatura ambiente fino a quando tutti i chorions sono staccati (20 – 40 min a seconda della temperatura ambiente). Lavare con Danieau 30% per rimuovere sia pronase e chorions e conservare gli embrioni a temperatura ambiente fino all'ulteriore utilizzo.
- Preparazione di piatti fondo riutilizzabile vetro rivestito di poli-L-lisina
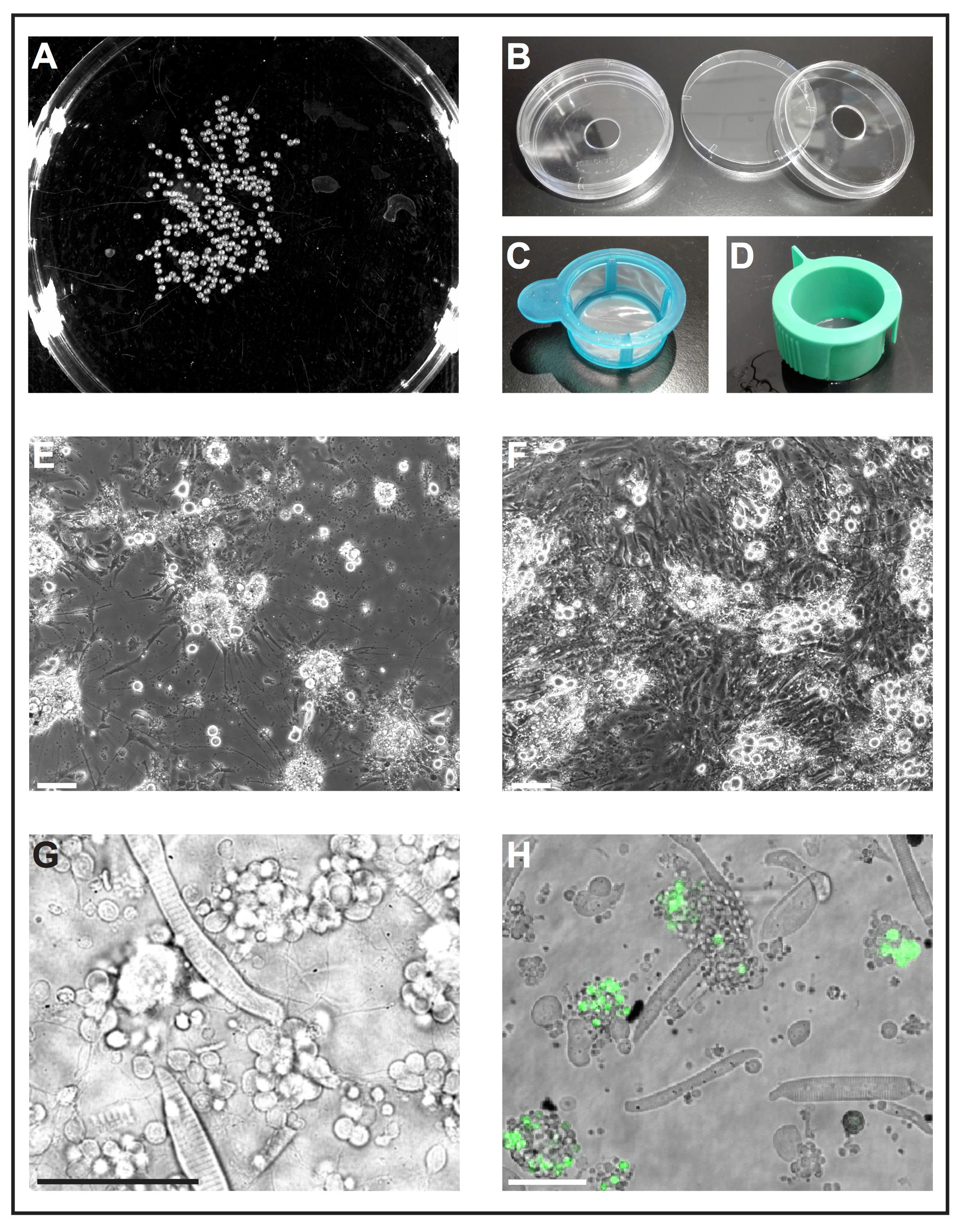
Nota: Piatti di fondo di vetro disponibili in commercio sono progettati per solo uso singolo e sono costosi. Nella procedura seguente viene descritto come preparare piatti di fondo di vetro self-made riutilizzabili da materiali di laboratorio standard.- Praticare un foro con un diametro di 10 mm nella parte inferiore delle piastre di coltura cellulare standard (diametro 6 cm, plastica) (Figura 1B). Lavare il piatto fondo accuratamente con acqua corrente per rimuovere la polvere di foratura.
- Spalmare grasso di silicone intorno al foro sul lato inferiore del piatto fondo e allegare un vetrino coprioggetti utilizzando il grasso come colla. Assicurarsi che il grasso sigilla il divario tra il fondo piatto e il vetrino coprioggetti.
Nota: Assicurarsi che lo spessore delle lamelle usate è appropriato per l'applicazione di imaging più tardi. - Lavare i self-made vetro fondo piatti accuratamente, ma con attenzione, con acqua fredda e sapone. Sciacquare i piatti di vetro inferiore tre volte con acqua deionizzata per rimuovere il sapone. Asciugare all'aria coperchi piatto e piatto fondo e memorizzarli in una scatola pulita fino al successivo utilizzo.
- Il giorno della preparazione di cultura (giorno 4, Vedi 1.1.4): inumidire la parte interna di entrambi piatto coperchi e piatto fondo con etanolo al 70% (v/v). Posizionare coperchi piatto e piatto fondo lato interno rivolto verso l'alto in un banco di lavoro sterile con flusso laminare e ai raggi UV. Asciugare all'aria fino a quando l'etanolo viene evaporato, quindi applicare la luce UV per 20 min. Dopo questo trattamento, i piatti sono assemblati e considerati come sterile.
- Per il rivestimento, Pipettare 200 µ l di poli-L-lisina (0,1 mg/mL) al centro di ogni piatto fondo di vetro e diffondere il liquido il coprivetrino rompendo la tensione superficiale con un puntale. Lasciare asciugare per 60 minuti, quindi lavare 1x con sterile 1x tampone fosfato salino (PBS). Rimuovere il liquido. Mantenere i piatti sotto la panca fino al successivo utilizzo.
Nota: Altri rivestimenti possono essere testati secondo lo scopo dell'esperimento. Abbiamo trovato poli-L-lisina sufficiente per sostenere la crescita dei neuroni, mentre trattati plastica senza qualsiasi ulteriore rivestimento sembrava essere favorevole per la crescita del fibroblasto-come delle cellule (Figura 1E, F).
Nota: Piatti di fondo di vetro self-made possono essere utilizzati molte volte. Per scambiare il vetrino coprioggetto, lavare con acqua calda di rubinetto, attentamente staccare il vetrino coprioggetto e rimuovere il grasso rimanente con etanolo al 70% (v/v) e sapone.
- Preparazione e placcatura di cellule primarie
- Il giorno della preparazione di cultura (giorno 4, Vedi 1.1.4): trasferire gli embrioni in una piastra di coltura sterile cella (diametro 6 cm) utilizzando una pipetta di Pasteur plastica fresca. Rimuovere il liquido graduale fino a quando tutti gli embrioni sono raccolti in una grande goccia con un diametro di circa 2 cm o meno.
- Mettere il piatto con gli embrioni nel banco di lavoro sterile e aggiungere CO2-mezzo indipendente (supplementato con 10% (v/v) filtrato siero bovino, 1 x della glutamina e dell'1,2% (v/v) 10.000 U penicillina-streptomicina; media con tutti i supplementi è in quanto segue indicato come "terreno di coltura cellulare") fino a quando il piatto è riempito a metà.
Nota: Alternative a CO2-mezzo indipendente può essere testato secondo lo scopo dell'esperimento, come ad esempio neurobasal medio, DMEM medio o L-15 medio di Leibovitz. CO2-mezzo indipendente e mezzo L-15 di Leibovitz hanno il vantaggio di non richiedere un incubatore a CO2 . - Per rimuovere il tuorlo, pipettare embrioni su e giù utilizzando un 200 µ l-puntale. Deyolking successo può essere riconosciuto dall'opacità del mezzo.
- Riempire una piastra di coltura cellulare con etanolo al 70% (v/v) e un altro piatto di cultura cellulare con terreno di coltura di cellule fresche. Utilizzare un cut-off 1.000 µ l-puntale per trasferire gli embrioni in un colino di cella sterile con manico (40 µm; Figura 1). Prendere il filtro dalla maniglia e immergerlo nel piatto con etanolo affinché tutti gli embrioni sono sommerse per 5 s. immediatamente in seguito, immergere il filtro con embrioni nel piatto con terreno di coltura di cellule fresche.
Nota: Filtri di cella possono essere riutilizzati più volte. Pulire con una spazzola morbida sotto l'acqua del rubinetto, negozio loro in etanolo al 70% e secco e UV-ossequio li sotto il lavoro sterile panca direttamente prima dell'uso (veda inoltre 1.2.4). - Trasferire gli embrioni in provette sterili da 1,5 mL (circa 100 embrioni in un tubo). Aggiungere collagenosi (tipo 2) diluita in mezzo di coltura cellulare a una concentrazione finale di 4 mg/mL in un volume totale di 1 mL. Incubare le provette con embrioni in un rotatore tubo verticale con 30 giri / min per 45 min a temperatura ambiente.
- Dissociare rimanente cella ciuffi pipettando su e giù la miscela di embrione-collagenosi con una punta di 1.000 µ l-pipetta. Quindi filtrare la sospensione cellulare attraverso un filtro sterile cella con sfiato slot (40 µm; Figura 1) in una provetta conica da 50 mL. Risciacquare il filtro con circa 10 mL di terreno di coltura di cellule fresche.
- Pellet di cellule mediante centrifugazione per 3 min a 180 x g. Il pellet può essere quasi invisibile. Con attenzione rimuovere il supernatante e risospendere le cellule in 200 µ l terreno di coltura di cellule fresche per 30 embrioni originalmente usati.
Nota: Per ottenere un pellet visibile, si consiglia di iniziare con un minimo di 100 embrioni. - Dispensare 200 µ l di sospensione cellulare ottenuta al passaggio 1.3.7 direttamente sulla zona di vetro di un piatto fondo di vetro rivestiti con poli-L-lisina self made (Vedi 1.2). Incubare per 60 min a temperatura ambiente sotto il banco di lavoro sterile. Aggiungere 6 mL di terreno di coltura di cellule fresche e incubare le cellule primarie a 28 ° C.
- Eseguire l'applicazione di imaging desiderato utilizzando un microscopio invertito a 28 ° C. Scambiare il mezzo di coltura cellulare ogni giorno. Culture possono essere utilizzate per l'imaging per diversi giorni dopo il placcaggio (dap).

Figura 1: cellula primaria coltura degli embrioni di zebrafish. (A) immagine in bianco e nero di 1 dap embrioni, che possono essere elaborati da uno strumento software per analizzare il numero di embrioni. (B) Cell piastre di coltura (diametro 6 cm) con un foro (diametro 10 mm) vengono utilizzati per preparare piatti di fondo di vetro self-made riutilizzabili. (C) filtri cella (40 µm) con una semplice maniglia sono utilizzati come "guadino" a tuffo deyolked embrioni in etanolo e di trasferire rapidamente al terreno di coltura di cellule fresche. (D) Cell colini (40 µm) con scanalature di ventilazione vengono utilizzati per filtrare le cellule dopo dissociazione collagenosi-mediata. (E) dopo 5 dap, cellule primarie seminate su vetro rivestito con poli-L-lisina soprattutto formare neuroni con estensioni pronunciate. Barra della scala = 100 µm. (F) dopo 5 dap su plastica trattata senza rivestimento, fibroblasto-come le cellule invadono la cultura. Barra della scala = 100 µm. (E) e (F) sono stati acquisiti da un microscopio epifluorescente. (G) luce trasmessa l'immagine di cellule primarie derivate dallo zebrafish wild-type 1 dap. Miociti striati e ammassi di neuroni estendendo i processi sottili possono essere facilmente osservate. Barra della scala = 50 µm. (H) cellule coltivate della linea transgenica Tg (ptf1a: eGFP) jh1, che esprime eGFP in progenitori neurali di principalmente i neuroni GABAergici in del hindbrain e un subset di cellule retiniche popolazioni29, 30 , 31. barra della scala = 50 µm. (G) e (H) sono stati acquisiti da un laser confocale scansione microscopio utilizzando i piatti di fondo di vetro fatti come illustrato in (B). Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}
2. transfezione di primario delle cellule con DNA plasmidico
- Risospendere filtrato e pellettate cellule ottenute fase 1.3.7 in PBS 1x invece di mezzo di coltura cellulare. Macchia di un'aliquota della sospensione delle cellule con Trypan Blue per determinare il numero di cellulare in un conteggio di camera9.
- Mescolare 0,5 milioni di cellule con 10 µ g ultra-puro DNA plasmidico in una provetta di reazione da 1,5 mL e regolare il volume totale a 100 µ l con PBS 1X.
Nota: Per i nostri esperimenti, abbiamo usato principalmente costrutti di espressione basati sul plasmide pCS2 +10. Espressione di open reading frame clonato nel polylinker di pCS2 + è guidato dal promotore onnipresente del citomegalovirus umano (promotore CMV). Altri costrutti di espressione e promotori possono essere testati (veda inoltre rappresentante risultati e Figura 2 e Figura 3). - Mix di trasferimento del DNA delle cellule immediatamente per una cuvetta di elettroporazione (0,4 cm), disponga la provetta in un dispositivo di elettroporazione ed electroporate con le seguenti impostazioni: impulso una tantum, decadimento esponenziale, 280 V, 950 µF.
- Direttamente dopo l'elettroporazione, trasferire la miscela di DNA delle cellule in una provetta di reazione da 1,5 mL con 300 µ l di terreno di coltura di cellule fresche.
- Piastra 200 µ l di sospensione cellulare come descritto in 1.3.8 e procedere come descritto nella 1.3.9 a seconda del costrutto di espressione usata, espressione di proteine fluorescenti può essere rilevabili dopo poche ore o il giorno successivo.
3. colorazione di cellule primarie fisso
Nota: Strutture subcellulari possono anche essere visualizzati immunostaining classico invece di utilizzare i reporter di proteina di fusione fluorescenti. Per cellule primarie di zebrafish, usiamo il seguente protocollo standard esemplare macchia nucleo, F-actina e tubulina acetilata con marcatori fluorescenti.
- Cellule di piastra su vetrini rivestiti di poli-L-lisina collocati nella cella cultura piatto o pozzetti piastra come descritto in precedenza (Vedi 1.3.8).
- Per il fissaggio, rimuovere il mezzo e coprire le cellule con paraformaldeide al 4% in 1X PBS. Incubare le cellule per 10 min a 4 ° C in un agitatore. Lavare le cellule x 3 per 5 minuti ciascuno con 1x PBS a temperatura ambiente. Assicurarsi che il PBS 1x copre le cellule completamente ed eseguire la procedura di lavaggio su un agitatore.
- Per bloccare e per permeabilize le celle fisse, coprire le cellule con 1x PBS contenente latte scremato 5% e lo 0,3% Triton X-100. Posizionare le cellule per 10 min a temperatura ambiente in un agitatore. Lavare le cellule come descritto al punto 3.2.
- Per assegnare un'etichetta tubulina acetilata, un indicatore per gli assoni11, diluire l'anticorpo primario 1:2,000 in 1X PBS contenente 1% di latte scremato. Coprire le cellule con questa soluzione e incubare a loro tutta la notte a 4 ° C in un agitatore. Il giorno dopo, lavare le cellule come descritto al punto 3.2.
- Diluire l'anticorpo secondario coniugato con la fluorochrom verde della fluorescina isotiocianato di fluoresceina (FITC) 1: 100 in PBS 1x contenente 1% di latte scremato e incubare le cellule con questa soluzione per 1 h a temperatura ambiente al buio (coprire il piatto per esempio con una scatola o foglio di alluminio) su un agitatore. Lavare le cellule come descritto al punto 3.2.
- Per macchiare simultaneamente il citoscheletro di actina e nuclei, incubare le cellule in PBS 1X completati con falloidina12 coniugati con un fluorocromo rosso (01:50) e 4', 6-diamidino-2-phenylindole (DAPI)13 (100 ng/mL) per 10 min a camera temperatura al buio su un agitatore. Lavare le cellule come descritto al punto 3.2.
- Per preparare le celle per l'imaging, mettere un vettore di oggetti di vetro (vetrino) su una superficie pulita e posizionare sulla goccia di mezzo di montaggio su di esso. Prendere un coprioggetto con celle fisse e macchiate su un piatto con una pinzetta e posizionarlo sul drop con le cellule rivolto verso il vettore oggetto. Assicurarsi che tale mezzo di montaggio si estende su tutta l'area del vetrino coprioggetti. Lasciare asciugare al buio.
- Archivio fisso e montato le cellule al buio a 4 ° C fino a quando non viene eseguita l'applicazione di imaging desiderato.

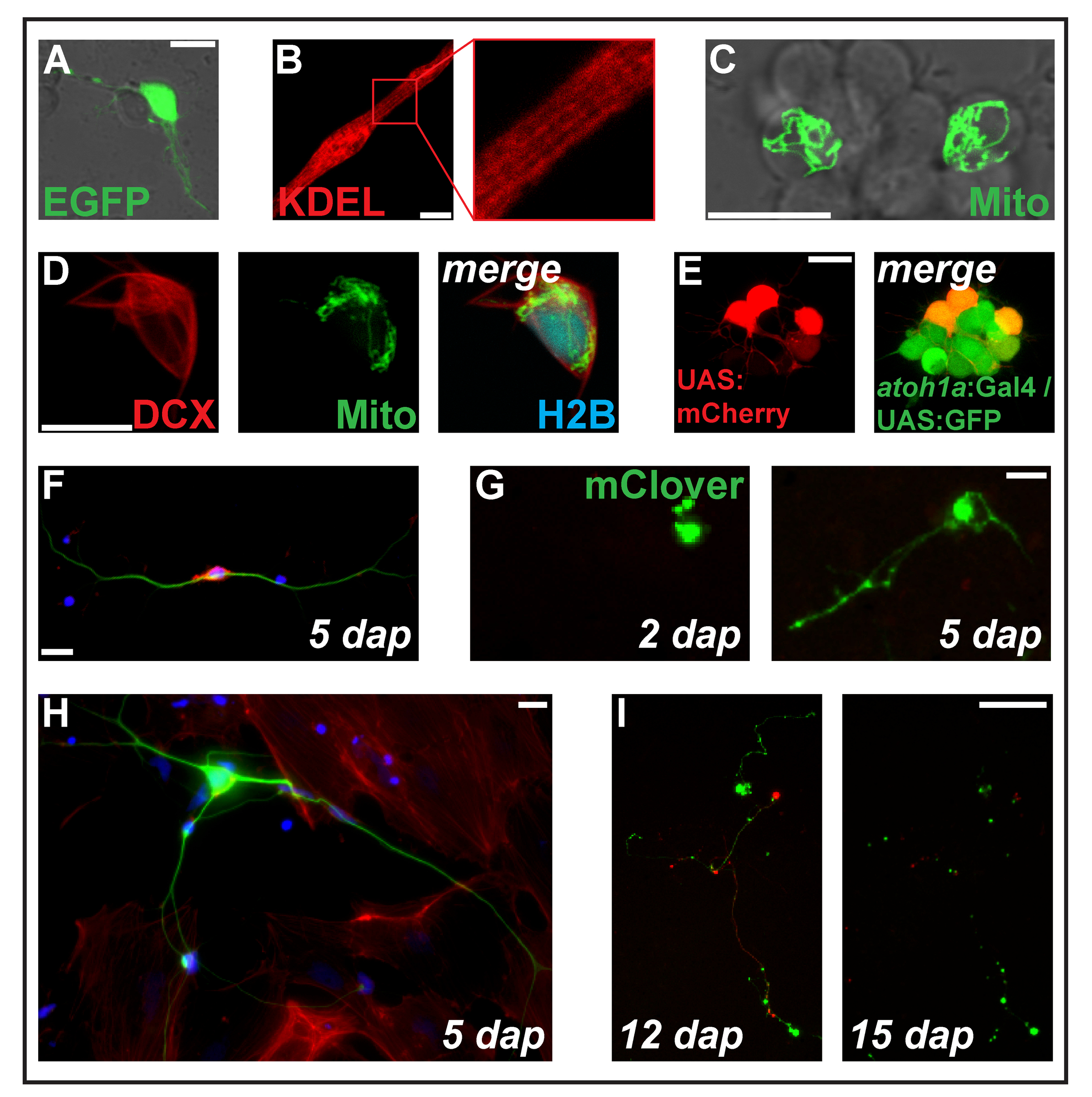
Figura 2: costrutti di transfezione di espressione mediante elettroporazione. (A) Putative neurone transfected con pz-eGFP 1 dap. (B) dei miociti striati (2 dap) che esprimono la proteina del reticolo endoplasmatico-mirati ss-RFP-KDEL. (C) due neuroni all'interno di un cluster neuronale transfettate con PC-MitoTag-YFP alle 2 dap. (D) Cell (dap 2) triple-transfected con pz-DCX-tdTomato, PC-MitoTag-YFP e pz-H2B-mseCFP. (E) pSK-UAS:mCherry individulamente in cellule primarie (1 dap) derivate da embrioni transgenici doppio che trasportano i transgeni Tg (atoh1a: Gal4TA4) hzm222 e Tg (4xUAS:KGFPGI) hzm332 conseguente espressione di GFP in progenitori neurali del hindbrain. Barre di scala = 10 µm. (A-E) sono stati acquisiti da un laser confocale microscopia utilizzando i piatti di fondo di vetro fatti come illustrato in Figura 1B. (F) colorazione fluorescente dei neuroni primari zebrafish fisso a 5 dap. Blu: DAPI (nucleo); Rosso: Falloidina (F-actina); Verde: Acetilato tubulina (neuroni). Barra della scala = 10 µm. (G) neurone-come cellule transfected con pz-mClover. Alle 2 dap, nessuna estensione è visibile. Alle 5 dap, ha formato una struttura del neurite. Barra della scala = 25 µm. (H) neurone dalla stessa preparazione della cella in (F), circondato da fibroblasti-come le cellule. Barra della scala = 10 µm. (mi) neurone derivate da un embrione transgenico che trasportano il transgene Tg (XITubb: DsRed) zf14828 transfected con pz-mClover. Tra 12 e 15 dap, i neurites subiscono la degenerazione voluminosa. Barra della scala = 100 µm. cellule indicate in (F-I) sono state seminate su vetro rivestito di poli-L-lisina (F, H) o plastica (G, I), coltivato nel mezzo di L-15 in presenza di 10% siero bovino del filtrato e il un neurone supplemento B-27 (diluito 01:50) e ripreso con un microscopio epifluorescente. Clicca qui per visualizzare una versione più grande di questa figura.
{kind=link}
4. preparazione di cellule primarie da cervello di Zebrafish adulto
-
Estrazione del cervello
- Selezionare un pesci adulti di almeno 90 giorni di età. Se lavoro con uno specifico tipo cellulare è necessario, scegliere una linea transgenica in cui espressione reporter fluorescente cellula-specifico vi permetterà di visualizzare le cellule desiderate.
- Mettere il pesce in un becher pieno con 200 mL di anestetico tricaina8 (0,2%) in Danieau 30%. Attendere che il pesce non si ferma. Immergere il pesce anestetizzato in un becher pieno di ghiaccio 200 mL di acqua per 15 minuti per eutanasia si.
- Riempire una scatola di Petri (diametro 6 cm) con etanolo al 70% (v/v). Tenere il pesce per la coda con un paio di pinzette e immergerlo nell'etanolo. Verificare il pesce sia completamente sommersa in etanolo per 5 s.
- Estrarre il cervello secondo il protocollo di Gupta e Mullins14 con i seguenti adattamenti: utilizzare solo strumenti sterilizzati nell'autoclave o confezionato sterilmente e sezionare la testa in PBS 1X sterile.
- Direttamente dopo l'estrazione, mettere il cervello in una capsula Petri (diametro 3 cm) riempito con PBS 1X sterile e spostare il piatto sotto un banco di lavoro sterile per la coltura cellulare.
-
Dissociazione di cervello e placcatura di cellule primarie
- Posizionare due insiemi delle pinzette sterili (in autoclave), una capsula di Petri sterile (diametro 6 cm) riempito con etanolo al 70% (v/v), una capsula di Petri sterile (diametro 10cm) riempito con il mezzo di L-15 di Leibovitz supplementato con 10% (v/v) filtrato siero bovino, B-27 (01:50) e 1,2% (v / v) 10.000 U penicillina-streptomicina e uno sterile cella filtro con manico (40 µm; Figura 1) sotto il banco pulito.
- Posto il filtro cella nella piastra di Petri riempito con etanolo e garantire il livello del liquido è di almeno 5 mm più in alto rispetto alla parte inferiore del filtro.
- Utilizzando il primo set di pinzette, trasferire il cervello nel filtro già inserito in etanolo e garantire che è completamente coperto dal liquido. Dopo 1 s, trasferimento del filtro con il cervello nella piastra di Petri contenente mezzo L-15 di Leibovitz con quanto sopra descritto i supplementi.
- Utilizzando il secondo set di pinzette, trasferire il cervello in una provetta di reazione sterile 1,5 mL riempito con il mezzo di L-15 di 500 µ l Leibovitz con quanto sopra descritto i supplementi. Aggiungere collagenosi (tipo 2) ad una concentrazione finale di 4 mg/mL in un volume totale di 1 mL.
- Incubare la provetta in un rotatore tubo verticale con 30 giri / min per 35 min a temperatura ambiente. Meccanicamente dissociare i grumi di tessuto rimanente pipettando su e giù utilizzando la punta di una pipetta di 1.000 µ l per aiutare il processo di dissociazione.
- Interrompere la dissociazione quando visibili particelle non rimangono in soluzione. Filtrare la sospensione cellulare attraverso un filtro sterile cella con sfiato slot (40 µm; Figura 1) in una provetta conica da 50 mL. Risciacquare il filtro con circa 10 mL di terreno di coltura di cellule fresche.
Nota: Quando avviene la sospensione unicellulare, il passo di filtrazione non è necessario come nel caso di dissociazione di embrioni. Il cervello è un tessuto molle e come tale è più incline a essere dissociato in modo omogeneo in sospensione unicellulare. - Pellet di cellule mediante centrifugazione per 5 min a 180 x g e risospendere il pellet cellulare in 1 mL di mezzo di L-15 di fresco Leibovitz con i supplementi di cui sopra descritti.
- Pipettare 500 µ l di sospensione cellulare (50% delle cellule ottenute) su un piatto fondo di self-made vetro rivestito di poli-L-lisina (Vedi 1.2) o in un pozzetto di una piastra a 24 pozzetti. Downscaling in caso di superfici più piccole (cioè, 125 µ l di soluzione per un pozzetto di una piastra a 96 pozzetti). Incubare per 60 min a temperatura ambiente sotto il banco di lavoro sterile. Quindi aggiungere il volume necessario di terreno nuovo per riempire il contenitore specifico e incubare le cellule primarie a 28 ° C.
- Eseguire l'applicazione di imaging desiderato utilizzando un microscopio invertito a 28 ° C. Culture possono essere utilizzate per l'imaging per diversi giorni dopo il placcaggio. Sostituire il 50% del mezzo su una base quotidiana.
Risultati
Figura 1 Mostra un'immagine di luce trasmessa di una cultura tipica derivata da embrioni di tipo selvaggio con miociti striati e mazzi delle cellule del neurone-come essere più abbondante. Per identificare più facilmente alcuni tipi di cellule, una linea transgenica con espressione di tipo specifico delle cellule di una proteina fluorescente può essere utilizzato (Figura 1 H).
Discussione
Qui, presentiamo due differenti protocolli di cellule di coltura primaria da 2 embrioni di zebrafish dpf o cervello adulto zebrafish.
La preparazione di colture cellulari primarie da 2 dpf zebrafish è relativamente facile da eseguire per qualcuno con esperienza in tecniche di coltura cellulare di base. Tuttavia, per ottenere risultati buoni e riproducibili, è cruciale un numero sufficiente di embrioni come materiale di partenza (100 è il minimo). Durante la raccolta degli embrioni, tutte le...
Divulgazioni
Gli autori non hanno nulla a rivelare.
Riconoscimenti
Ringraziamo T. Fritsch, A. Wolf-Asseburg, I. Linde e S.-M. Tokarski per eccellente cura degli animali e supporto tecnico. Siamo grati a tutti i membri del laboratorio Köster per discussioni intense e utili. Noi riconosciamo con gratitudine il finanziamento da parte del Deutsche Forschungsgemeinschaft (KO 1949/5-1) e lo stato federale della Bassa Sassonia, Niedersächsisches Vorab (VWZN2889).
Materiali
| Name | Company | Catalog Number | Comments |
| Fish lines | |||
| AB (wild-type) | established by Streisinger and colleagues, available from the Zebrafish International Resource Center (ZIRC) | ||
| Tg(ptf1a:eGFP)jh1 | stable transgenic line in which the enhancer of the zebrafish gene ptf1a drives expression of the fluorescent protein EGFP (Parsons et al., 2007) | ||
| Tg(XITubb:DsRed)zf148 | stable transgenic line in which the Xenopus neural-specific beta tubulin promoter drives expression of the fluorescent protein DsRed (Peri and Nüsslein-Volhard, 2008) | ||
| Name | Company | Catalog Number | Comments |
| Equipment | |||
| centrifuge | Eppendorf | model 5804 R | |
| ChemiDoc MP imaging system | BioRad | model XRS+, used to acquire black-and-white images of Petri dishes containing 1 da embryos | |
| confocal laser scanning microscope | Leica microsystems | model SP8, equipped with 28 °C temperature box and a 63X objective | |
| epifluorescent microscope | Leica microsystems | model DM5500B, equipped with 28 °C temperature box and a 40X objective | |
| Gene Pulser Xcell with capacitance extender | BioRad | 1652661 | electroporation device |
| Horizontal shaker | GFL | model 3011 | |
| incubator for cell culture (28 °C) | Memmert | model incubator I | |
| incubator for embryos (28 °C) | Heraeus | type B6120 | |
| light microscope | Zeiss | model TELAVAL 31 | |
| micro pipettes | Gilson | ||
| sterile work bench | Bio Base | with laminar flow and UV light | |
| tweezers | Dumont | Style 5, Inox | |
| vertical tube rotator | Labinco B.V. | model LD-79 | |
| Name | Company | Catalog Number | Comments |
| Software | |||
| Image Lab Software | BioRad | for the ChemiDoc MP imaging system from BioRad | |
| ImageJ | National Institutes of Health | used for counting 1 dpf embryos by applying the Count particles-tool to the respective black-and-white images; Rasband, W.S., ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA, https://imagej.nih.gov/ij/. (1997-2016). | |
| LAS X | Leica Microsystems | for both confocal and epifluorescent microscopes from Leica Microsystems | |
| Name | Company | Catalog Number | Comments |
| Plasmids | |||
| pCS-DCX-tdTomato | Köster Lab | # 1599 | based on the backbone pCS2+ (Rupp et al., 1994) |
| pCS-eGFP | Köster Lab | # 7 | based on the backbone pCS2+ (Rupp et al., 1994) |
| pCS-H2B-mseCFP | Köster Lab | # 2379 | based on the backbone pCS2+ (Rupp et al., 1994) |
| pCS-mClover | Köster Lab | # 3865 | based on the backbone pCS2+ (Rupp et al., 1994) |
| pCS-MitoTag-YFP | Köster Lab | # 2199 | based on the backbone pCS2+ (Rupp et al., 1994) |
| pCS-ss-RFP-KDEL | Köster Lab | # 4330 | based on the backbone pCS2+ (Rupp et al., 1994) |
| pCS-VAMP1-mCitrine | Köster Lab | # 2291 | based on the backbone pCS2+ (Rupp et al., 1994) |
| pSK-UAS:mCherry | Köster Lab | # 1062 | based on the pBluescript-backbone of Stratagene |
| Plasmid numbers refer to the database entries of the Köster lab. Plasmids are available upon request. | |||
| Name | Company | Catalog Number | Comments |
| Plastic and glass ware | |||
| BD Falcon Cell Strainer (40 µm) | FALCON | REF 352340 | distributed by BD Bioscience, used as “landing net” to dip deyolked embryos into ethanol and to transfer them quickly to fresh cell culture medium |
| 1.5 mL reaction tubes | Sarstedt | 72690550 | |
| 24-well plate | Sarstedt | 83.3922 | |
| 50 mL falconic tube | Sarstedt | 62.547.004 | |
| 96-well plate | Sarstedt | 83.3924.005 | |
| EasyStrainer (40 µm) | Greiner Bio-One | 542 040 | with venting slots; used to filter cells after collagenase-mediated dissociation |
| electroporation cuvette (0.4 cm) | Kisker | 4905022 | |
| glass coverslips | Heinz Herenz Medizinalbedarf GmbH | 1051201 | |
| Microscope slides | Thermo Fisher Scientific (Menzel Gläser) | 631-0845 | |
| Neubauer chamber | Henneberg-Sander GmbH | 9020-01 | |
| Pasteur pipettes (plastic; 3 mL) | A. Hartenstein | PP05 | |
| Petri dishes (plastic; diameter 10 cm) | Sarstedt | 821473 | for zebrafish embryos |
| pipette tips | Sarstedt | Blue (1000 µl): 70762; Yellow (200 µl): 70760002; White (10 µL): 701116 | |
| sterile cell culture dishes (plastic; diameter 3 cm) | TPP Techno Plastic Products AG | 93040 | |
| sterile cell culture dishes (plastic; diameter 6 cm) | Sarstedt | 72690550 | |
| sterile Petri dishes (plastic; diameter 10 cm) | Sarstedt | 83.3902 | for brain dissection |
| Name | Company | Catalog Number | Comments |
| Chemicals and Reagents | |||
| sodium chloride | Roth | 0601.1 | |
| 4 % paraformaldehyde in 1x PBS | Sigma-Aldrich | 16005 | |
| 4',6-diamidino-2-phenylindole (DAPI) | Thermo Fisher Scientific | D1306 | |
| calcium nitrate tetrahydrate | Sigma-Aldrich | C1396 | |
| ethanol p.a. 100% | Sigma-Aldrich | 46139 | |
| goat α-mouse IgG (Fc specific) FITC conjugated | Thermo Fisher Scientific | 31547 | |
| HEPES | Roth | 9105.4 | |
| high vacuum grease | DOW CORNING | 3826-50 | silicon grease used for self-made glass bottom dishes |
| magnesium sulfate heptahydrate | Merck | 105886 | |
| methylene blue | Serva | 29198.01 | |
| Monoclonal Anti-Tubulin, Acetylated antibody | Sigma-Aldrich | T6793 | |
| Aqua-Poly/Mount (mounting medium) | Polyscience | 18606 | |
| poly-L-lysine | Biochrom | L 7240 | |
| potasssion chloride | Merck | 104938 | |
| Skim milk | Roth | 68514-61-4 | |
| Texas Red-X Phalloidin | Thermo Fisher Scientific | T7471 | |
| Tricaine | Sigma-Aldrich | E10521 | Synonym: Ethyl 3-aminobenzoate methanesulfonate |
| Triton X-100 | BioRad | 1610407 | |
| Trypan Blue | Gibco by Life Technologies | 15250061 | |
| Name | Company | Catalog Number | Comments |
| Enzymes | |||
| collagenase (Type 2) | Thermo Fisher Scientific | 17101015 | dissolve powder in cell culture medium (8 mg/mL) and sterile-filter the solution, store aliquots at -20 °C |
| pronase (from Streptomyces griseus) | Roche | 11459643001 | distributed by Sigma-Aldrich, dissolve in 30% Danieau (10 mg/mL) and store aliquots at -20 °C |
| Name | Company | Catalog Number | Comments |
| Medium and solutions for cell culture | |||
| 1x PBS (Dulbecco's Phosphate Buffered Saline) | Gibco by Life Technologies | 14190-169 | distributed by Thermo Fisher Scientific |
| CO2-independent medium | Gibco by Life Technologies | 18045054 | distributed by Thermo Fisher Scientific |
| filtrated bovine serum (FBS) | PAN-Biotech | individual batch | |
| glutamine 100x | Gibco by Life Technologies | 25030081 | distributed by Thermo Fisher Scientific |
| Leibovitz's L-15 medium | Gibco by Life Technologies | 11415049 | distributed by Thermo Fisher Scientific |
| PenStrep (10,000 U/mL) | Gibco by Life Technologies | 15140148 | distributed by Thermo Fisher Scientific |
Riferimenti
- Ablain, J., Zon, L. I. Of fish and men: using zebrafish to fight human diseases. Trends in Cell Biology. 23, 584-586 (2013).
- Sassen, W. A., Köster, R. A molecular toolbox for genetic manipulation of zebrafish. Advances in Genomics and Genetics. , 151 (2015).
- Scheer, N., Campos-Ortega, J. A. Use of the Gal4-UAS technique for targeted gene expression in the zebrafish. Mechanisms of Development. 80, 153-158 (1999).
- Köster, R. W., Fraser, S. E. Tracing transgene expression in living zebrafish embryos. Developmental Biology. 233, 329-346 (2001).
- Driever, W., Rangini, Z. Characterization of a cell line derived from zebrafish (Brachydanio rerio) embryos. In Vitro Cellular & Developmental Biology - Animal. 29A, 749-754 (1993).
- Badakov, R., Jaźwińska, A. Efficient transfection of primary zebrafish fibroblasts by nucleofection. Cytotechnology. 51, 105-110 (2006).
- Senghaas, N., Köster, R. W. Culturing and transfecting zebrafish PAC2 fibroblast cells. Cold Spring Harbor Protocols. , (2009).
- Westerfield, M. . The zebrafish book. A guide for the laboratory use of zebrafish (Danio rerio). , (2007).
- Basic methods in cellular and molecular biology. Using a hemacytometer to count cells. Journal of Visualized Experiments Available from: https://www.jove.com/science-education/5048/using-a-hemacytometer-to-count-cells (2017)
- Rupp, R. A., Snider, L., Weintraub, H. Xenopus embryos regulate the nuclear localization of XMyoD. Genes & Development. 8, 1311-1323 (1994).
- Piperno, G., Fuller, M. T. Monoclonal antibodies specific for an acetylated form of alpha-tubulin recognize the antigen in cilia and flagella from a variety of organisms. Journal of Cell Biology. 101 (6), 2085-2094 (1985).
- Barden, J. A., Miki, M., Hambly, B. D., Dos Remedios, C. G. Localization of the phalloidin and nucleotide-binding sites on actin. European Journal of Biochemistry. 162 (3), 583-588 (1987).
- Kapuscinski, J. DAPI: a DNA-specific fluorescent probe. Biotechnic & Histochemistry. 70 (5), 220-233 (1995).
- Gupta, T., Mullins, M. C. Dissection of organs from the adult zebrafish. Journal of Visualized Experiments. 37, E1717 (2010).
- Chalfie, M., Tu, Y., Euskirchen, G., Ward, W. W., Prasher, D. C. Green fluorescent protein as a marker for gene expression. Science. 263, 802-805 (1994).
- Stornaiuolo, M. KDEL and KKXX retrieval signals appended to the same reporter protein determine different trafficking between endoplasmic reticulum, intermediate compartment, and Golgi complex. Molecular Biology of the Cell. 14, 889-902 (2003).
- Lithgow, T. Targeting of proteins to mitochondria. FEBS Letters. 476, 22-26 (2000).
- Nagai, T., Ibata, K., Park, E. S., Kubota, M., Mikoshiba, K., Miyawaki, A. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nature Biotechnology. 20, 87-90 (2002).
- Sassen, W. A., Lehne, F., Russo, G., Wargenau, S., Dübel, S., Köster, R. W. Embryonic zebrafish primary cell culture for transfection and live cellular and subcellular imaging. Developmental Biology. 430, 18-31 (2017).
- Horesh, D., et al. Doublecortin, a stabilizer of microtubules. Human Molecular Genetics. 8, 1599-1610 (1999).
- Shaner, N. C., Campbell, R. E., Steinbach, P. A., Giepmans, B. N. G., Palmer, A. E., Tsien, R. Y. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nature Biotechnology. 22, 1567-1572 (2004).
- Distel, M., Hocking, J. C., Volkmann, K., Köster, R. W. The centrosome neither persistently leads migration nor determines the site of axonogenesis in migrating neurons in vivo. Journal of Cell Biology. 191, 875-890 (2010).
- Matsuda, T., Miyawaki, A., Nagai, T. Direct measurement of protein dynamics inside cells using a rationally designed photoconvertible protein. Nature Methods. 5, 339-345 (2008).
- Archer, B. T., Ozçelik, T., Jahn, R., Francke, U., Südhof, T. C. Structures and chromosomal localizations of two human genes encoding synaptobrevins 1 and 2. Journal of Biological Chemistry. 265, 17267-17273 (1990).
- Griesbeck, O., Baird, G. S., Campbell, R. E., Zacharias, D. A., Tsien, R. Y. Reducing the environmental sensitivity of yellow fluorescent protein. Mechanism and applications. Journal of Biological Chemistry. 276, 29188-29194 (2001).
- Shaner, N. C., et al. A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nature Methods. 10, 407-409 (2013).
- Campbell, R. E., et al. A monomeric red fluorescent protein. Procedings of the National Academy of Sciences of the United States of America. 99, 7877-7882 (2002).
- Peri, F., Nüsslein-Volhard, C. Live imaging of neuronal degradation by microglia reveals a role for v0-ATPase a1 in phagosomal fusion in vivo. Cell. 133, 916-927 (2008).
- Godinho, L., et al. Targeting of amacrine cell neurites to appropriate synaptic laminae in the developing zebrafish retina. Development. 132, 5069-5079 (2005).
- Jusuf, P. R., Harris, W. A. Ptf1a is expressed transiently in all types of amacrine cells in the embryonic zebrafish retina. Neural Development. 4, 34 (2009).
- Kani, S., et al. Proneural gene-linked neurogenesis in zebrafish cerebellum. Developmental Biology. 343, 1-17 (2010).
- Distel, M., Wullimann, M. F., Köster, R. W. Optimized Gal4 genetics for permanent gene expression mapping in zebrafish. Procedings of the National Academy of Sciences of the United States of America. 106, 13365-13370 (2009).
- Choorapoikayil, S., Overvoorde, J., den Hertog, J. Deriving cell lines from zebrafish embryos and tumors. Zebrafish. 10, 316-332 (2013).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati