
このコンテンツを視聴するには、JoVE 購読が必要です。 サインイン又は無料トライアルを申し込む。
Method Article
選択したプロウイルス統合サイトの HIV-1 感染の Jurkat レポーターのモデルを生成する CRISPR ベース Cas9 ゲノム工学
要約
プロウイルス選択したゲノム サイト統合を要約 HIV-1 感染の新しい体外モデルの生成のゲノム工学ワークフローを提案します。記者の HIV 由来のターゲットは、CRISPR Cas9 媒介、サイト固有のゲノム操作によって促進されます。単一細胞クローン生成、スクリーニング、および正しいターゲットの検証のための詳しいプロトコルを提供しています。
要約
ひと免疫不全ウイルス (HIV) は、再発のサイトやゲノムのホット スポットでホスト細胞のゲノムに非ランダムにプロウイルス DNA を統合します。ここで CRISPR ベース Cas9 ゲノム工学技術を使用して選ばれたゲノム統合サイトと HIV 感染の新規の in vitro モデルの生成の詳細なプロトコルを紹介します。この方法では、選択の記者のシーケンスに統合できます、選択したターゲットを絞った genomic 位置臨床的に関連する統合サイトを反映しています。
プロトコルでは、HIV 由来の記者の設計およびターゲット サイトと gRNA シーケンスの選択を説明します。両手を相同性ターゲット ベクトルが構築され、Jurkat T 細胞にトランスフェクションします。ターゲット ・ サイトで Cas9 を介した二重鎖切断によって促進される相同組み換えによる記者シーケンスは選択したゲノム サイト向けです。単一細胞のクローンを生成およびフローサイトメトリー PCR によって対象化、イベント上映します。選択されたクローンが展開し、PCR、シーケンシング、および南にしみが付くことによって検証は正しいターゲットします。CRISPR Cas9 を介したゲノム工学の潜在的なターゲットを離れてイベントが分析されます。
このプロトコルは、新しい細胞培養システムを用いた臨床的に関連する統合サイトでそのモデルの HIV 感染を生成できます。正しい記者シーケンス統合の単一細胞クローンの生成と検証は時間がかかるが、結果として得られる系統、機能的プロウイルス統合サイトの選択を分析するための強力なツールです。
概要
感染宿主ゲノムにプロウイルス DNA の統合は、ひと免疫不全ウイルス (HIV) のライフ サイクルの重要なステップです。次の統合、HIV はメモリー cd4 陽性 T 細胞など寿命の長い cd 4 + T 細胞のサブセットの遅延を確立することによって永続化します。HIV の統合は、非ランダムな1,2に表示されます。急性と慢性感染した人2,3,4 の統合サイトのシーケンスを通していくつかの研究で反復的統合のプロウイルス DNA ゲノム ホット スポットの数が検出されました、5,6,7,8。興味深いことに、これらの統合サイトのいくつかで同じ軌跡がリカレント サイトの統合も積極的にクローン性増殖1に影響を与える可能性があります考えにつながる、感染細胞の大部分で検出されました。
再発統合サイトの意義の理解を進めるためプロウイルス統合サイトの選択肢を検討する必要があります。ただし、いくつかの技術的な側面を妨げる勉強 HIV 統合サイトの選択と結果。JLat 細胞株は臨床的再発統合サイト9を反映していないような広く HIV 待ち時間の細胞モデルを使用しました。プライマリ患者由来細胞の研究、一方で、配列による統合サイト風景の説明を有効にするが、機能解析を可能にしません。我々 の知識に十分な実験モデルは機能的選択した臨床的に関連する統合サイトを分析する利用可能なありません。
CRISPR ベース Cas9 ゲノム工学技術を使用して HIV 感染のための新しいモデルを生成するための詳細なワークフローをご紹介します。ここ記載されているワークフローは、モデル選択した統合サイトで genomically 統合プロウイルス記者を運ぶ HIV 感染 T 細胞由来のレポーター細胞ラインを生成する使用できます。彼らしたがってプロウイルス統合サイトによる hiv 感染生物学への影響し、provirus が別の治療戦略 (待機時間逆転エージェントによる,誘導など) に応答する方法を探索する新しいツールとして提供しています。本手法は、CRISPR ベース Cas9 ゲノム工学、ターゲット ・ サイトで、Cas9 ヌクレアーゼ誘起の二重鎖切断によって促進される相同組み換えによるシーケンス レポーターの統合の利点を使用します。ターゲット ・ サイトの統合には、HIV 感染者と Cas9 を介したゲノム工学のための適したの PAM モチーフの存在に関する研究から説明再発統合サイトへの近さによって判断されます。
模範的な結果、BTB および CNC の相同性転写レギュレータ 2 のコード BACH2 遺伝子座に注力します。抗レトロ ウイルス療法に慢性的な HIV 感染、個人で BACH2 は統合された HIV-1 シーケンス3,6,7,8,10の濃縮を示す遺伝子座であります。BACH2 イントロン 5 の 2 つの特定のサイトにしようと思いましたが HIV 由来 1 長いターミナル リピート (左から右)、tdTomato コーディング シーケンスとウシ成長ホルモン (BGH) 起こる信号 (PA) から成る最小 HIV 由来の記者をしました。提案するプロトコルは、Jurkat 細胞、ヒト cd4 T 細胞由来の懸濁液セル行が線を使用して他のセルおよびそれに応じて適応プロトコルに最適です。相同性の腕、CRISPR-Cas9-介したレポーターのターゲットに選ばれたゲノム サイト、世代クローン系統の選抜に、包括的な目標点のベクターの構築対象サイトの選択の詳細なワークフローを提案する検証新しく生成された、対象となるレポーター細胞。
プロトコル
1. ゲノム工学とベクトル (テレビ) の設計をターゲットに戦略をターゲット
注:ゲノム工学の最初のステップは、選択と CRISPR Cas9 媒介をターゲットに必要なツールの生成を含みます。ゲノム統合サイトの軌跡の選択は、ターゲットのセル型の選択と統合のため HIV 由来の記者の設計この手順を付けます。このプロトコルは、Jurkat 標的細胞への HIV LTR_tdTomato_BGH PA 最小限記者ターゲットについて説明します。CRISPR ベース Cas9 ターゲット、世代、スクリーニング、クローン系統の検証のワークフローのフローチャートは、図 1に描かれています。記載されているターゲット戦略s. 化膿Cas9 を使用して、選択した統合サイトで gRNA 監督 dsDNA 改を生成する (SpCas9)。相同組換えを介して選択された genomic 位置に記者が対象とし、いわゆる 5' と 3' の相同性 (HA) の腕11が並ぶ記者シーケンスを含む非線形ターゲット ベクトル (テレビ) を提供します。
-
ターゲットの軌跡、gRNA、およびベクター デザインをターゲットの選択
- 個々 の科学的な質問に基づいて対象とする thegenomic の軌跡を選択します。Hiv 感染の再発統合サイトの公開されたリストは、異なる患者で発見した使用研究2,3,4,5,6,7、8ガイドライン。インシリコ抽出対象 (完全な遺伝子のシーケンス) またはゲノムの少なくとも 5 kb 必要な genomic 位置のゲノム シーケンス UCSC のゲノムのブラウザー (http:// genome.edu.ucsc.edu) を使用しています。
- 選択ガイド Rna (gRNAs) 20 E カリカリ webtool (http://www.e-crisp.org) を使用して選択された genomic 位置のターゲットのための nt。
- 生物として「ホモ ・ サピエンス GRCh38」を選択します。1.1.1 手順で抽出した目的の genomic 位置をカバーするゲノム シーケンスの入力 2,000 bp。
- 中規模アプリケーション設定 (任意の PAM、任意 5' 基本、ターゲットを容認のミスマッチ、イントロン/CPG の島が除外されます) を使用して gRNA の検索を開始します。可能な gRNA デザイン一覧は、高い特異性と効率のためのスコアを下げることからランキング表示されます。
- できれば特異性と効率性の高いスコアを示していますし、対象とする目的の genomic 位置にできるだけ近く、gRNA を選択します。
注:目的の genomic 位置への近さと具体的かつ効率的な gRNA の設計間の妥協は発見されていません。
- NCBI blast ブラウザー (https://blast.ncbi.nlm.nih.gov) を使用して gRNA 結合部位の一意性をチェックする参照ゲノムに対して選択した gRNA シーケンスを爆発します。
- ゲノムとして「人間」を選択します。クエリ シーケンスとして gRNA シーケンスを入力します。「類似性の高いシーケンス」を選択 (メガブラスト) プログラムとして。GRNA シーケンスが一意であることを確認します。そうでない場合は、再びステップ 1.1.2.3 とブラストからの別 gRNA を選んだ。
- GRNA シーケンスを選択すると、それに応じて 5' と 3' HA として 1.1.1 手順で抽出したゲノム配列からインシリコ1,000 bp 上流と下流の gRNA シーケンスを選択します。
注: 、GRNAs 選択したゲノム統合サイトの軌跡に相同であるべきし、protospacer の隣接するモチーフに隣接 (PAM; 例えば、 SpCas9 の NGG) (図 2 a)。テレビには、5' と 3' HAs が並ぶの記者のシーケンスが含まれています。上流と下流の gRNA シーケンス11カバー 1000 bp があります。完全 gRNA シーケンスは、HA には含めない。最大 5 の重複 nt は受け入れられる (図 2 a)。
-
GRNA とターゲット ベクトルの生成
注:ベクター方式、図 2 bを参照してください。- SpCas9 と gRNA の発現用ベクターを生成するには、元 SpCas9 と RNA (sgRNA) の 1 つのガイドの両方同時に表せることのバックボーンとして、pX330-U6-Chimeric_BB-cBh-hSpCas9 を使用します。バックボーンに gRNA シーケンスをクローン、BbsI 制限サイト12を使用します。
- テレビを生成する、(, pMK や cDNA3.1 などのバックボーンとして高いコピーのプラスミッドを選択します。
- まず、記者を組み立てる (このプロトコルで: LTR_tdTomato_BGH PA) ギブソン アセンブリ clong によって構造物のバックボーンに13クローン商業アセンブリを使用してキット、および 5' と 3' 制限のサイトをご紹介 (例えば、 5' パーチと 3'SmaI) それに続く制限消化がのクローニングのため。
- 対象となるセル型のゲノム DNA (gDNA) から 1.1.4 で選んだ HA フラグメントの 1000 bp を増幅 (このプロトコルで: Jurkat 細胞) 活動を校正と DNA ポリメラーゼを用いた (表 1と2の PCR 食材を参照してくださいと循環の条件)。その後、記者が並ぶ各 HA (5' 3' HA の両端に SmaI、両端 HA のパーチ) の 5' と 3' 端に制限のサイトをご紹介します。
- 順番にクローンは既に14,15をクローニング制限酵素による記者 (1.2.2.1 の手順で生成された) を含む構造物のバックボーンに持っています。まず、クローンで 5' HA 3' HA でパーチ制限のサイトを使用して、クローン、SmaI 制限のサイトを使用して。
注:テレビ バックボーンに追加蛍光レポーターがある場合不要なバックボーン統合フローサイトメトリーによって評価される (3.2.2 および 3.2.3 の手順を参照してください)。PCR を用いたバックボーン統合を評価する必要がありますテレビのバックボーンに蛍光レポーターが含まれていない場合 (手順 3.2.8 参照)。

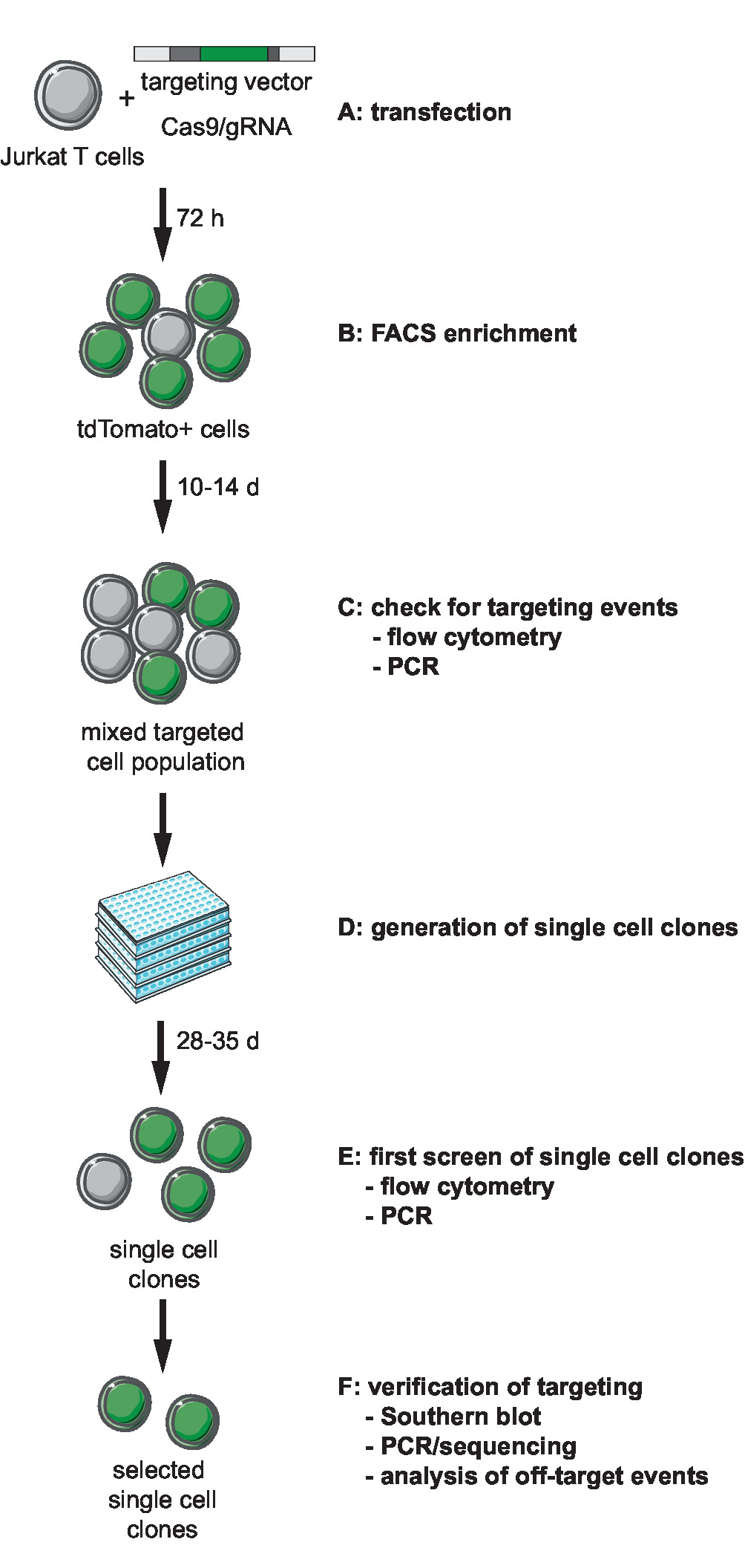
図 1: CRISPR Cas9 仲介されたターゲット、生成、およびワークフロー クローン記者の選択行に定義された統合サイト。(A) 目標点のベクターを生成し、目標点のベクターと Cas9/gRNA 発現プラスミド Jurkat T 細胞を変換します。(B) エンリッチ FACS による transfected セル 72 h ポスト トランスフェクションします。(C) 許可しなさいセル 10 ~ 14 日の成長ターゲットに PCR によるイベントの発生を確認およびフローサイトメトリーします。(D) 生成希釈させクローンを制限することによって単一細胞クローン成長 3 週間。(E) PCR による正しいターゲットのためのクローンを画面し、96 ウェル フォーマットでフローサイトメトリーします。選択されたクローンを展開します。(F) は南しみ、PCR やシーケンシング、および Cas9 a エンドヌクレアーゼ活性のターゲットをイベントの分析によって選択されたクローンのターゲットが正しいを確認します。この図の拡大版を表示するのにはここをクリックしてください。
{kind=link}

図 2: 戦略とベクトル デザインを対象とします。(、) の gRNA と相同性武器の選択。20 nt gRNA は選ばれたゲノムのターゲット ・ サイトに相同と腕 1,000 bp アップを補完するものと、gRNA の下流、gRNA シーケンスを含める必要がありますいない PAM. 相同に隣接して位置しています。(b) ベクトルと gRNA/Cas9 ベクトルをターゲットの模式図。ターゲットのベクトルは、5' と 3' ホモロジー腕が並ぶは、選ばれた記者のシーケンスで構成されます。GRNA/Cas9 ベクターは pX330 U6 Chimeric_BB cBh hSpCas9 バックボーンに基づいています。(c) の相同組み換えによるターゲットの模式図。目標点のベクターと guideRNA/Cas9 ベクトルは Jurkat 細胞に導入しました。Cas9 はゲノムのターゲット ・ サイトで二重鎖切断を仲介する (によって示される *) と相同組換えとゲノムのターゲット遺伝子座に記者シーケンスの統合が容易になります。この図の拡大版を表示するのにはここをクリックしてください。
{kind=link}
2 Jurkat 細胞のターゲット CRISPR ・ Cas9-ベース
-
Jurkat 細胞のトランスフェクション
- プレート 1.25 x 106 Jurkat T トランスフェクション前に 24 h は 10% (v/v) 牛胎児血清 (FCS) と 4 mM L-グルタミン [と呼ばれる「RPMI w.o. 抗生物質 (AB)」]/6 よく細胞培養プレートのウェルに添加 RPMI 1640 の 2.5 mL のセルします。単一ターゲットの実験の 1 つの完全な 6 ウェル プレートを準備 (すなわち, 6 井戸細胞懸濁液 2.5 mL をそれぞれ)。
- 次の日に transfect 円形テレビと pX330-U6-Chimeric_BB-cBh-hSpCas9/gRNA Jurkat 細胞のトランスフェクション試薬特定を使用して細胞が共同。
- 反応管でよく混ぜる商業 RPMI 培地の 250 μ L に低下した血清中濃度 (血清の 50% 削減を RPMI) の transfection のために最適化された円形のテレビの 2 μ g と pX330-U6-Chimeric_BB-cBh-hSpCas9/gRNA/ウェルの 2 μ g を追加します。
- 管の壁に触れることがなく DNA/メディアにゆっくりとトランスフェクション試薬の 12 μ L を追加し、旋回します。混合物は 15 分間インキュベートし、滴下 1 つよくセルの追加をしましょう。37 ° C と 5% の CO2のセルを孵化させなさい。
注:トランスフェクション反応の準備をスケール アップすることができます。トランスフェクション後、中型の変更は不要です。
-
蛍光活性化セル (FACS) を並べ替えして transfected セルの濃縮
- 72 時間後のトランスフェクション プール transfected セル カウントする方法、FACS による濃縮のための準備します。4 分の 300 x gと部屋の温度 (RT) で遠心分離 50 mL の円錐管の細胞を収集、PBS で 1 回洗浄、遠心分離機の再度、1 x 10 FACS バッファー (1% の FCS + 1 mM EDTA を添加した PBS) の適切な量の餌を中断7細胞/mL、最後に FACS チューブに移すと。
- FACS のセルを対象し、テレビ (例えばこのプロトコルで, tdTomato) の蛍光レポーターを表すものをソートします。RPMI 1640 年 10 %fcs、4 ミリメートル L-グルタミンと 50 U/mL ペニシリンとストレプトマイシン ("RPMI w/AB"と呼ばれる) を添加した細胞を収集します。
- 後 FACS の並べ替え、並べ替えられたセルに AB の w/RPMI 20 mL を追加することによって一度セルを洗浄してルートを再懸濁しますで 4 分の 300 × gで遠心分離機の AB の w/RPMI の適切な量の細胞ペレットとプレート細胞培養プレートのウェル 1 個のセル、セル番号ポスト FACS によると適切なボリューム。
注:文化は、細胞死のかなりのレベルを最初の週後 (80-90% まで) を対象に観察されているように少量 (例, 24 ウェル) でセルを並べ替えます。 - 75 cm ² 細胞培養用フラスコに 1 x 106セル/ml の密度まで混合ターゲット セル人口を拡大します。10-14 日ポスト FACS 並べ替えの周りがかかります。
-
混合ターゲット セル人口のフローサイトメトリーによるイベントをターゲットの確認
- 拡張 (ときセル 75 cm2細胞培養用フラスコに 1 x 106セル/ml の密度に達している) の 10-14 日後混合の 1 x 10 の6セルのプレート 2 つの井戸は 12 ウェルの細胞培養で AB の w/RPMI 1 mL の細胞集団を対象としました。プレート。
- レポーターの HIV LTR を誘発する (テレビの世代は 1.2.2.1 の手順で説明します). 50 ng/mL ホルボール 12-ミリスタート 13-アセタート (PMA) を追加することで井戸の 1 つで、1 μ M イオノマイシン (PMA Iono と呼ばれる)。非誘導制御として 2 番目の井戸のセルを使用します。誘導と 24 h の非誘導細胞を培養します。
- (各) 非誘導と誘導の細胞の細胞懸濁液 0.5 mL を取る、pbs は、一度洗って、FACS バッファーの 200 μ L のそれぞれを中断します。
- 100,000 細胞をフローサイトメトリーで分析します。前後脇の散布図、サイズに基づいて実行可能な単一セルをゲートし、蛍光レポーター遺伝子発現を解析します。
注: (10-14 日ポスト FACS 並べ替え) この段階ではトランスフェクションによって蛍光レポーターの一過性の発現を検出できなく必要があります。この時点で蛍光式記者シーケンスのゲノムの統合を示します。
-
ターゲット混合ターゲット細胞集団のゲノム DNA の PCR によるイベントの検出
注:デザイン 2 プライマーの 5' 統合 (int.) ジャンクションと 3' int 型ジャンクションの特定、PCR によってターゲットのイベントを検出します。5' int 型ジャンクション PCR、前方のプライマーをバインドする必要があります 5' HA とレポーター (P1 と P2図 3 aでのプライマー) の LTR の逆プライマーの上流。3' int. 接合用プライマー PCR では 100-200 bp 下流の 3' HA (プライマー P3 と P4 の図3 a) レポーターの PA からスパンする必要があります。プライマー P1 および P4 も混合対象人口で対象外の対立遺伝子の増幅のためになるでしょう。回路図、図 3 aを参照してください。- 2.2.4 のステップから混合ターゲット セル人口の細胞懸濁液 2 mL から gDNA を準備します。製造元のプロトコルに従って gDNA 抽出キットを使用します。コントロールとして非標的細胞の gDNA を準備しています。
- Int 型ジャンクション PCRs を実行 (5' と 3' int 型ジャンクション、P1 ・ P2 ・ P3 ・ P4 のプライマーそれぞれ) をターゲットとした遺伝子 PCR (プライマー P1 ・ P4) 高忠実度 DNA ポリメラーゼを使用して、(表 3および4 PCR 成分とサイクリング条件参照).5 μ L の 1.5% アガロース/泰ゲルの PCR の製品を分析します。
注: GDNA をターゲットとした細胞 (マイナス コントロール) の中に検出されている特定の PCR の製品を守らなければ混合対象人口にゲノム エンジニア リングを受けているセルがある場合。非標的遺伝子 PCR、両方対象と非対象のセル (ゲノム P1、P4 のプライマーの肯定的な制御) の同じサイズの製品を守らなければならない 1 つ。バンドは認められなかった、または (例えばを通して DMSO または Mg2 +の増加量を追加)、PCR のバッファーを変更するサイクル数の増加やポリメラーゼによって PCR サイクリング条件を変更することを検討してください。
3. 正しい対象系統およびスクリーニングの生成
注:フローサイトメトリーおよび PCR (セクション 2.2-2.4) による混合ターゲット セル人口のターゲット イベントの確認後単一細胞のクローンを生成 (期間: 28 〜 35 日) と記者のシーケンスの正しい統合のための画面。
- 希釈めっきによる単一細胞クローンの生成
- Jurkat 付きメディアに事前の準備: 1 x 10 の6セル/mL、300 x gで 5 分間遠心する成長健康、未処理の Jurkat T 細胞から AB 中 w/RPMI 離陸し、0.22 μ m シリンジ フィルター ユニットを使用して上清をフィルター処理します。
注:短期的なストレージ 4 ° c または-20 ° c 1 週間以上ストレージで馴化培地を保ちます。馴化培地希釈めっき前の 20 に 30 mL を準備します。 - 2.2.4 ステップ 10-14 日後膨張から標的細胞をカウントし、AB の w/RPMI で 1 × 105セル/mL の濃度に希釈します。1 x 10 の5セル/mL の溶液を 100 μ l 添加し 1,000 セル/ml の濃度を達成するために媒体の 9.9 mL で希釈します。1,000 セル/ml 溶液 1 mL をとり、100 細胞/mL の濃度を達成するために媒体の 9 mL で希釈します。
- ウェルあたり 1 つ含むのセルと 2 細胞/ウェル プレート 96年ウェルをプレートします。ウェルあたり 1 セルの 100 細胞/mL の溶液の 1 mL を取り出して軽く混ぜて馴化培地 5 mL、4 mL 滅菌試薬リザーバーで新鮮な媒体の。
- ウェルあたり 2 セルの 100 セル/ml 溶液 2 mL を取るし、馴化培地 5 mL と新鮮な培地 3 mL を混ぜて優しく。マルチ チャンネル ピペットを使用してあたりそれぞれセル希釈の 100 μ L の丸底プレート 96年ウェル プレート。
注:構築をターゲットあたり 10 の 96 ウェル プレート 5、スクリーニングのためのクローンを得るのに十分です。 - 96 ウェル プレートをスタック、3 mL の各ウェルに PBS を含む 6 ウェル プレートと各スタックをカバー、3 週間 5% CO2加湿細胞文化のインキュベーターで 37 ° C で版を孵化させなさい。
注:この時間の間に細胞培養培地を変更しないでください。週に一度か二度以上インキュベーターを開かないでください。最良の結果は、開いている水の貯水池でインキュベーターで観察されます。 - インキュベーションの 3 週間後に、光顕 (4 倍) を使用して成長したコロニーの存在を視覚的に確認し、井戸の底上の点として表示されているので栽培植民地と井戸をマークします。
- 1 つ AB w/RPMI の 100 μ L/ウェルの 96 ウェル丸底プレートを準備します。優しくマークの細胞を再懸濁しますピペッティングでよく。既に AB、w/RPMI の 100 μ L を含む新しい 96 ウェル プレートの 1 つのウェルに細胞懸濁液を 100 μ l 添加を転送し、ピペットで穏やかに混合します。第 2 空皿を複製する 96 ウェル丸底プレートにこの細胞懸濁液を 100 μ l 添加を転送します。
- 成長したコロニーのすべてのマークされた井戸を続行します。RPMI AB 中 w/200 μ L で空白のすべての井戸を埋めます。37 ° C、5% で版を孵化させなさい CO2.
注:これらのプレートのいずれかを務める単一細胞クローン (「株式板」) の拡大や、その他検診「重複プレート」。
- Jurkat 付きメディアに事前の準備: 1 x 10 の6セル/mL、300 x gで 5 分間遠心する成長健康、未処理の Jurkat T 細胞から AB 中 w/RPMI 離陸し、0.22 μ m シリンジ フィルター ユニットを使用して上清をフィルター処理します。
- フローサイトメトリーおよび PCR による単一細胞クローンのスクリーニング
注:単一細胞クローンを展開すると、PCR (手順 3.2.4–3.2.12) による記者シーケンスの存在とフローサイトメトリー (手順 3.2.2–3.2.3) による蛍光レポーターの発現の画面単一細胞クローンにステップ 3.1.8 から重複するプレートを使用(図 3)。- 重複板 24 〜 48 時間インキュベートし、プレートを再び複製をしましょう。これを行うには、各ウェルに 100 μ L AB w/RPMI を追加、ピペットで穏やかに混合、マルチ チャンネル ピペットを使用して新しい 96 ウェル丸底プレートに 100 μ L を転送します。流れ cytometry スクリーニングと PCR ベースのスクリーニングのための他の 1 つのプレートを使用します。
- 流れの cytometry の検診 PMA Iono で細胞を刺激します。0.1 μ l Ionomocin (1 mM ストック)、PMA の 0.1 μ L のマスター ミックスを準備 (50 μ g/μ L 在庫) し井戸の数で AB の w/RPMI の 4.8 μ L、5 μ L/ウェル マスター ミックスを追加。
注:誘導正常にクローン、LTR は転写活性サイレントかもしれない蛍光レポーターが表記されていないためを識別する必要があります。 - セル 24 時間インキュベートし、手順 2.3.3 で説明したフローサイトメトリー用セルを準備しなさいゲートの任意実行可能な単一セルは、前方と脇の散布図のサイズに基づいて、蛍光レポーター遺伝子発現のフローサイトメトリーによる分析 (たとえば、結果が図 3 cを参照)。テレビ バックボーンにプロモーターと 2 番目蛍光レポーターが含まれているかどうか (例えば, GFP)、またバックボーン記者式のいずれかのクローンを画面 (ステップ 1.2.2 と説明、次の注を参照)。
注:バックボーン記者式バックボーン シーケンスの不要な統合を示します。 - 2 つ目の重複したプレートでクローンが十分に成長している一度 (96 ウェル プレートの複製後通常 24 〜 48 時間) gDNA PCR のスクリーニングのための細胞溶解液を準備します。細胞ペレットを乱すことがなく慎重に右で 300 x gで 10 分間遠心するプレートは上澄みを脱ぐ。
注:マルチ チャンネル ピペットの lysates と PCR 反応の準備のためのすべての手順を実行できます。 - 穏やかなピペッティング室温 300 × gで 5 分間プレートを遠心分離して 100 μ L の PBS のセルを洗浄します。PBS を脱いで、換散バッファーの 200 μ L を追加 [200 mM の NaCl 100 mM トリス塩酸 pH 8 〜 8.5、5 ミリメートルの EDTA、0.1 %sds のプロティナーゼ K の 250-1,000 μ g/mL を追加 (凍結乾燥粉末、重量を量るたて)] ウェルあたり。ピペットで穏やかに混合し、懸濁液を新しい PCR プレートに転送します。
- パラフィン フィルム プレート シールし、たちに 55 ° C で 1 時間インキュベートします。10 分 (3,000 x g) スピンに最大速度で細胞の残骸を遠心し、上清を新しい PCR プレートに転送。
注:それ以上の使用まで 4 ° C でこの段階で板のセル lysates を格納できます。 - DH2O の 110 μ L、96 ウェル PCR プレートを準備し、細胞溶解液の 10 μ L を追加 (1:12 希釈)。セル lysates は粘性とピペットで移しなさいすることは困難かもしれない。少なくとも使用 20 μ L ピペット先端。
- 99 ° c、たちで 10 分間インキュベーションによってプロテイナーゼ K を不活性化します。その後 PCR のスクリーニングの不活化および希薄化後のセル lysates を使用します。
- PCR (P5 および P6) 選ばれた記者シーケンスに基づいて記者シーケンスの 500-800 bp を増幅するためのスクリーニングのためのプライマーを設計します。ポジティブ コントロール PCR の野生型、非ターゲット genomic 位置 (NUP188遺伝子) の 630 を増幅するプライマー P7 と P8 bp を使用する (図 3 cと表 5)。テレビ バックボーン シーケンス (バックボーン PCR) の不明確な統合のための制御としてテレビ バックボーンの 500-600 bp を増幅 3 プライマー対を設計します。
- スクリーニング、制御、および PCR のバックボーン、商業 PCR マスター ミックスを使用 (表 6および7 PCR 成分とサイクリング条件参照)。2 μ L の希釈と不活化ライセート ステップ テンプレートとして 3.2.8 に用意され、96-well フォーマットの PCR の拡大の 38、40 のサイクルの PCR を実行を使用します。
- 5 μ L の 1.5% アガロース/泰ゲルの PCR の製品を分析します。
注:コントロール PCR、630 の特定のバンドのセル lysates の品質が PCR のために十分であることを確認するすべてのサンプルは、bp を守らなければなりません。スクリーニングの PCR (プライマー デザインに応じて 500-800 bp) で特定の周波数帯は、記者シーケンスの統合を示します。バックボーン PCR (500-600 bp、プライマー設計によって) のための特定のバンドを示します (たとえば、結果参照図 3 c) テレビ バックボーン シーケンスの不要な統合。 - フローサイトメトリー (ステップ 3.2.3) と PCR ベースのスクリーニング (ステップ 3.2.12) の結果を結合します。PCR とポジティブ コントロール PCR およびフローサイトメトリーの PMA Iono 誘導後の蛍光レポーターの発現スクリーニングで PCR の製品の適切なサイズを表示するクローンを選択します。バックボーン PCR で PCR の製品やテレビ バックボーンでエンコードされた蛍光タンパク質、テレビ バックボーン シーケンスの不特定の統合を示す式を示すクローンを除外します。
- 徐々 にコート t75 フラスコ細胞培養フラスコ形式をすべて 2 〜 3 日新鮮な媒体を追加することによって達成するまで大きいよくフォーマット (48/24/12/6-よく) 96 ウェル株式板から選択したクローンを展開します。1 x 10 の5と 1 x 106セル/mL の細胞密度を維持します。
- 展開時にクローンの細胞株を準備することを確認: セルをカウント、常温 5 分間 300 × g で遠心分離機、上澄みを廃棄し、FCS + 5 x 106セル/mL で 10 %dmso に優しく細胞を中断します。セラム チューブで 1 ° C/分 80 ° C に細胞を凍結する使用低温凍結コンテナー因数。長期保存液 N2に転送します。
注:調製された gDNA 南しみが付くこと (セクション 3.4 を参照) によるターゲット設定の検証のための拡張中にコート t75 フラスコ細胞培養用フラスコ (すなわち,約 1 × 10 の7セル) を保持することをお勧めします。
- 選択されたクローンの PCR/シーケンス統合サイトの検証
注:選択されたクローンの 5' と 3' の int 型接合、PCR 増幅し、提出サンガーを確認するシーケンスをターゲット DNA シーケンス レベルで修正します。- Jurkat 野生型細胞商業 gDNA 抽出キットを使用して、選択されたクローンの gDNA を準備します。
- プライマー記者と 5' 5' int 型ジャンクション (P1 と P2 のプライマー) の河の上流 5' 端と 3' 端記者と 3' 3' int 型ジャンクション (P3 と P4 のプライマー) の河の下流ステップ 2.4 で説明されているようにバインドを使用します。記者インテグラル (図 4 a) のない対立遺伝子にターゲットを絞った統合サイトを増幅するのにプライマー P1 および P4 を使用します。
- テンプレートとして gDNA の 100-200 ng を PCR 反応を準備し、活動を校正のポリメラーゼを用いた PCR を行う (表 1および PCR 成分とサイクリング条件2を参照)。
注:バンドが認められなかった場合は、または (たとえば、追加した DMSO または Mg2 +の増加量)、PCR バッファーを変更するサイクル数を増やすことによって PCR の循環状態の変更を検討またはポリメラーゼを変更することによって。 - 5 μ L の 1.5% アガロース/泰ゲルの PCR の製品を分析します。正しいバンドのサイズが認められた場合商業キットを使用して残りの PCR の製品の浄化、サンガーの対象シーケンス。予期されるシーケンスとそれらを合わせて、5' int 型ジャンクション、3' int 型ジャンクション、ばれレポーターなしの対立遺伝子の標的となるサイトのシーケンスを確認します。
注:記者が統合されていない相同遺伝子はヌクレオチドの挿入または削除 (図 4 a) など、ターゲット ・ サイトでの変更の Cas9 を介したそう表示されます。 - 配置後正しい int 型ジャンクションのシーケンスを表示するクローン、全体対象の記者を増幅 PCR を実行し、サンガー、インテグラルの正しい順序を確認するシーケンス。
- 選択されたクローンのターゲットの検証のため南しみ分析
注:選択されたクローンの南しみ分析正しいターゲットの設定を確認し、対象となる統合サイトで発生した Cas9 を介する組み換えイベントを除外する必要があります。- 適切な gDNA 消化のための戦略を開発し、実験を開始する前にデザインをプローブします。
- 対象サイトで 2 に 10 kb の長さの適切なフラグメントを生成する gDNA 制限の制限酵素を選択します。Asp718、 Bamこんにちは、 Bglなど、特定の制限の酵素エコRV、ハインドIII下士官 BglII、私私は、 PstI, II、 Pvu Scaステュー、私、私とSst私は、正常に高分子量 gDNA を消化のため使用されています。
- 2 つの異なる南プローブの設計: レポーターのプローブとゲノム プローブ。レポーターのプローブは、記者 (すなわち, tdTomato 特定プローブ) 内のシーケンスに交配します。(重複していない) が、近くに、ゲノムのプローブが交配ゲノム領域に 1 HA。
- ゲノム プローブ結合によって検出される gDNA 消化によって生成されたフラグメントの長さ (2 kb 以上) 対象と対象外の対立遺伝子 (図 4 b) から異なるゲノムのプローブを選択します。プローブ長さ 400 に 1,000 bp をお勧めします。
- 2 つの必要なプローブを増幅する PCR プライマーを設計します。高忠実度 DNA ポリメラーゼを使用してテレビ テンプレートからレポーターのプローブを増幅 ( 3および表4 PCR 成分とサイクリング条件を参照)。
- 活動を校正と DNA ポリメラーゼを用いた商業 gDNA 抽出キットを使用した野生型 Jurkat gDNA からゲノムのプローブを増幅 (表 1および PCR 成分とサイクリング条件2を参照)。泰/agarose のゲルの PCR の製品を浄化し、製造元の指示に従って商業ゲル抽出キットを使用してフラグメントを抽出します。
- 野生型 Jurkat 細胞およびステップ 3.2.14 から選択されたクローンの 1 x 10 の7セルからの高分子量 gDNA を抽出します。
- RT で 5 分間 300 × gで遠心分離によって細胞のペレット、PBS で 1 回洗浄および換散バッファーの 4 mL にペレットを中断 [200 mM の NaCL、100 mM トリス塩酸 pH 8、5 ミリメートルの EDTA、0.1 %sds; 250-1,000 μ g/mL, プロテイナーゼ K (凍結乾燥粉末を追加、挽きたてで重量を量る)]。O/n 55 ° c、卓上 thermomixer で 350 rpm で揺れを孵化させなさい。
- イソプロパノールの 4 つの mL を追加し、混ぜるインバージョンによる 10 倍から 20 倍。GDNA が白い沈殿として表示されます。常温 (5 〜 10 分) 乾燥の 70 %etoh、750 μ L で新興によって洗い、ガラス ピペットの先に沈殿した gDNA をスプールします。
- 1 x TE バッファー (10 mM トリス塩酸 pH 8.0、1 mM EDTA) 500 μ L を含む 1.5 mL の反応管に沈殿物を流すし、o/n 350 rpm で揺れ、4 ° C での溶解に任せます。この段階から gDNA の任意のピペッティング ワイドボア剪断を避けるためにヒントとされるべきであります。
注:高分子量 gDNA の準備、南しみ分析のため不可欠であり市販 gDNA 準備キットは適していません。
- 選択されたクローンの選択された制限の酵素と野生型 Jurkat 細胞 gDNA のダイジェスト (2 回) 15 μ g (手順 3.4.1.1 参照) の酵素 (20 単位/μ L) 6 μ L で 60 μ L 反応: 最初に、DNA を追加消化バッファーと ddH2O、孵化させなさい o/n 37 ° C で、酵素を追加し、特定の酵素消化温 o/n を孵化させなさい。消化された gDNA の 15 の ug は南部のプローブごとに必要です。
- 1% のアガロース/TAE のゲルの電気泳動分析に 60 μ L 制限のダイジェストの 7 μ L を使用します。塗抹標本では、完全な消化力およびサザンブロット解析用 DNA 良質をことを示します。
- 1:10 を追加することによって残りの制限のダイジェストを沈殿させる 3 M 酢酸ナトリウムと 2 つの容積 100% エタノール、-80 ° C および 4 ° C で 15,600 x gで 30 分間遠心で 1 時間インキュベートし、
- 上澄みを廃棄し、ペレットを 70 %etoh。15,600 x gで 4 ° C で 15 分間遠心、上清を破棄、ペレットの常温では、簡単に乾燥し、ddH2o. の 20 μ L に溶解
- 1% アガロース/TAE のあぶらとりのゲルは、レーン当たり消化 gDNA の 20 uL の読み込みを実行します。60 V、400 で 2 h のためのゲルを実行 mA。
注: Agarose のゲルと実行時間/電圧の割合はステップ 3.4.1.1 で計算された南しみの検出のための期待されるフラグメント サイズに応じて調整する場合があります。南しみ分析の次の手順は、補足議定書 (手順 1 に 18) で詳細に説明します。次の手順で構成: ナイロン膜、放射性プローブの生成、プローブの交配、オートラジオグラフィー フィルムの開発にしみが付くこと、しみが付くゲルの洗浄します。南の戦略によると予想されるパターンとステップ 18 (補足議定書) でオートラジオグラフィー開発後得られた帯状の模様を比較 (たとえば、結果図 4 bを参照)。
- 適切な gDNA 消化のための戦略を開発し、実験を開始する前にデザインをプローブします。
- オフのターゲット イベントの分析
注: CRISPR Cas9 を介したゲノム工学がオフのターゲット効果、最高位の PCR 増幅 10 in silicoを生成できますので-オフのターゲット サイトのクローンを選択し、サンガーを予測した配列。- CCTop16 (http://crispr.cos.uni-heidelberg.de) を使用して最高ランク 10 のリストを生成するインシリコ予測オフのターゲット シーケンス。
- クエリ シーケンスとしてターゲット用の PAM を含む gRNA シーケンスを入力します。オフのターゲット予測の参照として"NGG"PAM と「ヒトゲノム」を選択します。
- 設定とターゲットの「4」に最大合計不一致 PAM がない gRNA の長さに長さのサイトします。出力ファイルは、それぞれ gRNA のゲノムのオフのターゲット サイトのランク一覧が提供されます。
- インシリコ上流と下流 UCSC のゲノムのブラウザー (http://genome.edu.ucsc.edu) と的外れ CCTop 結果リストからヒットの位置を使用して 10 最高位オフのターゲット ヒットのそれぞれのゲノム シーケンス 500 bp を抽出します。
- ごとに分析するターゲット サイトをオフに、PCR プライマー対を設計 600 に 700 のフラグメントを増幅予測オフのターゲット サイトを含む長さの bp。
- 選択されたクローンと商業 gDNA 抽出キットを用いた Jurkat 野生型細胞から gDNA を抽出します。オフのターゲット サイトごとにアクティビティを校正と DNA ポリメラーゼを用いた PCR を行う (表 1と2 PCR 成分とサイクリング条件参照) 野生型とそれぞれクローン由来 gDNA。
- 5 μ L の 1.5% アガロース/泰ゲルの PCR の製品を分析します。正しいバンドのサイズが認められた場合市販 PCR 精製キットを使用して残りの PCR の製品の浄化、サンガーの対象シーケンス。Jurkat 細胞でオフ対象のサイトとターゲットのクローンのシーケンスを比較します。
- CCTop16 (http://crispr.cos.uni-heidelberg.de) を使用して最高ランク 10 のリストを生成するインシリコ予測オフのターゲット シーケンス。
結果
この代表的な実験では、我々 は LTR、tdTomato コーディング シーケンス、および BACH2 遺伝子17のイントロンが 5 で 2 つの座位 polyA 信号系列から成る最小 HIV-1 由来の記者を対象とする分野します。HIV 感染患者2,4,5,6から主な T 細胞のさまざまな研究は、?...
ディスカッション
ここでは、選ばれたプロウイルス統合サイト CRISPR ベース Cas9 ゲノム工学の応用と HIV 由来 1 Jurkat 記者のモデルを生成するプロトコルについて述べる。
プロトコルのいくつかのポイントでは、計画段階で細心の注意が必要です。いくつかの遺伝子座は他の人よりもターゲットに簡単かもしれませんので対象となる軌跡を注意深く選択する必要があります最初に、(例,
開示事項
著者が明らかに何もありません。
謝辞
テクニカル サポート、ブリッタ Weseloh とベッティーナのアベルに感謝します。我々 もありがとうアルネ Düsedau、Jana Hennesen (流れ cytometry 技術プラットフォーム ハインリッヒ Pette Institut) テクニカル サポート。
資料
| Name | Company | Catalog Number | Comments |
| pX330-U6-Chimeric_BB-cBh-hSpCas9 | Addgene | 42230 | vector for expression of SpCas9 and gRNA |
| pMK | GeneArt | mammalian expression vector for cloning | |
| cDNA3.1 | Invitrogen | V79020 | mammalian expression vector for cloning |
| BbsI | New England Biolabs | R0539S | restriction enzyme |
| NEBuilder Hifi DNA Assembly Cloning Kit | New England Biolabs | E5520S | Assembly cloning kit used for target vector generation |
| TaqPlus Precision PCR System | Agilent Technologies | 600210 | DNA polymerase with proofreading activity used for amplification of homology arms (step 1.2.2.2), verification of integration site and reporter sequence (step 3.3.3 and 3.3.5), generation of genomic probe for Southern blot (step 3.4.1.5) and analysis of off-target events (step 3.5.4) |
| 96-well tissue culture plate (round-bottom) | TPP | 92097 | tissue culture plates for dilution plating |
| Phusion High-Fidelity DNA polymerase | New England Biolabs | M0530 L | DNA polymerase used for detection of targeting events (step 2.4.2) and generation ofreporter-specific probe for Southern blot (step 3.4.1.4) |
| Dimethyl sulfoxide (DMSO) | Sigma-Aldrich | D9170 | dimethyl sulfoxide as PCR additive |
| Magnesium Chloride (MgCl2) Solution | New England Biolabs | B9021S | MgCl2 solution as PCR additive |
| Deoxynucleotide (dNTP) Solution Mix | New England Biolabs | N0447S | dNTP mixture with 10 mM of each nt for PCR reactions |
| 5PRIME HotMasterMix | 5PRIME | 2200400 | ready-to-use PCR mix used for screening PCR (step 3.2.11) |
| QIAamp DNA blood mini kit | Qiagen | 51106 | DNA isolation and purification kit |
| QIAquick PCR Purification Kit | Qiagen | 28106 | PCR Purification Kit |
| RPMI 1640 without glutamine | Lonza | BE12-167F | cell culture medium |
| Fetal Bovine Serum South Africa Charge | PAN Biotech | P123002 | cell culture medium supplement |
| L-glutamine | Biochrom | K 0282 | cell culture medium supplement |
| Penicillin/Streptomycin 10.000 U/mL/ 10.000 µg/mL | Biochrom | A 2212 | cell culture medium supplement |
| Gibco Opti-MEM Reduced Serum Media | Thermo Fisher Scientific | 31985062 | cell culture medium with reduced serum concentration optimized for transfection |
| TransIT-Jurkat | Mirus Bio | MIR2125 | transfection reagent |
| phorbol 12-myristate 13-acetate | Sigma-Aldrich | P8139-1MG | cell culture reagent |
| Ionomycin | Sigma-Aldrich | I0634-1MG | cell culture reagent |
| Syringe-driven filter unit, PES membrane, 0,22 µm | Millex | SLGP033RB | filter unit for sterile filtration |
| Heracell 150i incubator | Thermo Fisher Scientific | 51026280 | tissue culture incubator |
| Amershan Hybond-N+ | GE Healthcare | RPN1520B | positively charged nylon membrane for DNA and RNA blotting |
| Stratalinker 1800 | Stratagene | 400072 | UV crosslinker |
| High Prime | Roche | 11585592001 | kit for labeling of DNA with radioactive dCTP using random oligonucleotides as primers |
| illustra ProbeQuant G-50 Micro Columns | GE Healthcare | 28-9034-08 | chromatography spin-columns for purification of labeled DNA |
参考文献
- Hughes, S. H., Coffin, J. M. What Integration Sites Tell Us about HIV Persistence. Cell Host and Microbe. 19 (5), 588-598 (2016).
- Marini, B., Kertesz-Farkas, A., et al. Nuclear architecture dictates HIV-1 integration site selection. Nature. 521 (7551), 227-231 (2015).
- Cesana, D., Santoni de Sio, F. R., et al. HIV-1-mediated insertional activation of STAT5B and BACH2 trigger viral reservoir in T regulatory cells. Nature Communications. 8 (1), 498 (2017).
- Cohn, L. B., Silva, I. T., et al. HIV-1 Integration Landscape during Latent and Active Infection. Cell. 160 (3), 420-432 (2015).
- Han, Y., Lassen, K., et al. Resting CD4+ T cells from human immunodeficiency virus type 1 (HIV-1)-infected individuals carry integrated HIV-1 genomes within actively transcribed host genes. Journal of Virology. 78 (12), 6122-6133 (2004).
- Ikeda, T., Shibata, J., Yoshimura, K., Koito, A., Matsushita, S. Recurrent HIV-1 integration at the BACH2 locus in resting CD4+ T cell populations during effective highly active antiretroviral therapy. The Journal of Infectious Diseases. 195 (5), 716-725 (2007).
- Wagner, T. A., Mclaughlin, S., et al. Proliferation of cells with HIV integrated into cancer genes contributes to persistent infection. Science. 345 (6196), 570-573 (2014).
- Maldarelli, F., Wu, X., et al. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science. 345 (6193), 179-183 (2014).
- Jordan, A., Bisgrove, D., Verdin, E. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. The EMBO Journal. 22 (8), 1868-1877 (2003).
- Mack, K. D., Jin, X., et al. HIV insertions within and proximal to host cell genes are a common finding in tissues containing high levels of HIV DNA and macrophage-associated p24 antigen expression. Journal of Acquired Immune Deficiency Syndromes. 33 (3), 308-320 (2003).
- Byrne, S. M., Ortiz, L., Mali, P., Aach, J., Church, G. M. Multi-kilobase homozygous targeted gene replacement in human induced pluripotent stem cells. Nucleic Acids Research. 43 (3), 1-12 (2014).
- CRISPR Genome Engineering Toolbox: Target Sequence Cloning Protocol. Addgene website Available from: https://www.addgene.org/static/cms/filer_public/e6/5a/e65a9ef8-c8ac-4f88-98da-3b7d7960394c/zhang-lab-general-cloning-protocol.pdf (2013)
- Gibson Assembly Protocol. Addgene website Available from: https://www.addgene.org/protocols/gibson-assembly/ (2009)
- Addgene Plasmid Cloning by PCR. Addgene website Available from: https://www.addgene.org/protocols/pcr-cloning/ (2014)
- Addgene Plasmid Cloning by Restriction Enzyme Digest (aka Subcloning). Addgene website Available from: https://www.addgene.org/protocols/subcloning/ (2013)
- Stemmer, M., Thumberger, T., Del Sol Keyer, M., Wittbrodt, J., Mateo, J. L. CCTop: An intuitive, flexible and reliable CRISPR-Cas9 target prediction tool. Public Library of Science (PLoS) ONE. 10 (4), (2015).
- Lange, U. C., Bialek, J. K., Walther, T., Hauber, J. Pinpointing recurrent proviral integration sites in new models for latent HIV-1 infection. Virus Research. 249, (2018).
- Bialek, J. K., Dunay, G. A., et al. Targeted HIV-1 Latency Reversal Using CRISPR-Cas9-Derived Transcriptional Activator Systems. PloS ONE. 11 (6), e0158294 (2016).
- Lee, C. M., Davis, T. H., Bao, G. Examination of CRISPR-Cas9 design tools and the effect of target site accessibility on Cas9 activity. Experimental Physiology. 103 (4), 456-460 (2018).
- Jensen, K. T., Fløe, L., et al. Chromatin accessibility and guide sequence secondary structure affect CRISPR-Cas9 gene editing efficiency. FEBS Letters. 591 (13), 1892-1901 (2017).
- Simonetti, F. R., Sobolewski, M. D., et al. Clonally expanded CD4 + T cells can produce infectious HIV-1 in vivo. Proceedings of the National Academy of Sciences. 113 (7), 1883-1888 (2016).
- Chen, H. C., Martinez, J. P., Zorita, E., Meyerhans, A., Filion, G. J. Position effects influence HIV latency reversal. Nature Structural and Molecular Biology. 24 (1), 47-54 (2017).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved