
Для просмотра этого контента требуется подписка на Jove Войдите в систему или начните бесплатную пробную версию.
Method Article
Основанный ТРИФОСФАТЫ Cas9 генома инженерии для создания модели Jurkat репортер для инфекции ВИЧ-1 с выбранной Proviral интеграции сайтов
В этой статье
Резюме
Мы представляем генома инженерных рабочих процессов для создания новых моделей в пробирке для инфекции ВИЧ-1, что итог proviral интеграции на отдельных участках генома. Нападения на журналистов ВИЧ производный облегчается манипуляция ТРИФОСФАТЫ-Cas9-опосредованной, участкам генома. Предоставляются подробные протоколы для одной ячейки клонов поколения, скрининг и правильной ориентации проверки.
Аннотация
Вирусом иммунодефицита человека (ВИЧ) не случайно интегрирует его proviral ДНК генома клетки хоста на текущих сайтах и геномных горячих точках. Здесь мы представляем подробный протокол для поколения роман в пробирке моделей для ВИЧ-инфекции с выбранной геномной интеграции сайтов с использованием генома, основанный ТРИФОСФАТЫ-Cas9 техника. С помощью этого метода репортер последовательность выбора могут быть интегрированы в целенаправленной, выбранной геномной локус, отражающие клинически значимых интеграции сайтов.
В протоколе описаны дизайн ВИЧ производные репортер и выбор целевого сайта и gRNA последовательности. Ориентации вектора с гомологии оружия построен и transfected в клетки Jurkat T. Репортер последовательность ориентирован на выбранный сайт геномной гомологичная рекомбинация, при содействии Cas9-опосредованной двухручьевой перерыв на целевом сайте. Клоны одной ячейки создаются и экранируется проточной цитометрии и ПЦР для ориентации событий. Выбранный клоны затем расширяется, и правильной ориентации подтверждена ПЦР, последовательности и Южный blotting. Проанализированы потенциальные пробить события ТРИФОСФАТЫ-Cas9-опосредованной генома техники.
Используя этот протокол, Роман клетки культуры системы, модель ВИЧ-инфекции в клинически значимых интеграции сайты могут быть созданы. Хотя поколение одной ячейки клонов и проверка правильного репортер последовательность интеграции является длительным, результирующий клоновых линий являются мощными инструментами, функционально анализировать выбор сайта proviral интеграции.
Введение
Интеграция proviral ДНК в геноме хоста после инфекции является важным шагом в жизненном цикле вируса иммунодефицита человека (ВИЧ). После интеграции ВИЧ сохраняется путем установления задержку в долгоживущих подмножеств клеток CD4 + T например CD4 + Т-клеток памяти. Интеграции ВИЧ, по-видимому, не случайные1,2. Ряд очагов геномной с периодически комплексной proviral ДНК была обнаружена в нескольких исследованиях через последовательность интеграции сайтов в остро и хронически инфицированных лиц2,3,4 ,5,6,,78. Интересно, что в некоторых из этих сайтов интеграции, же Локус был обнаружен в большой доли инфицированных клеток, приводит к идее, что интеграции на текущих сайтах может положительно повлиять на клоновых расширения1.
Для продвижения нашего понимания значимости периодических интеграции сайтов, необходимо изучить выбор сайта proviral интеграции. Однако, несколько технических аспектов препятствуют изучению интеграции ВИЧ сайт выбор и последствия. Широко используется модели культуры клеток для задержки ВИЧ как JLat клеточных линий не отражают клинически значимых периодически интеграции сайтов9. Исследования на первичной клетки пациента производные, с одной стороны, включить описание интеграции сайта ландшафта путем sequencing но не позволяют для функционального анализа. Насколько нам известно не адекватные Экспериментальная модель доступна для функционально анализировать выбранный клинически значимых интеграции сайтов.
Здесь мы представляем подробный рабочий процесс для создания новых моделей для ВИЧ-инфекции с использованием генома, основанный ТРИФОСФАТЫ-Cas9 техника. Рабочий процесс описан здесь может использоваться для создания Т клеток, полученных репортер клеточных линий, которые моделируют ВИЧ-инфекции, перевозящих genomically комплексных proviral репортер на сайте выбранного интеграции. Таким образом, они выступают в качестве новых инструментов для изучения влияния на сайте proviral интеграции биологии ВИЧ и как Провирус реагирует на различные лечения (например, inducibility, задержка обратного агентами). Наш метод использует преимущества инженерства основанный ТРИФОСФАТЫ-Cas9 генома, в котором интеграции репортер последовательности гомологичная рекомбинация облегчается Cas9 Нуклеаза индуцированной двухручьевой перерыв на целевом сайте. Целевые объекты для интеграции выбираются по близости от описанных периодических интеграции сайтов от исследований на ВИЧ инфицированных лиц и наличие подходящих PAM мотивы для машиностроения Cas9-опосредованной генома.
В наши образцовые результаты мы сосредоточились на BACH2 Локус гена, который коды для BTB и CNC гомологии transcriptional регулятор 2. В хронически ВИЧ инфицированных людей на антиретровирусной терапии BACH2 является одним из локусов, показаны обогащения комплексных ВИЧ-1 последовательности3,6,,78,10. Мы выбрали минимальная ВИЧ производные репортер, состоящий из ВИЧ-1-производные длинные концевые повторить (LTR), tdTomato кодирующая последовательность и гормон роста крупного рогатого скота (BGH) сплайсингу сигнала (ПА), которые мы избрали двух определенных сайтов в BACH2 Интрон 5. Представленные протокол оптимизирован для Jurkat клеток, человека CD4 + Т клеток, полученных подвеска клеток линии, но другие ячейки, строки могут быть использованы и протокола, адаптированными таким образом. Мы представляем подробную рабочего процесса для выбора целевого сайта, строительство целевой вектора с гомологии оружия, ТРИФОСФАТЫ-Cas9-опосредованной против репортера в выбранный геномной сайт, поколения и выбор клоновых линий и всеобъемлющей проверки созданный, целенаправленных репортер клеточных линий.
протокол
1. Ориентация стратегии для генома инженерных и ориентации дизайн вектор (ТВ)
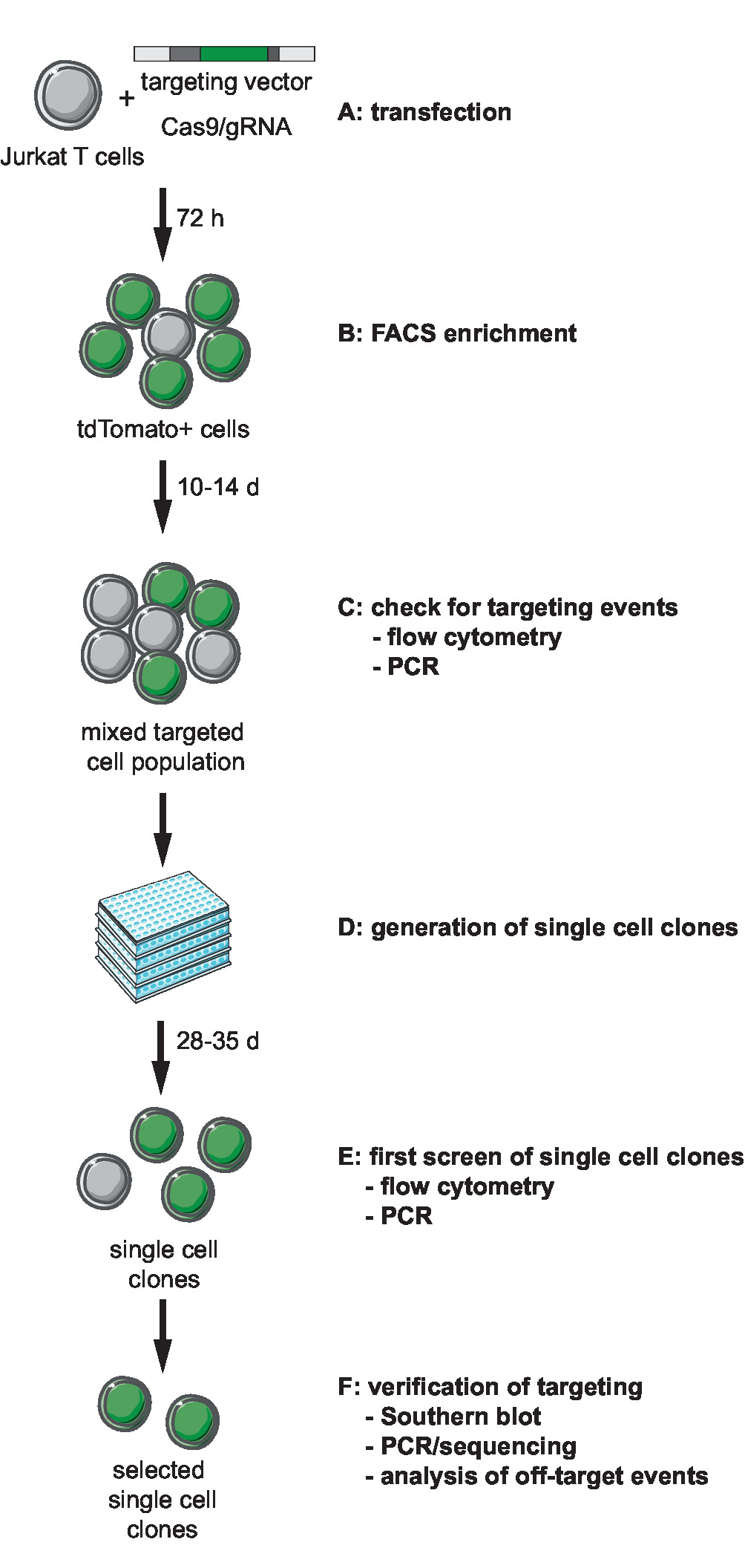
Примечание: Первый шаг генома техники включает в себя подбор и создание необходимых инструментов для ТРИФОСФАТЫ-Cas9-опосредованной ориентации. Выбор геномной интеграции сайта локус, выбор типа клеток для ориентации и дизайн ВИЧ производные репортер для интеграции должно предшествовать этот шаг. Этот протокол описывает ориентации минимальной репортер ВИЧ-LTR_tdTomato_BGH-PA в Jurkat клетки-мишени. На рисунке 1изображена схема рабочего процесса на основе ТРИФОСФАТЫ-Cas9 ориентации, поколение, отбора и проверки клоновых линий. Описывается стратегия нацеленности использует Cas9 S. pyogenes (SpCas9) для создания направленных gRNA dsDNA перерывы на сайте выбранного интеграции. Репортер затем направлена в выбранной геномной Локус через гомологичная рекомбинация, предоставляя-линеаризованных ориентации вектора (ТВ), содержащий последовательность репортер, обрамленная так называемые 5' и 3' гомологии оружия (HA)11.
-
Выбор целевых локус, gRNA и ориентации вектора дизайн
- Выберите thegenomic Локус направлены на основании индивидуальных научный вопрос. Использование опубликованные списки сайтов периодических интеграции ВИЧ по направлению пациентов в различных исследованиях2,3,4,5,6,,78 как Руководящий принцип. В silico экстракта геномные последовательности желаемого геномной Локус целевых (последовательность гена полной) или по крайней мере 5 КБ геномные последовательности с помощью браузера геноме UCSC (http:// genome.edu.ucsc.edu).
- Выберите руководство РНК (оформления) 20 nt для нападения на выбранной геномной локус, используя E-ХРУСТЯЩИЙ webtool (http://www.e-crisp.org).
- Выберите «Homo sapiens GRCh38» организма. Bp ввода 2000 геномные последовательности, охватывающих желаемого геномной локус, добываемый в шаге 1.1.1.
- Начните поиск gRNA с использованием среднего приложения параметров (Пэм, любые 5' база,-целей терпеть несоответствия, и исключаются интронов/CPG острова). С возможным gRNA конструкции появится список, ранжирование от самого высокого к снижению баллов для специфики и эффективности.
- Выберите gRNA, который желательно показывает высокий балл для специфики и эффективности и как можно ближе к желаемой геномной Локус направлены.
Примечание: Должен быть найден компромисс между близость к желаемой геномной Локус и разработке конкретных и эффективных gRNA.
- Доменная выбранной gRNA последовательность против геном ссылку, с помощью браузера взрыва NCBI (https://blast.ncbi.nlm.nih.gov) для проверки уникальности gRNA привязки сайта.
- Выберите «человека» как генома. Ввод gRNA последовательности как последовательности запроса. Выберите «очень похожие последовательности» (Мегавзрыв) как программы. Убедитесь, что последовательность gRNA является уникальным. Если нет, выбрал другой gRNA из шага 1.1.2.3 и взрыв снова.
- После того, как выбран gRNA последовательности, выберите в silico 1000 bp вверх и вниз по течению gRNA последовательности геномные последовательности, добываемый в шаге 1.1.1 5' и 3' ха соответственно.
Примечание: Оформления должен быть гомологичных Локус сайт выбранной геномной интеграции и расположен рядом с protospacer рядом мотив (Пэм, например, NGG для SpCas9) (рис. 2a). Телевизор содержит последовательность репортер, которая является 5' и 3', обрамленная HAs. Вверх и вниз по течению от последовательности gRNA11имеет крышка 1000 bp. Полный gRNA последовательности не должны включаться в HA. Дублирование до 5 nt является приемлемым (рис. 2a).
-
Поколение gRNA и ориентации векторов
Примечание: Для векторных систем, см. Рисунок 2b.- Для создания вектора выражения SpCas9 и gRNA, используйте pX330-U6-Chimeric_BB-cBh-hSpCas9 как основой, из которой может быть одновременно выражено SpCas9 и единого руководства РНК (sgRNA). Чтобы клонировать gRNA последовательность в позвоночник, используйте BbsI ограничения сайтов12.
- Для создания ТВ, выберите высокий копировать плазмиду как основу (например, ПМК, или cDNA3.1).
- Во-первых, собрать репортер (в настоящем протоколе: LTR_tdTomato_BGH-PA) в основу конструкции, Гибсон Ассамблея clong13 использование коммерческой сборки клонирования комплект и ввести 5' и 3' фланговые ограничения сайтов (например, 5' Голд и 3' Ссии) для последующего ограничения пищеварение клонирования HAs.
- Усилить bp 1000 ха фрагментов, выбранный на шаге 1.1.4 от геномной ДНК (геномная ДНК) ячейки типа направлены (в настоящем протоколе: клетки Jurkat) с помощью ДНК-полимеразы с корректура деятельности (см. таблицы 1 и 2 для ПЦР ингредиентов и Велоспорт условия). Затем ввести репортер фланговые ограничения сайтов на 5' и 3' концах каждого га (Голд на 5' ха на обоих концах, ссии на 3' ха на обоих концах).
- Последовательно клон имеет в конструкции позвоночника, уже содержащий репортер (созданного на шаге 1.2.2.1) путем клонирования14,15энзима ограничения. Во-первых, клон в 5' ха с помощью Голд ограничения сайтов, затем клонировать в 3' ха с помощью ссии места ограничения.
Примечание: Если ТВ позвоночника содержит дополнительные флуоресцентные репортер, нежелательные основой интеграции можно оценить по проточной цитометрии (см. шаги 3.2.2 и 3.2.3). Если телевизор позвоночника содержит не флуоресцентные репортер, основой интеграции должна оцениваться с помощью ПЦР (см. шаг 3.2.8).

Рисунок 1: рабочий процесс для ТРИФОСФАТЫ-Cas9-опосредованной ориентации, поколения и выбор клоновых репортер линий с определенными интеграции сайта. (A) создания целевого вектора и передают Jurkat Т-клеток с целевой вектора и Cas9/gRNA выражение плазмиды. (B) обогащения трансфекции пост 72 h transfected клеток, СУИМ. (C) пусть клетки растут на 10-14 дней и подтвердить возникновение ориентации события по ПЦР и проточная цитометрия. (D) создания одной ячейки клонов, ограничивая разрежения и пусть клоны расти на 3 недели. (E) экран клонов для правильной ориентации методом ПЦР и проточная цитометрия в 96-луночных формате. Разверните узел выбранного клонов. (F) убедитесь в правильной ориентации в отдельных клонов, Южная помарка, ПЦР и секвенирования и анализ событий пробить активности эндонуклеазы Cas9. Пожалуйста, нажмите здесь, чтобы посмотреть большую версию этой фигуры.
{kind=link}

Рисунок 2: ориентация стратегии и векторный дизайн. () gRNA и выбор оружия гомологии. 20 nt gRNA гомологичных выбранной геномной целевой сайт и расположенный рядом PAM. гомологии оружия являются взаимодополняющими-1000 bp вверх и вниз по течению от gRNA и не должно включать gRNA последовательности. (b) схемы ориентации вектора и gRNA/Cas9 вектора. Ориентации вектора состоит из выбранной репортер последовательности, которая является 5' и 3', обрамленная гомологии оружия. GRNA/Cas9 вектор основан на магистрали pX330-U6-Chimeric_BB-cBh-hSpCas9. (c) схема ориентации гомологичная рекомбинация. Целевой вектор и guideRNA/Cas9 вектор являются transfected в Jurkat клетки. Cas9 является посредником разрыв двойной нити в геномной целевого сайта (обозначается *) и содействует интеграции репортер последовательности в геномной целевой Локус гомологичная рекомбинация. Пожалуйста, нажмите здесь, чтобы посмотреть большую версию этой фигуры.
{kind=link}
2. ТРИФОСФАТЫ Cas9-нацеливание на основе Jurkat клеток
-
Transfection клетки Jurkat
- за 24 часа до трансфекции, плита 1,25 х 106 Jurkat T клеток в 2,5 мл RPMI 1640 дополнены с 10% (v/v) плода телячьей сыворотки (FCS) и 4 мм L-глютамин [именуется как «RPMI дней антибиотики (AB)»] за хорошо плиты культуры клеток 6-хорошо. Для одного эксперимента таргетинга, подготовить один полный 6-ну пластины (т.е., 6 скважин с 2,5 мл суспензии клеток).
- На следующий день, совместно transfect клетки с круговой ТВ и pX330-U6-Chimeric_BB-cBh-hSpCas9/gRNA, с помощью конкретного трансфекции реагент для Jurkat клеток.
- Добавьте 2 мкг круговой телевизора и 2 мкг pX330-U6-Chimeric_BB-cBh-hSpCas9/gRNA за хорошо 250 мкл коммерческих RPMI среды с ограниченной сывороточной концентрации, оптимизированный для transfection (RPMI с 50% сокращения в сыворотке) в реакции трубки и хорошо смешивают.
- Медленно добавьте 12 мкл Реагента трансфекции ДНК/средний без касатьться стенке трубки и вихрем. Пусть смесь Инкубируйте 15 мин и добавить каплям одной скважиной клеток. Инкубируйте клетки при 37 ° C и 5% CO2.
Примечание: Подготовку трансфекции реакции можно вычислить по маштабу. После трансфекции требуется не среднего изменения.
-
Обогащение transfected клеток путем активации флуоресценции клеток, сортируя (FACS)
- 72 ч после трансфекции, бассейн transfected клеток, считать их и подготовить для обогащения СУИМ. Собрать клетки в 50 мл Конические трубки, центрифуги на 300 x g и комнатной температуры (RT) 4 мин, вымыть клетки один раз в PBS, центрифуга снова, приостановить Пелле в соответствующее количество СУИМ буфера (PBS с FCS 1% + 1 мм ЭДТА) 1 x 10 7 клеток/мл и наконец в трубку СУИМ.
- При условии клетки СУИМ и отсортировать те, которые выражают флуоресцентные корреспондент tv (например, tdTomato в этом протоколе). Соберите клетки в RPMI 1640 с 10% FCS, 4 мм L-глютамином и 50 ед/мл пенициллина и стрептомицина (упоминаемые как «RPMI ж / AB»).
- После сортировки СУИМ, вымыть клетки один раз путем добавления 20 мл RPMI ж / AB для сортировки клеток и центрифуги на 300 x g 4 мин на RT. Ресуспензируйте Пелле клеток в соответствующее количество RPMI ж / AB и плиты клетки в одной скважиной плиты культуры клеток с соответствующий объем согласно ячейки номер пост СУИМ.
Примечание: Культура сортировать клетки в небольшом объеме (например, 24-ну), как значительного уровня гибели клеток наблюдалось в течение первой недели после ориентации (до 80-90%). - Разверните смешанные целевые клетки населения до плотности 1 х 106 клеток/мл в колбе культуры клеток 75 см². Это займет около 10-14 дней пост СУИМ сортировки.
-
Подтверждения целевых событий подачей cytometry в смешанной целевой популяции клеток
- После 10-14 дней расширения (когда клетки достигли плотности 1 х 106 клеток/мл в колбе культуры клеток 75 см2 ) два лунках с 1 x 106 клеток смешанной целевой популяции клеток в 1 мл RPMI ж / AB в культуре клеток 12-хорошо пластины.
- Побудить ВИЧ LTR репортер (поколения телевизора описано в шаге 1.2.2.1.) в одном из колодцев, добавляя 50 нг/мл phorbol 12-миристат 13-ацетат (PMA) и 1 мкм Ionomycin (именуемый PMA-ионосферы). Использование клеток в второй хорошо как элемент-индуцированной. Культура как искусственных, так и клетки-индуцированной за 24 ч.
- Взять 0,5 мл суспензии клеток-индуцированной и индуцированных клеток (каждый), мыть их раз в PBS и приостановить в 200 мкл буфера СУИМ.
- Анализировать 100000 клеток проточной цитометрии. Ворота жизнеспособных клеток одного, на основе размера в прямых и сбоку разброс и анализировать флуоресцентные репортер экспрессии генов.
Примечание: На этом этапе (10-14 дней пост СУИМ сортировки), переходных выражение флуоресцентные репортер, transfection больше не должны поддаваться обнаружению. Флуоресцентный выражение в этот момент времени указывает геномной интеграции репортер последовательности.
-
Обнаружение целевых событий методом ПЦР на геномной ДНК смешанной целевой популяции клеток
Примечание: Для выявления ориентации событий через PCR, дизайн двух грунтовка пар для интеграции (внутр.)-Джанкшн 5' и 3' int. Джанкшен. Для 5' int. перекрестка PCR, вперед грунт должно связывать вверх по течению от 5' ха и обратный грунт в ЛТР репортер (праймеры P1 и P2 на рисунке 3a). Грунтовка пара для int. перекрестка 3' ПЦР должен охватывать от ПА репортер до 100 – 200 bp вниз по течению от 3' ха (праймеры P3 и P4 на рисунке 3a). Праймеры P1 и P4 будет также служить для усиления непромысловых аллеля в смешанной целевой популяции. Схема см. рис. 3а.- Подготовьте геномная ДНК от 2 мл суспензии клеток населения смешанной целевой ячейки из шага 2.2.4. Использование набора извлечения геномная ДНК согласно протоколу производителя. Затем подготовьте геномная ДНК клеток, не являющихся объектом как элемент управления.
- Выполнять int. Джанкшен ДЗП (праймеры P1/P2 и P3/P4 для 5' и 3' int. junction, соответственно) и не ориентированные аллеля ПЦР (праймеры P1/P4) с использованием высокоточных ДНК-полимеразы (см. таблицы 3 и 4 для ПЦР ингредиентов и Велоспорт условий) . Анализ 5 мкл продуктов ПЦР на геле агарозы/TAE 1,5%.
Примечание: Если смешанное население целевых содержит клетки, которые претерпели генома инженерии, конкретный продукт PCR должна соблюдаться это не обнаружено в геномная ДНК клеток, не являющихся объектом (отрицательный контроль). Для непромысловых аллеля PCR один должен соблюдать продукт такого же размера как целевые и нецелевые ячеек (положительный контроль за геномной P1 и P4 грунты). Если наблюдаются не полос, рассмотрите возможность изменения Велоспорт условия PCR, увеличивая количество циклов или изменяя буфер ПЦР (например, путем добавления ДМСО или увеличение количества мг2 +), или путем изменения полимеразы.
3. поколение клоновых линий и скрининга для правильной ориентации
Примечание: После подтверждения ориентации событий в популяцию смешанных целевых клеток проточной цитометрии и ПЦР (разделы 2.2-2.4), создания одной ячейки клонов (продолжительность: 28-35 дней) и экран для правильной интеграции репортер последовательности.
- Поколение одной ячейки клонов путем разбавления плакировка
- Подготовить Jurkat кондиционером средний заранее: взлет RPMI ж / AB среднего от здоровых, неочищенные Jurkat Т-клеток, выросло до 1 x 106 клеток/мл, центрифуги для 5 мин на 300 x gи фильтровать супернатанта, с помощью фильтра шприц 0,22 мкм.
Примечание: Держать кондиционером среднего при 4 ° C для краткосрочного хранения или при-20 ° C для хранения более 1 недели. Подготовка 20-30 мл кондиционером среды до обшивки разрежения. - Граф целевые клетки от шага 2.2.4 через 10 – 14 дней после расширения и развести их в RPMI ж / AB в концентрации 1 х 105 кл/мл. Взять 100 мкл 1 х 105 клеток/мл раствора и разбавляют 9.9 мл среды для достижения концентрации 1000 клеток/мл. Возьмите 1 мл 1000 клеток/мл раствора и разбавляют 9 мл среды для достижения концентрации 100 клеток/мл.
- Плита 96-луночных пластины, содержащие 1 ячейку, в колодец и 2 за хорошо. Для 1 ячейки в колодец взять 1 мл 100 клеток/мл раствора и перемешать аккуратно с 5 кондиционером средних и 4 мл свежей среды в стерильных реагент водохранилище.
- Для 2 ячеек на колодец Возьмите 2 мл 100 клеток/мл раствора и перемешать аккуратно с 5 кондиционером среднего и 3 мл свежей среды. Пластина 96-луночных раунд нижней пластины с 100 мкл соответствующих клеток разрежения на хорошо с помощью многоканальные пипетки.
Примечание: 5-10 96-луночных пластины в ориентации конструкции достаточно получить клоны для скрининга. - Стек 96-луночных пластины, охватывают каждого стека с 6-ну пластины, содержащие 3 мл PBS в каждой скважине и инкубировать пластины при 37 ° C в инкубатор культуры увлажненные клеток с 5% CO2 на 3 недели.
Примечание: Не изменяйте среднего культуры клеток в это время. Не открывайте инкубатора более чем один раз или два раза в неделю. Лучшие результаты наблюдаются в инкубаторы с открытой воды водохранилища. - После 3 недели инкубации визуально подтвердить наличие выросли колоний, с использованием световой микроскопии (4 X увеличение) и Марк скважин с выросли колоний, чтобы они были видны как точки в нижней части скважины.
- Подготовьте один 96-луночных раунд-днище с 100 мкл RPMI ж / AB в колодец. Нежно ресуспензируйте клетки заметное, закупорить. Перевести 100 мкл суспензии клеток в одной скважиной новой 96-луночных пластины, уже содержащей 100 мкл RPMI ж / AB, а затем осторожно перемешать, закупорить. Перевести 100 мкл этой суспензии клеток в второй пустой 96-луночных раунд-днище дублировать пластину.
- Продолжите с все отмеченные скважин с выросли колоний. Заполните все пустые скважины с 200 мкл RPMI ж / AB среднего. Инкубировать пластин при 37° C и 5% CO2.
Примечание: Один из этих плит будет служить «дублировать пластина» для скрининга для расширения одной ячейки клонов («фондовый Латы») и другие.
- Подготовить Jurkat кондиционером средний заранее: взлет RPMI ж / AB среднего от здоровых, неочищенные Jurkat Т-клеток, выросло до 1 x 106 клеток/мл, центрифуги для 5 мин на 300 x gи фильтровать супернатанта, с помощью фильтра шприц 0,22 мкм.
- Скрининг из одной ячейки клонов проточной цитометрии и ПЦР
Примечание: В то время как расширение одной ячейки клонов, используйте пластину дубликаты из шага 3.1.8 для экрана одной ячейки клонов присутствие репортер последовательности методом ПЦР (3.2.4–3.2.12 меры) и выражение флуоресцентные репортер подачей cytometry (шаги 3.2.2–3.2.3) (Рис. 3 c).- Пусть пластину дублировать Проинкубируйте 24-48 ч и дублировать пластину снова. Чтобы сделать это, 100 мкл RPMI ж / AB в каждый хорошо, осторожно перемешать, закупорить и передать новой 96-луночных раунд-днище с помощью многоканального накапайте 100 µL. Использование одной пластиной для потока цитометрии скрининг и другой для скрининга на основе ПЦР.
- Для потока цитометрии скрининга, стимулируют клетки с PMA-ионосферы. Подготовить mastermix 0.1 мкл Ionomocin (1 мм запасов), 0.1 мкл PMA (50 мкг/мкл акции), и 4,8 мкл RPMI ж / AB за количество скважин, затем добавить 5 мкл mastermix за хорошо.
Примечание: Индукции необходимо успешно идентифицировать клоны, где LTR может быть транскрипционно молчание и поэтому не выражается флуоресцентные репортер. - Пусть клетки Инкубируйте 24 h и подготовить клетки для проточной цитометрии, как описано в шаге 2.3.3. Ворота любой жизнеспособной сингл клетки, на основе размера в прямых и сбоку точечной и анализ экспрессии генов флуоресцентные репортер подачей cytometry (например результаты, см. рис. 3 c). Если телевизор позвоночника содержит второй флуоресцентные репортер с промоутером (например, GFP), экран любой клоны для позвоночника репортер выражение также (см. шаг 1.2.2 и следующее примечание для объяснения).
Примечание: Позвоночника репортер выражение указывает нежелательных интеграции позвоночника последовательностей. - После того, как клоны в пластину второй дубликат выросли достаточно (обычно 24-48 ч после дублирования 96-луночных плиты), подготовить lysates клетки содержащие геномная ДНК для PCR скрининг. Центрифуга пластину для 10 мин на 300 x g на RT. тщательно снять супернатант не нарушая Пелле ячейки.
Примечание: Все шаги для подготовки лизатов и ПЦР-реакции могут быть выполнены с многоканальные пипетки. - Вымыть клетки с 100 мкл PBS нежный закупорить и центрифугирование пластину для 5 мин на 300 x g на RT. Взлет PBS и 200 мкл буфера lysis [200 мм NaCl, 100 мм трис-HCl рН 8-8,5, 5 мм ЭДТА, 0.1% SDS; затем добавить 250-1000 мкг/мл протеиназы K (лиофилизированный порошок, весят в свежую)] в колодец. Осторожно перемешать, закупорить и передачи подвеска к новой плите ПЦР.
- Печать пластину с парафином пленкой и инкубировать 1 час при температуре 55 ° C в Термоциклер. Центрифуга с максимальной скоростью за 10 мин (3000 x g) спина вниз ячейки мусор и передачи супернатант новой ПЦР-планшете.
Примечание: Lysates клетки в пластины могут храниться на данном этапе на 4 ° C до дальнейшего использования. - Подготовить 96-луночных ПЦР плита с 110 мкл dH2O и 10 мкл lysate клетки (1:12 разрежения). Lysates клетки могут быть вязкой и трудно пипетки. Используйте по крайней мере 20 мкл накапайте советы.
- Инактивирует протеиназы K по инкубации за 10 мин на 99 ° C в Термоциклер. Впоследствии используют инактивированные и разбавленных lysates клетки для PCR скрининг.
- Дизайн праймеров для скрининга ПЦР (P5 и P6) на основе выбранной репортер последовательности для того чтобы усилить 500 – 800 bp репортер последовательности. Для позитивного управления PCR, используйте bp грунтовки P7 и P8, которые усиливают 630 одичал тип, без целенаправленных геномной локус (NUP188 ген) (рис. 3 c и 5 таблицы). Дизайн третьей пары грунтовка, что усиливает 500 – 600 ВР ТВ позвоночника как элемент управления для неспецифической интеграции ТВ позвоночника последовательностей (костяк ПЦР).
- Для проверки, контроля и магистральной PCR, использовать коммерческие ПЦР mastermix (см. таблицы 6 и 7 для ПЦР ингредиентов и Велоспорт условий). Используйте 2 мкл разбавленного и инактивированных lysate подготовлен на шаге 3.2.8 как шаблон и запустить ПЦР для 38-40 циклов амплификации PCR в 96-луночных формате.
- Анализ 5 мкл продуктов ПЦР на геле агарозы/TAE 1,5%.
Примечание: Для управления PCR, конкретные группы 630 bp должны соблюдаться для каждого образца, подтверждающее, что качество lysates клетки является адекватной для ПЦР. Конкретные группы в скрининг ПЦР (500 – 800 bp в зависимости от конструкции праймера) показывает интеграцию репортер последовательности. Конкретные группы для позвоночника ПЦР (500 – 600 bp, в зависимости от конструкции праймера) показывает нежелательные интеграции ТВ позвоночника последовательностей (например результаты, см. рис. 3 c). - Объедините результаты проточной цитометрии (шаг 3.2.3) и на основе ПЦР скрининга (шаг 3.2.12). Выберите клоны, которые показывают правильные размеры продуктов PCR скрининг ПЦР и позитивного управления ПЦР и выражение флуоресцентные репортер после индукции с PMA-ионосферы в проточной цитометрии. Исключите клоны, которые показывают любой продукт PCR в основу ПЦР или выражение ТВ позвоночника кодировке флуоресцентный белок, указанием неспецифических интеграции ТВ позвоночника последовательности.
- Постепенно раскройте отдельных клонов из 96-луночных запасов пластины больше хорошо форматов (48/24/12/6-ну) до достижения формат колбу культуры клеток T75, добавляя свежий среднего каждые 2-3 дней. Поддерживать плотность ячеек между 1 x 10-5 и 1 х 106 клеток/мл.
- Убедитесь, что подготовка запасов ячейки клонов во время расширения: подсчитать количество ячеек, центрифуги на 300 g x 5 мин на RT, удалить супернатант и приостановить клетки мягко в FCS + 10% ДМСО на 5 х 106 клеток/мл. Алиготе в криогенных флаконов и использования крио замораживание контейнер заморозить клетки до 80 ° C со скоростью 1 ° C/мин. Для длительного хранения передачи их в жидкости N2.
Примечание: Это целесообразно сохранить колбу культуры клеток T75 (т.е., около 1 x 107 ячеек) во время расширения в подготовке геномная ДНК для проверки ориентации, Южный blotting (см. раздел 3.4).
- Проверка интеграции сайтов по ПЦР/последовательности в отдельных клонов
Примечание: 5' и 3' int. узлов выбранного клоны являются ПЦР усиливается и представлен к Сэнгер последовательности для проверки исправления, ориентация на уровне последовательности ДНК.- Подготовьте геномная ДНК выбранного клоны и Jurkat одичал тип клеток с помощью комплекта извлечения коммерческих геномная ДНК.
- Используйте грунт пары привязки ' конец 5 репортера и вверх по течению от 5' HA для 5' int. Джанкшен (праймеры P1 и P2) и 3' конца репортер и вниз по течению от 3' HA для 3' int. Джанкшен (праймеры P3 и P4), как описано в шаге 2.4. Использование грунтовки P1 и P4 для того чтобы усилить целенаправленной интеграции сайта на аллеля без репортер автоконтроля (рис. 4a).
- Подготовить реакций PCR с 100 – 200 ng геномная ДНК как шаблон и выполнить используя корректура активность полимеразы PCR (см. таблицы 1 и 2 для ПЦР ингредиентов и Велоспорт условий).
Примечание: Если наблюдаются не полос, рассмотрите возможность изменения Велоспорт условия PCR, увеличивая количество циклов или изменяя буфер ПЦР (например, путем добавления ДМСО или увеличение количества мг2 +), или путем изменения полимеразы. - Анализ 5 мкл продуктов ПЦР на геле агарозы/TAE 1,5%. Если соблюдаются правильные группы размеров, очистит оставшиеся ПЦР продукта с использованием коммерческих комплект и подвергнуть его Сэнгер последовательности. Проверка последовательности 5' int.-Джанкшн, 3' int. Джанкшен и целевой сайт аллеля без репортер укрепились, совместив их с ожидаемой последовательности.
Примечание: Гомологичных аллеля, где репортер не включил вероятно покажет Cas9-опосредованной изменения на конечном сайте, например нуклеотидов вставки или удаления (рис. 4a). - Для клонов, которые показывают правильное int. последовательности перехода после выравнивания, выполнять ПЦР, усиливая весь целевой репортер и подвергнуть его Сэнгер последовательности для проверки правильной последовательности укрепились.
- Южная помарка анализ для проверки ориентации в отдельных клонов
Примечание: Для проверки правильной ориентации и исключить Cas9-опосредованной рекомбинации события, которые произошли на сайте целенаправленной интеграции требуется Южная помарка анализ отдельных клонов.- Разработать стратегию для соответствующих геномная ДНК переваривания и зонд дизайн до начала эксперимента.
- Выберите энзима ограничения для ограничения геномная ДНК, который генерирует соответствующие фрагменты длиной от 2 до 10 КБ на целевой сайт. Некоторые энзимы ограничения, например Asp718, BamПривет, BglI, II, Bgl экоRV, III Хинд, Ncoя, Pstя, PvuII, Scaя, Стю, и SST Я успешно использовался для переваривания геномная ДНК высокой молекулярной массой.
- Дизайн два разных Южный зондов: репортер конкретной зонда и геномная зонд. Репортер конкретных зонд скрестил последовательность в пределах репортер (т.е., tdTomato конкретных зонд). Геномный зонд скрестил геномной регион близко к (но не перекрывающихся) один га.
- Выберите геномной зонд, чтобы фрагмент, порожденных пищеварение геномная ДНК, которые будут обнаружены геномные зонд привязки отличается в длину (более чем 2 КБ) от целевые и нецелевые аллели (рис. 4b). Длина зонда 400 до 1000 порекомендованное bp.
- Праймеры PCR дизайн для того чтобы усилить две необходимые датчики. Усилить репортер конкретных зонд из ТВ шаблона с помощью высокоточных ДНК-полимеразы (см. таблицы 3 и 4 для ПЦР ингредиентов и Велоспорт условий).
- Усилить геномной зонд из геномная ДНК одичал типа Jurkat, подготовленных с комплектом извлечения коммерческой геномная ДНК с помощью ДНК-полимеразы с корректура деятельности (см. таблицы 1 и 2 для ПЦР ингредиентов и Велоспорт условий). Очистить продуктов ПЦР на геле агарозы/TAE и извлекать фрагменты с помощью набора извлечения коммерческих гель согласно инструкциям производителя.
- Экстракт геномная ДНК высокой молекулярной массой от 1 x 107 клеток одичал тип Jurkat клеток и отдельных клонов из шага 3.2.14.
- Пелле клетки центрифугированием при 300 x g 5 мин на RT, мыть раз с PBS и приостановить гранулы в 4 мл буфера lysis [200 мм NaCL, 100 мм трис-HCl рН 8, 5 мм ЭДТА, 0.1% SDS; затем добавить 250-1000 мкг/мл протеиназы K (лиофилизированный порошок весят свежую)]. Инкубируйте o/n при 55 ° C, тряски на 350 об/мин в настольная thermomixer.
- Добавить 4 мл изопропанола и перемешать путем инверсии 10-20 раз. Геномная ДНК должны стать видимыми как Белый преципитат. Катушка осажденный геномная ДНК на кончик тонкой стеклянной пипетки, мыть, возникающих в 750 мкл 70% EtOH и пусть сухой на RT (5-10 мин).
- Пролить осадок в трубу реакции 1,5 мл, содержащих 500 мкл буфера TE 1 x (10 мм трис-HCl рН 8,0, ЭДТА 1 мм) и оставить раствориться o/n на 4 ° C, тряски на 350 об/мин. Любые закупорить геномная ДНК из этой стадии должно быть сделано с wide родила советы, чтобы избежать сдвига.
Примечание: Подготовка высокой молекулярной массой геномная ДНК имеет важное значение для анализа Южная помарка, и коммерчески доступных геномная ДНК подготовки наборы не подходят.
- Дайджест (два раза) 15 мкг геномная ДНК выбранного клоны и одичал тип Jurkat ячеек с выбранной энзима ограничения (см. шаг 3.4.1.1) в 60 мкл реакции с 6 мкл фермента (20 единиц/мкл): во-первых, добавить ДНК, буфер, пищеварение и ddH2O, инкубировать o/n при 37 ° C , затем добавить фермента и инкубировать o/n при температуре пищеварение конкретного фермента. 15 ug переваривается геномная ДНК требуется на юге зонд.
- Используйте 7 мкл дайджест ограничение 60 мкл аналитического гель-электрофорез на геле агарозы/ТЭ 1%. Мазок показывает полный пищеварение и хорошее качество ДНК для анализа Южная помарка.
- Осадок оставшиеся ограничения дайджест, добавив 1:10 ацетат натрия 3 М и 100% 2 тома EtOH, затем инкубировать 1 час при-80 ° C и центрифуги для 30 мин на 15600 x g при 4 ° C.
- Отменить супернатант и помыть лепешка с 70% EtOH. Центрифуга для 15 мин на 15600 x g при 4 ° C, отбросить супернатанта, пусть гранулы сухого кратко на RT и растворяют в 20 мкл ddH2O.
- Запустите blotting гель 1% агарозном/ТЭ, загрузки 20 uL переваривается геномная ДНК в переулок. Побегите гель 2 h на 60 V, 400 мА.
Примечание: Процент агарозы гель и работает время/напряжения может корректироваться согласно размер ожидаемых фрагментов для обнаружения Южная помарка, рассчитанные на шаге 3.4.1.1. Следующие шаги Южная помарка анализа описаны подробно в дополнительный протокол (шаги 1-18). Эти шаги включают: мытье blotting геля, промокательной на мембраны нейлона, поколение радиоактивных зонд, зонд гибридизации и развития авторадиография фильма. Сравните полученные диапазонов шаблон после авторадиография развития в шаг 18 (дополнительный протокол) с ожидаемым шаблоном согласно южной части стратегии (например результаты, см. рис. 4b).
- Разработать стратегию для соответствующих геномная ДНК переваривания и зонд дизайн до начала эксперимента.
- Анализ событий, мимо
Примечание: Так как ТРИФОСФАТЫ-Cas9-опосредованной генома инженерия может генерировать пробить эффекты, ПЦР усилить десять высокий рейтинг в silico-предсказал пробить сайты в выбранные клоны и подвергает их Сэнгер последовательности.- Использование CCTop16 (http://crispr.cos.uni-heidelberg.de) для создания списка из десяти сильнейших в silico предсказал пробить последовательности.
- Входной последовательности gRNA, включая PAM, применявшихся для нанесения ударов в последовательности запроса. Выберите «Нг» как Пэм и «Геном человека» как ссылка-целевого прогноза.
- Задать максимальное общее несоответствия «4» и целевого сайта длина длина gRNA без PAM. Выходной файл обеспечит занимает список геномной пробить сайтов для соответствующих gRNA.
- В silico экстракт геномные последовательности 500 bp вверх и вниз по течению от каждого из десяти сильнейших пробить хитов с помощью геноме UCSC Browser (http://genome.edu.ucsc.edu) и положение пробить хит из списка результатов CCTop.
- Для каждого целевого сайтов необходимо проанализировать, Дизайн праймера PCR пару, которая усиливает фрагмент 600 до 700 bp в длину, включая прогнозируемые-целевого сайта.
- Извлечь геномная ДНК из выбранного клоны и Jurkat одичал тип ячейки, используя набор для извлечения коммерческих геномная ДНК. Для каждого-целевого сайта, выполнить используя корректура активность ДНК-полимеразы PCR (см. таблицы 1 и 2 для ПЦР ингредиентов и Велоспорт условий) на дикого типа и соответствующих производных клон геномная ДНК.
- Анализ 5 мкл продуктов ПЦР на геле агарозы/TAE 1,5%. Если соблюдаются правильные группы размеров, очистит оставшиеся ПЦР продукта с использованием коммерческих комплект очистки ПЦР и подвергнуть его Сэнгер последовательности. Сравнение последовательности-целевых сайтов в Jurkat клетках и целевых клонов.
- Использование CCTop16 (http://crispr.cos.uni-heidelberg.de) для создания списка из десяти сильнейших в silico предсказал пробить последовательности.
Результаты
В этот представитель эксперимента мы выбрали для минимальной ВИЧ-1-производные репортер, состоящий из ЛТР, tdTomato кодирования последовательность и последовательность поля сигнала для двух локусов в Интрон 5 гена BACH217. Локусов для ориентации были выбраны по ?...
Обсуждение
Здесь мы описываем протокол для создания ВИЧ-1-производные Jurkat репортер модели с выбранной proviral интеграции сайтов применения ТРИФОСФАТЫ-Cas9-основанные геном инженерии.
Несколько точек протокола требуют пристального внимания на этапе планирования. Во-первых, локус напра?...
Раскрытие информации
Авторы не имеют ничего сообщать.
Благодарности
Мы благодарим Бритта Weseloh и Bettina Абель для оказания технической помощи. Мы также благодарим Арне Düsedau и Яна Hennesen (поток цитометрии технологическая платформа, Генриха Pette Institut) для технической поддержки.
Материалы
| Name | Company | Catalog Number | Comments |
| pX330-U6-Chimeric_BB-cBh-hSpCas9 | Addgene | 42230 | vector for expression of SpCas9 and gRNA |
| pMK | GeneArt | mammalian expression vector for cloning | |
| cDNA3.1 | Invitrogen | V79020 | mammalian expression vector for cloning |
| BbsI | New England Biolabs | R0539S | restriction enzyme |
| NEBuilder Hifi DNA Assembly Cloning Kit | New England Biolabs | E5520S | Assembly cloning kit used for target vector generation |
| TaqPlus Precision PCR System | Agilent Technologies | 600210 | DNA polymerase with proofreading activity used for amplification of homology arms (step 1.2.2.2), verification of integration site and reporter sequence (step 3.3.3 and 3.3.5), generation of genomic probe for Southern blot (step 3.4.1.5) and analysis of off-target events (step 3.5.4) |
| 96-well tissue culture plate (round-bottom) | TPP | 92097 | tissue culture plates for dilution plating |
| Phusion High-Fidelity DNA polymerase | New England Biolabs | M0530 L | DNA polymerase used for detection of targeting events (step 2.4.2) and generation ofreporter-specific probe for Southern blot (step 3.4.1.4) |
| Dimethyl sulfoxide (DMSO) | Sigma-Aldrich | D9170 | dimethyl sulfoxide as PCR additive |
| Magnesium Chloride (MgCl2) Solution | New England Biolabs | B9021S | MgCl2 solution as PCR additive |
| Deoxynucleotide (dNTP) Solution Mix | New England Biolabs | N0447S | dNTP mixture with 10 mM of each nt for PCR reactions |
| 5PRIME HotMasterMix | 5PRIME | 2200400 | ready-to-use PCR mix used for screening PCR (step 3.2.11) |
| QIAamp DNA blood mini kit | Qiagen | 51106 | DNA isolation and purification kit |
| QIAquick PCR Purification Kit | Qiagen | 28106 | PCR Purification Kit |
| RPMI 1640 without glutamine | Lonza | BE12-167F | cell culture medium |
| Fetal Bovine Serum South Africa Charge | PAN Biotech | P123002 | cell culture medium supplement |
| L-glutamine | Biochrom | K 0282 | cell culture medium supplement |
| Penicillin/Streptomycin 10.000 U/mL/ 10.000 µg/mL | Biochrom | A 2212 | cell culture medium supplement |
| Gibco Opti-MEM Reduced Serum Media | Thermo Fisher Scientific | 31985062 | cell culture medium with reduced serum concentration optimized for transfection |
| TransIT-Jurkat | Mirus Bio | MIR2125 | transfection reagent |
| phorbol 12-myristate 13-acetate | Sigma-Aldrich | P8139-1MG | cell culture reagent |
| Ionomycin | Sigma-Aldrich | I0634-1MG | cell culture reagent |
| Syringe-driven filter unit, PES membrane, 0,22 µm | Millex | SLGP033RB | filter unit for sterile filtration |
| Heracell 150i incubator | Thermo Fisher Scientific | 51026280 | tissue culture incubator |
| Amershan Hybond-N+ | GE Healthcare | RPN1520B | positively charged nylon membrane for DNA and RNA blotting |
| Stratalinker 1800 | Stratagene | 400072 | UV crosslinker |
| High Prime | Roche | 11585592001 | kit for labeling of DNA with radioactive dCTP using random oligonucleotides as primers |
| illustra ProbeQuant G-50 Micro Columns | GE Healthcare | 28-9034-08 | chromatography spin-columns for purification of labeled DNA |
Ссылки
- Hughes, S. H., Coffin, J. M. What Integration Sites Tell Us about HIV Persistence. Cell Host and Microbe. 19 (5), 588-598 (2016).
- Marini, B., Kertesz-Farkas, A., et al. Nuclear architecture dictates HIV-1 integration site selection. Nature. 521 (7551), 227-231 (2015).
- Cesana, D., Santoni de Sio, F. R., et al. HIV-1-mediated insertional activation of STAT5B and BACH2 trigger viral reservoir in T regulatory cells. Nature Communications. 8 (1), 498 (2017).
- Cohn, L. B., Silva, I. T., et al. HIV-1 Integration Landscape during Latent and Active Infection. Cell. 160 (3), 420-432 (2015).
- Han, Y., Lassen, K., et al. Resting CD4+ T cells from human immunodeficiency virus type 1 (HIV-1)-infected individuals carry integrated HIV-1 genomes within actively transcribed host genes. Journal of Virology. 78 (12), 6122-6133 (2004).
- Ikeda, T., Shibata, J., Yoshimura, K., Koito, A., Matsushita, S. Recurrent HIV-1 integration at the BACH2 locus in resting CD4+ T cell populations during effective highly active antiretroviral therapy. The Journal of Infectious Diseases. 195 (5), 716-725 (2007).
- Wagner, T. A., Mclaughlin, S., et al. Proliferation of cells with HIV integrated into cancer genes contributes to persistent infection. Science. 345 (6196), 570-573 (2014).
- Maldarelli, F., Wu, X., et al. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science. 345 (6193), 179-183 (2014).
- Jordan, A., Bisgrove, D., Verdin, E. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. The EMBO Journal. 22 (8), 1868-1877 (2003).
- Mack, K. D., Jin, X., et al. HIV insertions within and proximal to host cell genes are a common finding in tissues containing high levels of HIV DNA and macrophage-associated p24 antigen expression. Journal of Acquired Immune Deficiency Syndromes. 33 (3), 308-320 (2003).
- Byrne, S. M., Ortiz, L., Mali, P., Aach, J., Church, G. M. Multi-kilobase homozygous targeted gene replacement in human induced pluripotent stem cells. Nucleic Acids Research. 43 (3), 1-12 (2014).
- CRISPR Genome Engineering Toolbox: Target Sequence Cloning Protocol. Addgene website Available from: https://www.addgene.org/static/cms/filer_public/e6/5a/e65a9ef8-c8ac-4f88-98da-3b7d7960394c/zhang-lab-general-cloning-protocol.pdf (2013)
- Gibson Assembly Protocol. Addgene website Available from: https://www.addgene.org/protocols/gibson-assembly/ (2009)
- Addgene Plasmid Cloning by PCR. Addgene website Available from: https://www.addgene.org/protocols/pcr-cloning/ (2014)
- Addgene Plasmid Cloning by Restriction Enzyme Digest (aka Subcloning). Addgene website Available from: https://www.addgene.org/protocols/subcloning/ (2013)
- Stemmer, M., Thumberger, T., Del Sol Keyer, M., Wittbrodt, J., Mateo, J. L. CCTop: An intuitive, flexible and reliable CRISPR-Cas9 target prediction tool. Public Library of Science (PLoS) ONE. 10 (4), (2015).
- Lange, U. C., Bialek, J. K., Walther, T., Hauber, J. Pinpointing recurrent proviral integration sites in new models for latent HIV-1 infection. Virus Research. 249, (2018).
- Bialek, J. K., Dunay, G. A., et al. Targeted HIV-1 Latency Reversal Using CRISPR-Cas9-Derived Transcriptional Activator Systems. PloS ONE. 11 (6), e0158294 (2016).
- Lee, C. M., Davis, T. H., Bao, G. Examination of CRISPR-Cas9 design tools and the effect of target site accessibility on Cas9 activity. Experimental Physiology. 103 (4), 456-460 (2018).
- Jensen, K. T., Fløe, L., et al. Chromatin accessibility and guide sequence secondary structure affect CRISPR-Cas9 gene editing efficiency. FEBS Letters. 591 (13), 1892-1901 (2017).
- Simonetti, F. R., Sobolewski, M. D., et al. Clonally expanded CD4 + T cells can produce infectious HIV-1 in vivo. Proceedings of the National Academy of Sciences. 113 (7), 1883-1888 (2016).
- Chen, H. C., Martinez, J. P., Zorita, E., Meyerhans, A., Filion, G. J. Position effects influence HIV latency reversal. Nature Structural and Molecular Biology. 24 (1), 47-54 (2017).
Перепечатки и разрешения
Запросить разрешение на использование текста или рисунков этого JoVE статьи
Запросить разрешениеThis article has been published
Video Coming Soon
Авторские права © 2025 MyJoVE Corporation. Все права защищены