
Method Article
小脳スライス培養におけるex vivo髄鞘形成および再髄鞘形成は、発生および疾患関連操作の定量的モデルとして
要約
提示されているのは、マウス小脳スライス培養物を使用した脱髄および再髄鞘形成のex vivo定量モデルのプロトコルです。この方法は、in vitroシステムの化学的、遺伝的、および環境的適合性を維持しながら、無傷の組織にCNS細胞タイプを完全に補完するin vivoモデルを厳密に再現します。
要約
in vitroおよびin vivoでの髄鞘形成の研究には、多くの課題があります。オリゴデンドロサイト前駆細胞(OPC)のin vitroでの分化は、スケーラブルではありますが、軸索髄鞘形成を再現しません。OPC-ニューロン共培養およびOPCファイバー培養は、in vitro髄鞘形成の検査を可能にしますが、アストロサイトやミクログリアなど、in vivoに存在する追加の細胞タイプを欠いています。しかし、in vivoマウスモデルは、化学的、環境的、遺伝子的操作にあまり適しておらず、はるかに労働集約的です。ここでは、1)発生性髄鞘形成の研究、2)脱髄と再髄鞘形成のモデル化、3)トランスレーショナル研究の実施に役立つex vivoマウス小脳スライス培養(CSC)定量システムについて説明します。小脳と後脳の矢状切片は、出生後(P)0〜2マウスから単離され、その後、12日間生体外で骨髄を形成します。この期間中、スライスは、発生性髄鞘形成への影響をテストするための化合物の追加など、さまざまな方法で操作できます。さらに、組織を電子顕微鏡で固定して、ミエリンの超微細構造と圧縮を評価することができます。疾患をモデル化するために、CSCを急性低酸素症にさらして髄鞘形成不全を誘発することができます。これらの外植片における脱髄は、リゾレシチンによっても誘発される可能性があり、これにより再髄鞘形成を促進する因子の同定が可能になります。化学的および環境的修飾の他に、CSCはトランスジェニックマウスから単離することができ、Ad-Creアデノウイルスおよびタモキシフェンによって誘導される遺伝子操作に反応します。したがって、小脳スライス培養物は、髄鞘形成を反復するための迅速で再現性があり、定量化可能なモデルです。
概要
軸索の髄鞘形成は、活動電位の迅速な伝播、塩性伝導1として知られるメカニズムを可能にします。ミエリンの重要性は、多発性硬化症(MS)などの脱髄疾患によって強調されており、これには視力喪失、認知障害、麻痺など、さまざまな衰弱症状が含まれます。多発性硬化症の治療法はなく、現在の治療法は末梢免疫細胞を標的とすることで疾患の進行を制限することに焦点を当てています。多発性硬化症および関連疾患の障害は、再髄鞘形成の失敗と進行性の神経変性によって引き起こされると考えられています。特に、進行性のMSでは、脱髄、萎縮、および軸索の喪失が観察されます2,3。したがって、再髄鞘形成の促進は、現在の治療と並行して実施され、追加の治療効果をもたらす可能性のある有望な戦略を表しています。
中枢神経系(CNS)では、髄鞘形成はオリゴデンドロサイトとして知られる特殊なグリア細胞によって行われます。オリゴデンドロサイト前駆細胞(OPC)は、軸索に接触するプロセスの成長、形態学的複雑さの増加、ミエリン膜の拡張、そして最後にミエリン鞘の圧縮4など、一連の高度に調整されたステップを通じて、成熟した有髄オリゴデンドロサイトに分化します。したがって、オリゴデンドロサイトとニューロンの間の相互作用は非常に密接です。ニューロンとオリゴデンドロサイトとの間の相互相互作用も、CNS2の健康と維持に必要です。軸索活動は髄鞘形成を刺激する役割を果たし、グリア神経栄養因子はニューロンの完全性をサポートします。CNSにおけるグリア-グリアクロストークの重要性もますます認識されるようになってきています5,6,7,8。例えば、アストロサイト因子は、OPCの分化や白質路の維持に影響を与える可能性があります。ミクログリアは、OPCの分化を調節する役割も果たすだけでなく、再髄鞘形成プロセスの重要なステップであるミエリン破片の除去にも役割を果たします。細胞自律性因子を同定し、脱髄および再髄鞘形成における他のCNS細胞タイプの影響を理解することは、脱髄および髄鞘形成不全疾患の治療法を開発する上で非常に貴重です。
ここでは、マウス小脳スライス培養物(CSC)を使用して、無傷の中枢神経系組織の操作と定量を可能にするex vivoシステムについて説明します。CSCを使用すると、免疫染色や電子顕微鏡法11,12,13など、in vivo研究で伝統的に使用されてきた方法を用いて、リゾレシチン9,10による脱髄誘導後の髄鞘形成、または再髄鞘形成を測定することができます。リゾレシチンは、ミエリンとオリゴデンドロサイトを急速に失わせる膜破壊化学物質です。注意すべき潜在的な注意点は、リゾレシチンが病変領域に近い他の細胞タイプの減少ももたらす可能性があることです。しかし、in vivo実験とは異なり、小脳の矢状切片は、化合物を添加することで容易に操作したり、Ad-Creアデノウイルスを使用して遺伝子組み換えしたりすることができます。この方法はまた、トランスジェニックマウスまたは低酸素症11,12,13,14などの環境侮辱を受けたマウスからの単離された組織の操作も可能にする。したがって、CSCモデルは、オリゴデンドロサイトの機能に対するさまざまなCNS細胞タイプの寄与を統合しながら、発生性髄鞘形成の研究、疾患のモデリング、および髄鞘形成を促進または阻害する因子の特定を可能にします。
プロトコル
すべての動物実験は、ジェネンテックの動物管理・使用委員会によって承認・承認されています。
1. 解剖用培地と消耗品の準備 (~30–45分)
- 表1に詳述されているように、フィルタースライス培地(SCM)および解剖培地(DM)を調製し、滅菌します。4°Cで保存してください。 髄鞘形成アッセイで試験中の因子がある場合は、使用直前にそれらをSCMに追加してください。
- 1 mLのSCMを6ウェルプレートの各ウェルにピペットで移します。
- 滅菌鉗子を使用して、各ウェルに器官型インサートを配置し、気泡がメンブレンの下に閉じ込められないようにします。7.5% CO2 インキュベーターで37°Cの培地でプレートを温めます。
2. 解剖エリアの準備 (5 ~ 10 分)
- 滅菌済みの層流フードまたはベンチトップで、手順のすべてのステップで適切な無菌技術を使用して手順を実行します。70%エタノールですべての領域をきれいに拭きます。
- ティッシュチョッパーに新しいブレードとシリコンカッティングステージを配置します。切断ステージの下に~300 μLの滅菌水または70%エタノールをピペットで固定し、所定の位置に留まるようにします。
- 綿棒を使用して、ティッシュチョッパーブレードとカッティングステージを70%エタノールで優しく拭きます。ティッシュチョッパーを使用する前に乾かしてください。
- すべての解剖ツールに70%エタノールをスプレーし、解剖前に乾かします。
- 10cmのシャーレを2つ用意します:1つは15〜20mLのDM入り、もう1つは10mLのSCM入りです。氷の上に保存します。解剖しないときは、すべてのメディアとペトリ皿を氷の上に置いてください。
3. 小脳切片培養解剖 (子犬あたり 15 ~ 20 分)
- DMを搭載したペトリ皿を解剖顕微鏡に置きます。
- 出生後P0〜2日目のマウスの子犬を、鋭利なハサミで迅速に斬首することにより安楽死させます。
- DMを入れたシャーレに頭部を切解顕微鏡で入れ、血液を透明にします。
- 細いハサミを使用して、頭蓋骨の基部の各横端に1回ずつ切ります。
- DMで頭を逆さまに置いた状態で、#5/45鉗子を使用して頭蓋骨の下側をしっかりと押し下げ、損傷していない後脳を頭蓋骨の穴に押し込みます。
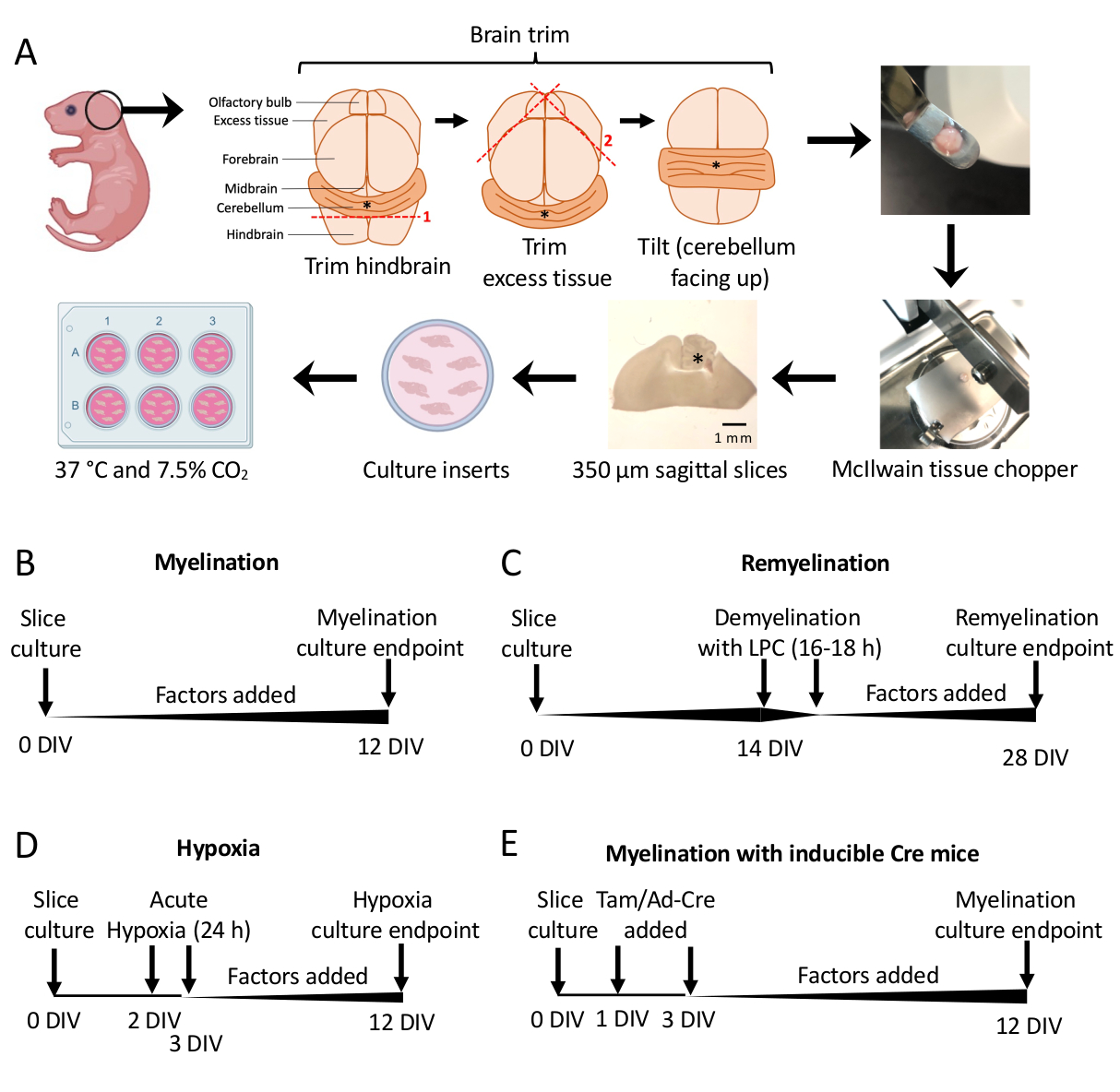
- 解剖顕微鏡下で、2つの#11メスを使用して余分な組織を切り取ります。小脳が後脳の根底にある部分に付着したままであることを確認してください。詳細な図については、 図 1A を参照してください。
- へらを使用して、小脳を組織チョッパーの切断段階に移し、組織を後脳の吻側面と後脳の尾側面に置き、研究者に面します。脳の内側面が組織チョッパーブレードと正確に平行であることを確認してください。
- P200ピペットを使用して、組織の周りの余分な培地を取り除き、まだ湿っているが浮いたり液体に囲まれたりしていないことを確認します。
- DMを含むペトリ皿をSCMを含むシャーレと解剖顕微鏡で交換します。
- ティッシュチョッパーを使用して、組織を350 μmの厚さにスライスします。
注意: 最適な切断条件を確保するためには、ブレードの速度(例:1秒あたり約1カット)と力を最適化する必要があります。まれに、ティッシュがブレードに引っかかることがあります。ティッシュ切片の損失を最小限に抑えるために、親指を電源ボタンに置いたままにして、必要に応じてティッシュチョッパーをすばやくオフにします。 - 100μLのSCMを組織の下に静かにピペットで入れ、スライスが浮くようにします。スライスした組織をへらを使用して、解剖顕微鏡下でSCMを備えたシャーレに移します。
- 綿棒を使用して、ティッシュチョッパーブレードとカッティングステージを70%エタノールで優しく拭きます。
- メス(鈍い面)を使用して、解剖顕微鏡下でスライスを静かに分離します。メスとヘラを使用して、スライスをSCMで6ウェルプレートのメンブレンに移します。
注:最も内側の切片は、多くの場合、最高の髄鞘形成と組織の完全性を持っています。平均して、1つのマウスの脳から4〜6スライスを解剖できます。 - スライスをSCMで37°C、7.5%CO2中でインキュベートします。
4. 文化とメディアの変化 (~15-30分)
- SCMは一日おきに交換してください。
- 新しい6ウェルプレートの各ウェルに新鮮な培地1 mLをピペットで移し、インキュベーターで温めます。滅菌鉗子を使用して、メンブレンを新しいプレートに移し、気泡が下にないことを確認します。プレートをインキュベーターに戻します。
5. 標準的な髄鞘形成と再髄鞘形成
- 標準的な髄鞘形成プロトコル(図1B)。
- セクション3で0〜12日in vitro(DIV)で解剖した培養スライス。髄鞘形成に対する何らかの因子の影響が試験されている場合は、培養時に培地に添加し(0 DIV)、培地を交換するたびに補充してください。
- 12 DIV (ステップ 7.1) で固定し、不完全髄鞘形成 11,12,13 に最適化された時点です。これにより、培地に添加された因子による髄鞘形成の増強効果または阻害効果を検出することができます。100 ng/mL のアクチビン A を添加した場合の代表的な陽性結果を図 2 に示します。
- 再髄鞘形成プロトコル(図1C)
- セクション5.1に記載されている標準的な髄鞘形成プロトコルに従い、スライスが完全に有髄化される時点である14 DIVまでは追加の要素はありません11,12,13。
- 表1に詳述されているように、リゾレシチンストック(125 mg / mL)を調製します。.SCM中の0.5%リゾレシチン1mLを新しい6ウェルプレートの各ウェルにピペットで移し、インキュベーターで温めます。
- メンブレンをリゾレシチンプレートに移し、インキュベーターに一晩16〜18時間置きます。
- 翌日、新しいSCMで新しいプレートを準備し、必要に応じて再髄鞘形成効果について試験する因子を追加します。インキュベーターで温かいプレート。
- 培地を交換する場合は、ウェルの側面にあるメンブレンを軽くたたいて、すべてのリゾレシチン培地が除去されていることを確認します。各メディア交換でテストされる係数を補充します(セクション4)。
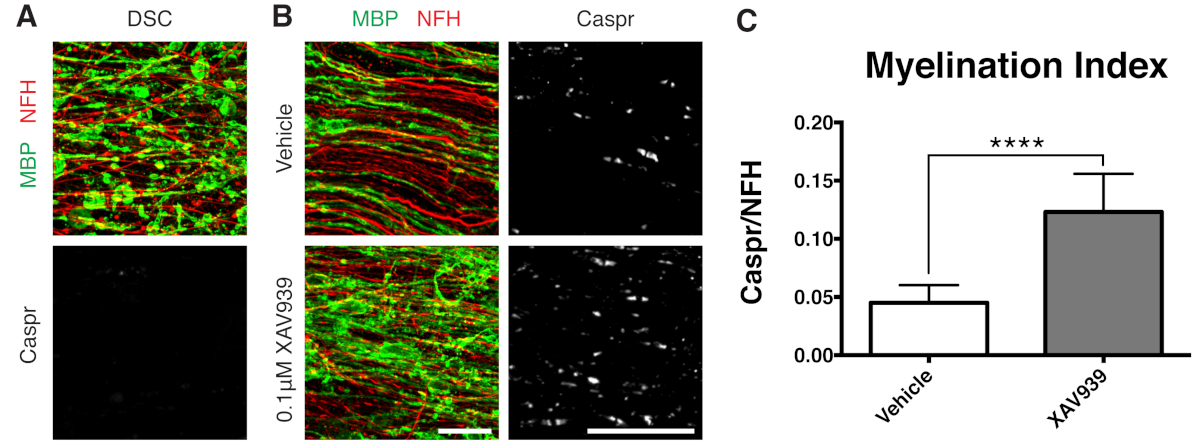
- スライスを不完全な再髄鞘形成に最適化された時点である 28 DIV (ステップ 7.1) で固定します。これにより、培地に付加された因子による再髄鞘形成の増強またはブロッキングの検出が可能になります。0.1 μM XAV939を添加した代表的な陽性結果を 図3に示します。
6. 髄鞘形成および再髄鞘形成プロトコルのバリエーション
- 低酸素症誘発性髄鞘形成不全プロトコル(図1D)11,13。
注:これは、セクション5.1で提示された標準的な髄鞘形成プロトコルのバリエーションとして提示されています。- 因子なしでSCMのプレートスライス。
- スライスプレートを低酸素インキュベーター(2%FiO2)に2〜3 DIVの24時間移します。
- 翌日、新しいSCMで新しいプレートを準備し、必要に応じて髄鞘形成の可能性についてテストされている因子を追加します。インキュベーターで温かいプレート。
- 低酸素状態で培養した後、培地を交換し(セクション4)、培養物を標準的な培養インキュベーター(37°C、7.5%CO2)に戻します。テスト対象のファクターは、その後のメディア交換のたびに補充します (セクション 4)。
- 12 DIV(ステップ7.1)で修正します。
- 遺伝子操作プロトコル(図1E)13
- セクション3のように、トランスジェニックマウスから組織を分離して解剖します。
- 誘導性トランスジェニックマウス系統では、タモキシフェン(100 nM、髄鞘形成またはOPC分化を誘導しない用量)またはAd-Creアデノウイルスを、1 DIVおよび3 DIVで行われる培地交換中にSCMに追加できます。
- タモキシフェンまたはAd-Creアデノウイルスによる遺伝子操作に続いて、セクション4のように隔日で培地交換を続けます(必要に応じて因子を追加します)。
注:1匹のマウスからのすべてのスライスは1つの膜に収まり、解剖後のジェノタイピングが可能になり、トランスジェニックマウスでの実験が容易になります。
7. 組織処理と分析
- メンブレンを4%パラホルムアルデヒドに室温(RT)で1時間穏やかに浸して、スライスを固定します。
- リン酸緩衝生理食塩水(PBS)に浸して、穏やかにすすいでください。次に、PBSの6ウェルプレートに沈めます。
注:プロトコルはここで一時停止できます。固定スライスは、4°CのPBSで最大1か月間保存できます。スライスを保管しない場合は、先に進む前にPBSで2回5分間すすいでください。 - メスを使用してリングからメンブレンを切り取り、6ウェルプレートで染色を進めます。スライスを上(膜面を下にして)に保管し、染色してスライドにマウントしてください。
- ブロック溶液(3%熱不活化ウマ血清、2%ウシ血清アルブミン、およびPBSで希釈した0.25%Triton-X 100)でRTで1時間ブロックします。
注:抗体染色に抗原賦活化が必要な場合(例:CC1/Olig2)、ガラスシャーレで行うことができます。 - プレートをパラフィンフィルムで包み、ブロッキング溶液で希釈した一次抗体で4°Cで一晩インキュベートします。
- ブロッキング溶液で3回洗浄します:1)短時間すすぎ、2)15分間の洗浄、3)1時間の洗浄。短時間のすすぎの場合は、小さなペトリ皿に溶液を入れ、鉗子を使用して膜を静かに沈めてから、新鮮な溶液に移します。長時間の洗浄(15分および1時間)の場合は、メンブレンを6ウェルプレートの溶液に沈め、穏やかなロッカーの上に置きます。
- PBSで1:500に希釈した二次抗体で、室温で2時間、光で覆ってインキュベートします。
- PBSで3回洗ってください:1)短時間のすすぎ、2)15分の洗浄、3)1時間の洗浄。
- DAPI染色が必要な場合は、1:1,000 DAPI溶液に室温で7分間浸漬し、短時間すすぎた後、PBSで15分間洗浄します。
- 二重蒸留水ですすいでください。
- スライスを上に向けて(膜側を下にして)、スライドにメンブレンを取り付けます。
注:マウントされたスライスは4°Cで保存できます。 共焦点顕微鏡でのイメージングは、メンブレンが不透明でイメージングが困難になる前の1週間以内に行うのが最適です。 - 共焦点顕微鏡を用いた画像スライスおよび前述の11,12,13の定量化。
結果
P0-2マウス由来の小脳切片培養物(図1A)を用いて、髄鞘形成を研究し、0-12 DIVからの種々の因子の添加による影響を評価しました(図1B)。再髄鞘形成を研究するために、14 DIVのスライス培養物を最初にリゾレシチンで脱髄し、試験した因子を用いて培養中でさらに14日間再髄鞘化させ(図1C)、その後、再髄鞘形成を定量化しました。髄鞘形成に対する低酸素の影響も、スライス培養物を2%FiO2低酸素インキュベーターに2〜3 DIVの24時間置くことによって研究されました(図1D)。最後に、トランスジェニックマウスに由来するスライス培養物を使用して、遺伝子ノックアウトが髄鞘形成に及ぼす影響を研究しました。このシステムでは、1 DIV および 3 DIV にタモキシフェンまたは Ad-Cre アデノウイルスを添加し、12 DIV で分析用に固定したスライス培養物により、Cre 組換えを誘導しました(図 1E)。組織を固定した後、免疫染色(図2、図3)および電子顕微鏡11,12,13により、脱髄および再髄鞘形成の定量化を行った。
示されている代表的なデータ(図2、図3)は、スライス培養における髄鞘形成と再髄鞘形成のダイナミックレンジを示しています。髄鞘形成指数は、コンパクトミエリンの間接的な読み取りであるパラノダルマーカーであるCasprと、軸索を染色するニューロフィラメントタンパク質H(NFH)の比率によって定量化されました。この髄鞘形成指数は、電子顕微鏡法およびナトリウムチャネル染色12によるコンパクトなミエリンの形成を表すことが確認されている。リゾレシチンによる脱髄9,10は、Caspr陽性パラノードの完全な消失(髄鞘形成指数= 0、図3A)とコンパクトミエリンの喪失をもたらし、これは再髄鞘形成中に回復しました11,12。
髄鞘形成で陽性の結果を示すために、図1Bのタイムラインに従ってスライスをアクチビンAで培養しました。アクチビンAは、オリゴデンドロサイト上のアクチビン受容体を関与させ、オリゴデンドロサイトの分化とミエリンの圧縮を促進します15。代表的なデータによると、髄鞘形成中に100 ng/mLのアクチビンAでスライスを処理すると、髄鞘形成指数が高かったことが示されています(図2A、B)。これと一致して、アクチビンA処理は、CC1陽性成熟オリゴデンドロサイトとOlig2+オリゴデンドロサイト系統細胞の比率によって定量化されるように、OPC分化を促進しました11,13(図2C、D)。
タンキラーゼの低分子阻害剤であるXAV939は、オリゴデンドロサイト11のAxin2レベルの安定化により、髄鞘形成と再髄鞘形成を促進することが示されています。リゾレシチン処理後、断片化されたミエリン塩基性タンパク質(MBP)染色とCaspr陽性パラノードの欠如により、スライス培養物での脱髄が可視化および定量されました(図3A)。再髄鞘形成(15-28 DIV)中に0.1 μM XAV939で処理すると、CasprとNFH染色の比で定量化されるように、ビヒクルコントロールと比較して髄鞘形成指数が有意に増加しました(図3B、C)。したがって、スライス培養モデルを使用して、無傷組織におけるOPCの分化、髄鞘形成、および再髄鞘形成への影響を調べ、定量化することができます。

図1:解剖プロセスの主要なステップとさまざまなスライス培養プロトコルのタイムラインを示す小脳スライス培養の概略図。(A)スライス培養物を生成するステップの描写:P0–2マウスの仔から脳を解剖し、脳をトリミングし(図を参照)、組織チョッパーで脳を切断して350μmの矢状スライスを生成し、スライスを6ウェルディッシュの器官型培養インサートに配置し、37°Cおよび7.5%CO2でインキュベートします。*は小脳を示します。(B)髄鞘スライス培養のタイムライン。スライス培養物を12日間インキュベートした後、分析のために固定しました。(C)スライス培養物の再ミエリン化のタイムライン。スライスを14 DIVでリゾレシチンで16〜18時間処理して完全な脱髄を誘導し、分析のために固定する前に28 DIVまで再髄鞘化させました。(D)低酸素性傷害後の髄鞘形成のタイムライン。スライスは、2〜3 DIVの間で24時間急性低酸素性傷害にさらされ、髄鞘形成不全を引き起こしました。(E)遺伝子改変マウスからのCSCのタイムライン。タモキシフェン(Tam、100nM)またはAd-Creウイルスを1 DIVおよび3 DIVで添加して遺伝子改変を誘導し、スライスを12 DIVで分析しました。この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図2:アクチビンAが髄鞘形成とOPCの分化を促進することを示す代表的なデータ。 (A)100 ng/mLアクチビンA処理による髄鞘形成(MBP)およびパラノード(Caspr)の増加は、スライス培養物の免疫染色によって示されます。(B)髄鞘形成指数の定量化(Caspr+パラノードで染色された面積とNFH+軸索で染色された面積の比率)。(C)OPC分化の定量化(CC1+オリゴデンドロサイトとOlig2+オリゴデンドロサイト系統細胞の比率)。(D)CC1/Olig2染色の代表的な画像。表示されている値は、平均 + 標準偏差です。P<0.0001;対応のない T 検定。スケールバー:A = 25 μm、D = 50 μm。 この 図の拡大版を表示するには、ここをクリックしてください。
{kind=link}

図3:XAV939が再髄鞘形成を促進することを示す代表的なデータ。(A)断片化されたMBP染色による脱髄と、リゾレシチンで処理したスライス培養物におけるCasprパラノードの欠如を示す画像。(B)スライス培養物の免疫染色によって示された、リゾレシチン誘発性脱髄後の0.1 μM XAV939処理による再髄鞘形成(MBP)およびパラノード(Caspr)の増加。(C)髄鞘形成指数の定量化(Caspr+パラノードで染色された面積とNFH+軸索で染色された面積の比率)。表示されている値は、平均 + 標準偏差です。P<0.0001;対応のない T 検定。スケールバー = 25 μm。 この図の拡大版を表示するには、ここをクリックしてください。
{kind=link}
| スライス培地(SCM) | |
| 容積 | 試薬 |
| 100ミリリットル | 最小必須培地(MEM)、HEPES、グルタミンなし |
| 50ミリリットル | 熱不活化ウマ血清 |
| 50ミリリットル | アールのバランスの取れた塩溶液 |
| 2ミリリットル | ペニシリン-ストレプトマイシン 10,000 U/mL |
| 2ミリリットル | GlutaMAXサプリメント |
| 2888μL | 45%グルコース溶液 |
| 1 mLの | 菌類ゾーン |
| 0.22μmのフィルターで滅菌フィルターを洗浄し、4°Cの冷蔵庫で最大2週間保存します。 | |
| 解剖メディア(DM) | |
| 容積 | 試薬 |
| 100ミリリットル | MEM、HEPES、アールの塩と |
| 1 mLの | ペニシリン-ストレプトマイシン 10,000 U/mL |
| 0.22μmのフィルターで滅菌フィルターを敷き詰め、4°Cの冷蔵庫で最大2ヶ月間保存します。 | |
| リゾレシチンストック(125 mg / mL) | |
| 量 | 試薬 |
| 100ミリグラム | リゾレシチン |
| 0.8ミリリットル | 滅菌PBS |
| 100mgのリゾレシチンを0.8mLの滅菌PBSに溶解します。.80 μLのストック溶液を-20°Cで保存します。 | |
| 使用前に、80 μLのアリコートを解凍し、20 mLのSCM(SCM中の0.5%リゾレシチン)に溶解し、37°C、7.5%CO2インキュベーターで温めてください。必要に応じて、ボルテックスしてリゾレシチンをSCMに溶解します。 | |
表1:スライス培養培地(SCM)、解剖培地(DM)、およびリゾレシチン溶液を作成するためのコンポーネントとプロトコルのリスト。
ディスカッション
このプロトコルは、in vitroモデルのシンプルさでin vivo細胞組成を再現する器官型小脳スライス培養モデルについて説明しています。このプロトコルは、人間の病状のより代表的なモデルになるためにさらに開発される可能性があります。CSCは、力による組織損傷、ミエリン特異的抗体による損傷、末梢免疫細胞の添加によるMSのオリゴデンドロサイト損傷など、疾患特異的な損傷のモデルとして開発できる可能性があります。このプロトコールは、脳および脊髄16,17の他の部分から解剖された組織に対しても最適化することができる。このプロトコルは髄鞘形成の組織学的定量に焦点を当てていますが、CSCはさまざまな実験エンドポイントを調べるための便利なモデルです。これらの培養物は、シングルセルRNAシーケンシングによるさまざまな細胞タイプの特性評価、電子顕微鏡による軸索およびミエリンシースの超微細構造の評価、タイムラプスイメージングによるOPCダイナミクスの研究に使用できます。免疫染色は、トランスジェニックマウスレポーターライン18,19を使用してバイパスすることもできる。最後に、マウスの子犬の異なる年齢、培養中の時間、または切片の厚さを使用して、CSCを異なる疾患メカニズムをモデル化するように適応させることができる10,17,18,19,20,23。
スライス培養は、髄鞘形成および再髄鞘形成に影響を与える因子の試験および定量化には理想的ですが、特定の状況におけるCNSの発現には制限があります。P 0-2脳に由来するCSCは、発達の初期段階にある脳に由来し、老化または神経変性脳との類似性は限られています。アルツハイマー病21や統合失調症22などの中枢神経系疾患におけるミエリンの重要性に対する認識が高まっていることを考えると、髄鞘形成を定量化し特徴付ける成人モデルまたは老化モデルが必要とされている。他のプロトコルは、まだ発達段階にあるものの、古いげっ歯類から組織を単離する発表されています10,18,19,23。さらに、脳の脊髄や他の白質路と比較して、小脳のオリゴデンドロサイトとOPCには内因性および外因性の違いがあるかもしれません。また、CSCは、細胞ベースのin vitroシステムよりも、大規模なCRISPRや低分子スクリーニングには適していません。最初の組織スライスにより、脳に常在する自然免疫細胞(すなわち、ミクログリアおよびアストロサイト)の活性化もあり、これはシステム24にとって重要な注意点である。最後に、末梢免疫細胞はMSの病理学において大きな役割を果たします25。CSCは、これらの細胞が外因的に追加されない限り末梢細胞を欠いているため、炎症性中枢神経系環境の理想的なモデルではありません。
このプロトコルの解剖部分は、おそらく最も重要な部分です。解剖手順は、組織やその下にある構造を傷つけないように、細心の注意を払って行う必要があります。ティッシュチョッパーブレードに沿って脳を適切に位置合わせすることで、組織や細胞へのダメージを最小限に抑えることができます。さらに、スライスの慎重な解剖と分離、およびそれらを培養のための膜への移動が重要です。このプロトコルで概説されている時間枠は、記載されている研究に対して最適化されていますが、さまざまな研究で最適な結果を得るために調整が必要な場合があります。このプロトコルは、発達性髄鞘形成とミエリン修復を研究したい人に役立ちます。
注目すべきは、多発性硬化症の治療法がないことです。現在の治療法は適応免疫系を弱めるのに非常に効果的ですが、現在の治療法で進行を止めることはできません。再髄鞘形成の失敗とそれに続く神経変性が、MS2の進行の根底にあると考えられています。慢性多発性硬化症病変におけるOPCの存在は、ミエリン修復の失敗がOPC分化の停止によるものである可能性があることを示唆しています。CSCの使用は、MSの進行を逆転させ、機能を回復させるのに役立つ可能性のあるミエリン修復療法の発見の道を開きます。ミエリン修復療法は、脱髄路が自発運動機能を阻害する脊髄損傷患者の回復にも役立つ可能性があります26。したがって、CSCの重要性は、in vivo動物モデルと比較して比較的高いスループットで哺乳類の脱髄および再髄鞘形成に影響を与える因子を特定するための適合性にあります。多くの研究で、分化に影響を与える化合物のスクリーニングに一次OPCが使用されていますが、一次OPCの生成と単離には、面倒で逐次的な免疫パニングが必要です27。さらに、細胞ベースのアッセイは、in vivoに存在する細胞タイプの多様性と相互作用を再現しません。マウスの子犬からCSCを生成することは、高価な機器や消耗品を必要としない、髄鞘形成と再髄鞘形成を研究するための迅速で費用対効果の高いモデルです。したがって、小脳スライス培養物は、生体外で髄鞘形成を再現するための非常に貴重な定量的モデルであり、創薬と基礎科学研究を可能にします。
開示事項
筆者は、ロシュ・グループ傘下のジェネンテック社の社員です。
謝辞
著者は、この記事に対する建設的なコメントと意見を提供してくれた Yun-An Shen、Roxanne Kyauk、Chris Bohlen に感謝します。さらに、著者らは、以前に公開された11,12,13の関連方法論の初期開発におけるCharles ffrench-Constant、Andrew Jarjour、Veronique Miron、およびDavid Rowitchからの貢献を認めています。
資料
| Name | Company | Catalog Number | Comments |
| #5/45 Forceps | Fine Science Tools | 11251-35 | Dissection tools |
| 32% Paraformaldehyde | Electron Microscopy Sciences | 50-980-495 | Fixative for staining |
| 45% glucose solution | Sigma | G8769 | Slice culture media component |
| 6-well tissue culture plates | Corning | 3516 | Plates for cultures |
| Alexa Fluor Goat anti-chicken 647 | Thermo Fisher Scientific | A21449 | Secondary staining antibody |
| Alexa Fluor Goat anti-mouse 647 | Thermo Fisher Scientific | A21236 | Secondary staining antibody |
| Alexa Fluor Goat anti-rabbit 555 | Thermo Fisher Scientific | A11006 | Secondary staining antibody |
| Alexa Fluor Goat anti-rat 488 | Thermo Fisher Scientific | A21428 | Secondary staining antibody |
| Blades, Double-edge, PTFE coated Stainless steel | Ted Pella | 121-6 | Chopping blades |
| Bovine Serum Albumin (BSA) | Sigma | A9418 | Blocking solution component |
| Chicken anti-NFH antibody | Encor Biotech | CPCA-NF-H | Staining antibody |
| Confocal Microscope | Zeiss | LSM780 | Confocal for imaging |
| Corning 250 mL vacuum filter/storage bottle system, 0.22 µm pore | Corning | 431096 | For media preparation |
| Earl's balanced salt solution | Sigma | E2888 | Slice culture media component |
| Feather surgical blade | Feather | 2976#11 | Dissection tools |
| Fungizone | Thermo Fisher Scientific | 15290-18 | Dissection media component |
| GlutaMAX supplement | Thermo Fisher Scientific | 35050-061 | Slice culture media component |
| Heat-inactivated horse serum | Thermo Fisher Scientific | 26050-88 | Slice culture media component |
| Lysolecithin | Sigma | L4129 | |
| Lysophosphatidylcholine from egg yolk | Sigma | L4129 | To induce demyelination |
| McIlwain Tissue Chopper | Ted Pella | 10180 | Tissue chopper |
| MEM, Hepes, no glutamine | Thermo Fisher Scientific | 12360-038 | Slice culture media component |
| MEM, Hepes, with Earle's salts | Sigma | M7278 | Dissection media component |
| Metal spatula | Fisher Scientific | 470149-442 | Dissection tools - bend flat end at angle for use |
| Millicell Culture Plate Insert 30mm Organotypic PTFE | Fisher | PICM0RG50 | Culture insert |
| Mouse anti-CC1 antibody | Millipore | OP80-100UG | Staining antibody |
| Noyes spring scissors | Fine Science Tools | 15012-12 | Dissection tools |
| PBS pH 7.4 | Gibco | 10010023 | For dilution of fixative |
| Penicillin-streptomycin 10,000U/mL | Thermo Fisher Scientific | 15140-122 | Slice culture media component |
| Rabbit anti-Caspr antibody | Abcam | ab34151 | Staining antibody |
| Rabbit anti-Olig2 antibody | Millipore | AB9610 | Staining antibody |
| Rat anti-MBP antibody | Serotec | MCA409S | Staining antibody |
| Scalpel handle #3 | Fine Science Tools | 10003-12 | Dissection tools |
| Silicone rubber foam sheet | MSC Industrial Direct Co | 31939176 | Cutting base |
| Stereo Microscope | Olympus Life Sciences | SZ61 | Microscope for dissection |
| Student surgical scissors | Fine Science Tools | 91401-12 | Dissection tools |
参考文献
- Purves, D., Augustine, G. J., Fitzpatrick, D. Increased Conduction Velocity as a Result of Myelination. Neuroscience 2nd edition. , (2001).
- Dutta, R., Trapp, B. D. Mechanisms of neuronal dysfunction and degeneration in multiple sclerosis. Progress in Neurobiology. 93 (1), 1-12 (2011).
- Trapp, B. D., Nave, K. A. Multiple sclerosis: an immune or neurodegenerative disorder?. Annual Review of Neuroscience. 31, 247-269 (2008).
- Bradl, M., Lassmann, H. Oligodendrocytes: biology and pathology. Acta Neuropathologica. 119 (1), 37-53 (2010).
- Domingues, H. S., Portugal, C. C., Socodato, R., Relvas, J. B. Oligodendrocyte, Astrocyte, and Microglia Crosstalk in Myelin Development, Damage, and Repair. Frontiers in Cell and Developmental Biology. 4, 71 (2016).
- Nave, K. A., Werner, H. B. Myelination of the nervous system: mechanisms and functions. Annual Review of Cell and Developmental Biology. 30, 503-533 (2014).
- Franklin, R. J. M., Ffrench-Constant, C. Regenerating CNS myelin – from mechanisms to experimental medicines. Nature Reviews Neuroscience. 18 (12), 753-769 (2017).
- Molina-Gonzalez, I., Miron, V. E. Astrocytes in myelination and remyelination. Neuroscience Letters. 713, 134532 (2019).
- Keough, M. B., Jensen, S. K., Yong, V. W. Experimental demyelination and remyelination of murine spinal cord by focal injection of lysolecithin. Journal of Visualized Experiments. (97), e52679 (2015).
- Birgbauer, E., Rao, T. S., Webb, M. Lysolecithin induces demyelination in vitro in a cerebellar slice culture system. Journal of Neuroscience Research. 78, 157-166 (2004).
- Fancy, S. P., et al. Axin2 as regulatory and therapeutic target in newborn brain injury and remyelination. Nature Neuroscience. 14 (8), 1009-1016 (2011).
- Yuen, T. J., et al. Identification of endothelin-2 as an inflammatory factor that promotes CNS remyelination. Brain. 136, 1035-1047 (2013).
- Yuen, T. J., et al. Oligodendrocyte-encoded HIF function couples postnatal myelination and white matter angiogenesis. Cell. 158 (2), 383-396 (2014).
- Stokes, C. E., Murphy, D., Paton, J. F., Kasparov, S. Dynamics of a transgene expression in acute rat brain slices transfected with adenoviral vectors. Experimental Physiology. 88, 459-466 (2003).
- Dillenburg, A., et al. Activin receptors regulate the oligodendrocyte lineage in health and disease. Acta Neuropathologica. 135 (6), 887-906 (2018).
- Dean, J. M., et al. An organotypic slice culture model of chronic white matter injury with maturation arrest of oligodendrocyte progenitors. Molecular Neurodegeneration. 6, 46 (2011).
- Zhang, H., Jarjour, A. A., Boyd, A., Williams, A. Central nervous system remyelination in culture--a tool for multiple sclerosis research. Experimental Neurology. 230 (1), 138-148 (2011).
- Hill, R. A., Medved, J., Patel, K. D., Nishiyama, A. Organotypic slice cultures to study oligodendrocyte dynamics and myelination. Journal of Visualized Experiments. (90), e51835 (2014).
- Sherafat, A., Hill, R. A., Nishiyama, A. Organotypic Slice Cultures to Study Oligodendrocyte Proliferation, Fate, and Myelination. Molecular Biology (Clifton, N.J.). 1791, 145-156 (2018).
- Croft, C. L., Noble, W. Preparation of organotypic brain slice cultures for the study of Alzheimer's disease. F1000Research. 7, 592 (2018).
- Nasrabady, S. E., Rizvi, B., Goldman, J. E., Brickman, A. M. White matter changes in Alzheimer's disease: a focus on myelin and oligodendrocytes. Acta Neuropathologica Communications. 6 (1), 22 (2019).
- Flynn, S. W., et al. Abnormalities of myelination in schizophrenia detected in vivo with MRI, and post-mortem with analysis of oligodendrocyte proteins. Molecular Psychiatry. 38, 811-820 (2003).
- Thetiot, M., Ronzano, R., Aigrot, M. S., Lubetzki, C., Desmazières, A. Preparation and Immunostaining of Myelinating Organotypic Cerebellar Slice Cultures. Journal of Visualized Experiments. (145), e59163 (2019).
- Gerlach, J., Donkels, C., Münzner, G., Haas, C. A. Persistent Gliosis Interferes with Neurogenesis in Organotypic Hippocampal Slice Cultures. Frontiers in Cellular Neuroscience. 10, 131 (2016).
- Reich, D. S., Lucchinette, C. F., Calabresi, P. A. Multiple Sclerosis. New England Journal of Medicine. 378 (2), 169-180 (2018).
- Jeffery, N. D., Blakemore, W. F. Locomotor deficits induced by experimental spinal cord demyelination are abolished by spontaneous remyelination. Brain. 120, 27-37 (1997).
- Emery, B., Dugas, J. C. Purification of oligodendrocyte lineage cells from mouse cortices by immunopanning. Cold Spring Harbor Protocols. , 854-868 (2013).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved