
Method Article
電子顕微鏡での生体試料の良好な超構造保存のためのサンドイッチ凍結装置を用いた急速凍結
要約
ここでは、細菌、酵母、培養細胞、単離細胞、動物およびヒト組織、ウイルスを含む生物学的標本の迅速凍結にサンドイッチ凍結装置を使用する方法を示す。また、急速凍結後の超薄切片用試料の調製方法もご紹介します。
要約
細胞や組織の超構造を観察するために化学固定が使用されています。しかし、この方法は細胞の超構造を十分に維持しません。通常、細胞内容物のアーティファクトと抽出が観察される。急速な凍結は細胞構造の保存のためのよりよい代替である。生きた酵母や細菌のサンドイッチ凍結後に凍結置換が続き、細胞の絶妙な自然な超構造を観察するために使用されています。近年、グルタルアルデヒド固定培養細胞やヒト組織のサンドイッチ凍結は、細胞や組織の超構造を明らかにするためにも使用されている。
これらの研究はこれまで手作りのサンドイッチ凍結装置で行われており、他の研究室での研究への応用は限られている。新しいサンドイッチ冷凍装置が最近製造され、現在市販されています。本論文は、細菌、酵母、培養細胞、単離細胞、動物およびヒト組織、ウイルスを含む生物学的標本の迅速凍結にサンドイッチ凍結装置を使用する方法を示す。また、急速凍結後の超薄切片用試料の調製、凍結置換、樹脂埋め込み、ブロックのトリミング、超薄切片の切断、切片の回収、染色、および支持フィルムによるグリッドのカバーの手順も示されています。
概要
電子顕微鏡は、細胞の超構造を研究するための強力なツールです。従来の脱水処理による化学固定は、細胞や組織の超構造を観察するために用いられてきた。しかし、この方法は細胞の超構造を十分に保存しておらず、アーチファクトおよび細胞内容物の抽出は通常観察される。細胞および組織の急速凍結および凍結置換は、細胞構造の保存のためのより良い選択肢である。
凍結細胞を急速に凍結する方法は3つ:1)突当凍結は、プロパンなどの冷却されたクライオゲンに試料を突っ込んで行われ、1950年代初期から使用された。2)冷たい金属ブロック凍結は、液体窒素または液体ヘリウム3、4で冷却された金属ブロックに細胞および組織を急速に叩くことによって行われる。そして3)高圧凍結は、高圧5、6、7の下で液体窒素で細胞および組織を凍結することによって行われる。
サンドイッチ凍結は、2つの銅ディスクの間に薄い生物材料を挟み、液体プロパン8、9、10に突っ込んで急速に凍結することによって行われるプランジ凍結の一種である。この方法では、極めて薄い試料(数マイクロメートル厚)が、両側からの熱伝導性のよい金属を用いて、凍結剤で急速に冷却される。これにより、試料から熱を効果的に除去し、氷晶損傷を受けずに細胞を安定的に凍結することが可能となる。サンドイッチ凍結は、生きている酵母および細菌細胞の凍結置換に続いて、細胞10、11、12、13、14、15、16の自然な超構造を明らかにする。
近年、グルタルアルデヒド固定微生物17、18、19、20、21、22、23、24、培養細胞25、26、27、ヒト細胞および組織1、28の透明な細胞画像を保存するのに有用であることが分かった。.これらの研究は手作りのサンドイッチ凍結装置29を用いて行われており、他の研究室での他の研究への応用は限られているが、新しいサンドイッチ凍結装置(SFD)は28で製造され、現在市販されている。
本論文は、細菌、酵母、培養細胞、単離細胞、動物およびヒト組織、ウイルスを含む生物学的標本の迅速凍結にSFDを使用する方法を示す。また、急速凍結後の超薄切片用の試料の調製、凍結置換、樹脂埋め込み、ブロックのトリミング、超薄切片の切断、切片の回収、染色、および支持フィルム付きグリッドのカバーの手順も示されています。
プロトコル
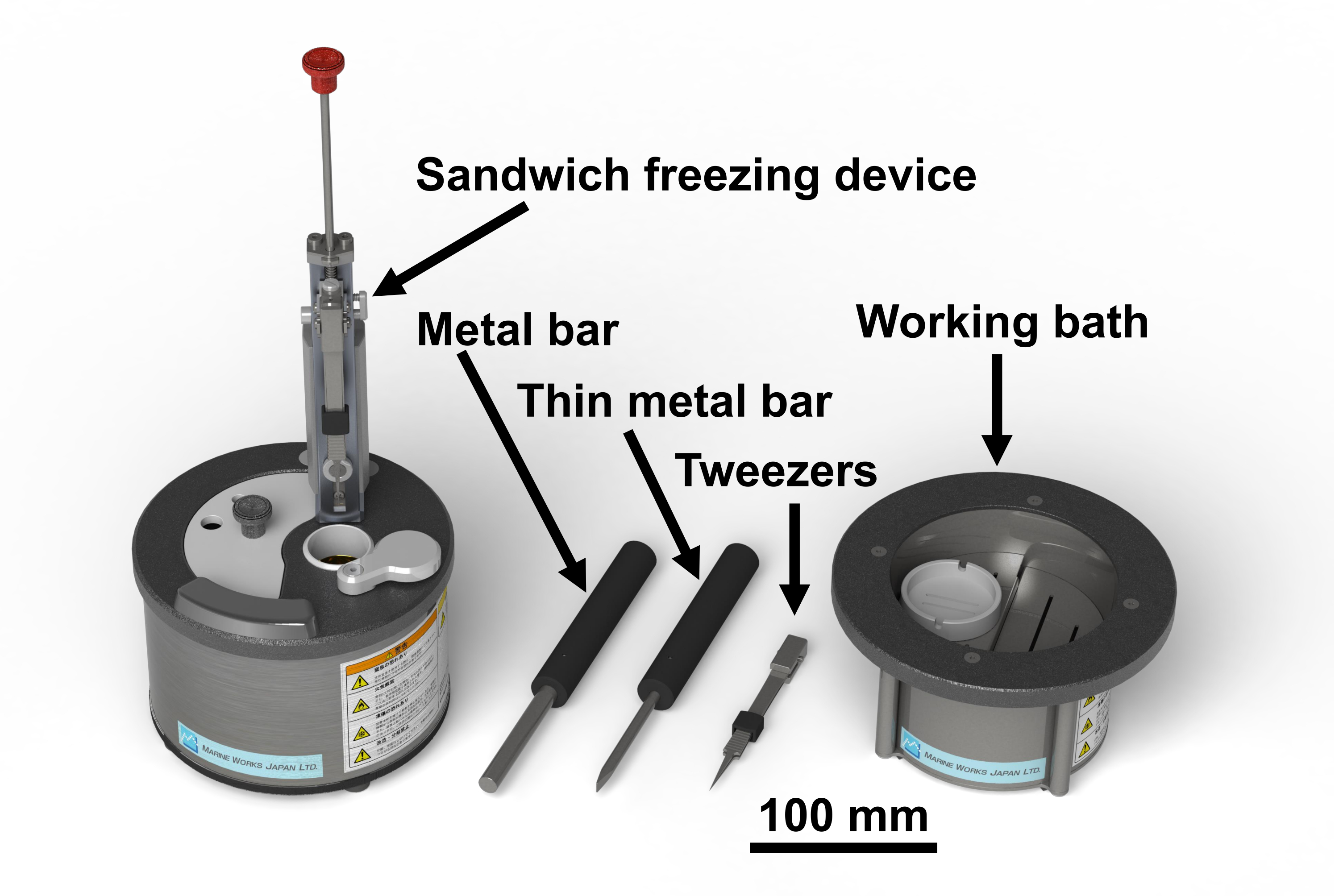
注:研究のヒトサンプルの議定書は、千葉大学医学研究科生物医学研究倫理委員会(3085)によって承認されました。四酸化オスミウムは危険な化学物質です。それはヒュームフードに手袋を着用して扱われるべきです。図1はサンドイッチ凍結装置と必要な工具28を示す。図2は、サンドイッチ凍結実験を行うために必要な材料を示す。ガラスバイアルは、オスミウム四酸化オスミウムを含むアセトンで満たされ、使用するまで-80°Cに保たれます(図2B)。銅ディスクは直径3mmで、穴がなく、片面に文字があり、市販されている(図2C)。
1. 凍結置換のための細胞懸濁液の急速凍結
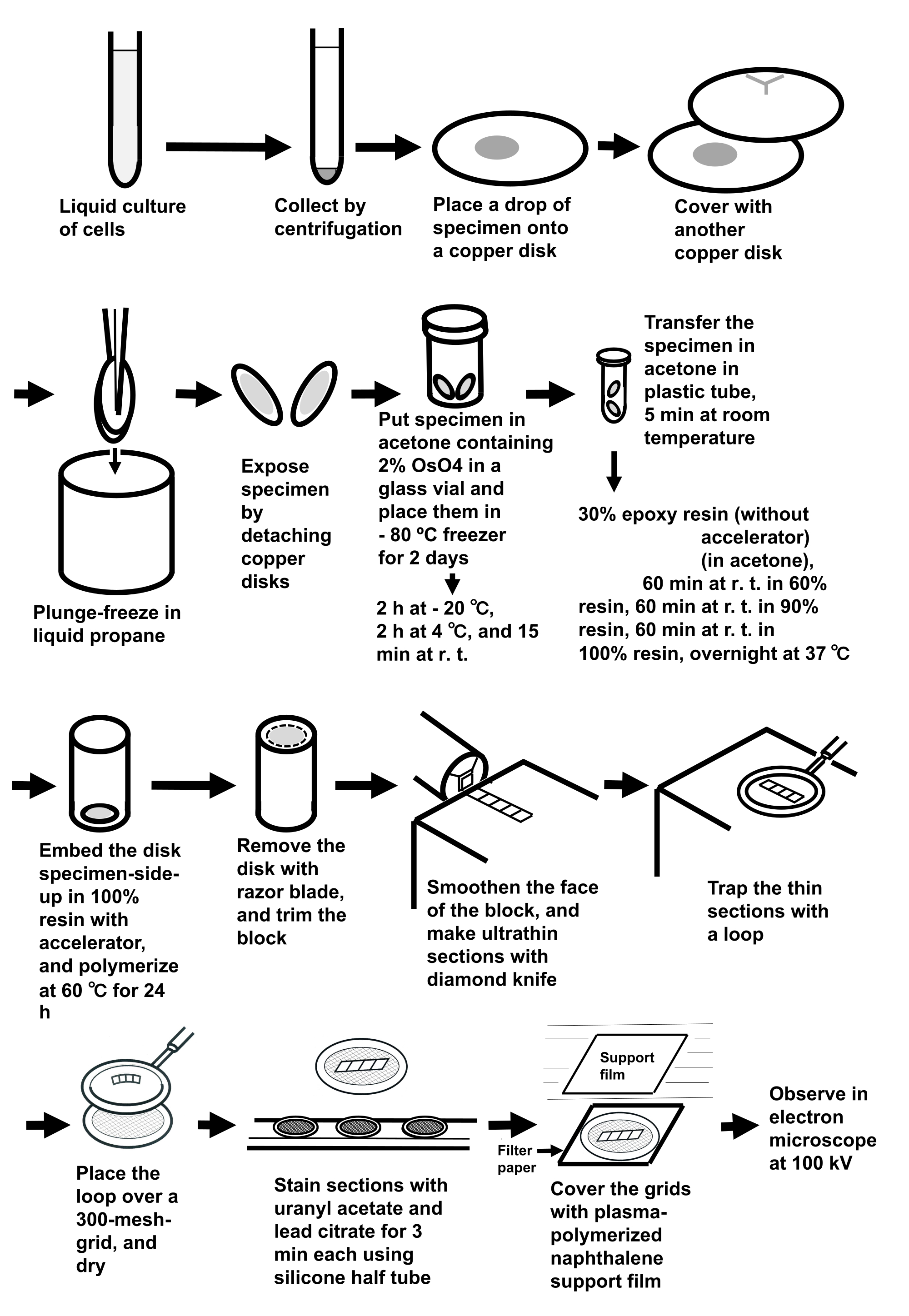
注: 手順全体を図 3に示します。
- 細胞
- 細菌、酵母(図2A)、培養細胞、および分離細胞の細胞懸濁液を使用してサンドイッチ凍結を行う。
注:リビングとグルタルアルデヒド固定細胞の両方を使用することができます28.
- 細菌、酵母(図2A)、培養細胞、および分離細胞の細胞懸濁液を使用してサンドイッチ凍結を行う。
- 液体プロパンの調製
注:液体窒素を扱う際には、手袋とゴーグルを使用してください。プロパンは爆発的であるため、同じ部屋で火災を使用しないように注意する必要があり、窓を開けておく必要があります。- SFDの液体窒素容器を液体窒素で満たします(図4A)。液体プロパン容器を、微細なノズルを用いてプロパンガスを導入して液体プロパンを充填する(図4B,C)。冷却された金属棒を使用してプロパンの凝固を加速する(図1および図4D、E)。
- 銅ディスクの準備
- ノーレター側を上にしてスライドガラスに銅ディスクを配置し(図2D)、10Paでのグロー放電で処理し、400ボルト、1mA(図4F、G)でイオンスパッタ装置30を用いてディスク表面親水性にする。
- 細胞懸濁液のサンドイッチとプランジ・フリーズ
- セル懸濁液を2mL遠心管(図5A)に移し、遠心分離機を室温で10sの2,900×g(図5B,C)に移す。 上清を取り除き、ペレットを吊り下げ、厚い懸濁液を得る(説明参照)。
- 銅ディスク上に少量のセル懸濁液(約0.02 μL)を置き(図5D-F)、別の銅ディスク(図5G)で覆い、ピンセットでディスクを拾います(図5H)。
注:細胞懸濁液の約0.02μLを測定するには、体型顕微鏡で懸濁液の0.1μL滴を観察し、この体積の1/5分 の1の液滴に分けます。 - 薄い金属棒で固体プロパンの中央に井戸を作る(図5I)。ピンセットをSFDにセットし、装置のインジェクションボタンを押して急速にフリーズします(図5J、K)。
注:試料を乾燥させず、2つのディスクを完全に互いに覆わないよう注意してください(そうでない場合は、次のステップで取り外すのが非常に困難になります)。
- 動物およびヒト組織のサンドイッチとプランジ凍結
- 動物およびヒト組織(0.5mm x 0.5mm x 1.5mm)を2.5%グルタルアルデヒド-0.1 Mリン酸緩衝液(pH 7.4)に固定(図6A、B)に使用する。それらを立体顕微鏡下でカミソリの刃で0.1〜0.2-mmの厚いセクションにスライスします(図6C,D)。
- 銅ディスク上に小さな滴(約0.02 μL)のグルタルアルデヒド溶液を配置します(図6E、F)。次に、ピンセットを使用して、銅ディスク上のグルタルアルデヒドに組織片を配置し(図6G,H)、別の銅ディスクで覆います(図6I-K)。
- セクション1.4で説明されているように、SFDの溶融プロパン内の組織とディスクを迅速に凍結します (図5J,K)。
注意:グルタルアルデヒドは危険な化学物質であるため、ヒュームフードに手袋を着用して処理する必要があります。組織は、銅ディスク上に置かれたときに緩衝液で洗浄すべきではないが、グルタルアルデヒドは凍結防止効果を有するのでグルタルアルデヒド溶液に浸漬し続ける。
- 動物およびヒト組織(0.5mm x 0.5mm x 1.5mm)を2.5%グルタルアルデヒド-0.1 Mリン酸緩衝液(pH 7.4)に固定(図6A、B)に使用する。それらを立体顕微鏡下でカミソリの刃で0.1〜0.2-mmの厚いセクションにスライスします(図6C,D)。
- オスミウムアセトンによる凍結置換
- 作業浴場の液体窒素にディスクを移します(図7A,B)。液体窒素で冷却したピンセットのペアを使用して、互いにディスクを取り外して標本を露出する(図7C-E)。
- 細胞を含むディスクをガラスバイアル(図7F)に入れ、2%オスミウム四酸化スメキシドを含む1mLのアセトン(図2B)を液体窒素に入れ、固化させた(図4H)。
- ディスクを深い冷凍庫に移し、細胞の凍結置換のために2〜4日間-80°Cに保ちます(図3)。使用したピンセットを室温で水に浸し、以下の標本の凍結のために温めます(図7G)。
注:試料を取り扱うピンセットは、急速に凍結する前に試料を凍結し、氷の結晶の形成につながる可能性があるため、暖かい(室温)にする必要があります。
- サンプルの温暖化と埋め込み
- 試験片を室温(-20°Cで2時間、4°Cで2h、室温で15分)徐々に、図3と図8Aで1mL充填された2mLプラスチックチューブにディスクを移します(図3および図8B,C)。
- 攪拌機を用いて使い捨てプラスチック容器に試薬を混合してエポキシ樹脂を調製する(図8D-F)。
- ステップ1.7.1で連続して30%樹脂(アセトン)、60%樹脂、および90%樹脂を室温で1時間交換する。その後、90%樹脂を100%樹脂と一晩37°Cで交換します。最後に、試料を100%樹脂(図8G-J)にシリコン埋め込み金型に埋め込み、60°Cで24時間重合する(図3)。
注:試料は、(断面を容易にするために)全体の手順を通して銅ディスクに取り付けられたままにする必要があります。細胞への樹脂の浸透にほとんど寄与しないため、埋め込みプロセス中に回転または揺れ装置の使用はお勧めできません。また、装置からの振動は、銅ディスクから切り離すために標本を引き起こすことがある。一晩37°Cでインキュベーションすると、熱エネルギーによる樹脂の細胞への浸透が加速します(加速器が含まれていないため樹脂は重合しません)。
- 標本ブロックのトリミング
- シリコン埋め込み金型から重合ブロックを取り出します(図9A)。ブロックに標本番号を書く(図9B)。
- カミソリの刃(図9C,D)でブロックから銅ディスクを取り出し、立体顕微鏡31の下で超音波トリミングブレード(図9E)とカミソリブレード(図9F-H)を使用して、ブロック表面に埋め込まれた試料を0.7mm x 0.7mmにトリミングする。
- ブロックをウルトラミクロトームの検体ホルダー(図9I)にセットし、ダイヤモンドのトリミングナイフでブロックの表面を滑らかに切ります(図9J、K)。
- 超薄切片の切断
- ブロックを超ミクロトーム(図10A)31から取り出し、それを立体顕微鏡にセットし、さらにかみそり刃で0.2mm x 0.3mmにトリミングする(図10B、C)。
- グリッドにネオプレンを適用して接着剤を作ります(図10D)。ブロックを超ミクロトームに戻し、プラスチックカバー(図10E)31でウルトラミクロトームを覆い、50-70 nm-厚いセクションをカットします(図10F-H)。
- ループを使用してセクションを取り出し(図10I)、ネオプレンで処理した300または400メッシュ銅格線に取り付け、それらを乾燥させます(図10J)。
- 電子顕微鏡下での切片の染色と観察
- 半シリコン管の溝に切片を設けてグリッドを(図11A)31、ウラニル酢酸溶液に浸漬し、それぞれ3分間クエン酸鉛を浸して染色する(図11B-E)32。
- 酵母および真菌標本の場合、グリッドをフィルター紙(4mm x 4 mm)に置き、ピンセットで拾い上げ、水面にプラズマポリマー化されたナフタレンフィルム33でそれらを覆う(図11F、G)。グリッドを電子顕微鏡に挿入し、100kV(図11H,I)で観察します。
2. ウイルスと高分子の急速凍結
- 液体エタンの調製
注:液体窒素を扱う際には、手袋とゴーグルを使用してください。エタンは爆発的であるため、同じ部屋で火災を使用しないように注意する必要があり、窓を開けておく必要があります。エタンは、プロパンが蒸発しない間に電子顕微鏡で蒸発するので使用される。- 液体窒素でSFDの液体窒素容器を充填します。微細なノズルを通してエタンガスを導入して、液体エタン容器にエタンを充填します。
- マイクログリッドと試料の調製と迅速な凍結
- マイクログリッドの両方の面を、イオンスパッタ装置30を用いてグロー放電(10 Pa,400 V,1 mA)で処理することで親水性にする。
- SFDにマイクログリッドをセットし、マイクログリッドにウイルスまたは高分子懸濁液(1mgタンパク質/mL)の2μLを適用します。濾紙を用いて余分な液体を除去し、装置の注入ボタンを押してマイクログリッドを迅速に凍結する。
- 凍結移動ホルダーにおける冷凍マイクログリッドの設定と電子顕微鏡観察
- 凍結したマイクログリッドを液体窒素で移動し、あらかじめ液体窒素温度で冷却したクライオ転写ホルダーにセットし、低温34で電子顕微鏡で観察する。
結果
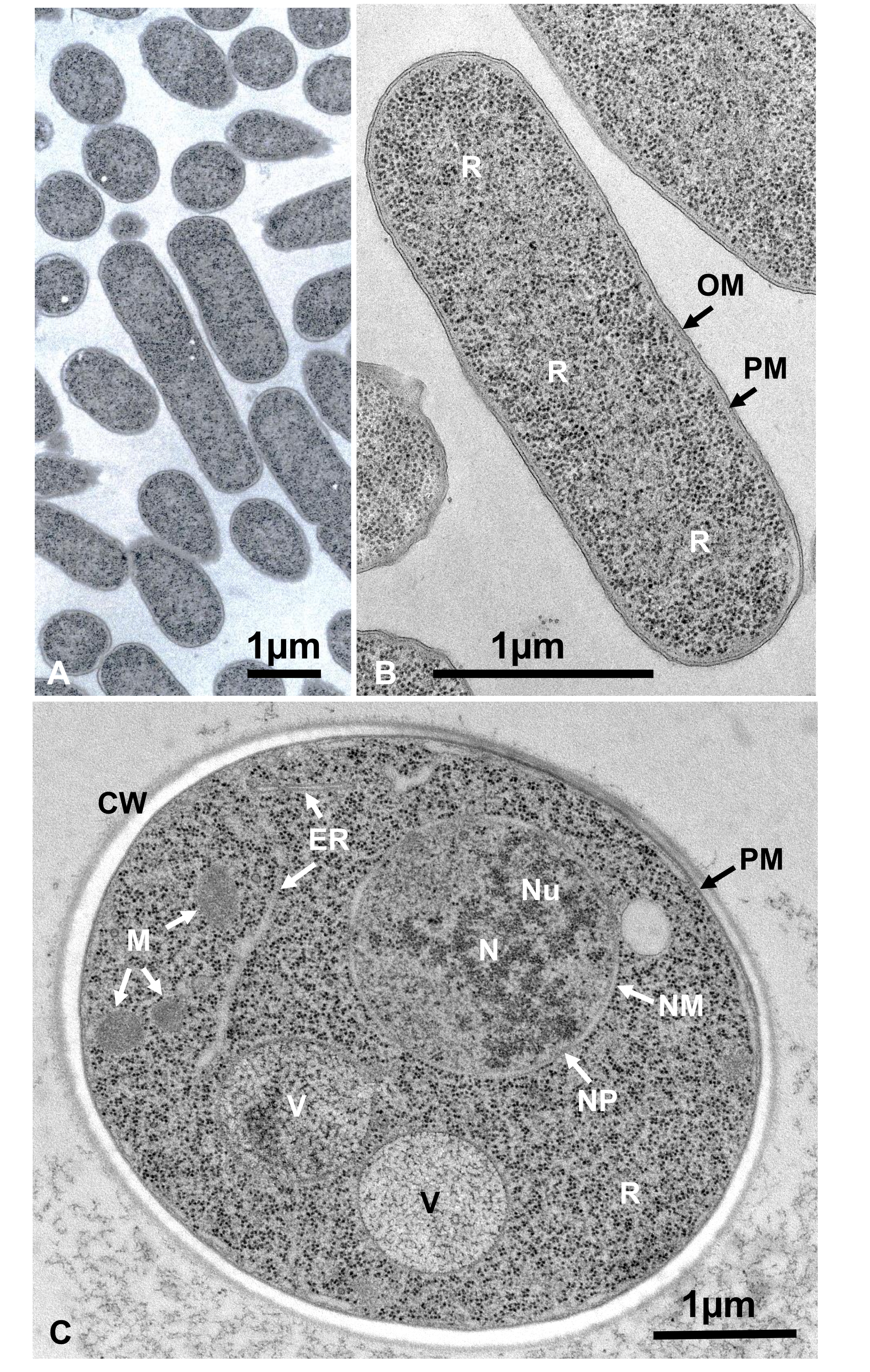
懸濁液中の微生物の生きた細胞を、遠心分離により収集し、2つの銅ディスクに挟み込み、SFDで急速に凍結し、凍結置換、エポキシ樹脂に埋め込まれ、超薄切除、染色、および電子顕微鏡下で観察した。図12は、大腸菌の超薄切片(バクテリア、図12A、B)16およびサッカロミセス・セレビシエ(酵母、図12C)15を示す。 画像は非常に明確であり、自然形態を示していることに注意してください。
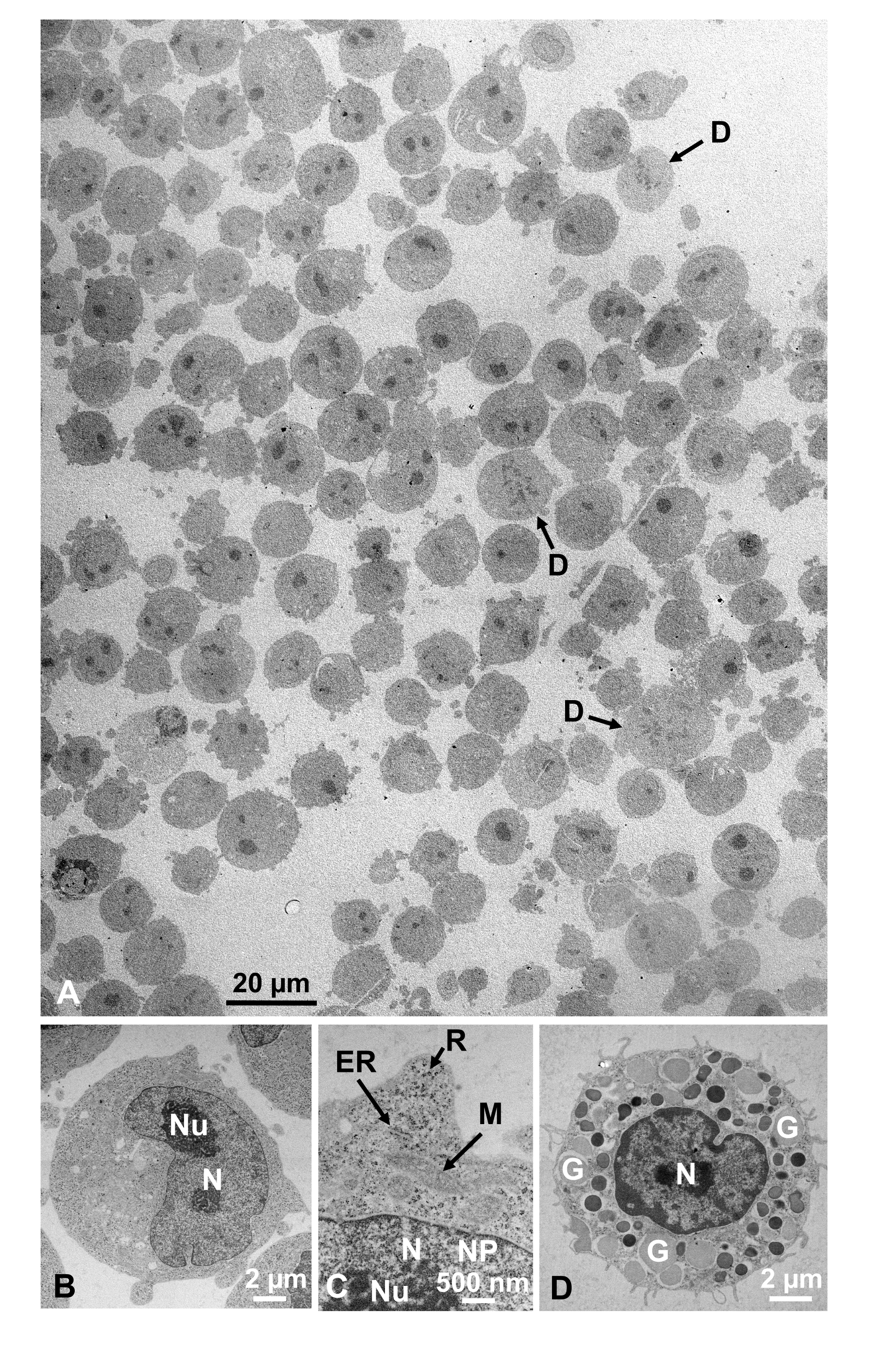
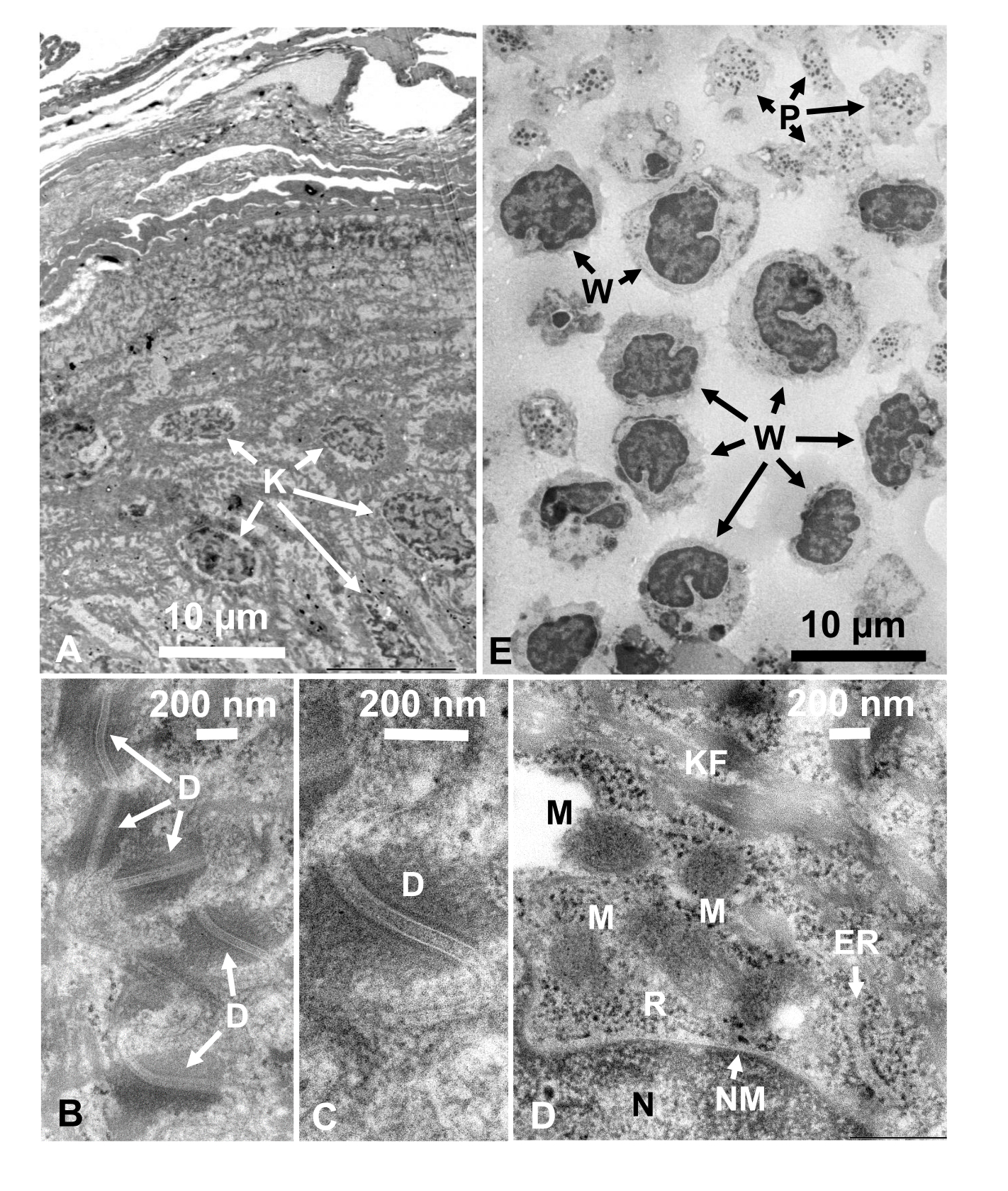
培養細胞および単離された動物細胞のグルタルアルデヒド固定細胞懸濁液を、SFDで回収し、迅速に凍結し、凍結置換し、上記の手順に従うことにより電子顕微鏡で観察した。図13は、培養細胞の超薄切片(図13A-C)1、28およびマウス腹腔からの単離細胞(図13D)28を示す。図14は、ヒト皮膚の超薄部分(図14A-D)及びバフィーコート(図14E)1を示す。画像も非常に明確で自然な形態を示していることに注意してください。図15は、SFDで急速に凍結し、クライオ電子顕微鏡34で観察されたB型肝炎ウイルスコア粒子を示す。他の細胞と同様に、画像は非常に明確であり、自然形態を示しています。

図1:サンドイッチ凍結装置28 と必要なツールを クリック して、この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図2:サンドイッチ凍結実験を行うために必要な材料。 スケールバー=10μm(B)2%オスミウム四酸化とアセトンの1 mLを含むガラスバイアル(10 mL)。(C) 文字のない表面(左)と文字(右)の表面を示す銅ディスク。スケールバー= 3 mm走査型電子顕微鏡。(D) 文字なしの面を持つ銅ディスクを、両面粘着テープ(*)でガラススライド上に置いた。スケールバー= 3 mm((E)両刃のカミソリと、動物や人間の組織をスライスするための壊れた両刃のカミソリ、トリミングブロック用の片刃のカミソリ、動物や人間の組織をスライスするための細断されたボード。(F)押し出されたポリスチレンフォームを持つピンセットは、寒さから指を保護します。この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図3:サンドイッチ凍結法を用いた細胞懸濁液の試料調製 省略形: r.t. = 室温。 この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図4:液体プロパン、銅ディスク、および固定剤の調製(A)をサンドイッチ凍結装置の液体窒素容器に注ぎ込んだ。(B)プロパンガスを微細なノズルを通して液体プロパン容器に導入した。(C) 液体プロパン (矢印).(D) 液体プロパンの固化を促進するために液体プロパンを冷却するために金属棒(矢印)を使用した。(E) 固化プロパン (矢印).(F)のイオンスパッタ装置(矢印)を用いて、銅ディスクをグロー放電で親水性にする。(G)グロー放電。(H)ガラスバイアルを作業浴中の液体窒素に入れる。この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図5:サンドイッチ凍結装置を用いた細胞懸濁液(酵母)の急速凍結(A)遠心管への酵母培養の移入(B)マイクロ遠心分離機。(C) 遠心管のサッカロミセス・セレビシエのペレット(矢印)。(D)遠心管からマイクロピペットを用いて検体を移す。(E)銅ディスクに試料を置く。(F) 銅ディスク上の標本の小さな滴(矢印)。(G)別の銅ディスクで標本を覆う。(H) ピンセットで2枚のディスクを拾う。(I)薄い金属棒を使って固体プロパンで井戸を作る。(J)注射ボタンを押して検体の凍結を凍結する。(K)凍結が完了しました。この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図6:ヒト組織(皮膚)の標本を準備する。(A, B)ペトリ皿のグルタルアルデヒドで固定されたヒト皮膚組織。スケールバー=5mm(C、D)組織(矢印)は、細断された板に2つの両刃のカミソリを使用してスライスした。 (E, F)銅ディスク上にグルタルアルデヒド溶液(矢印)の小さな滴を置いた。(G, H)ピンセットを使用して銅ディスクに皮膚組織の一部を置いた。(I) 銅ディスクを皮膚組織で覆うために別のディスクを使用した。(J)挟んだディスクはピンセットで拾われた。(K)挟んだディスクはピンセットで優しく保持されていました。組織を破砕しないように、先端(矢印)の間のギャップを維持することに注意してください。この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図7:試料を液体窒素に移し、ディスクを取り外す。 (B) 液体窒素中のディスク (矢印)。(C)ピンセットを液体窒素に入れ、作業浴中で冷却した。(D, E)ピンセットを使用して銅ディスク(矢印)を取り外す。(F)ピンセットでガラスバイアルにディスク(矢印)を転送する。(G) 次の標本を凍結するために水にピンセットを温める。この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図8: 試料の温めと埋め込み. (A)ガラスバイアル内の試料を室温で(B)ピンセット付きの2mLプラスチックチューブにディスク(矢印)を移す。(C)プラスチックチューブ中のアセトンの銅ディスク(矢印)。(D) インジェクションチューブ(矢印)で樹脂を測定する。(E) 使い捨てカップ(矢印)に樹脂を移す。(F)撹拌機を用いて樹脂を混合する。(G)シリコン埋め込み金型の穴に少量の樹脂を入れた。(H)グリッド内の過剰樹脂を濾紙で除去した。(I, J)試料を有する銅ディスクは、試料側を上にして埋め込み金型の穴の底に配置した。 この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図9:試料ブロックをトリミングする(A)埋め込み金型から重合ブロックを取り出す。(B)標本番号がブロックに書き込まれた。(C, D)銅ディスクはカミソリでブロックから取り除かれました。(D)=1mm(E-G)のスケールバーは、試料を超音波切断刃とカミソリブレードでトリミングした。スケールバー(G)=1mm(H)の高倍率(G)を表示する。個々の明るいスポットはセルです。細胞を単一層10に埋め込んだ。スケールバー=10μm(I-K)ブロックの表面をダイヤモンドトリミングナイフで滑らかに切った。(K)= 1 mmの縮尺バーをクリックして、この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図10: 超薄切片を切断する. (A) 標本ブロックをミクロトーム31から取り外した。(B, C)標本はさらにカミソリの刃でトリミングされた。(C)=0.5mmのスケールバー(D)ネオプレンは、それらを固執させるためにグリッドに適用されました。(E)超薄切除中の気流を避けるために、超ミクロトームはプラスチックで覆われていました。(F)ダイヤモンドナイフボートは水で満たされていた。(G, H)超薄切片を70nmの厚さに切断した。(H)= 1 mmのスケールバー (I, J) セクションをループを使用して取り出し、乾燥させた。この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図11:染色部(A)は、半シリコンチューブの溝にグリッドを配置した。(B)ウラニル酢酸塩に浸漬して染色した。(C)使用した酢酸ウラニルを自己シール性ワックス状フィルム上に回収した。(D)グリッドをウォータージェットで洗浄し、その後、鉛クエン酸に浸した。(F)プラズマ重合されたナフタレンフィルムを水上に浮かべ、(G)4mm×4mmのフィルターペーパーに配置された格子を覆う。(H)グリッドを標本ホルダーに入れ、電子顕微鏡で観察した。この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図12:大腸菌(細菌、A、B)およびサッカロミセスセレビシエ(酵母、C)の超薄切片。(A)検体は密に均一に埋め込まれ、変形を示さないことに注意してください。(B, C)膜構造は、明確で滑らかな形態を示し、リボソームは、すべての粒子を列挙することができる十分に明確である16.(C)酵母核と液胞は、自然形態である可能性のある真の円状を示す。ミトコンドリアのマトリックスは、電子密度の高い外観を示し、これは急速な凍結によって固定されている生きた細胞の特徴であり得る。スケールバー= 1 μm略語: CW = 細胞壁;ER = 小胞子;NM = 核膜;NP = 核毛穴;OM = 外膜;PM = 形質膜;R = リボソーム;N = 核;M = ミトコンドリア。(B)は許可を得て山田ら16から再現される。この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図13:ウルトラ薄いセクション(A-C)K562培養細胞。(D) マウスから孤立したマストセル。(A)検体は密に均一に埋め込まれ、変形を示さないことに注意してください。スケールバー=20μm(B-D)高倍率では、核、核小胞、核膜、核孔、小胞体、ミトコンドリア、リボソーム、顆粒が明確に観察される。スケールバー= 2 μm(B),500 nm(C),2 μm(D).略語: N = 核;Nu = ヌクレオラス;NM =核膜、NP =核毛穴;ER = 小胞子;M = ミトコンドリア;R = リボソーム;G = 顆粒;D = セルを分割します。(A)は許可を得て山口ら1から複製される。(B-D)は許可を得て山口ら28から再現しています。この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図14:ウルトラ薄い部分(A -D)人間の皮膚と(E)バフィーコート。(A-E)組織および細胞画像は明確で自然であり、断面は非常に薄い(50 nm)が良好なコントラストを示していることに注意してください。ミトコンドリアのマトリックスは、急速に凍結した生細胞の緻密なミトコンドリア行列(D)のものと類似した緻密な外観を示す。スケールバー = 10 μm(A, E),200 nm (B-D).略語: D = デスモソーム;ER = 小胞子;K = 角質細胞;KF = ケラチン繊維;M = ミトコンドリア;N = 核;NM = 核膜;P = 血小板;R = リボソーム;W = 白血球。許可を得て山口ら28から再現。この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}

図15:B型肝炎ウイルス(HBV)コア粒子はサンドイッチ凍結装置を用いて液体エタンで急速に凍結し、クライオ電子顕微鏡で観察した。 球状の中空粒子は、HBVコア粒子である。スケール バー = 100 nm。略語: HBV = B型肝炎ウイルス;I = 氷。許可を得て山口ら28 から再現。 この図の大きなバージョンを表示するには、ここをクリックしてください。
{kind=link}
ディスカッション
以下の議論は、1,000以上のサンプルに対する120以上のサンドイッチ凍結凍結置換実験と、36年間に行われた75以上のサンプルに対する70以上のプランジ凍結凍結-凍結電子顕微鏡実験に基づいています。
サンドイッチ凍結による良好な凍結の成功率
良好な凍結を達成する成功率は、サンプルに依存します。YPD培地で培養したサッカロミセスセレビシエ(酵母)細胞(1%酵母エキス、2%ペプトン、2%デキストロース)は、氷晶形成なしで良好な凍結のためにほぼ100%の成功を与えた10、11、15、35、36。他の酵母種,シゾサッカロミセス37, 38,39, クリプトコッカス14,40,41,42,43,44,45 ,46,47,48, 49,フサリウム50,51,52,オーレオバシジウム53,カンジダ54,55,フェロミセス56,アスペルギルス57,トリコスポロンも良好な凍結を示した。マイコバクテリウム58、59および大腸菌16を含む細菌も、良好な凍結を示した。培養細胞および単離された動物細胞は、1、25、26、27、60の両方の生きているおよびグルタルアルデヒド固定細胞の良好な凍結を示した。グルタルアルデヒド固定動物およびヒト組織を0.1〜0.2mmの厚さにスライスした場合も、ほとんどの時間1,28の良好な凍結を示した。
良好な凍結のための条件
適切な増殖段階と条件のセルのみを使用してください。培養中の細胞は指数相であるべきである。銅ディスクに濃縮サンプル(S.cerevisiae、3-5×109セル/mLの0.02 μL)の細胞懸濁液を非常に少量塗布します。グルタルアルデヒド固定の動物やヒト組織のスライスも非常に小さくする必要があります(好ましくは0.3 mm x 0.3 mm x 0.1 mm)。厚さ0.1mmの厚い組織スライスを切断することは困難であるため、多くの組織をスライスし、薄くて半透明のスライスを選択します。迅速に、しかし慎重に作業し、サンプルを乾燥させないでください。ピンセットで積み重ねられた銅ディスクを拾う際には、細胞や組織を粉砕しないようにディスクを強く押しすぎないでください。検体の負荷は、正常な凍結のための最も重要なステップであり、必要な条件は、高圧凍結のための良好な凍結のための条件と同じです。読者はマクドナルド61による優れたレビューを参照してください。
その他のアプリケーション
本論文では、細菌、酵母、培養細胞、単離された動物細胞、ヒト組織、ウイルス粒子の電子顕微鏡写真を紹介する。グルタルアルデヒド固定海洋藻類の良好な凍結を観察した。しかし、生きた淡水緑藻の細胞構造は氷結晶形成によって破壊された。20%のウシ血清アルブミン(BSA)と細胞を混合すると、超構造が氷結晶損傷なしで十分に保存されていることを保証した。20%BSAの使用は、キノコの茎細胞の超構造を維持するためにも有益であった。20%BSAを適用することにより植物細胞および組織の凍結に関する実験が進行中である。サンドイッチ凍結凍結置換試料の走査電子顕微鏡は試みられていないが、よく保存された細胞構造の観察は、以前9に報告されている。
サンドイッチの凍結方法に関する注意
細胞の近くからネイティブな超構造は、生きている細胞の急速な凍結および凍結置換によって最もよく観察される。SFDによる氷の結晶形成は、細胞の厚みを≤30μm1に制限することで回避できる。グルタルアルデヒド固定は細胞構造をより硬くし、生細胞の収集および遠心分離の際に可能な超構造変化を防ぐため、グルタルアルデヒドを有する組織を保持することはしばしば懸濁培養細胞を観察するための細胞構造のよりよい保存をもたらす。グルタルアルデヒド固定は、高圧凍結(HPF)法と同様に、0.2mm1まで凍結深度を延長することを可能にする。そのため、HPFマシンは、動物およびヒト組織の深い凍結のためのSFDに置き換えることができます。
グルタルアルデヒド固定組織は28年以上保存できるため、ユーザーの都合に応じてサンドイッチの凍結が行えます。グルタルアルデヒドで組織を固定すると、組織が固定化してより硬くなるため、組織の切断も容易になります。HPF機とは異なり、SFDは、クライオ電子顕微鏡用のウイルスの迅速な凍結や細菌や真核細胞に使用できます。さらに、HPFマシンと比較して、SFDは小型で、携帯用で、安価で、より多くの実験室で取得することができます。SFDのこれらの機能が、より多くの研究所が研究目標を達成するのに役立つことを願っています28.
細胞の自然形態の特徴
細胞構造は、外膜の膜構造(図12A、B)、血漿膜(図12B、C、および、図12B、C)の外観を示す場合、自然な状態にある。図 13A-D;そして図14E)、核エンベロープ(図12C、図13B-D、および図14D-D)、ミトコンドリア(図12C、図13C、および図14D)、および空胞(図12C)が滑らかな輪郭を示す。核と液胞はほぼ円形である(図12C)。リボソームは、直径が20nmの明確な電子密な外観を示す(図12B、C;図 13C;および図 14D)。細胞質は電子ルーセントである(図12B、C;図 13C;および図 14D)。
細胞形態に及ぼすグルタルアルデヒド固定の効果
グルタルアルデヒド固定は、氷結晶のない超構造を得るためにサンドイッチ凍結前に動物またはヒト組織に対して行われた。この方法で得られた顕微鏡写真は、生体組織の急速凍結によって得られたものと同様の鮮明な画像を示す(図14)1。酵母細胞、深海微生物、培養細胞に関する研究は、超構造の変形が主に室温1、17、18、28におけるエタノールによるオスミウム四酸化ス固定および脱水によるものであることを示している。また、グルタルアルデヒド固定は培養線維芽細胞におけるアクチンの組織を破壊しないが、室温でアセトンまたはエタノールによる四酸化オスミウム固定および脱水はアクチン組織62を破壊すると報告されている。
したがって、細胞形態に対するグルタルアルデヒド固定の効果に関する詳細な研究を行う必要があります。大野は、生きている組織が血液供給を止めずに急速に凍結する in vivo クライオフィクセション法を開発した。組織を凍結置換し、エポキシ樹脂に埋め込み、超薄切片を観察した。電子顕微鏡画像は、化学固定-従来の脱水によって得られたものと比較して、生きている組織の最もネイティブな超構造と新鮮な未固定組織の急速な凍結によって示された。したがって、グルタルアルデヒド固定凍結置換(本方法)と インビボで の凍結置換によって得られた超構造を比較してグルタルアルデヒド固定の効果を調べるのが興味深い場合がある。
環境への配慮と実験効率の向上
樹脂の置換には2mLプラスチックチューブを使用しています。希釈樹脂の1mLは、各置換ステップに十分である。使用されたプラスチックチューブは、各実験の後に廃棄されてもよい。これにより、樹脂置換に使用するガラスバイアルの洗浄に要する時間と労力を節約できます。さらに、ウラニル酢酸溶液は、セクション染色32のために繰り返し使用することができる。セクションを染色した後、ウラニル酢酸溶液を保存し、再利用することができます。酢酸ウラニルは放射性物質であるため、再利用は廃棄物の発生を回避し、環境の保護に貢献します。
プラズマ重合ナフタレンフィルム
プラズマ重合されたナフタレンフィルムは、ナフタレンガスから作製された3次元重合炭素膜であり、グロー放電33の下でプラズマ重合により作られる。膜は電子の爆撃および化学薬品に対して弾力性があり、非常にきれいで、電子に対して透明で、平坦な表面および非晶質構造を有する。従って、市販されているプラズマ重合ナフタレンフィルムは優れており、支持膜として推奨される。
開示事項
著者らは利益相反を宣言しない。
謝辞
何一つ
資料
| Name | Company | Catalog Number | Comments |
| Sandwich Freezing Device | Marine works Japan, Ltd, Yokosuka, Japan | MW-SFD-01 | with metal bar, thin metal bar, tweezers, and working bath |
| 10 mL glass vials | - | Scintillation counter vials for fixative | |
| Acetone | - | ||
| Osmium tetroxide | Nisshin EM Co. Ltd., Tokyo | 3004 | 0.1 g |
| Deep freezer | Sanyo Co. Ltd., Osaka | MDF-C8V1 | |
| Copper disk | Nisshin EM Co. Ltd., Tokyo | - | Refer to this paper |
| Slide glass | - | ||
| Double-sided adhesive tape | - | ||
| Single-edged razor blade | Nisshin EM Co. Ltd., Tokyo | - | Feather, FAS-10 |
| Double-edged razor blade | Nisshin EM Co. Ltd., Tokyo | - | Feather, FA-10 |
| Shredded board | Nisshin EM Co. Ltd., Tokyo | 428 | |
| Tweezers | Nisshin EM Co. Ltd., Tokyo | - | Several pairs |
| Tweezers with polystyrene foam | - | One pair | |
| Glutaraldehyde | Nisshin EM Co. Ltd., Tokyo | 3052 | |
| Liquid nitrogen | - | ||
| Propane gas | - | Cryogen | |
| Ion sputter apparatus | Hitachi high technologies, Tokyo | Hitachi E102 | |
| Micropipette | - | For 1 mL, 200 μL, and 2 μL | |
| Microcentrifuge | Tomy digital biology Co. Ltd., Tokyo | Capsulefuge, PMC-060 | |
| Stereomicroscope | Nikon Co. Ltd., Tokyo et al. | - | SMZ 645 |
| LED illumination for stereomicroscope | Nikon Co. Ltd., Tokyo et al. | SM-LW 61 Ji | |
| Disposable plastic container | - | 50 mL and 200 mL | |
| Ethane gas | - | Cryogen | |
| 2 mL Eppendorf tubes | - | For embedding | |
| Disposable plastic syringes | - | 1 mL, 5 mL, 10 mL, and 20 mL | |
| Magnetic stirrer | - | ||
| Epoxy resin | Nisshin EM Co. Ltd., Tokyo | 340 | Quetol 812 set |
| Silicon embedding mold | Nisshin EM Co. Ltd., Tokyo | 4217 | 7 mm in diameter, 13 mm deep |
| Incubater | - | For 37 °C and 60 °C | |
| Trimming stage | Sunmag Co. Ltd., Tokyo | - | Tilting mechanism equipped, Refer to this paper |
| LED illumination for trimming stage | Sunmag Co. Ltd., Tokyo | - | Refer to this paper |
| Ultrasonic Trimming Blade | Nisshin EM Co. Ltd., Tokyo | 5240 | EM-240, Refer to this paper |
| Ultramicrotome | Leica Microsystems, Vienna | Ultracut S | |
| Grids | Nisshin EM Co. Ltd., Tokyo | 2633, 2634 | 300 mesh, 400 mesh |
| 0.5% Neoprene W solution | Nisshin EM Co. Ltd., Tokyo | 605 | |
| Perfect Loop | Nisshin EM Co. Ltd., Tokyo | 2351 | Fot retrieving sections |
| Half Tube for section staining | Nisshin EM Co. Ltd., Tokyo | 463 | Refer to this pape |
| Super Support Film | Nisshin EM Co. Ltd, Tokyo | 647 | |
| Syringe filter | Toyo Roshi Kaisha, Ltd., Tokyo | DISMIC-03CP | Cellulose acetate, 0.45 μm |
| Transmission electron microscope | JEOL Co. Ltd., Tokyo | JEM-1400 |
参考文献
- Yamaguchi, M., et al. Good ultrastructural preservation of human tissues and cultured cells by glutaraldehyde fixation, sandwich freezing, and freeze-substitution. Cytologia. 85 (1), 15-26 (2020).
- Gilkey, J. C., Staehelin, L. A. Advances in ultrarapid freezing for the preservation of cellular ultrastructure. Journal of Electron Microscopy Technique. 3 (2), 177-210 (1986).
- Van Harreveld, A., Crowell, J. Electron microscopy after rapid freezing on a metal surface and substitution fixation. The Anatomical Record. 149, 381-385 (1964).
- Aoki, N., Ito, M., Ejiri, S., Ozawa, H. Ultrastructure of human skin by a rapid freezing technique: structural preservation and antigenicity. Journal of Investigative Dermatology. 102 (3), 354-361 (1994).
- Moor, H., Steinbrecht, R. A., Zierold, K. Theory and practice of high pressure freezing. Cryotechniques in Biological Electron Microscopy. , 175-191 (1987).
- McDonald, K. L., Morphew, M., Vertkade, P., Muller-Reichert, T. Recent advances in high-pressure freezing: Equipment-and specimen-loading methods. Methods in Molecular Biology. 369, 143-173 (2007).
- Sosinsky, G. E., et al. The combination of chemical fixation procedures with high pressure freezing and freeze substitution preserves highly labile tissue ultrastructure for electron tomography applications. Journal of Structural Biology. 161 (3), 359-371 (2008).
- Costello, M. J. Ultra-rapid freezing of the biological samples. Scanning Electron Microscopy. , 361-370 (1980).
- Baba, M., Osumi, M. Transmission and scanning electron microscopic examination of intracellular organelles in freeze-substituted Kloeckera and Saccharomyces cerevisiae yeast cells. Journal of Electron Microscopy Technique. 5 (3), 249-261 (1987).
- Yamaguchi, M., Okada, H., Namiki, Y. Smart specimen preparation for freeze-substitution and serial ultrathin sectioning of yeast cells. Journal of Electron Microscopy. 58 (4), 261-266 (2009).
- Yamaguchi, M., et al. Electron microscopy of hepatitis B virus core antigen expressing yeast cells by freeze-substitution fixation. European Journal of Cell Biology. 47 (1), 138-143 (1988).
- Baba, M., Takeshige, K., Baba, N., Ohsumi, Y. Ultrastructural analysis of the autophagic process in yeast: detection of autophagosomes and their characterization. Journal of Cell Biology. 124 (6), 903-913 (1994).
- Yamaguchi, M., et al. The spindle pole body duplicates in early G1 phase in a pathogenic yeast Exophiala dermatitidis: an ultrastructural study. Experimental Cell Research. 279 (1), 71-79 (2002).
- Yamaguchi, M., Biswas, S. K., Ohkusu, M., Takeo, K. Dynamics of the spindle pole body of the pathogenic yeast Cryptococcus neoformans examined by freeze-substitution electron microscopy. FEMS Microbiology Letters. 296 (2), 257-265 (2009).
- Yamaguchi, M., et al. Structome of Saccharomyces cerevisiae determined by freeze-substitution and serial ultrathin sectioning electron microscopy. Journal of Electron Microscopy. 60 (5), 321-335 (2011).
- Yamada, H., et al. Structome analysis of Escherichia coli cells by serial ultrathin sectioning reveals the precise cell profiles and the ribosome density. Microscopy. 66 (4), 283-294 (2017).
- Yamaguchi, M., Ohkusu, M., Sameshima, M., Kawamoto, S. Safe specimen preparation for electron microscopy of pathogenic fungi by freeze-substitution after glutaraldehyde fixation. Japanese Journal of Medical Mycology. 46 (3), 187-192 (2005).
- Yamaguchi, M., et al. Improved preservation of fine structure of deep-sea microorganisms by freeze-substitution after glutaraldehyde fixation. Journal of Electron Microscopy. 60 (4), 283-287 (2011).
- Yamaguchi, M., et al. Prokaryote or eukaryote? A unique microorganism from the deep-sea. Journal of Electron Microscopy. 61 (6), 423-431 (2012).
- Yamada, H., Chikamatsu, K., Aono, A., Mitarai, S. Pre-fixation of virulent Mycobacterium tuberculosis with glutaraldehyde preserves exquisite ultrastructure on transmission electron microscopy through cryofixation and freeze-substitution with osmium-acetone at ultralow temperature. Journal of Microbiological Methods. 96, 50-55 (2014).
- Yamaguchi, M. An electron microscopic study of microorganisms: from influenza virus to deep-sea microorganisms. JSM Mycotoxins. 65 (2), 81-99 (2015).
- Yamaguchi, M., et al. High-voltage electron microscopy tomography and structome analysis of unique spiral bacteria from the deep sea. Microscopy. 65 (4), 363-369 (2016).
- Yamaguchi, M., Yamada, H., Uematsu, K., Horinouchi, Y., Chibana, H. Electron microscopy and structome analysis of unique amorphous bacteria from the deep sea. Cytologia. 83 (3), 337-342 (2018).
- Yamaguchi, M., Yamada, H., Chibana, H. Deep-sea bacteria harboring bacterial endosymbionts in a cytoplasm?: 3D electron microscopy by serial ultrathin sectioning of freeze-substituted specimen. Cytologia. 85 (3), 209-211 (2020).
- Yamaguchi, M., Takahashi-Nakaguchi, A., Aida, Y., Sato-Okamoto, M., Chibana, H. Convenient method for better preservation of fine structures of cultured macrophages and engulfed yeast cells by freeze-substitution fixation. Microscopy. 66 (3), 209-211 (2017).
- Aoki, S., et al. Shift in energy metabolism caused by glucocorticoids enhances the effect of cytotoxic anticancer drugs against acute lymphoblastic leukemia cells. Oncotarget. 8 (55), 94271-94285 (2017).
- Hirao, T., et al. Altered intracellular signaling by imatinib increases the anticancer effects of tyrosine kinase inhibitors in chronic myelogenous leukemia cells. Cancer Science. 109 (1), 121-131 (2018).
- Yamaguchi, M., et al. Sandwich freezing device for rapid freezing of viruses, bacteria, yeast, cultured cells, and animal and human tissues in electron microscopy. Microscopy. 70 (2), 215-223 (2021).
- Yamaguchi, M. Troubleshooting in specimen preparation of microorganisms. Kenbikyo. 42, 26-28 (2007).
- Yamaguchi, M., Aoyama, T., Yamada, N., Chibana, H. Quantitative measurement of hydrophilicity/hydrophobicity of the plasma-polymerized naphthalene film (Super support film) and other support films and grids in electron microscopy. Microscopy. 65 (55), 444-450 (2016).
- Yamaguchi, M., Chibana, H. A method for obtaining serial ultrathin sections of microorganisms in transmission electron microscopy. Journal of Visualized Experiments: JoVE. (131), e56235 (2018).
- Yamaguchi, M., Shimizu, M., Yamaguchi, T., Ohkusu, M., Kawamoto, S. Repeated use of uranyl acetate solution for section staining in transmission electron microscopy. Plant Morphology. 17 (1), 57-59 (2005).
- Yamaguchi, M., Tanaka, A., Suzuki, T. A support film of plasma-polymerized naphthalene for electron microscopy: method of preparation and application. Journal of Electron Microscopy. 41 (1), 7-13 (1992).
- Yamaguchi, M., et al. Cryo-electron microscopy of hepatitis B virus core particles produced by transformed yeast: comparison with negative staining and ultrathin sectioning. Journal of Electron Microscopy. 37 (6), 337-341 (1988).
- Yamaguchi, M., et al. Dynamics of hepatitis B virus core antigen in a transformed yeast cell: analysis with an inducible system. Journal of Electron Microscopy. 43 (6), 386-393 (1994).
- Yamaguchi, M., Miyatsu, T., Mizokami, H., Matsuoka, L., Takeo, K. Translocation of hepatitis B virus core particles through nuclear pores in transformed yeast cells. Journal of Electron Microscopy. 45 (4), 321-324 (1996).
- Sipiczki, M., Takeo, K., Yamaguchi, M., Yoshida, S., Miklos, I. Environmentally controlled dimorphic cycle in fission yeast. Microbiology. 144, 1319-1330 (1998).
- Sipiczki, M., et al. Role of cell shape in determination of the division plane in Schizosaccharomyces pombe: random orientation of septa in spherical cells. Journal of Bacteriology. 182 (6), 1693-1701 (2000).
- Encz, i. K., Yamaguchi, M., Sipiczki, M. Morphology transition genes in the dimorphic fission yeast Schizosaccharomyces japonicus. Antonie van Leeuwenhoek. 92 (2), 143-154 (2007).
- Kopecka, M., et al. Microtubules and actin cytoskeleton in Cryptococcus neoformans compared with ascomycetous budding and fission yeasts. European Journal of Cell Biology. 80 (4), 303-311 (2001).
- Yamaguchi, M., Biswas, S. K., Kita, S., Aikawa, E., Takeo, K. Electron microscopy of pathogenic yeasts Cryptococcus neoformans and Exophiala dermatitidis by high-pressure freezing. Journal of Electron Microscopy. 51 (1), 21-27 (2002).
- Ikeda, R., et al. Contribution of the mannan backbone of cryptococcal glucuroxylomannan and a glycolytic enzyme of Staphylococcus aureus to contact-mediated killing of Cryptococcus neoformans. Journal of Bacteriology. 189 (13), 4815-4826 (2007).
- Yamaguchi, M., et al. The spindle pole body of the pathogenic yeast Cryptococcus neoformans: variation in morphology and positional relationship to the nucleolus and the bud in interphase cells. Journal of Electron Microscopy. 59 (2), 165-172 (2010).
- Kozubowski, L., et al. Ordered kinetochore assembly in the human pathogenic basidiomycetous yeast Cryptococcus neoformans. MBio. 4 (5), 00614 (2013).
- Stepanova, A. A., Yamaguchi, M., Chibana, H., Vasilyeva, N. V. Ultrastructural aspects of cell components migration during budding in the yeast Cryptococcus leurentii. Problems in Medical Mycology. 18 (3), 24-29 (2016).
- Ohkusu, M., et al. Cellular and nuclear characteristics of Exophiala dermatitidis. Studies in Mycology. 43 (43), 143-150 (1999).
- Yamaguchi, M., Biswas, S. K., Suzuki, Y., Furukawa, H., Takeo, K. Three-dimensional reconstruction of a pathogenic yeast Exophiala dermatitidis cell by freeze-substitution and serial sectioning electron microscopy. FEMS Microbiology Letters. 219 (1), 17-21 (2003).
- Yamaguchi, M., et al. The spindle pole body of the pathogenic yeast Exophiala dermatitidis: variation in morphology and positional relationship to the nucleolus and the bud in interphase cells. European Journal of Cell Biology. 82 (10), 531-538 (2003).
- Biswas, S. K., Yamaguchi, M., Naoe, N., Takashima, T., Takeo, K. Quantitative three-dimensional structural analysis of Exophiala dermatitidis yeast cells by freeze-substitution and serial ultrathin sectioning. Journal of Electron Microscopy. 52 (2), 133-143 (2003).
- Takaya, N., et al. Cytochrome P450nor, a novel class of mitochondrial cytochrome P450 involved in nitrate respiration in the fungus Fusarium oxysporum. Archives of Biochemistry and Biophysics. 372 (2), 340-346 (1999).
- Zhou, Z., et al. Ammonia fermentation, a novel anoxic metabolism of nitrate by fungi. Journal of Biological Chemistry. 277 (3), 1892-1896 (2002).
- Takasaki, K., et al. Fungal ammonia fermentation, a novel metabolic mechanism that couples the dissimilatory and assimilatory pathways of both nitrate and ethanol. Role of acetyl CoA synthetase in anaerobic ATP synthesis. Journal of Biological Chemistry. 279 (13), 12414-12420 (2004).
- Kopecka, M., et al. Analysis of microtubules and F-actin structures in hyphae and conidia development in opportunistic human pathogenic black yeast Aureobasidium pullulans. Microbiology. 149, 865-876 (2003).
- Ueno, K., Namiki, Y., Mitani, H., Yamaguchi, M., Chibana, H. Differential cell wall remodeling of two chitin synthase deletants Δchs3A and Δchs3B in the pathogenic yeast Candida glabrata. FEMS Yeast Research. 11 (5), 398-407 (2011).
- Ikezaki, S., et al. Mild heat stress affects on the cell wall structure in Candida albicans biofilm. Medical Mycology Journal. 60 (2), 29-37 (2019).
- Gabriel, M., et al. The cytoskeleton in the unique cell reproduction by conidiogenesis of the long-neck yeast Fellomyces (Sterigmatomyces) fuzhouensis. Protoplasma. 229 (1), 33-44 (2006).
- Yoshimi, A., et al. Functional analysis of the α-1,3-glucan synthase genes agsA and agsB in Aspergillus nidulans: agsB is the major α-1,3-glucan synthase in this fungus. PLoS One. 8 (1), 54893 (2013).
- Yamada, H., Mitarai, S., Chikamatsu, K., Mizuno, K., Yamaguchi, M. Novel freeze-substitution electron microscopy provides new aspects of virulent Mycobacterium tuberculosis with visualization of the outer membrane and satisfying biosafety requirements. Journal of Microbiological Methods. 80 (1), 14-18 (2010).
- Yamada, H., Yamaguchi, M., Chikamatsu, K., Aono, A., Mitarai, S. Structome analysis of virulent Mycobacterium tuberculosis, which survives with only 700 ribosomes at density per 0.1 fl cytoplasm. PLoS One. 10 (1), 0117109 (2015).
- Shiratori, R., et al. Glycolytic suppression dramatically changes the intracellular metabolic profile of multiple cancer cell lines in a mitochondrial metabolism-dependent manner. Scientfic Reports. 9 (1), 18699 (2019).
- McDonald, K. L. Out with the old and in with the new: rapid specimen preparation procedures for electron microscopy of sectioned biological material. Protoplasma. 251 (2), 429-448 (2014).
- Small, J. V. Organization of actin in the leading edge of cultured cells: influence of osmium tetroxide and dehydration on the ultrastructure of actin meshworks. Journal of Cell Biology. 91 (3), 695-705 (1981).
- Ohno, S., Terada, N., Fujii, Y., Ueda, H., Takayama, I. Dynamic structure of glomerular capillary loop as revealed by an in vivo cryotechnique. Virchows Archiv. 427 (5), 519-527 (1996).
転載および許可
このJoVE論文のテキスト又は図を再利用するための許可を申請します
許可を申請さらに記事を探す
This article has been published
Video Coming Soon
Copyright © 2023 MyJoVE Corporation. All rights reserved