
Method Article
Enriquecimento de uma única etapa de um histone TAP-Tagged deacetilase do fungo filamentoso Aspergillus nidulans para o ensaio de atividade enzimática
Neste Artigo
Resumo
As deacetilases da histona da classe 1 (HDACs) como RpdA ganharam a importância como alvos potenciais para tratar infecções fungosas. Aqui nós apresentamos um protocolo para o enriquecimento específico de rpda Tap-Tagged combinado com um ensaio da atividade de HDAC que permita in vitro o teste da eficácia de inibidores da deacetilase do histona.
Resumo
As deacetilases da histona da classe 1 (HDACs) como RpdA ganharam a importância como alvos potenciais para o tratamento de infecções fungosas e para a mineração do genoma de Metabolites secundários fungosos. A triagem de inibidores, no entanto, requer atividades enzimáticas purificadas. Como as deacetilases da classe 1 exercem sua função como complexos Multiproteicos, elas geralmente não são ativas quando expressas como polipeptídeos únicos em bactérias. Conseqüentemente, os complexos endógenos precisam de ser isolados, que, quando as técnicas convencionais como a cromatografia da troca iônica e da exclusão do tamanho são aplicadas, são trabalhoso e demoram. A purificação em tandem da afinidade foi desenvolvida como uma ferramenta para enriquecer complexos da multiproteína das pilhas e girou assim para fora para ser ideal para a isolação de enzimas endógenas. Aqui nós fornecemos um protocolo detalhado para o enriquecimento da único-etapa de complexos ativos de RpdA através da primeira etapa da purificação de RpdA C-terminally da torneira-etiquetada de Aspergillus nidulans. Os complexos purified podem então ser usados para a seleção subseqüente do inibidor que aplica um ensaio do desacetilase. O enriquecimento protéico, juntamente com o ensaio de atividade enzimática, pode ser completado em dois dias.
Introdução
As deacetilases de histona (HDACs) são enzimas hidrolíticas dependentes de Zn2 +capazes de remover grupos acetil de resíduos de lisina de histonas e outras proteínas. Baseado na similaridade da seqüência, os HDACs são agrupados em diversas classes1. Recentemente, a classe fúngica 1 HDAC RpdA, um ortolog de levedura de padeiro (Saccharomyces cerevisiae) Rpd3p, mostrou-se essencial para o patógeno fúngico oportunista Aspergillus fumigatus2. Conseqüentemente, RpdA ganhou a importância como um alvo potencial para tratar infecções fungosas2. A avaliação da atividade da deacetilase in vitro é importante para a caracterização de propriedades enzimáticas e permite determinar a eficácia de novas substâncias para o desenvolvimento de inibidores. Embora a expressão de recombinação de uma versão Codon-aperfeiçoada de HDAC1 humano em Escherichia coli seja relatada recentemente3, as tentativas de expressar o rpda full-length neste anfitrião falharam4. Além disso, como os HDACs de classe fúngica 1, como RpdA e HosA, exercem sua função como complexos Multiproteicos, é favorável ao uso de complexos endógenos nativos para estudos de inibidores enzimáticos. Entretanto, devido aos fatores de inibição e à presença de HDACs diferentes em lysates fungosos, a atividade catalítica medida em extratos inteiros da proteína é relativamente baixa e não pode ser atribuída às enzimas individuais. Além disso, estudos prévios no fungo filamentoso a. nidulans identificaram uma classe 2 HDAC, hdaa, como a deacetilase predominante em frações cromatográficas de extratos fúngicos. Assim, várias etapas de purificação cromatográfica convencional são necessárias para separar a atividade não HdaA de cepas fúngicas4. A introdução da estratégia de purificação de afinidade em tandem (TAP) em a. nidulans5 tem facilitado significativamente o enriquecimento de atividades específicas de deacetilase. A tag original da TAP é composta por dois domínios de proteína A e um peptídeo de ligação de calmodulina (CBP) separado por um local de clivagem de protease do vírus do tabaco etch (TEV)6. Isso permite a purificação nativa e eluição de proteínas marcadas, incluindo seus parceiros de interação7. Ao usar as proteínas enriquecidas para ensaios de atividade, a eluição leve em condições nativas pela clivagem da protease é uma característica importante da purificação da tag TAP. Uma proteína GFP-etiquetada, por exemplo, pode ser enriquecida por anticorpos imobilizados também, entretanto, não pode ser eluída circunstâncias nativas.
Aqui nós fornecemos um protocolo detalhado para o enriquecimento da único-etapa de complexos ativos de RpdA através da primeira etapa da purificação de RpdA C-terminally da torneira-etiquetada de a. nidulans (separação de IgG e clivagem de TeV) para a seleção subseqüente do inibidor que aplica um ensaio histona deacetilase. Como provado ser suficiente, o enriquecimento da afinidade foi restrito a apenas uma etapa da purificação também porque a atividade enzimática foi reduzida significativamente após a purificação da torneira em dois passos quando comparada à purificação de IgG sozinho.
No entanto, o protocolo introduzido deve ser também aplicável para o enriquecimento de outras enzimas marcadas envolvidas na regulação da cromatina, tais como acetiltransferases, metiltransferases e demethylases. Acrescentando a segunda etapa de purificação do protocolo TAP, as proteínas copurificadas com as iscas marcadas podem ser consideradas como parceiras complexas de complexos enzimáticos (novos).
Protocolo
1. cultivo de A. nidulans
- Preparação do inóculo
Nota: todas as etapas aparte da incubação devem ser executadas um armário laminar do fluxo.- Risca a estirpe TAP (por exemplo , Tib 32.12) do estoque de glicerol (-80 ° c) para o meio mínimo de glicose/XILOSE (gxmm; por litro: 10,0 g de glicose, 0,5 g de xilose, 10 ml de solução de tartarato de di-amônio de 1 M, 20 ml de solução salina 50 × [por 26,0 litro KCl, 26,0 g MgSO4 · 7 H2O, 76,0 g KH2po4, 1 ml de clorofórmio], 1 ml de oligoelementos de 1.000 × Hutner8) incluindo 1,5% (p/v) Agar e os suplementos necessários conforme descrito por Todd et al. 20079. Incubar durante 2 – 3 dias a 37 ° c.
Nota: TIB 32.1 contém uma fusão Rpda:: TAP controlada por um promotor xilose inducible, xylp(p)10. É por isso que a xilose está incluída na receita acima. Omitir xilose quando as cepas crescentes expressando construções que não estão Xylp(p) controle. - Prepare um tubo de centrifugação estéril de 1,5 ml com 1,5 ml de solução de suspensão conídios estéril (CSS; 0,9% (p/v) NaCl, 0, 1% (v/v) Polissorbato 80).
- Molhe um laço de inoculação descartável em CSS, raspe os conídios da placa da etapa 1.1.1 e suspenda no tubo da etapa 1.1.2.
- Transferir 150 μl cada uma de suspensão conídios para 10 25 cm2 frascos de cultura de células com tampão de ventilação contendo Agar gxmm incluindo suplementos e 200 μL de CSS.
- Espalhe a suspensão conídios dentro dos frascos usando um laço estéril da inoculação e incubar os frascos em 37 ° c por 2 – 3 dias.
- Para colher os conídios, use 10 mL de CSS por balão. Despeje a solução em um balão, feche firmemente o balão com a tampa de rosca fornecida e agite vigorosamente o balão.
- Após molhar a superfície fúngica, raspar completamente os conídios remanescentes com um loop de inoculação estéril.
- Passe os conídios através dos filtros da pilha do μm 40 coloc em um tubo estéril do centrifugador de 50 mL e colete a suspensão de cinco frascos em um tubo.
- Centrifugue o tubo a 1.000 × g durante 10 min.
- Decantar o sobrenadante e ressuspender o pellet em 10 mL de CSS por tubo.
- Colete ambas as suspensões em um tubo, enxague o tubo vazio com 40 mL de CSS, adicione à suspensão e centrifugue conforme descrito na etapa 1.1.9.
- Decantar o sobrenadante e suspender a pelota conídios em 4 ml de CSS.
- Prepare duas diluições de série 1:50 da suspensão conídios e determine o número de conídios na suspensão 1:2, 500-diluída resultante com uma câmara de contagem como descrito11.
- Risca a estirpe TAP (por exemplo , Tib 32.12) do estoque de glicerol (-80 ° c) para o meio mínimo de glicose/XILOSE (gxmm; por litro: 10,0 g de glicose, 0,5 g de xilose, 10 ml de solução de tartarato de di-amônio de 1 M, 20 ml de solução salina 50 × [por 26,0 litro KCl, 26,0 g MgSO4 · 7 H2O, 76,0 g KH2po4, 1 ml de clorofórmio], 1 ml de oligoelementos de 1.000 × Hutner8) incluindo 1,5% (p/v) Agar e os suplementos necessários conforme descrito por Todd et al. 20079. Incubar durante 2 – 3 dias a 37 ° c.
- Crescimento e colheita de micélio
- Enquanto trabalhava um gabinete de fluxo laminar, inocular 4 – 6 frascos cônicos de um litro cada contendo 250 mL de GXMM, incluindo suplementos adequados a uma densidade de 5 × 106 conídios/mL e incubar a 180 rpm a 37 ° c por 14 – 16 h.
- Coloque o pano de queijo em um funil em cima de um balão e filtre o micélio através do pano. Lave brevemente com água desionizada.
- Retire o máximo de humidade possível apertando o micélio preso no pano de queijo entre as primeiras mãos e depois as toalhas de papel.
- Transfira o micélios secado como folhas lisas a um copo plástico com uma tampa de parafuso e um congelamento instantâneo com nitrogênio líquido. Isso garante uma alta relação área/volume para o seguinte processo de liofilização.
- Conservar o micélio congelado a-80 ° c antes da liofilização.
Observação: o protocolo pode ser pausado aqui. - Lyophilize o micélios durante a noite.
- Pare o processo de secagem por congelamento quando a temperatura de micélio permanecer constante (18 – 24 h). Retire os copos e sele imediatamente com as tampas de rosca fornecidas.
Nota: quando firmemente selado, micélio liofilizado pode ser armazenado por várias semanas na RT.
2. enriquecimento de uma única etapa da TAP-Tagged HDAC (adaptado de Bayram et al. 2012)12
- Preparação de buffers e soluções
Nota: Adicione 2-mercaptoethanol (EtSH) e inibidores de protease aos bufferes diretamente antes do uso. Filtre todos os buffers utilizados para cromatografia através de membranas de nitrocelulose de 0,22 μm para evitar a introdução de impurezas/contaminações na resina de cromatografia. As instruções nos passos abaixo referem-se à preparação de 1 L de cada tampão. Armazene os buffers a 4 ° c.- Tampão de extração (B250): 250 mM NaCl, 100 mM Tris-HCl pH 7,5 (RT), 0,1% (v/v) TX-100, 5 mM EtSH.
- Dissolva 12,35 g de Tris-HCl, 2,62 g de tris (base livre) e 13,2 g de NaCl em 800 mL de água desionizada e adicione 10 mL de uma solução TX-100 de 10% (v/v).
- Verifique o pH na RT e ajuste para pH 7,5 com NaOH ou HCl [5 M], se necessário.
- Fazer até 1 L e filtrar através de uma membrana de nitrocelulose de 0,22 μm.
- Adicionar 35 μL de EtSH por 100 mL de tampão (concentração final de 5 mM) directamente antes da utilização.
- Tampão de lavagem 250 (WB250): 250 mM NaCl, 40 mM Tris-HCl pH 8,0 (RT), 0,1% (v/v) TX-100, 5 mM EtSH.
- Prepare WB250 como descrito para B250 (2.1.1) mas com 3,59 g de Tris-HCl, e 2, 8 g de tris (base livre).
- Tampão de lavagem 150 (WB150): 150 mM NaCl, 40 mM Tris-HCl pH 8,0 (RT), 0,1% (v/v) TX-100, 5 mM EtSH.
- Prepare WB150 conforme descrito para B250 (2.1.1), mas com 3,59 g de Tris-HCl, 2, 8 g de tris (base livre) e 8,77 g de NaCl.
- Tampão de equilíbrio de TEV (TEB): 150 mM NaCl, 40 mM Tris-HCl pH 8,0 (RT), 0,5 mM EDTA, 0,1% (v/v) TX-100, 5 mM EtSH.
- Mesmos que WB150 mas adicionam o EDTA à concentração final de 0,5 milímetros.
- Tampão do clivagem de TeV (TCB): 150 mm NaCl, 40 mm Tris-HCl pH 8,0 (RT), 0,5 mm EDTA, 0,1% (v/v) TX-100, 10% (v/v) glicerol, 5 mm etsh.
- Prepare-se conforme descrito na etapa 2.1.1 com 3,59 g de Tris-HCl, 2, 8 g de tris (base livre) e 8,77 g de NaCl e adicione 100 mL de glicerol antes de ajustar o volume para 1 L.
- 50 cocktail de inibidor de protease ×: dissolva 1 comprimido de um cocktail disponível comercialmente de inibidores da protease em 1 mL de água e armazene a-20 ° c.
- TBS-T: 50 mM Tris-HCl pH 7,6 (RT), 0,9% (p/v) NaCl, 0,1% (v/v) polissorbato 20.
- Prepare-se conforme descrito na etapa 2.1.1. Use 6, 6 g de Tris-HCl, 1,40 g de tris (base livre), 9 g de NaCl e 1 mL de polissorbato 20.
- 5 × LSB (tampão da amostra de Laemmli): 315 mM Tris-HCl pH 6,8 (RT), 25% (v/v) EtSH, 50% (v/v) glicerol, 10% (w/v) SDS, 0, 5% (w/v) azul de bromofenol.
- Tampão de extração (B250): 250 mM NaCl, 100 mM Tris-HCl pH 7,5 (RT), 0,1% (v/v) TX-100, 5 mM EtSH.
- Preparação de extrato proteico
- Adicionar 1,5 g de micélio liofilizado e uma bola de moagem no pote de moagem de um moinho de bolas.
Nota: o micélios fresco pôde ser usado também. Ao fazê-lo, seque o micélio tão completamente quanto possível antes de congelar e certifique-se de precool os frascos de moagem em nitrogênio líquido para moer micélio fresco. - Moer micélio em pó a 25 Hz por 30 s.
Nota: nos casos em que não há máquina de moagem disponível, moer micélio para um pó usando um almofariz e pilão, como descrito por Bayram et al. doze anos. - Transfira o pó micelial para um tubo centrífugo de 15 mL.
- Incline o tubo de centrífuga incluindo micélio moído para permitir a subsequente mistura de micélio com tampão.
- Adicione 6 mL de gelo-frio B250 incluindo 1 × coquetel inibidor de protease por grama de pó micelial e misture com uma pequena espátula até que a homogeneização completa do extrato bruto seja alcançada. Ao utilizar micélio fresco, consulte Bayram et al. 12 para atingir a biomassa direita para a relação tampão de extração.
- Mantenha o tubo no gelo por 5 min.
- Coloque o tubo e um tubo de equilíbrio em uma centrífuga e gire em 40.000 × g para ≥ 20 min a 4 ° c. Realize a equilibração da resina de IgG (etapa 2,3) durante a centrifugação.
- Após centrifugação, retire 10 μL do sobrenadante para análise SDS-PAGE. Colocar a amostra num tubo de 1,5 mL contendo 40 μL de água e 12,5 μL de 5 × LSB; referido como "ex" na Figura 1.
- Retire cuidadosamente o sobrenadante (lisado limpo) utilizando uma pipeta serológica e transfira para a coluna que contém as esferas de IgG equilibradas (passo 2.3) e feche firmemente com a tampa de fim fornecida.
- Adicionar 1,5 g de micélio liofilizado e uma bola de moagem no pote de moagem de um moinho de bolas.
- Equilibração da resina de IgG (executada durante a etapa 2.2.7)
- Prepare uma coluna de cromatografia descartável de 10 mL e pipeta 300 μL de resina IgG bem ressuspendida (50% de chorume) na coluna. Encha acima a coluna a 10 mL com B250 e deixe o buffer fluir completamente pela gravidade.
- Adicione 1 mL de B250 incluindo 1 × coquetel inibidor da protease e deixe fluir. Conecte a parte inferior da coluna.
- Purificação em lote da TAP-Tagged HDAC
- Incubar a coluna cromatográfica contendo os grânulos equilibrados e o lisado limpo do passo 2.2.9 num misturador rotativo a 10 RPM a 4 ° c durante 2 – 4 h.
- Após a vinculação do lote, remova a tampa e abra a coluna na parte inferior para coletar o fluxo.
- Tome uma amostra para a análise SDS-PAGE como descrito na etapa 2.2.8; referido como "FT" na Figura 1.
- Lave os grânulos com 10 mL de WB250. Primeiramente, use 1 ml do amortecedor para remover os grânulos prendidos da tampa da coluna usando um pipetador e transfira esta suspensão em um flush na resina assentada para permitir o ressuspensão dos grânulos. Em seguida, encha a coluna para a parte superior, feche usando uma tampa de pilha e conecte-se a uma bomba peristáltica. Ajuste um caudal de aproximadamente 1 – 5 mL/min. impeça que a resina seque.
- Repita o passo 2.4.4 três vezes para um total de quatro lavagens com WB250.
- Lave os grânulos três vezes com 10 mL de TEB como descrito no passo 2.4.4.
- Feche a coluna de cromatografia na parte inferior, Ressuspender os grânulos de IgG em 1 mL de TCB e adicionar 20 μL de 50 × cocktail de inibidor da protease, bem como 10 ΜL de TeV (~ 1 mg/ml de estoque, variante mutante S219V produzida internamente).
- Tampe a coluna e incubar num misturador rotativo a 10 RPM a 4 ° c durante a noite para elute os complexos proteicos ligados através do HDAC etiquetado.
Nota: Alternativamente, realize a digestão TEV a uma temperatura elevada (16 – 25 ° c), o que reduz o tempo de reacção, no entanto, está a aumentar o risco de degradação das proteínas. - No dia seguinte, abra a coluna e colete o eluato em um tubo de centrífuga de 2 mL. Use 0,7 mL de TCB para remover os grânulos da tampa e enxague a parede da coluna.
- Coloque a coluna no tubo de 2 mL aberto a partir da etapa anterior que se coloca dentro de um tubo centrífugo de 50 mL.
- Transfira este conjunto em uma centrifugação da parte superior de tabela e gire-a em 300 × g por 2 minutos. Este é o eluato de TEV.
Nota: ao executar a purificação de afinidade em tandem, use o eluato TeV como entrada para a etapa de afinidade de calmodulina. Você igualmente pode rachar o eluato de TeV e usar uma peça para a determinação da atividade de HDAC e a segunda parte para terminar a purificação da torneira. - Remova 50 μL do eluato de TEV e adicione 12,5 μL de 5 × LSB para SDS-PAGE (referido como "TE" nas figuras 1 e 2) e mantenha o eluato remanescente no gelo.
- Para avaliar a eficácia da eluição da protease, adicionar 2 mL de ácido acético a 5% (v/v) e incubar na RT durante 5 min. Novamente, use o primeiro mL para Ressuspender a resina.
- Colete o eluato de ácido e volte a tomar 50 μL de amostra e adicione 12,5 μL de 5 × LSB (referido como "AE" na Figura 1). LSB vai virar amarelo quando adicionado à solução ácida. Para neutralizar o ácido, adicione 10 M de NaOH em passos de 1 μL e misture bem até que a cor mude para azul novamente.
- Re-equilibrar a resina de IgG com TBS-T para neutralizar o ácido. Armazene a resina em TBS-T/20% (v/v) etanol a 4 ° c para reutilização com a mesma proteína marcada.
- Armazenamento de frações de eluição
- Aliquot o eluato em ~ 100 frações de μl, para evitar vários ciclos de congelamento-descongelação.
- Congelar alíquotas em nitrogênio líquido e manter a-80 ° c.
Nota: as amostras armazenadas desta forma serão estáveis durante meses sem perder a atividade enzimática.
3. análise de purificação por SDS-PAGE e western blotting
- Use protocolos padrão para a fundição de géis de poliacrilamida SDS ou use géis pré-moldados13. Amostras de gel de denature da seção anterior em 95 ° c por 5 min, centrifugar em ≥ 15.000 × g por 5 min, e carregar em géis de 12%. Os volumes de carregamento recomendados são fornecidos na legenda da Figura 1. Electrophorese as amostras em 1 × tampão running da sds-página da Tris-glicina em 180 constante de V por 60-70 minutos, até que o marcador azul do bromofenol do amortecedor do carregamento SDS-PAGE comece a migrar fora do gel.
- Use protocolos padrão para coloração de prata dos géis. Por exemplo, use o protocolo de Blum et al. 198714 que também é compatível com a análise de MS.
- Use protocolos padrão para western blotting15. Para a geração de resultados representativos abaixo, foi utilizado um sistema de blotting comercialmente disponível.
- Manchas de sonda com anticorpo anticalmodulina (anti-CBP) de proteína obrigatória comercialmente disponível em 5% (p/v) de leite em pó em TBS-T a 4 ° c durante a noite.
Nota: anticorpo anti-CBP é dirigido contra a parte da tag TAP ainda presente após clivagem TEV. - Use protocolos padrão para detecção e desenvolvimento de blots. Por exemplo, o Conjugado IgG-fosfatase alcalina anti-coelho e um substrato de desenvolvimento de cor BCIP/NBT foram utilizados para a geração de resultados representativos abaixo.
4. ensaio de deacetilase utilizando in vitro [3H] acetato-rotulado de aves reticulócitos histonas (adaptado de trojer et al. 2003)4
- Refira o protocolo de Kölle et al. 199816 para a preparação de [3H] acetato-rotulado histonas do reticulócitos da galinha.
- Por a condição do ensaio coloc três 1,5 tubos de centrifugação do mL no gelo para medidas em triplicates. Prepare também três tubos para o controle de fundo de buffer-only.
- Coloque 25 μL de WB150 em cada tubo e adicione 25 μL do eluato de TEV do passo 2.4.11 e mantenha-o no gelo.
- Pré-aqueça uma incubadora do tubo a 25 ° c.
- Use os intervalos de 15 s (relógio de parada) para a adição de 10 μL de histonas rotuladas [1,5 mg/mL] a cada tubo.
- Antes de iniciar o ensaio, tome até 10 μL das histonas de frango rotuladas com acetato de [3H] e inicie o relógio de parada.
- Cinco segundos após o início, adicione as histonas rotuladas, feche o tubo firmemente, vórtice, e colocá-lo na incubadora.
- Use intervalos de 15 s para a adição de 10 μL de histonas rotuladas e proceda conforme descrito na etapa anterior.
- Após 60 min de incubação adicionar 50 μL de solução de paragem (1 M HCl/0,4 M de ácido acético) para cada tubo em intervalos de 15 s; vórtice imediatamente. Após a adição da solução ácida, a mistura do ensaio é estável e pode ser mantida na RT até o próximo passo.
- Adicionar 800 μL de acetato de etila a cada tubo para extrair o ácido acético libertado [3H].
- Feche firmemente os tubos e vórtice cada tubo para 5 s.
- Coloque o tubo em uma microcentrífuga e gire em 10.000 × g por 10 min em RT.
- Entretanto, prepare um frasco de cintilação por amostra de ensaio e adicione 3 mL de cocktail de cintilação para amostras hidrofóbicas.
- Após centrifugação (passo 2,12), transfira cuidadosamente 600 μL da fase orgânica superior para os tubos de cintilação preparados e feche os tubos firmemente.
- Meça a radioatividade correspondente à atividade HDAC em um contador de cintilação líquida.
Resultados
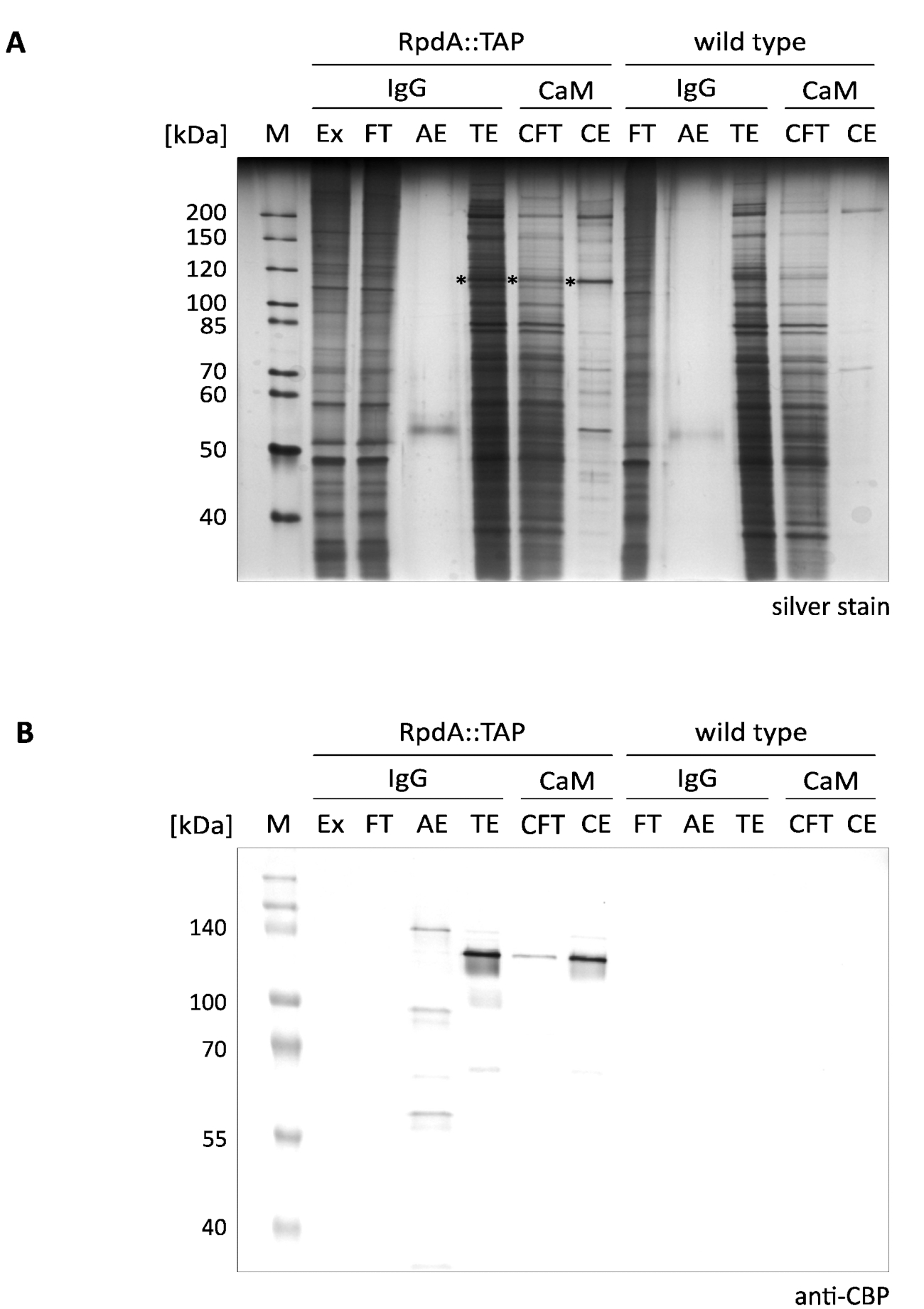
Um resultado típico do enriquecimento de um único passo apresentado de RpdA é mostrado na Figura 1 (referido como "IgG"). Por uma questão de completude, também incluímos frações de fluxo e eluição ("CFT" e "CE") ilustrando o segundo passo de purificação por uma resina de calmodulina ("CaM") conforme descrito12. O gel de prata manchado exibido (a) ilustra claramente a eficácia da primeira etapa de afinidade que é ainda mais aumentada ao executar a purificação em tandem. As proteínas mais proeminentes presentes no extrato proteico e o escoamento, entretanto, já estão esgotadas no eluato de TEV (TE). É importante notar que as frações de eluição TEV são > 100 × concentrado em comparação com o extrato e fluxo-através. A marca de asteriscos marcou RpdA (compare o painel B). O MW calculado de RpdA que inclui o CBP (RpdA:: CBP) é 82 kDa, entretanto, a proteína migra em um peso molecular aparente muito mais elevado de aproximado. 120 kDa. Esse fenômeno tem sido observado anteriormente e pode ser atribuído às propriedades específicas de seu C-Terminus4,17. O immunoblot (B) mostra os sinais fortes que migram em aproximado. 120 kDa que corresponde a CBP-Tagged full-length rpda (rpda:: CBP) no eluato de TeV ("te"), o fluxo-através do calmodulina ("CFT"), e eluato ("CE") frações. No eluato de ácido ("AE") um segundo sinal com um MW ligeiramente maior é visível. Isto representa a proporção de RpdA TAP-Tagged ligado à resina de IgG que não foi liberado pela clivagem de TEV. A diferença no tamanho corresponde a 16 kDa da proteína uma repetição de RpdA uncleaved:: torneira. Como esperado, nenhuma faixa poderia ser detectada pelo anticorpo anti-CBP nas frações de controle do tipo selvagem (painel B, "tipo selvagem").
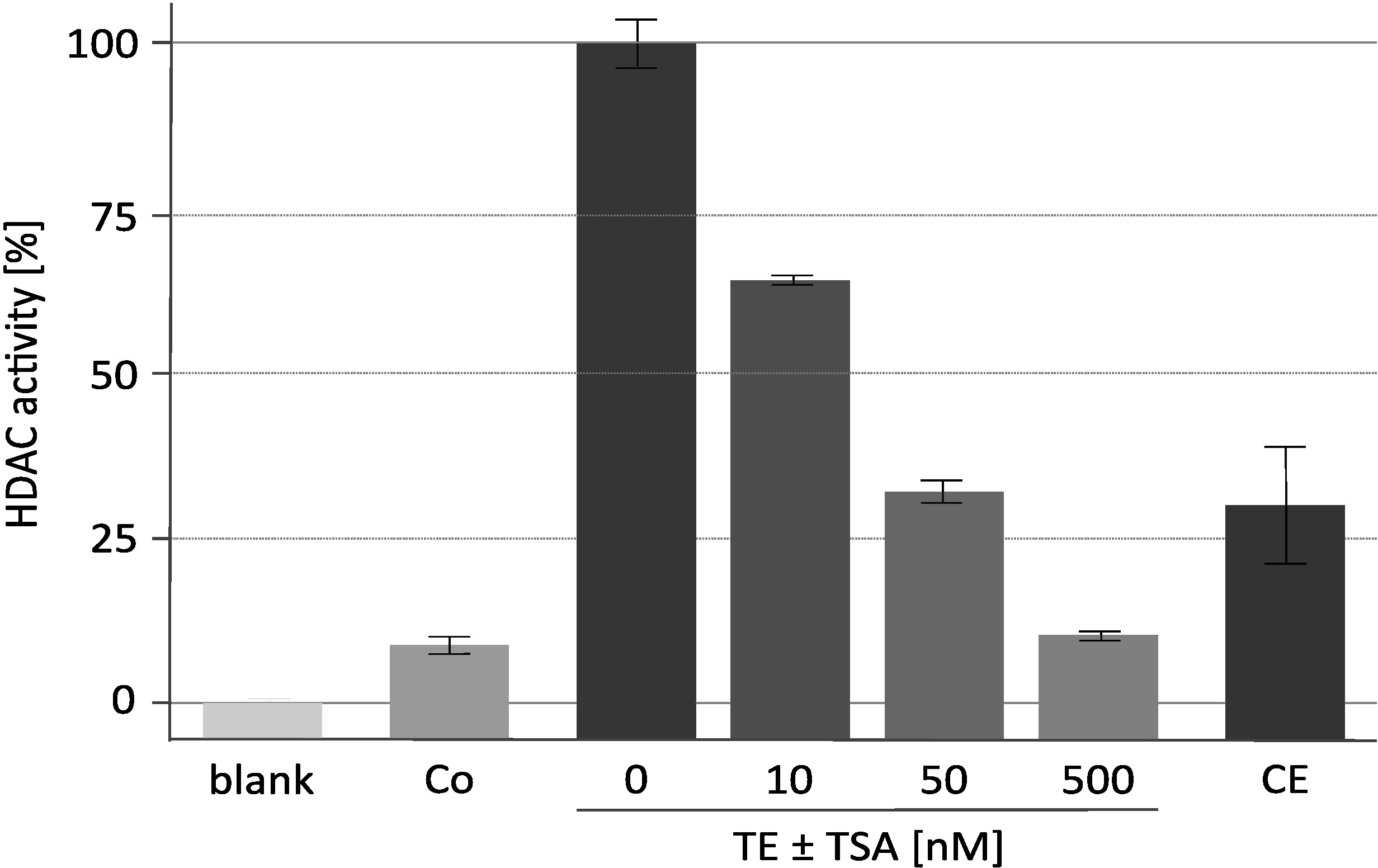
Os resultados de um ensaio representativo da atividade da desacetilase com o inibidor específico de HDAC tricostatina a (TSA) são mostrados na Figura 2. Este ensaio foi desenvolvido originalmente para plantas18 e foi usado igualmente para a seleção do inibidor de encontro aos desacetilases mamíferos19,20. As histonas utilizadas para o ensaio foram rotuladas e preparadas conforme descrito16. O efeito do aumento das concentrações de TSA na atividade catalítica do complexo RpdA enriquecido ("TE ± TSA") é mostrado. A sensibilidade da atividade confirma que os valores medidos do CPM são realmente devido a RpdA e não causados pela atividade inespecífica do protease. Esta é uma observação importante porque indica que o TEV, que está atual em uma concentração rather elevada, não interfere com o ensaio da atividade de HDAC. A fim atribuir a atividade de HDAC medida ao RpdA, uma estirpe do selvagem-tipo foi usada como o controle negativo ("co"). Como esperado, somente a atividade marginal de HDAC (aproximado. 5-10% de frações rpda-enriquecidas) foi detectada no eluato de TeV do tipo selvagem. Curiosamente, a atividade HDAC é significativamente reduzida após a segunda etapa de purificação de afinidade ("CE") quando comparada com o eluato de TEV.

Figura 1. A purificaçãode afinidade em tandem da TAP-Tagged RpdA. Um gel 10% de prata-manchado SDS-polyacrylamide (a) e um borrão ocidental sondada com o anticorpo do anti-CBP (B) são indicados. A rotulagem da pista e os volumes carregados são os seguintes: "M": 2 μl de 1:10 marcador de proteína não manchado diluído (mancha de prata), 3,5 μl de marcador de proteína Prestained (Western blot); "Ex": amostra de extrato proteico como preparada na etapa 2.2.8 (2 μl de diluição de 1:10, 5 μl); "FT": amostra de fluxo de resina IgG preparada na etapa 2.4.3 (2 μl de diluição de 1:10, 5 μl); "AE": eluato de ácido do passo 2.4.14 (10 μl, 10 μl); "TE": eluição de TEV da elução overnight pela clivagem de TEV, passo 2.4.12 (10 μl, 10 μl); "CFT": fluxo de calmodulina (20 μL, 20 μL); "CE": eluato de calmodulina (10 μL, 10 μL). O tamanho das proteínas do marcador selecionado é indicado no lado esquerdo dos painéis. Os volumes dados entre parênteses correspondem às cargas da amostra para a mancha de prata e o borrão ocidental, respectivamente. Asteriscos no gel manchado de prata indicam a proteína de fusão RpdA. O immunoblot (B) foi detectado com fosfatase alcalina usando o sistema de desenvolvimento da cor de BCIP/NBT. Estale por favor aqui para ver uma versão maior desta figura.
{kind=link}

Figura 2. Ensaio de atividade de HDAC concentrações crescentes de tricostatina A. A eficácia da inibição de RpdA foi testada com 25 μL de RpdA recombinante afinidade-purified ("TE") e 25 μL de 0, 10, 50, e 500 nanômetro do inibidor de HDAC TSA diluído no meio de RPMI-1640. O RPMI foi utilizado para avaliar a atividade de fundo ("blank"). As atividades do eluato final após a segunda etapa da afinidade do calmodulina ("CE") e de uma tensão untagged após a primeira etapa da purificação da afinidade (controle negativo, "co") são indicadas. As atividades são mostradas como percentagem de RpdA enriquecido sem TSA (100%, "TE"). As barras de erro indicam o desvio padrão de três repetições. Este número foi modificado de Bauer et al. 20162. Estale por favor aqui para ver uma versão maior desta figura.
{kind=link}
Discussão
Este protocolo descreve um enriquecimento da único-etapa de uma classe Tap-Tagged 1 HDAC do fungo filamentosos a. nidulans para a avaliação da atividade in vitro do desacetilase. A tag TAP foi originalmente introduzida no fermento de Baker para a identificação de parceiros de interação proteína-proteína da proteína etiquetada6. Subseqüentemente, o Tag era Codon-aperfeiçoado para seu uso em A. nidulans5. Aqui nós fornecemos um protocolo passo a passo direto para a aplicação da primeira etapa de purificação de afinidade da estratégia TAP para o enriquecimento em uma única etapa da classe 1 HDAC RpdA. A segunda etapa da purificação da afinidade aumenta claramente o nível de purificação, que é particularmente importante para a identificação de proteínas isca-interagindo. No entanto, apenas o primeiro passo é recomendado para o teste de atividade subsequente, uma vez que a falta de contaminação significativa após o primeiro passo foi confirmada por um experimento de controle usando uma estirpe de tipo selvagem. Além disso, a atividade eluída é consideravelmente mais elevada após o enriquecimento em uma única etapa, quando comparada com a da TAP completa. Além do que o rpda2, este protocolo foi usado igualmente com sucesso para purificação os complexos de a. nidulans da segunda classe 1 HDAC HosA21.
Devido às nossas observações de que os HDACs de classe fúngica 1 constroem complexos estáveis4, conseguimos modificar o protocolo de Bayram et al. 201212 que representa a base deste método. No entanto, alguns passos críticos têm de ser mencionados. A preparação de extratos proteicos altamente concentrados para garantir condições quase fisiológicas é fundamental para uma estabilidade complexa. Portanto, é importante ter em mente as recomendações dadas a respeito da relação tampão biomassa/extração. A este respeito, também é fundamental para usar bem moído fino pós micelial para garantir a extração adequada. Aqui, o uso de uma máquina de moagem é claramente vantajoso. Como mencionado na seção do protocolo, vale a pena tentar a etapa do TEV-Cleavage para 1-2 h na temperatura ambiente a fim acelerar a purificação. Isso foi testado para RpdA sem observar quaisquer efeitos deletérios sobre a estabilidade (observação inédita, Bauer I, 2018). Além, a recolocação de NP40 (usada no protocolo original) com TX-100 não pôde ser apropriada para outros complexos da proteína. Ao usar este método para a purificação de outras proteínas TAP-etiquetadas, uma deve igualmente consultar ao protocolo de Bayram, que contem um número de sugestões valiosas que puderam ser úteis para uma purificação suficiente de outros complexos da proteína12.
Além do aqui descrito TAP-método, outras marcas de afinidade e técnicas são comumente usados para o enriquecimento de uma única etapa de proteína de fungos filamentosos, incluindo his-e GFP-Tags. Entretanto, porque os HDACs da classe 1 são geralmente funcionais como complexos high-molecular-weight, as circunstâncias nativas da eluição são um pré-requisito para o enriquecimento de HDACs catalètica ativos. É importante ressaltar que muitas outras purificações de afinidade são realizadas em condições desfavoráveis. Por exemplo, o enriquecimento de HDACs através da GFP-armadilha, que é baseado na interação do antígeno-anticorpo, não é apropriado devido às condições de eluição ácida que interferem com a interação da proteína-proteína de complexos de HDAC acoplados às resinas. Além disso, as tentativas de purificar o seu-Tagged RpdA por cromatografia de afinidade quelato de metal22, resultou em uma perda significativa de atividade catalítica durante o procedimento de purificação, embora imidazol em vez de condições de pH baixo foi usado para eluição ( dados inéditos, Bauer, I, 2010).
Uma limitação do protocolo de ensaio enzimático descrito é o uso do substrato radioativo. Entretanto, os ensaios em uma base fluorescente foram desenvolvidos também23,24 e são comercialmente disponíveis. Estes ensaios são executados em placas de poço e, portanto, são adequados também para a triagem de alta taxa de transferência de inibidores HDAC. Nesse caso, seria necessário um upscale do procedimento apresentado.
As possíveis aplicações futuras deste protocolo incluem o enriquecimento de subcomplexos específicos de HDACs de classe 1 para avaliar os seus papéis fisiológicos específicos e/ou diferenças na sua susceptibilidade aos inibidores da HDAC. Ao estabelecer o método descrito para outras enzimas, é altamente recomendável realizar um experimento de controle negativo com uma cepa sem etiqueta. Isso garante a especificidade das atividades enzimáticas mensuradas e revelará contaminação por enzimas não especificamente ligadas.
A purificação e o ensaio da atividade descritos aqui podem ser executados no prazo de dois dias e as enzimas enriquecidas são estáveis por pelo menos diversos meses, quando armazenadas nas alíquotas em-80 ° c. Em conclusão, este protocolo fornece uma maneira relativamente simples e rentável de conseguir os complexos da classe 1 HDAC para a medida da atividade e a determinação da eficácia do inibidor.
Divulgações
Os autores não têm nada a divulgar.
Agradecimentos
Gostaríamos de agradecer a Petra Merschak, divisão de biologia molecular (Biocenter, universidade médica de Innsbruck), por sua ajuda e apoio em relação a este manuscrito. Além disso, gostaríamos de agradecer aos revisores por seus valiosos comentários.
Este trabalho foi financiado pelo fundo austríaco de ciência (P24803 para SG e P21087 para GB) e pelo financiamento intramural (MUI Start, ST201405031 to IB).
Materiais
| Name | Company | Catalog Number | Comments |
| 10 x SDS-PAGE running buffer | Novex | ||
| 2-mercaptoethanol (EtSH) | Roth | 4227 | |
| 25 cm2 cell culture flasks with vent cap | Sarstedt | 833910002 | For spore production |
| 47 mm vacuum filtration unit | Roth | EYA7.1 | |
| AccuFLEX LSC-8000 | HITACHI | – | Scintillation counter |
| Acetic acid | Roth | 7332 | |
| Anti-Calmodulin Binding Protein Epitope Tag Antibody | Millipore | 07-482 | Used at 1:1333 dilution |
| Anti-Rabbit IgG (whole molecule)–Alkaline Phosphatase (AP) antibody | Sigma-Aldrich | A3687 | |
| Ball mill | Retsch | 207450001 | Mixer Mill MM 400 |
| BCIP/NBT | Promega | S3771 | Color development substrate for AP |
| Cell strainer | Greiner | 542040 | |
| Cheese cloth for harvesting mycelia | BioRen | H0028 | Topfentuch |
| Dimethylsulfoxid (DMSO) | Roth | 4720 | |
| EDTA | Prolabo | 20309.296 | |
| Ethyl acetate | Scharlau | Ac0155 | |
| Freeze Dryer | LABCONCO | 7400030 | FreeZone Triad |
| Glycerol | Roth | 3783 | |
| HCl | Roth | 4625 | |
| IgG resin | GE Healthcare | 17-0969-01 | IgG Sepharose 6 Fast Flow |
| Inoculation loops | VWR | 612-2498 | |
| KOH | Merck | 5033 | |
| Laminar flow cabinet | Thermo Scientific | – | Hera Safe KS |
| Mixed Cellulose Esters Membrane Filters | Millipore | GSWP04700 | |
| NaCl | Roth | 3957 | |
| NaOH | Roth | 6771 | |
| Neubauer counting chamber improved | Roth | T728 | |
| Novex gel system | Thermo Scientific | For SDS-PAGE | |
| Novex Tris-glycine SDS running buffer (10X) | Thermo Scientific | LC2675 | Running buffer for SDS-PAGE |
| peqGold protein-marker II | VWR | 27-2010P | Protein ladder used for silver stain |
| Peristaltic Pump P-1 | GE Healthcare | 18111091 | |
| Pipette controller | Brand | 26302 | accu-jet pro |
| Poly-Prep chromatography columns | Bio-Rad | 731-1550 | |
| ProSieve QuadColor protein marker | Biozym | 193837 | Prestained protein ladder used for western blot |
| Protease inhibitor cocktail tablets | Sigma-Aldrich | 11873580001 | cOmplete, EDTA-free |
| Rotary mixer | ELMI | – | Intelli-Mixer RM-2 S |
| Rotiszint eco plus | Roth | 0016 | |
| RPMI-1640 | Sigma-Aldrich | R6504 | |
| Scintillation vials | Greiner | 619080 | |
| Sorvall Lynx 4000 | Thermo Scientific | ||
| Thermomixer comfort | Eppendorf | ||
| TIB32.1 | A. nidulans rpdA::TAP strain. Genotype: alcA(p)::rpdA; veA1; argB2; yA2; pIB32::argB; ArgB+; PyrG+ | ||
| Trans-Blot Turbo RTA Midi Nitrocellulose Transfer Kit | Bio-Rad | 1704271 | |
| Trans-Blot Turbo Transfer System | Bio-Rad | 1704150 | |
| Trichostatin A (TSA) | Sigma-Aldrich | T8552 | 5 mM stock in DMSO |
| Tris (free base) | Serva | 37190 | |
| Tris-HCl | Roth | 9090 | |
| Polysorbate 20 | Roth | 9127 | Tween 20 |
| Polysorbate 80 | Sigma-Aldrich | P1754 | Tween 80 |
| TX-100 | Acros Organics | 215682500 | Triton X-100, Octoxynol-9 detergent |
Referências
- Gregoretti, I. V., Lee, Y. -. M., Goodson, H. V. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. Journal of Molecular Biology. 338 (1), 17-31 (2004).
- Bauer, I., et al. A Class 1 Histone Deacetylase with Potential as an Antifungal Target. mBio. 7 (6), (2016).
- Stefan, A., et al. Purification of active recombinant human histone deacetylase 1 (HDAC1) overexpressed in Escherichia coli. Biotechnology Letters. 40 (9-10), 1355-1363 (2018).
- Trojer, P., et al. Histone deacetylases in fungi: novel members, new facts. Nucleic Acids Research. 31 (14), 3971-3981 (2003).
- Bayram, &. #. 2. 1. 4. ;., et al. VelB/VeA/LaeA complex coordinates light signal with fungal development and secondary metabolism. Science. 320 (5882), 1504-1506 (2008).
- Puig, O., et al. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods. 24 (3), 218-229 (2001).
- Rigaut, G., et al. A generic protein purification method for protein complex characterization and proteome exploration. Nature Biotechnology. 17 (10), 1030-1032 (1999).
- Hill, T. W., Kafer, E. Improved protocols for Aspergillus minimal medium: trace element and minimal medium salt stock solutions. Fungal Genetics Reports. 48, (2001).
- Todd, R. B., Davis, M. A., Hynes, M. J. Genetic manipulation of Aspergillus nidulans: meiotic progeny for genetic analysis and strain construction. Nature Protocols. 2 (4), 811-821 (2007).
- Zadra, I., Abt, B., Parson, W., Haas, H. xylP promoter-based expression system and its use for antisense downregulation of the Penicillium chrysogenum nitrogen regulator NRE. Applied and Environmental Microbiology. 66 (11), 4810-4816 (2000).
- Stolz, D. J., et al. Histological Quantification to Determine Lung Fungal Burden in Experimental Aspergillosis. Journal of Visualized Experiments. (133), (2018).
- Bayram, &. #. 2. 1. 4. ;., et al. Identification of protein complexes from filamentous fungi with tandem affinity purification. Methods in Molecular Biology. 944, 191-205 (2012).
- Laemmli, U. K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 227 (5259), 680-685 (1970).
- Blum, H., Beier, H., Gross, H. J. Improved silver staining of plant proteins, RNA and DNA in polyacrylamide gels. Electrophoresis. 8 (2), 93-99 (1987).
- Towbin, H., Staehelin, T., Gordon, J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Biotechnology. 24, 145-149 (1992).
- Kölle, D., et al. Biochemical methods for analysis of histone deacetylases. Methods. 15 (4), 323-331 (1998).
- Tribus, M., et al. A novel motif in fungal class 1 histone deacetylases is essential for growth and development of Aspergillus. Molecular Biology of the Cell. 21 (2), 345-353 (2010).
- Sendra, R., Rodrigo, I., Salvador, M. L., Franco, L. Characterization of pea histone deacetylases. Plant Molecular Biology. 11 (6), 857-866 (1988).
- Valente, S., et al. 1,3,4-Oxadiazole-containing histone deacetylase inhibitors: anticancer activities in cancer cells. Journal of Medicinal Chemistry. 57 (14), 6259-6265 (2014).
- Mai, A., et al. Synthesis and biological properties of novel, uracil-containing histone deacetylase inhibitors. Journal of Medicinal Chemistry. 49 (20), 6046-6056 (2006).
- Pidroni, A., et al. A Class 1 Histone Deacetylase as Major Regulator of Secondary Metabolite Production in Aspergillus nidulans. Frontiers in Microbiology. 9, 2212 (2018).
- Porath, J., Carlsson, J., Olsson, I., Belfrage, G. Metal chelate affinity chromatography, a new approach to protein fractionation. Nature. 258 (5536), 598-599 (1975).
- Wegener, D., Wirsching, F., Riester, D., Schwienhorst, A. A fluorogenic histone deacetylase assay well suited for high-throughput activity screening. Chemistry & Biology. 10 (1), 61-68 (2003).
- Peng, L., Yuan, Z., Seto, E. Histone deacetylase activity assay. Methods in Molecular Biology. 1288, 95-108 (2015).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados