Method Article
Análise genoma-em todo o DNA da metilação de DNA no câncer gastrointestinal
Neste Artigo
Resumo
Aqui, descrevemos um procedimento para análise genoma de metilação de DNA em cânceres gastrointestinais. O procedimento é de relevância para estudos que investigam relações entre padrões de metilação de genes e fatores que contribuem para a carcinogênese em cânceres gastrointestinais.
Resumo
A metilação do DNA é uma importante mudança epigenética que é biologicamente significativa e um foco frequente da pesquisa do câncer. A metilação de DNA em todo o genoma é uma medida útil para fornecer uma análise precisa do estado de metilação das malignidades gastrointestinais (GI). Dado os múltiplos usos translacionais potenciais da análise da metilação de DNA, os médicos praticantes e outros novos estudos de metilação de DNA precisam ser capazes de entender passo a passo como essas análises em todo o genoma são realizadas. O objetivo deste protocolo é fornecer uma descrição detalhada de como este método é usado para a identificação biomarcadora em malignidades gi. É importante ressaltar que descrevemos três etapas críticas necessárias para obter resultados precisos durante a análise em todo o genoma. De forma clara e concisa, esses três métodos são muitas vezes carentes e não perceptíveis para aqueles novos estudos epigenéticos. Foram utilizadas 48 amostras de uma malignidade gi (câncer gástrico) para destacar praticamente como a análise de metilação de DNA em todo o genoma pode ser realizada para as malignidades de GI.
Introdução
Epigenética refere-se a alterações hereitárias na função genética sem alteração da sequência de DNA1. Tais alterações podem ser devido à metilação do DNA, na qual grupos de metila em uma base de DNA podem alterar a expressão genética através de alterações na embalagem de cromatina. O desenvolvimento e a progressão do câncer podem ocorrer se esse efeito resultar na expressão alterada dos genes supressores tumorais2. Envelhecimento e inflamação crônica são as causas do câncer e as principais razões para alterações na metilação do DNA em humanos3,4,5. Consequentemente, isso permite a utilização da metilação do DNA como biomarcador no diagnóstico de câncer, e como alvo de tratamento e prevenção. Para detecção precoce e prognóstico de câncer, a metilação de DNA está sendo medida em amostras de tumor, sangue, urina e fezes6, enquanto agentes desmetiladores estão sendo usados para tratar leucemias como a síndrome mielodsplásica7.
A análise de metilação de DNA em todo o genoma utilizando uma plataforma de matriz para avaliação complexa da metilação de DNA em um lócus cpg individual no genoma humano pode ser utilizada para examinar o status de metilação de mais de 450.000 locais de CpG no DNA genômico8,9, que permite a exploração de epigenéticas cancerígenas (ver Tabela de Materiais). As tecnologias de sequenciamento de bissulfito de genoma inteiro (WGBS) mudaram nossas abordagens no campo da epigenética10,11. No entanto, existem algumas desvantagens para as tecnologias em termos de um custo substancial e tempo de processamento para análise epigenética de um grande número de amostras10,11. Portanto, a plataforma de matriz é mais viável para avaliação complexa da metilação de DNA no genoma humano. A disponibilidade de abordagens para análises de metilação em todo o genoma melhorou nos últimos anos e nos permite expandir nosso conhecimento de como a metilação do DNA contribui para o desenvolvimento e progressão do câncer12. Os recentes avanços nas abordagens da plataforma de microarray nos fornecem a lógica para a análise da metilação em todo o genoma para identificar uma nova assinatura epigenética em cânceres gastrointestinais13. O objetivo deste protocolo é fornecer uma descrição detalhada de como este método é usado para identificação de biomarcadores em malignidades gi.
Protocolo
Todos os procedimentos seguidos estavam de acordo com os padrões éticos do comitê de ética em pesquisa humana das instituições. O estudo foi aprovado pelo Conselho de Revisão Institucional do Hospital Shizuoka da Universidade de Juntendo, e o consentimento por escrito foi dispensado por causa do desenho retrospectivo.
1. Lavar os slides
- Prepare 10 μm de seções de parafina fixa-formalina não manchada (FFPE).
- Coloque slides em um suporte de lâmina de vidro: use cerca de 3-5 maiores lâminas transversais, a menos que o tecido seja mínimo, e mais slides sejam necessários.
- Encha o suporte de slides com xileno e certifique-se de que todo o tecido na lâmina está submerso. Deixe descansar por 15 minutos.
- Depois de 15 minutos, despeje o xileno com os slides segurados com uma ponta de pipeta para que os slides não caiam.
- Despeje mais xileno no mesmo nível de antes. Deixe descansar por mais 15 minutos.
- Depois de 15 minutos, despeje o xileno novamente.
- Encha o suporte de slides com 100% de etanol (EtOH), e certifique-se de que todo o tecido na lâmina está totalmente submerso. Deixe descansar por 3 minutos.
- Despeje o EtOH enquanto segura cuidadosamente os slides. Refil para o mesmo nível com mais EtOH. Deixe descansar por 2 minutos.
- Despeje novamente o EtOH e remova o slide. Coloque-os cuidadosamente de frente para cima em uma toalha de papel limpa para secar. Deixe descansar por 10 minutos.
2. Raspando os slides
- Prepare o tampão de digestão/lise com 650 mL de água tratada com dietilpiroto (DEPC), 100 mL de ácido etilenodiaminatotraacético (EDTA), 50 ml de cloridrato tris (Tris-HCL) 2 M pH 8,8, e 200 mL de sulfato de dodecil de sódio de 10% de sódio (SDS) (ver Tabela de Materiais).
- Encha um tubo de polipropileno de uso único de 1,5 mL com 80 μL de tampão de digestão/lise (ver Tabela de Materiais).
- Coloque uma ponta de pipeta limpa no tampão.
- Identificar tecidos cancerígenos da área tumoral mais adequados para macrodisseção de acordo com a seção manchada de H&E apropriada.
NOTA: A área tumoral para macrodisseção deve ser identificada preferencialmente por dois patologistas qualificados. - Tecido de câncer macrodissect baseado na seção manchada de H&E.
- Pegue uma lâmina de barbear limpa e raspe suavemente o tecido do câncer do slide, tentando mantê-lo inteiro para que seja mais fácil de trabalhar.
- Tire a ponta da pipeta do tubo de polipropileno de uso único contendo tampão e use-a para transferir o tecido sucateado para o frasco tampão (ver Tabela de Materiais).
NOTA: A ponta molhada deve atrair o tecido, o que facilita a transferência.
- Repita o passo 2.5 com o resto dos slides.
- Depois de todo o tecido estiver em um tubo de polipropileno de uso único, use a ponta para garantir que o tecido esteja totalmente submerso e não esteja preso à parede do tubo.
- Adicione 20 μL de uma protease serina relacionada à subtilisina ao frasco e filme suavemente para misturar (ver Tabela de Materiais).
- Coloque o frasco em um bloco de calor de 55 °C por pelo menos 4h ou durante a noite. Certifique-se de um pouco de vórtice depois de 2h.
3. Tratamento de bisulfito
- Utilize 45 μL da solução de tecido digerido como amostra.
- Realize o tratamento de bisullfita utilizando reagentes em um kit de conversão de bisullfita de acordo com as instruções do fabricante (ver Tabela de Materiais).
- Adicione 5 μL de tampão de diluição à amostra de DNA e incubar a 37 °C por 15 min (ver Tabela de Materiais).
- Enquanto as amostras estão incubando, prepare o reagente de conversão de bisulfato adicionando 750 μL de dH2O e 210 μL de tampão de diluição a um tubo de reagente de conversão ct (ver Tabela de Materiais). Misture os tubos por vórtice por 10 minutos.
- Após a incubação de 15 min, adicione 100 μL do reagente de conversão ct preparado a cada amostra e misture por inversão.
- Incubar as amostras no escuro a 50 °C por 12 a 16 h.
- Após a incubação, remova as amostras e coloque no gelo por 10 minutos.
- Adicione 400 μL de tampão de ligação e misture cada amostra por pipetação para cima e para baixo (ver Tabela de Materiais). Carregue cada amostra em uma coluna de spin e coloque a coluna em um tubo de coleta de 2 mL (ver Tabela de Materiais).
- Centrifugar cada amostra a toda velocidade (10.000 x g) por 1 min e descartar o fluxo.through.
- Adicione 200 μL de tampão de lavagem a cada coluna e gire a toda velocidade por 1 min, descartando o fluxo -through (ver Tabela de Materiais).
- Adicione 200 μL de tampão de dessulfonação a cada coluna e deixe a coluna ficar em temperatura ambiente por 15 minutos (ver Tabela de Materiais). Após a incubação, gire as colunas a toda velocidade por 1 min e descarte o fluxo.
- Adicione 200 μL de tampão de lavagem a cada coluna e gire a toda velocidade por 1 min (ver Tabela de Materiais).
- Repita este passo mais uma vez.
- Adicione 46 μL de dH2O a cada coluna e coloque cada coluna em um novo tubo de polipropileno de uso único de 1,5 mL (ver Tabela de Materiais). Gire cada tubo por 2 minutos para eluto o DNA.
- Remova cada coluna de giro do tubo de polipropileno de uso único e descarte (ver Tabela de Materiais). O DNA está pronto para a análise.
4. Plataforma de matriz para avaliação da metilação de DNA em um lócus CpG no genoma humano
- Avalie a qualidade do DNA (verificação de qualidade: QC) utilizando o ensaio FFPE QC em um instrumento de amplificação e detecção de PCR em tempo real, com análise de dados subsequente realizada de acordo com as instruções dos fabricantes (ver Tabela de Materiais).
NOTA: Amostras com valores ∆Cq inferiores aos 5.0 recomendados são processadas14. - Analisar amostras com uma plataforma de matriz para avaliação complexa da metilação de DNA para avaliar o estado de metilação de >450.000 sítios cpG no genoma (ver Tabela de Materiais). Folha de informações do produto, folha de dados e arquivos de produtos para a plataforma array estão disponíveis15.
NOTA: O ensaio é realizado de acordo com as instruções do fabricante para o materialFFPE 14. - Calcule a metilação de locus alta e baixa em uma ferramenta de análise de dados para uma plataforma de matriz como β-valor, com intervalo de 0 a 1, respectivamente (ver Tabela de Materiais). Use software comercialmente disponível (ver Tabela de Materiais) para calcular um valor de β.
- Importar dados gerados na plataforma de matriz para avaliação complexa da plataforma de metilação de DNA no ambiente de software R (R v.2.15.1) e processá-los usando uma ferramenta para analisar os arrays de metilação16,17.
NOTA: No mapa de calor, gerado por meio de uma ferramenta de análise de dados, as colunas são ordenadas por clustering não supervisionado, enquanto a ordem das linhas baseia-se na diminuição da significância da estatística t para metilação diferencial de cima para baixo. - Divida o mapa de aquecimento em grupos de metilação altos e baixos usando o primeiro diferencial no agrupamento não supervisionado.
NOTA: Para validação com PCR específico de metilação quantitativa (qMSP), escolha genes baseados em um valor de β maior em relação às ilhas CpG na região promotora, e devido à sua adequação para primer e design de sonda para qMSP.
5. PCR específico da metilação quantitativa (qMSP)
- Use o DNA modificado por bisulfita a partir da etapa 3.3 como modelo para PCR em tempo real baseado em fluorescência em qMSP para avaliar a metilação da região promotora em cada análise genética.
- Execute qMSP usando um instrumento PCR em tempo real de 96 poços (ver Tabela de Materiais).
- Verifique se há o status de metilação do promotor do gene alvo no DNA modificado por bisulfato usando primer avançado de 200 nM, primer reverso de 200 nM e sondas de 80 nM. Prepare o mix master com 16,6 mM (NH4)2SO4, 67 mM Tris pH 8.8, 10 mM β-mercaptoetanol, 10 nM fluoresceína, 0,166 mM de cada triphosfato desoxinucleotídeo e 0,04 U/μl de polimerase de DNA (ver Tabela de Materiais). O volume final de reação em cada poço para todos os ensaios é de 25 μL.
- Realizar o ciclismo de qMSP da seguinte forma: 95 °C para 5 minutos, seguido por 55 ciclos de 95 °C para 15 s, 60 °C para 1 min e 72 °C para 1 min.
NOTA: O gene alvo deve ser escolhido com base nos critérios de ter valores beta maiores, estar relacionado às ilhas CpG na região promotora e ser adequado para o projeto de primer e sonda para qMSP.
- Use o tratamento genômico humano com O Metilase CpG (M.SssI) como um controle de metilação positivo (ver Tabela de Materiais).
NOTA: A quantificação final da metilação é definida como o valor relativo da metilação (RMV), e calculada como 2–ΔΔCt para cada detecção de metilação replicada em comparação com a média de Ct para β-Actin (ACTB)18. As sequências de primer e sonda são mostradas na Tabela 1. Um Ct de 100 é usado para réplicas não detectadas, o que dá um valor de 2–ΔΔCt perto de zero. Utiliza-se a seguinte fórmula: média 2–ΔΔCt (RMV) = (2–ΔΔ Ct_replicate_1 + 2–ΔΔ Ct_replicate_2 + 2–ΔΔ Ct_replicate_3)/318.
Resultados
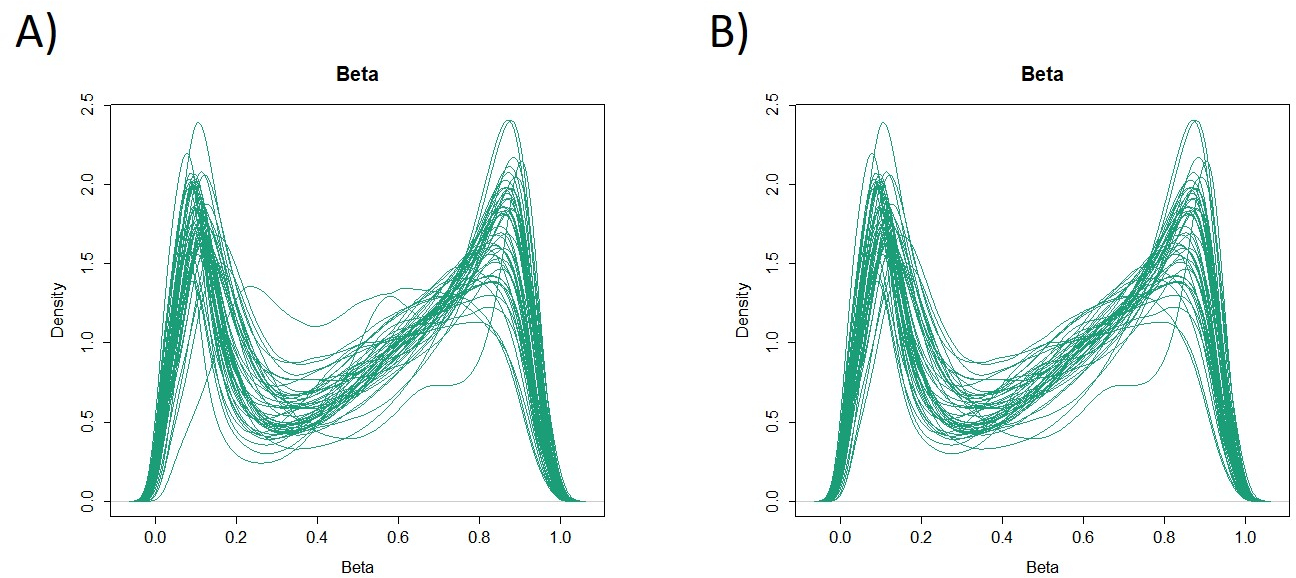
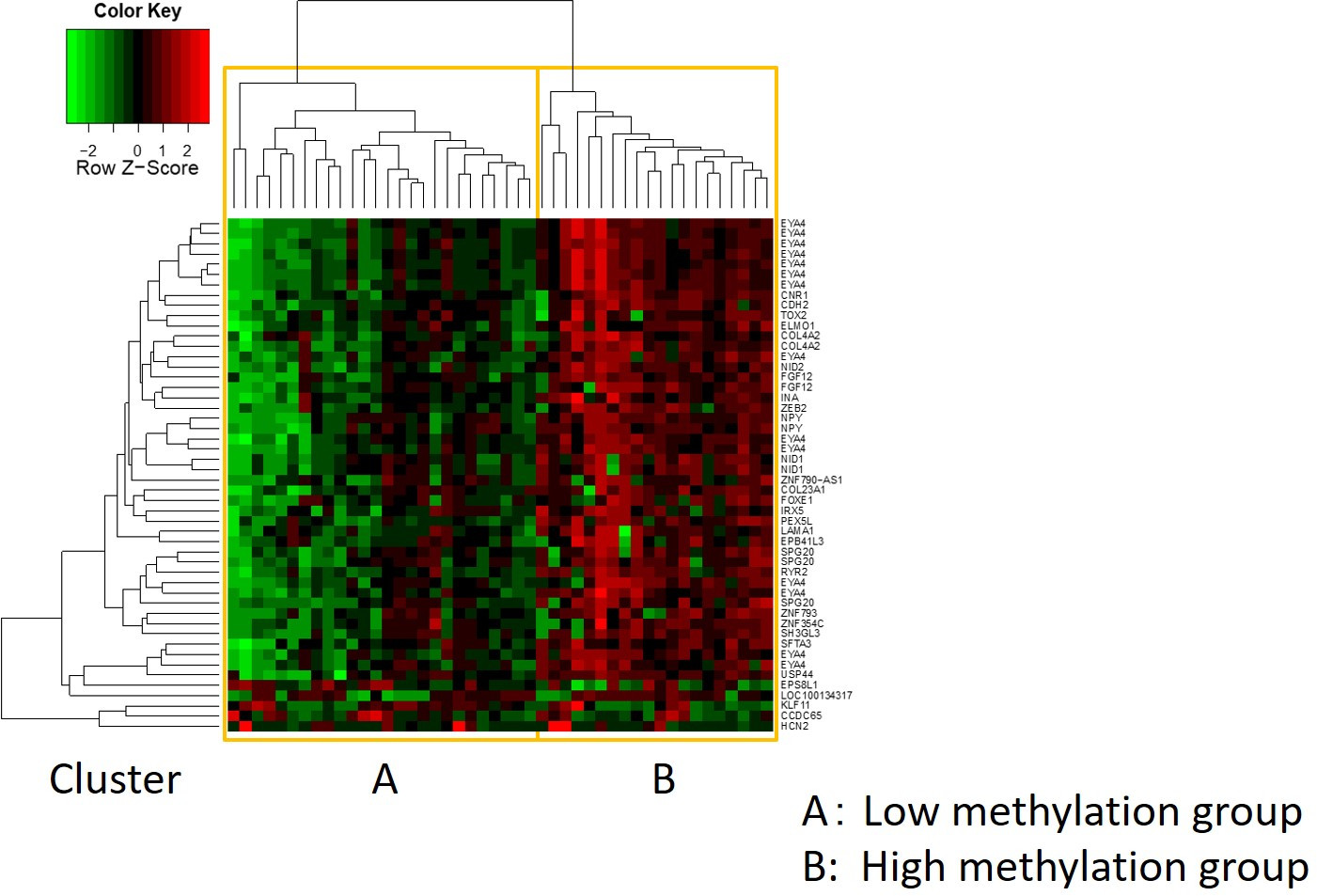
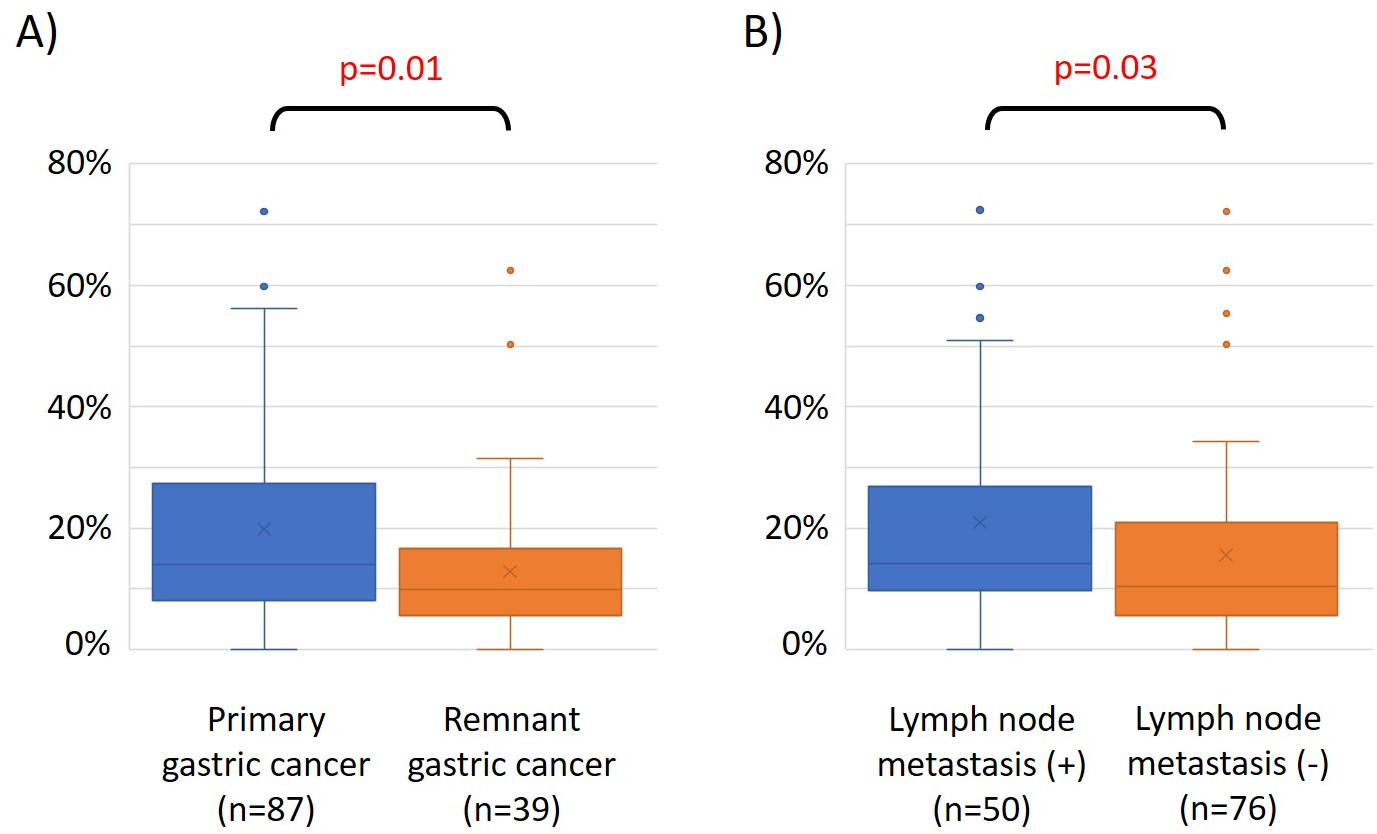
As características de 48 pacientes com câncer gástrico na coorte de formação são as seguintes (Tabela 2): a idade mediana dos pacientes foi de 74 anos (52 a 89 anos), e a coorte incluiu 38 homens (79,2%) e 10 mulheres (20,8%). Foram 35 pacientes (72,9%) com câncer gástrico primário e em 13 pacientes (27,1%) com câncer gástrico remanescente (câncer gástrico primário: primeira ocorrência de malignidade não metastática no estômago; câncer gástrico remanescente: câncer no estômago remanescente que se desenvolveu mais de 5 anos após a gastrectomia distal, independentemente da razão da ressecção original19). Foram 23 pacientes (47,9%) com metástase linfática e 25 pacientes (52,1%) Sem. Primeiro, todas as 48 amostras (a coorte de treinamento) foram carregadas para identificação de outliers(Figura 1A). Duas amostras deram picos superiores a dois desvios-padrão deslocados dos demais, e estas amostras foram removidas(Figura 1B). Portanto, 46 amostras foram agrupadas pela hipermetilação promotora de DNA. O mapa de calor resultante foi dividido em dois grupos baseados na alta e baixa metilação(Figura 2). Este mapa de calor permite a visualização das 50 principais sondas dentro de 1.500 bp do local de início transcricional (TSS) na análise diferencial de metilação. Os grupos de alta e baixa metilação diferem em fatores clinicopatológicos relacionados a um fenótipo maligno agressivo. Ou seja, o tipo de câncer (câncer gástrico primário: PGC) (p = 0,01, razão de chances = 9,09 (1,67-50,00)) e presença de metástase linfática (positiva) (p = 0,03, razão de chances = 6,82 (1,16-40,08)) emergiram como fatores preditivos independentes significativos quando os fatores clínicos foram utilizados como covariados na análise multivariada(Tabela 3). Finalmente, identificamos o gene EPB41L320,21 (sequências de primer e sonda mostradas na Tabela 1) como fortemente associado à codificação da coorte de treinamento em grupos de alta e baixa metilação na análise de microarray. Utilizando qMSP, foram validados os resultados da análise de microarray para EPB41L3 na coorte de teste (126 amostras). As características dos pacientes na coorte de teste são mostradas na Tabela 4. Os RMVs de EPB41L3 em tecidos PGC foram significativamente maiores do que os do câncer gástrico remanescente (RGC) na análise univariada (p = 0,01) (Figura 3A). Da mesma forma, os RMVs em amostras com metástase linfática foram significativamente maiores do que aqueles sem metástase linfática (p = 0,03) (Figura 3B). Dessa forma, a análise do genoma da metilação de DNA pode nos ajudar a identificar genes específicos para caracterizar determinado estado clínico em pacientes com malignidades gi.

Figura 1: Valores beta em 48 amostras (coorte de treinamento). Todas as 48 amostras (coorte de treinamento) foram carregadas e os outliers foram examinados(A). Duas amostras tinham picos que eram excepcionais, e estas foram removidas(B). Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 2: O mapa de calor resultante. As 46 amostras restantes foram agrupadas pela hipermetilação promotora de DNA. O mapa de calor foi dividido em grupos de alta e baixa metilação. Este mapa de calor permite a visualização das 50 principais sondas dentro de 1.500 bp do local de início transcricional (TSS) na análise diferencial de metilação. Clique aqui para ver uma versão maior desta figura.
{kind=link}

Figura 3: Valores relativos de metilação (RMVs) para EPB41L3 em câncer gástrico primário (PGC) vs. câncer gástrico remanescente (RGC), e em casos com e sem metástase do linfonodo. Os resultados da análise de microarray para EPB41L3 na coorte de teste (126 amostras) foram validados utilizando qMSP. (A) Na análise univariada, os RMVs de EPB41L3 em tecidos PGC foram significativamente maiores do que os do RGC (p = 0,01). (B) Da mesma forma, os RMVs em amostras com metástase do linfonodo foram significativamente maiores do que naqueles sem metástase linfática (p = 0,03). Clique aqui para ver uma versão maior desta figura.
{kind=link}
| Gene | Atacante 5' - 3' | Reverso 5' - 3' | Sonda |
| B-ACTIN | TAG GGA GTA TAT AGG TTG GGG AAG TT | AAC ACA CAA TAA CAA ACA CAA ATT CAC | \56-FAM\ TGT GGG GTG \ZEN\ GTG ATG GAG GAG GTT TAG \3IABkFQ\ |
| EPB41L3 | GGG ATA GTG GGG TTG ACG C | ATA AAA ATC CCG ACG AAC GA | AAA TTC GAA AAA CCG CGC GAC GCC GAA ACC A |
Tabela 1: Sequências de primer e sonda.
| Fatores clinicopatológicos | Variáveis | |
| Idade | 74 (52 - 89) * | |
| Gênero | Masculino / Feminino | 38 (79.2%) / 10 (20.8%) |
| Tipo | PGC / RGC | 35 (72.9%) / 13 (27.1%) |
| Metástase do nódulo linfático | (+) / (-) | 23 (47.9%) / 25 (52.1%) |
| PGC: Câncer gástrico primário, RGC: Câncer gástrico remanescente | ||
| * Mediana (mínimo-máximo) | ||
Tabela 2: As características em 48 pacientes com câncer gástrico na coorte de treinamento.
| Valor-P | Razão de chances | Intervalo de confiança de 95% | |
| Tipo (PGC) | 0.01 | 9.09 | 1.67 – 50.00 |
| Metástase do nódulo linfático (+) | 0.03 | 6.82 | 1.16 – 40.08 |
| PGC: Câncer gástrico primário | |||
Tabela 3: Fatores preditivos para o grupo de alta metilação (Cluster B).
| Fatores clinicopatológicos | Variáveis | |
| Idade | 71 (33 - 86) * | |
| Gênero | Masculino / Feminino | 96 (76.2%) / 30 (23.8%) |
| Tipo | PGC / RGC | 87 (69.0%) / 39 (31.0%) |
| Metástase do nódulo linfático | (+) / (-) | 50 (39.7%) / 76 (60.3%) |
| PGC: Câncer gástrico primário, RGC: Câncer gástrico remanescente | ||
| * Mediana (mínimo-máximo) | ||
Tabela 4: As características em 126 pacientes com câncer gástrico na coorte de teste.
Discussão
Existem três etapas críticas na obtenção de resultados precisos da análise do genoma da metilação de DNA. A primeira é a macrodisseção de uma área tumoral por, preferencialmente, dois patologistas qualificados com base em seções representativas manchadas de H&E. A macrodisseção imprecisa pode causar contaminação com tecidos não cancerígenos adjacentes, o que gera resultados não confiáveis; assim, é necessária uma macrodisseção cuidadosa. A segunda é a avaliação da qualidade do DNA (verificação de qualidade: QC). Amostras que falham no QC (∆Cq > 5.0) podem dar dados de má qualidade. Portanto, devem ser removidas amostras com ∆Cq > 5.0 e outras utilizadas. O terceiro passo é o cálculo do valor β, que é determinado por uma ferramenta de análise de dados para o software da plataforma de array como o sinal metilado / o sinal total (metilado + não-metilado)17. O valor β varia de 0 a 1 (ou 0%-100%), o que é simples de interpretar biologicamente17. O principal problema com esse valor são suas propriedades estatísticas pobres, uma vez que sua alta heteroscedasticidade implica que a variância entre as amostras nos extremos da faixa de metilação (β = 0 ou β = 1) é altamente reduzida17. Além disso, devido à má qualidade amostral, os valores de β podem não apresentar picos bifásicos reprodutíveis22, e amostras sem esses picos devem ser excluídas de um estudo posterior. Além disso, o gene-alvo deve ser escolhido com base nos critérios de ter valores beta maiores, estar relacionado às ilhas CpG na região promotora e ser adequado para o projeto de primer e sonda para qMSP.
A avaliação da metilação de DNA em um lócus cpg no genoma humano é realizada usando tecnologia baseada em microarray com um número fixo de sondas para levantamento de loci genômico específico. É o método mais utilizado em estudos de associação epigenome-wide (EWAS) devido ao seu baixo custo, pequena quantidade de DNA necessária e tempo de processamento de amostras marcadamente menor, o que permite o processamento de alta produtividade de muitas amostras clínicas23. No entanto, uma plataforma de matriz para avaliação complexa da metilação de DNA em um lócus cpg individual é limitada pelo número e especificidade das sondas para loci alterado epigeneticamente, o que impede a exploração de algumas regiões genômicas. O WGBS é geralmente visto como o método padrão-ouro devido ao seu espectro mais amplo de cobertura genômica10,11. No entanto, este método tem um custo substancial e um tempo de processamento relativamente longo para análise de um grande número de amostras10,11. Assim, nem sempre é viável. Em comparação, a plataforma de matriz para avaliação complexa da metilação de DNA em um lócus cpg individual no genoma humano é razoável para uso em termos de custo e cobertura genômica. Recentemente, os mais recentes chips de contas atualizados se prepararam para usar24. Esses ensaios podem nos ajudar a analisar locais de CpG medidos quase duplicados, que podem alcançar o estudo ideal de associação genoma (GWAS) para grandes populações de amostras.
Em resumo, a análise do genoma da metilação de DNA com a plataforma de matriz para avaliação complexa da metilação de DNA em um lócus cpg individual no genoma humano pode fornecer informações importantes sobre biomarcadores epigenéticos no câncer gastrointestinal. Comparado com o WGBS, este método é econômico e reduz o tempo de processamento de amostras. Portanto, este método de detecção de metilação de DNA em um lócus CpG é provável que seja amplamente utilizado em pesquisas de biomarcadores epigenéticos.
Divulgações
Os autores não têm nada a revelar.
Agradecimentos
Somos especialmente gratos a todos os membros do Departamento de Cirurgia, do Sidney Kimmel Comprehensive Cancer Center na Johns Hopkins University School of Medicine por discussões úteis e apoio técnico. Agradecemos também a Kristen Rodgers por generosas orientações técnicas sobre os procedimentos para tratamento de bisulfato e qMSP.
Materiais
| Name | Company | Catalog Number | Comments |
| (NH4)2SO4 | Sigma-Aldrich | 14148 | Step 5.2. |
| 10% Sodium dodecyl sulfate (SDS) | Quality Biological | 351-032-721 EA | Step 2.1. |
| 100 % Ethanol | Sigma-Aldrich | 24194 | Step 1.7. |
| ABI StepOnePlus Real-Time PCR System | Applied BioSystems | 4376600 | Step 5.2. 96-well Real-Time PCR instrument |
| CT conversion reagent | Zymo Research | D5001-1 | Step 3.2.3. |
| Deoxynucleotide triphosphate (dNTP) | Invitrogen | 10297-018 | Step 5.2. |
| DEPC-treated water | Quality Biological | 351-068-131 | Step 2.1. |
| Ethylenediaminetetraacetic acid (EDTA) | Corning | 46-034-CL | Step 2.1. |
| EZ DNA Methylation Kit | Zymo Research | D5002 | Step 3.2. |
| Fluorescein | Bio-Rad | #1708780 | Step 5.2. |
| GenomeStudio | omicX | OMICS_00854 | Step 4.3. Data analysis tool for an array platform as a β-value, with a range from 0 to 1 |
| Human genomic DNA | New England Bio Labs | N4002S | Step 5.3. |
| Infinium HD FFPE QC Kit | Illumina | WG-321-1001 | Step 4.1. FFPE QC assay on a real-time PCR amplification |
| Infinium HumanMethylation450 assay | Illumina | WG-314 | Step 4.2. Array platform for complex evaluation of DNA methylation to assess the methylation status of >450,000 CpG sites in the genome |
| LightCycler 480 | Roche | 5015278001 | Step 4.1. |
| M-Binding Buffer | Zymo Research | D5002-3 | Step 3.2.6. |
| M-Desulphonation Buffer | Zymo Research | D5002-5 | Step 3.2.9. |
| M-Dilution Buffer | Zymo Research | D5002-2 | Step 3.2.1. |
| Minfi package | Bioconductor | N/A | Step 4.4. |
| M-Wash Buffer | Zymo Research | D5002-4 | Step 3.2.10. |
| Platinum Taq polymerase | ThermoFisher Scientific | 10966-034 | Step 5.2. |
| Proteinase K | New England Biolabs | P8107S | Step 2.8. |
| Single-use polypropylene (Eppendorf) tube | Eppendorf | 24533495 | Step 2.5.2. |
| Tris hydrochloride (Tris-HCL) 2 M pH 8.8 | Quality Biological | 351-092-101 | Step 2.1. |
| Xylene | Sigma-Aldrich | 214736 | Step 1.3. |
| Zymo Spin 1 Column | Zymo Research | C1003 | Step 3.2.6. |
| β-Mercaptoethanol | Sigma-Aldrich | M3148 | Step 5.2. |
Referências
- Qiu, J. Epigenetics: unfinished symphony. Nature. 441 (7090), 143-145 (2006).
- Okugawa, Y., Grady, W. M., Goel, A. Epigenetic alterations in colorectal cancer: emerging biomarkers. Gastroenterology. 149 (5), 1204-1225 (2015).
- Ahuja, N., Li, Q., Mohan, A. L., Baylin, S. B., Issa, J. P. Aging and DNA methylation in colorectal mucosa and cancer. Cancer Research. 58 (23), 5489-5494 (1998).
- Hsieh, C. J., et al. Hypermethylation of the p16INK4a promoter in colectomy specimens of patients with long-standing and extensive ulcerative colitis. Cancer Research. 58, 3942-3945 (1998).
- Ushijima, T., Okochi-Takada, E. Aberrant methylations in cancer cells: where do they come from. Cancer Science. 96 (4), 206-211 (2005).
- Coppedè, F. Epigenetic biomarkers of colorectal cancer: Focus on DNA methylation. Cancer Letters. 342 (2), 238-247 (2014).
- Vasilatou, D., Papageorgiou, S. G., Dimitriadis, G., Pappa, V. Epigenetic alterations and microRNAs: new players in the pathogenesis of myelodysplastic syndromes. Epigenetics. 8 (6), 561-570 (2013).
- Ma, X., Wang, Y. W., Zhang, M. Q., Gazdar, A. F. DNA methylation data analysis and its application to cancer research. Epigenomics. 5 (3), 301-316 (2013).
- Morris, T. J., Beck, S. Analysis pipelines and packages for Infinium HumanMethylation450 BeadChip (450k) data. Methods. 72, 3-8 (2015).
- Rauluseviciute, I., Drabløs, F., Rye, M. B. DNA methylation data by sequencing: experimental approaches and recommendations for tools and pipelines for data analysis. Clinical Epigenetics. 11 (1), 193 (2019).
- Cazaly, E. Making sense of the epigenome using data integration approaches. Frontiers in Pharmacology. 10, 126 (2019).
- Leal, A., Sidransky, D., Brait, M. Tissue and cell-free DNA-based epigenomic approaches for cancer detection. Clinical Chemistry. 66 (1), 105-116 (2020).
- Song, W., Ren, J., Wang, W. J., Wang, C. T., Fu, T. Genome-wide methylation and expression profiling identify a novel epigenetic signature in gastrointestinal pan-adenocarcinomas. Epigenomics. 12 (11), (2020).
- Wong, E. M., et al. Tools for translational epigenetic studies involving formalin-fixed paraffin-embedded human tissue: applying the Infinium HumanMethyation450 Beadchip assay to large population-based studies. BMC Research Notes. 8, 543 (2015).
- . Illumina Available from: https://jp.support.illumina.com/downloads/infinium_humanmethylation450_product_files.html (2020)
- Fackler, M. J., et al. Genome-wide methylation analysis identifies genes specific to breast cancer hormone receptor status and risk of recurrence. Cancer Research. 71 (19), 6195-6207 (2011).
- Dedeurwaerder, S., et al. A comprehensive overview of Infinium HumanMethylation450 data processing. Briefings in Bioinformatics. 15 (6), 929-941 (2014).
- Hulbert, A., et al. Early detection of lung cancer using DNA promoter hypermethylation in plasma and sputum. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research. 23 (8), 1998-2005 (2017).
- Shimada, H., Fukagawa, T., Haga, Y., Oba, K. Does remnant gastric cancer really differ from primary gastric cancer? A systematic review of the literature by the Task Force of Japanese Gastric Cancer Association. Gastric Cancer: Official Journal of the International Gastric Cancer Association and the Japanese Gastric Cancer Association. 19 (2), 339-349 (2016).
- Eijsink, J. J., et al. Detection of cervical neoplasia by DNA methylation analysis in cervico-vaginal lavages, a feasibility study. Gynecologic Oncology. 120 (2), 280-283 (2011).
- Sugimoto, K., et al. DNA methylation genome-wide analysis in remnant and primary gastric cancers. Gastric Cancer: Official Journal of the International Gastric Cancer Association and the Japanese Gastric Cancer Association. 22 (6), 1109-1120 (2019).
- Wang, Z., Wu, X., Wang, Y. A framework for analyzing DNA methylation data from Illumina Infinium HumanMethylation450 BeadChip. BMC Bioinformatics. 19, 115 (2018).
- Teh, A. L., et al. Comparison of methyl-capture sequencing vs. Infinium 450K methylation array for methylome analysis in clinical samples. Epigenetics. 11 (1), 36-48 (2016).
- Zhou, W., Laird, P. W., Shen, H. Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. Nucleic Acids Research. 45 (4), 22 (2017).
Reimpressões e Permissões
Solicitar permissão para reutilizar o texto ou figuras deste artigo JoVE
Solicitar PermissãoThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. Todos os direitos reservados