Method Article
Determinazione delle interazioni proteina-ligando Utilizzo scansione differenziale Fluorimetria
In questo articolo
Riepilogo
Differential scanning fluorimetry is a widely used method for screening libraries of small molecules for interactions with proteins. Here, we present a straightforward method to extend these analyses to provide an estimate of the dissociation constant between a small molecule and its protein partner.
Abstract
Una vasta gamma di metodi attualmente disponibili per determinare la costante di dissociazione tra una proteina e interagendo piccole molecole. Tuttavia, la maggior parte di questi richiedono l'accesso alle attrezzature specialistiche, e spesso richiedono un certo grado di esperienza per stabilire efficacemente esperimenti affidabili e analizzare i dati. Fluorimetria a scansione differenziale (DSF) viene sempre più utilizzato come metodo affidabile per lo screening iniziale delle proteine per interagire piccole molecole, sia per identificare i partner fisiologiche o per la scoperta colpo. Questa tecnica ha il vantaggio che richiede solo una macchina PCR adatti per PCR quantitativa, e quindi idonea strumentazione è disponibile nella maggior parte delle istituzioni; un'eccellente gamma di protocolli sono già disponibili; e vi sono forti precedenti nella letteratura per molteplici utilizzi del metodo. Lavoro passato ha proposto diverse modalità di calcolo costanti di dissociazione dai dati DSF, ma questi sono matematicamente esigente. Qui, abbiamo DEMstrare un metodo per stimare le costanti di dissociazione da una moderata quantità di DSF dati sperimentali. Questi dati possono generalmente essere raccolti e analizzati in un solo giorno. Dimostriamo come i diversi modelli possono essere utilizzati per adattare i dati raccolti da semplici eventi vincolanti, e dove sono presenti siti di legame di cooperazione o indipendenti vincolante. Infine, presentiamo un esempio di analisi dei dati nel caso in cui non si applicano modelli standard. Questi metodi sono illustrati con i dati raccolti sulle proteine di controllo disponibili in commercio, e due proteine del nostro programma di ricerca. Nel complesso, il nostro metodo fornisce un modo semplice per i ricercatori di acquisire rapidamente una visione più interazioni proteina-ligando con DSF.
Introduzione
Tutte le proteine si legano, con affinità diverse, ad una vasta gamma di altre molecole di ioni semplici ad altre grandi macromolecole. In molti casi, le proteine si legano a molecole piccole parti come parte della loro normale funzionamento (ad esempio, una chinasi legame ATP). Altre interazioni possono essere estranei alla funzione, ma sono sperimentalmente utili come strumenti (ad esempio, piccole molecole che stabilizzano le proteine per migliorare il successo di cristallizzazione, o aiutare a mantenere le proteine in soluzione); mentre piccole molecole che si legano ai siti attivi e siti allosterici di proteine può agire come inibitori, e quindi modulare l'attività degli enzimi.
Vi è un'ampia gamma di tecniche che possono essere utilizzate per determinare l'affinità di proteine per molecole partner. Calorimetria isotermica di titolazione 1 è ampiamente considerato come un "gold-standard", in quanto fornisce informazioni sulle reazioni ricco, è l'etichetta libera, e ha limitato le opportunità per unrtifacts dell'esperimento. Tuttavia, nonostante i recenti miglioramenti nella sensibilità della strumentazione e automazione del set-up sperimentale, è ancora relativamente costoso in termini di fabbisogno proteico, ha al massimo un rendimento basso-medio, ed è più adatto alle interazioni con moderata a affinità alta (10 Nm a 100 mM K d) 2. Altra etichetta metodi gratuiti come risonanza plasmonica di superficie o doppio strato interferometria 3 offerta throughput più elevati, e hanno raggiunto la sensibilità per rilevare molecole più piccole a partire da 100 bis. Tuttavia, gli strumenti high throughput per questi metodi sono relativamente costosi, sono giustificati solo se ci sarà un continuo rendimento di progetti rilevanti, e quindi sono suscettibili di essere inaccessibile a molti laboratori universitari.
Fluorimetria differenziale a scansione (DSF, o thermofluor) è stato descritto nel 2001 4 come metodo per la scoperta di nuovi farmaci. In questo metod, proteine vengono incubati con un colorante fluorescente (stati usati coloranti inizialmente naftalen-solfonico), che ne modifica la fluorescenza dopo il legame alle regioni idrofobiche delle proteine. Il campione di proteina-colorante viene poi riscaldata, e la fluorescenza monitorato come aumenta il calore. Il dispiegamento della proteina, e l'esposizione di parti idrofobiche della proteina, dà luogo ad un pattern caratteristico della fluorescenza in funzione della temperatura (Figura 1A). L'esperimento può essere eseguita in piccoli volumi in qualsiasi strumento commerciale quantitativa PCR, e quindi in un singolo esperimento, un gran numero di campioni può essere simultaneamente testati (solitamente 48, 96 o 384 campioni, a seconda del modello di strumento). Esperimenti di solito può essere eseguita in circa un ora, fornendo la possibilità di alto rendimento analisi dei campioni 5.
Ulteriori miglioramenti della metodologia hanno visto l'adozione di coloranti con migliori proprietà spettrali 6,7 , strumenti generici per l'analisi dei dati, e suggerito protocolli per lo screening iniziale di 8,9. La gamma di applicazioni del metodo è stato esteso, con un focus particolare sulla creazione di condizioni ottimali per la preparazione e la conservazione delle proteine 10, e sull'identificazione di potenziali partner di legame per aiutare la cristallizzazione 11. Il relativamente alto rendimento del metodo, costo relativamente basso contenuto di proteine (~ 2 mg per reazione), e l'applicabilità di studiare molecole leganti deboli ha fatto DSF uno strumento prezioso per frammento basato progettazione di farmaci, soprattutto in un contesto accademico 12-14.
Nonostante l'ampia applicazione di DSF per studiare le interazioni proteina-ligando, pochi studi hanno descritto determinazione delle costanti di dissociazione da questi studi. Tuttavia, questi hanno la tendenza a produrre equazioni dettagliate che descrivono lo svolgersi della proteina, con molti parametri che devono essere installati i dati sparsi o in alcuni casi estimated 7,15-17. Questi metodi sono di particolare rilevanza nei casi difficili, come ad esempio i composti strettamente vincolanti, o proteine che mostrano le transizioni insolite. Tuttavia, per molti laboratori, queste analisi dettagliate sono troppo ingombranti per l'uso di routine. Proponiamo quindi trattamenti alternativi per diversi scenari, e dimostrare come questi possono essere usati per adattare i dati risultanti dalle diverse interazioni proteina-ligando. Il nostro metodo utilizza lo strumento StepOne qPCR, per la quale software di analisi dei dati su misura è disponibile; mentre questo accelera l'analisi dei dati, i risultati da altri strumenti possono essere elaborati con i metodi precedentemente pubblicati 9, e la stessa analisi a valle possono essere eseguite.
Protocollo
1 Determinazione di un valore approssimativo per la costante di dissociazione (cioè, entro un ordine di grandezza)
- Preparare l'impasto indicato nella tabella 1.
- Preparare scorte del ligando di interesse a più alta concentrazione disponibile, e poi a sei dieci diluizioni al raddoppio del presente. Qualora un approssimativo K d è noto dai dati precedenti, puntare ad avere almeno due concentrazioni al di sopra e al di sotto del K d.
- Aliquota 18 ml della miscela in otto pozzetti in un piatto qPCR. Aggiungere 2 ml di solvente per il primo pozzo. Aggiungere 2 ml di ciascun membro della serie di diluizioni ligando (punto 1.2) per un bene ciascuno dei rimanenti sette pozzi.
- Posizionare una guarnizione qPCR sopra la piastra. Per ottenere una buona tenuta della piastra, posizionare un applicatore mano (vedi tabelle di reagenti specifici) al centro della piastra. Smooth premuto il sigillo di lato e ripetere sull'altra metà dila piastra.
- Centrifugare la piastra a 500 xg per due minuti per rimuovere le bolle d'aria.
- Porre la piastra in uno strumento StepOne qPCR. Selezionare l'opzione "curva Melt", i filtri ROX, e scegliere la velocità di rampa veloce (questo fornisce una pausa di 2 min a 25 ° C, seguita da una rampa a 99 ° C oltre 40 minuti, e poi 2 minuti di pausa). Eseguire un denaturazione termica.
NOTA: I file di script per l'esecuzione di una corsa sono disponibili online all'indirizzo http: // www.exeter.ac.uk/biosciences/capsular. - Al termine della corsa dello strumento, fare clic sul pulsante "Analizza" sullo schermo. Salvare il file risultato.
- Aprire il software Protein Maiusc termica.
- Creare un nuovo studio; nella scheda delle proprietà, dare a questo un nome, e nella scheda Condizioni, dettaglio i ligandi.
- Passare alla scheda File Experiment, e importare il file salvato risultati (XXX.eds), e impostare il contenuto di ogni pozzetto (template files sono disponibili presso gli autori).
- Passare alla scheda Analisi, e premere il pulsante "Analizza".
NOTA: Questo analizzerà i risultati. E 'possibile esportare i risultati di ulteriori indagini con Excel utilizzando la scheda Esporta. I risultati vengono esportati in un formato scheda delineata. E 'meglio aprire il file esportato in Excel, e immediatamente salvare in formato Excel.
- Verificare che la proteina in presenza del solo solvente dà un risultato simile a quello mostrato nella Figura 1A. Avanti, esaminare le temperature di fusione osservate nei risultati nel riquadro "replicare". Assicurarsi che questa mostra un chiaro aumento della temperatura di fusione con l'aumento della concentrazione di ligando.
NOTA: Idealmente, questo fornirà un chiaro temperatura massima di fusione (supponendo che la proteina è completamente ligando legato), e un approssimativo K d dove la temperatura di fusione è a metà strada tra la proteina-ligando libero eil massimo.
2 set-up sperimentale per la determinazione della costante di dissociazione
- Preparare l'impasto indicato nella tabella 2 come master mix.
- Preparare le scorte di legante a quindici diverse concentrazioni, che saranno diluiti dieci volte l'esperimento finale. Idealmente, includere le concentrazioni di almeno due ordini di grandezza al di sopra e al di sotto della stima K d, e centrare le concentrazioni sul stimata K d. Focus su sette dei punti all'interno di un ordine di grandezza della stima K d, con altri quattro punti su entrambi i lati di questo; se vi è una scelta, includere più punti a valori che stanno saturando.
NOTA: Se necessario, è possibile modificare le condizioni sperimentali in modo tale che le scorte ligando sono al doppio della concentrazione sperimentale, dove ligando solubilità è limitante. - Aggiungere 120 ml di mastermescolare a otto pozzetti in una piastra ben fondo a U 96, a fungere da serbatoio per una comoda erogazione del master mix. Utilizzare una pipetta a 8 canali per erogare 18 microlitri in una colonna di una piastra PCR. Ripetere per altri cinque colonne, per un totale di 48 pozzetti riempiti in un modello 6 x 8 sulla piastra.
- Aggiungere 20 ml di stock ligando, o il solvente, pozzetti in una piastra ben U-fondo 96. Usando una pipetta a 8 canali, aspirare 2 ml di otto diversi stock ligando (o solvente). Aggiungi questi per una colonna della piastra PCR che era pieno di master mix al passo 2.3. Ripetere con gli stessi otto ligando / scorte solvente per due ulteriori colonne. Aspirare 2 ml di restanti otto titoli leganti o solventi, e aggiungere questi per una quarta colonna nel piatto. Ripetere questa operazione per altre due colonne. Questo darà campioni in triplicato per tutti i 16 campioni di legante e solvente.
- Inserite un sigillo qPCR sopra la piastra (vedi passo 1.4).
- Centrifugare la piastra a 500 xg perdue min.
- Posizionare la piastra nello strumento qPCR. Eseguire una denaturazione termica utilizzando i parametri specificati al punto 1.6.
- Al termine della corsa dello strumento, fare clic sul pulsante "Analizza" sullo schermo. Salvare il file risultato.
- Aprire il software Protein Maiusc termica. Creare un nuovo studio; nella scheda delle proprietà, dare a questo un nome, e nella scheda Condizioni, dettaglio i ligandi.
- Passare alla scheda File Experiment, e importare il file salvato risultati (XXX.eds), e impostare il contenuto di ciascun pozzetto.
NOTA: file di modello sono disponibili online all'indirizzo http: // www.exeter.ac.uk/biosciences/capsular. - Passare alla scheda Analisi, e premere il pulsante "Analizza".
- Selezionare la scheda "replicati" dal menu sul lato sinistro dello schermo per mostrare i risultati come triplica. Valutare l'affidabilità dei dati in base a come sono stretti i triplica. Should i triplicato mostrano scarsa riproducibilità, esaminare i dati grezzi da vicino.
- Analizzare i dati utilizzando sia il Boltzmann o metodi derivati per valutare la temperatura di fusione. Selezionare la scheda "Replicare risultati", e nella "Replica risultati complotto", spostare il "complotto:" tasto tra "Tm - Boltzmann" e "Tm - Derivata". Selezionare il metodo che conferisce maggiore riproducibilità per il campione. Esportare i risultati di ulteriori indagini con Excel utilizzando la scheda Esporta.
NOTA: Per i campioni che mostrano molteplici transizioni, è quasi sempre meglio usare il metodo Derivata in modalità multipla fusione. I risultati vengono esportati in un formato scheda delineata. E 'meglio aprire il file esportato in Excel, e immediatamente salvare in formato Excel. - Ripetere l'esperimento almeno due volte, tra cui una ripetizione in un giorno separato, al fine di garantire la riproducibilità dei risultati. In caso di analisi dei dati (vedere il punto 3 di seguito)indicano che il valore di K d è significativamente diverso alla stima originale, alterare le concentrazioni di ligando di conseguenza (vedi punto 2.2) per garantire una buona gamma di valori intorno K d.
3 Analisi dei dati per determinare la costante di dissociazione sotto denaturazione termica
- Creare una tabella in Excel delle concentrazioni di ligando e la temperatura di fusione.
- Aprire il software GraphPad Prism, e creare una tabella XY. Inserire i dati, utilizzando la colonna X per le concentrazioni di ligando e la colonna Y per la fusione risultati di temperatura.
NOTA: un esempio è mostrato Figura 1B. Uno script con equazioni precaricati, e le istruzioni alternative per l'utilizzo del pacchetto statistico SPSS, è disponibile online all'indirizzo http: // www.exeter.ac.uk/biosciences/capsular. - Nella scheda Analisi, selezionare l'opzione per cambiare il parametro di analisis (Ctrl + T). Per inserire il modello corretto, selezionare "Nuovo" e "Crea nuova equazione". Inserire l'equazione indicato nella Tabella 3 come "legame unico sito ligando".
NOTA: Un esempio di questi passi è mostrato in Figura 1C. Quando si utilizza lo script con le equazioni precaricati, l'equazione in questione può essere diretto scelti dalla lista piuttosto che inseriti. Derivazione di questa equazione è riportato in appendice. - Selezionare il "Regolamento per i valori iniziali" casella e inserire le regole per i valori iniziali come indicato nella tabella 3.
- Vincolare il parametro P, come "costante uguale a" e inserire la concentrazione finale di proteine (nelle stesse unità come il legante è dato in).
- Selezionare OK per eseguire l'analisi.
NOTA: Un esempio di questi passi è mostrato in Figura 1D. Il software grafica produce una figura che mostra i dati e l'adattamento al modello. Esempi di tanalisi hese sono riportati nei dati rappresentativi.
4. Montaggio dati a modelli cooperativi
Per inserire i dati di un modello cooperativo, scegliere tra una un semplice modello cooperativo, o un modello in cui sono definite due costanti di dissociazione separati. Il primo approccio è preferito nel caso di cooperatività negativa, o come un primo esame. Tuttavia, in linea di principio è meglio in caso di cooperatività positiva per modellare due diverse costanti di dissociazione 18. In questo caso, la modellazione può procedere assumendo o sequenziale legame di ligandi, o un legame indipendente di leganti.
- Seguire gli stessi passi iniziali come nel protocollo 3 Tuttavia, al punto 3.3, inserire una delle equazioni nella tabella 3 elencata come "Simple modello cooperativo", "sequenziale vincolante di due ligandi", o "Independent legame di due ligandi" 18.
- Selezionare le regole per i valori inizialiassociato a ciascuna di queste equazioni nella tabella 3.
- Esaminare l'adattamento del modello ai dati. Qualora i dati adattarsi male, prendere in considerazione un altro modello.
NOTA: è anche importante esaminare con attenzione il montaggio della temperatura di fusione di dati da parte del software Maiusc termica Protein (passo 2,9): a volte è necessario modificare i parametri qui per ottenere i migliori risultati. Un'altra considerazione è se la gamma di punti dati è l'ideale, e se ci sono punti anomali: o una serie limitata di dati su entrambi i lati di K d, o un unico punto anomalo (specialmente alle più alte concentrazioni di ligando), può influenzare in modo significativo i risultati. - Ripetere l'esperimento almeno due volte (vedi punto 2.12) per garantire la riproducibilità.
5. Montaggio dei dati per le curve che indicano Spostamenti binari in fusione Temperatura
Di tanto in tanto, piuttosto che una risposta graduata a ligando, le proteine sono stateosservato adottare una risposta binaria, dove campione legata sia chiaramente separata dalle campione non legato. Un esempio è fornito nei risultati rappresentativi (Figura 4). In questo caso, il montaggio delle temperature di fusione non fornirà una buona misura per K d.
- Esportare l'output di dati grezzi dal Proteine software Maiusc termica. Per ogni punto di temperatura, calcolare la fluorescenza media per il ligando zero e alte concentrazioni di ligando. Tabulare i risultati di ciascun punto dati accanto a questi.
NOTA: L'errore creato qui è inferiore errore nelle temperature di fusione a muro. - Aprire il pacchetto statistico SPSS. Copiare le temperature, le due serie di dati medi, e dati per ogni esperimento di una finestra di dati in SPSS. Nella scheda variabile, impostare il set di dati media per nessun ligando come "Low", e il set di dati media per la più alta concentrazione di ligando come "alto".
- Scaricare il file di sintassi disponibileonline all'indirizzo http: // www.exeter.ac.uk/biosciences/capsular . Selezionare "Esegui → Esegui tutti".
- Copiare la proporzione risultati legati ad una nuova cartella di lavoro di Excel, con le relative concentrazioni di ligando.
- Aprire il software GraphPad, e creare una tabella XY. Inserire i dati, utilizzando la colonna X per le concentrazioni di ligando e la colonna Y per la fusione risultati di temperatura. Nella scheda di analisi, selezionare "Parametri cambiamento di analisi". Per inserire il modello corretto, selezionare "Nuovo" e "Crea nuova equazione". Inserire l'equazione data nella tabella 3, indicato come "Analisi degli spostamenti binari a temperatura di fusione".
- Selezionare la casella "Norme per valori iniziali", e inserire le regole per i valori iniziali specificati nella tabella 3. Vincolare il parametro P, come "costante uguale a" e inserire la concentrazione finale di proteine (nelle stesse unità della ligand è dato in).
NOTA: esempi di completare queste scatole per il protocollo nella sezione 3 sono mostrati in figura 1C, D. - Se c'è una buona misura, i risultati possono essere migliorati estrapolando al risultato atteso a una concentrazione del ligando infinita. Dal modello della percentuale legata a ciascuna concentrazione ligando, esaminare il valore per il valore più alto di concentrazione di ligando. Se questa è la 0.99 o superiore, ulteriori analisi è improbabile per migliorare i risultati.
- Se la proporzione è minore di 0,99, un passaggio aggiuntivo è richiesto per correggere gli effetti della proteina non legata nel più alto campione concentrazione del ligando. Al punto 5.2, scrivere la percentuale di legante legato al più alto punto di concentrazione del ligando (dal punto 5.7) nella cella R2 (una cella diversa può essere utilizzato, e R2 sostituito opportunamente nell'equazione nella tabella 3). Creare una colonna supplementare dopo la media dei risultati più alti di concentrazione ligando. Negli abeticellule T, copiare l'equazione elencati nella Tabella 3 come "estrapolazione all'infinito concentrazione di ligando". Copia questa formula per le celle rimanenti in questa colonna.
NOTA: Questo calcolo elimina l'effetto della proteina non legata a più alta concentrazione di ligando. La differenza tra la proteina ligando libero e la più alta concentrazione ligando viene moltiplicato per il reciproco della proporzione vincolato alla massima concentrazione ligando per fornire la differenza attesa tra stati proteiche completamente legati e non legati ad ogni punto di temperatura. Questa differenza viene aggiunto o sottratto dallo stato non legato per dare la fluorescenza previsto per la proteina legante completamente legata. - Sostituire la colonna di concentrazione massima ligando nella scheda tecnica SPSS con questa nuova rubrica, e ripetere il raccordo dei dati.
NOTA: i passaggi 5,7-5,9 può avere bisogno di essere ripetuta se il modello suggerisce un ulteriore cambiamento significativo della percentuale legata ai concentrazi massime ligando(se questo è il caso, probabilmente sarebbe l'ideale per ripetere l'esperimento con un più alto punto di concentrazione del ligando incluso). - Nei casi in cui la proteina mostra un comportamento cooperativo traslazione binaria e display, l'equazione suggerito nel passaggio 5.5 dovrebbero essere sostituite con quelle dal punto 4.1. I parametri "bottom" "Top" e dovrebbero essere sostituiti da 1 e 0, rispettivamente.
- Ripetere l'esperimento almeno due volte (vedi punto 2.12) per garantire la riproducibilità.
Risultati
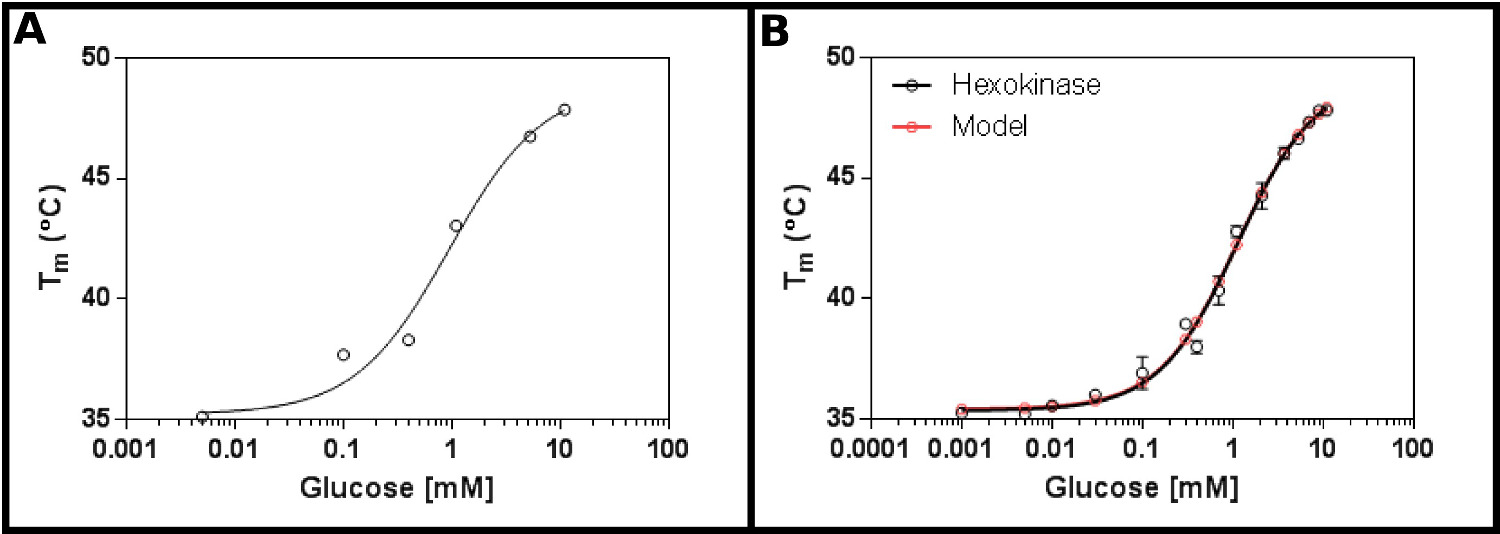
Un substrato di prova eccellente per questo metodo è esochinasi. Questo ha il vantaggio di essere facilmente reperibili in commercio, e avendo due substrati che si trovano nella maggior parte dei laboratori, e che forniscono risultati chiari e riproducibili nel test. Uno schermo concentrazione iniziale (protocollo 1), utilizzando esochinasi e glucosio (Figura 2A), suggerisce che la probabilità K d sarà nel da 0.2 a 1,7 mm. Pertanto, è stato eseguito uno schermo più grande (protocollo 2), in base alle concentrazioni indicate nella Tabella 4 I risultati (Figura 2B) mostrano un buon adattamento all'equazione legame singolo sito ligando (sezione protocollo 3.3) [9], e ha dato un K d di 1,2 ± 0,1 mm.
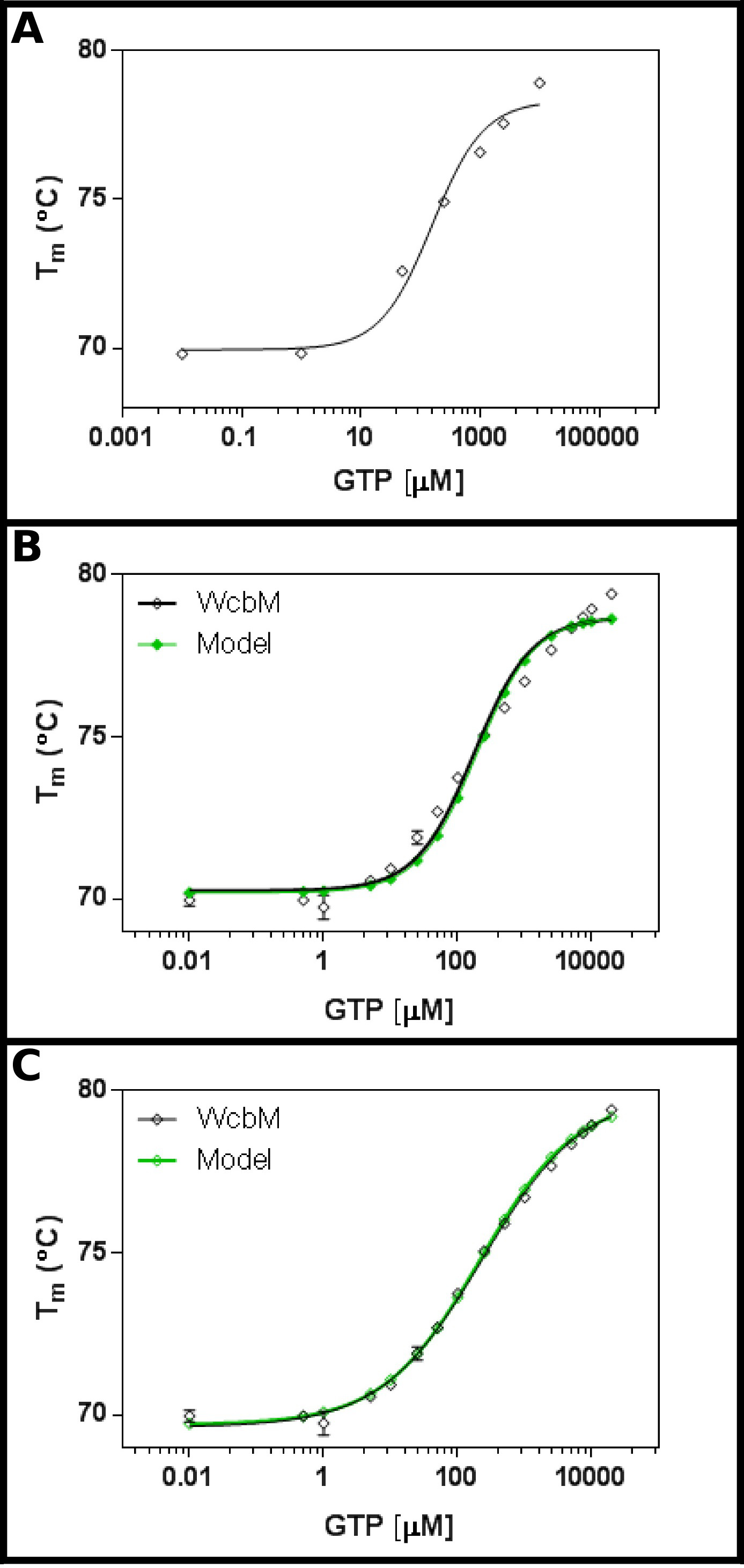
Il putativo transferasi heptose-guanyl WCBM 19,20 mostra un forte cambiamento termico legame GTP (Figura 3A). Una schermata iniziale suggerito che il Kd sarebbe nella gamma di circa 100 mM. Pertanto, uno schermo pieno è stato istituito, con le concentrazioni indicate nella tabella 5 Montaggio dei risultati di equazione 3.3 ha mostrato una misura ragionevole (R 2 di 0,981; Figura 3B). .Tuttavia, C'è una differenza evidente tra i dati e la modello, suggerendo che è necessaria una equazione diversa. Ricerca della banca dati Protein 21 con la sequenza WCBM ha dimostrato che le omologhi più vicini per i quali le strutture sono state determinate formare dimeri. I dati sono stati quindi analizzati utilizzando le tre equazioni per la cooperativa, sequenziale, e un legame indipendente dei due ligandi (Protocollo 4). Le statistiche di montaggio di un modello cooperativo ha dato un valore di R 2 di 0.998 e la deviazione standard dei residui (Sy.x) di 0,215, mentre entrambi i modelli vincolanti sequenziali e indipendenti, hanno un valore di R 2 di 0,992 e un Sy.x di 0.480 e 0,461 rispettivamente. Ciò suggerisce che il modellodando il migliore adattamento ai dati è stato il modello cooperativo: qui, è stato osservato un K ½ di 230 ± 10 micron, con un valore n di 0,52 ± 0,02 (Figura 3C). Questo indicava un cooperatività negativa per l'associazione. Si noti che un K ½ stato utilizzato in questo caso piuttosto che K d, come unità per K d sarebbe la poco soddisfacente 0,52 mM.
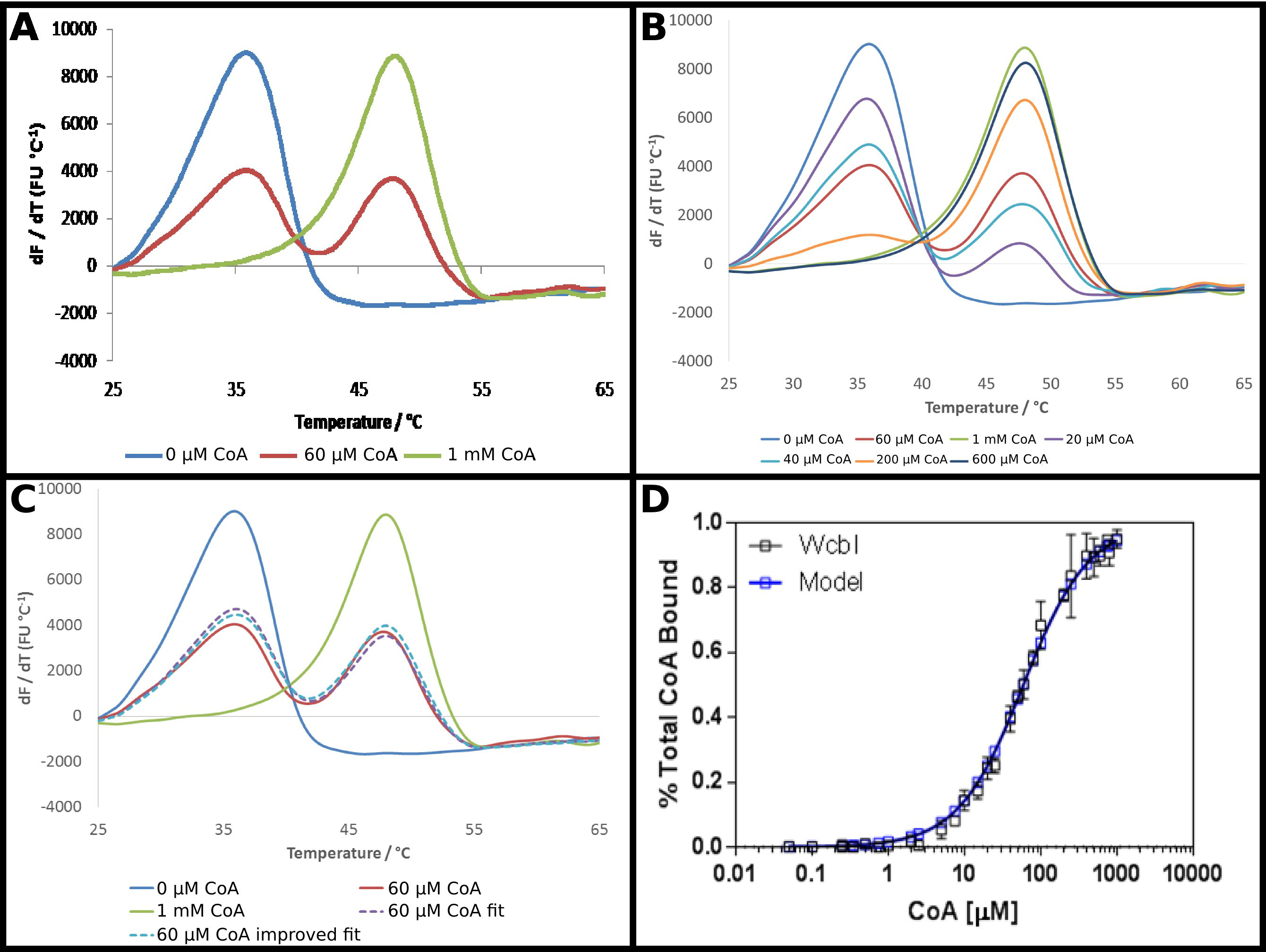
Il Pil-6-desossi-β-D-manno putativo -heptopyranose 2 O -acetylase, WcbI 22, mostra un risultato piuttosto inusuale in fluorimetria differenziale a scansione. In assenza di ligandi, mostra una denaturazione chiaro e semplice (Figura 4A). Coenzima A (CoA) è stato identificato come un ligando di questa proteina mediante DSF, e l'affinità della proteina per questo partner è stato studiato come descritto nel protocollo. In presenza di elevate concentrazioni di CoA, un forte shift ad una temperatura più elevata si osserva, con una variazione nella temperatura di fusione di 15 ° C. Tuttavia, a concentrazioni intermedie, piuttosto che un passaggio a una fusione monofasica ad una temperatura di fusione intermedia, WcbI mostrato una fusione bifasico, con la proteina che sembra fondere sia a temperatura libera-ligando, o la temperatura di fusione completamente legato (Figura 4A) . Le proporzioni delle due specie alterati in modo dose-dipendente, con concentrazioni crescenti di substrato aumentare la percentuale che fuso alla temperatura più alta (Figura 4B). Analisi diretta di questi dati è stato impegnativo: raccordo per l'equazione di Boltzmann ha dato molto poveri attacchi, mentre i metodi derivati evidenziato che due eventi di fusione stavano accadendo, ma non aiutano a dimostrare un cambiamento con l'aumento della concentrazione di ligando.
Un approccio meno convenzionale per analizzare questi dati è stato quindi adottato (protocollo 5). La fluorescenza drisultati erivative senza ligando e presso la più alta concentrazione del ligando sono stati presi come rappresenta sostanzialmente tutte le proteine della temperatura di fusione più bassa, o lo stato di temperatura di fusione più elevata. I dati derivati rimanenti sono stati montati in ciascun punto come la somma di una proporzione di ciascuno di questi due stati, con la proporzione sommate per unità (Figura 4C). I dati ottenuti sono stati poi montati come prima ottenere una apparente K d, utilizzando le stesse equazioni come prima. Ciò ha evidenziato che il punto ligando "alta" è probabile che sia solo il 95% ligando legato. I dati sono stati poi estrapolati a una previsione del risultato per una proteina legata al 100%, ed i dati raccordo ripetute invia un apparente Kd di 58 ± 2 mM. Questo ha fornito una misura eccellente dei risultati sperimentali al modello di legame (Figura 4D).
fo: content-width = "5in" src = "/ files / ftp_upload / 51809 / 51809fig1highres.jpg" width = "500" />
Figura 1 Esempi di esperimento di set-up e di analisi. (A) Esempio di forma atteso di un profilo di denaturazione termica (tratta da dati di lievito esochinasi). La caratteristica forma dei dati grezzi mostra un progressivo aumento della fluorescenza ad un massimo, seguito da un declino superficiale (discusso più in dettaglio nel 9). Questo è accompagnato da un singolo picco nella prima derivata della fluorescenza. (B) Esempio di inserimento dati in GraphPad. Concentrazione Ligand è dato sul l'asse X, e osservò temperature di fusione in Y. (C) Esempio di definizione dell'equazione in GraphPad. (D) Esempi di impostare correttamente i valori iniziali delle variabili, e di fissare la concentrazione di proteine, per consentire una corretta determinazione della costante di dissociazione.m / files / ftp_upload / 51809 / 51809fig1highres.jpg "target =" _blank "> Clicca qui per visualizzare una versione più grande di questa figura.

. Figura 2 Interazione di esochinasi con glucosio misurata mediante fluorimetria a scansione differenziale (A) Un esperimento iniziale testare una vasta gamma di concentrazioni di glucosio suggerisce che il K d è probabile che sia nel range di 0,2 -. 1,7 mm (B) A dettagliato esperimento, prova di 16 concentrazioni di glucosio, permette la determinazione della apparente K d come 1,12 ± 0,05 mm. I dati si adatta molto bene al modello per un singolo evento vincolante (con il fondo (T1) e superiore (T2) temperature raccordo a 35,4 ± 0,2 ° C e 49,3 ± 0,5 ° C, rispettivamente). Notare chequesti dati sono stati raccolti in presenza di 10 mM MgCl 2. Queste immagini sono state preparate utilizzando GraphPad. Cliccate qui per vedere una versione più grande di questa figura.
{kind=link}

Figura 3 Interazione di WCBM con GTP rivela un anti-cooperativa vincolante. (A) Un esperimento iniziale testare una vasta gamma di concentrazioni di GTP suggerisce che il K d è probabile che sia nel range di 200 -. 500 mM (B) Un esperimento dettagliata, testando 16 concentrazioni di GTP, suggerisce un valore per l'apparente K d di 120 ± 20 micron. Tuttavia, quando una scala logaritmica viene utilizzata per l'asse x, vi è una significativa discrezionalitàPancy tra il modello e dati. (C) Analisi degli stessi dati con un modello cooperativo mostra una misura eccellente per i dati in cui viene utilizzato un semplice modello cooperativo. Qui, un K ½ di 230 ± 20 micron è stato determinato, con il coefficiente di cooperatività n = 0,52 ± 0,02 (con il fondo (T1) e superiore (T2) temperature raccordo a 69.63 ± 0.06 ° C e 79,9 ± 0,1 ° C, rispettivamente). Come WCBM sembra essere dimerica, questo implica che l'enzima è perfettamente anticooperative nel suo legame al GTP. Queste immagini sono state preparate utilizzando GraphPad. Cliccate qui per vedere una versione più grande di questa figura.
{kind=link}

Figura 4 WcbI mostra un modello di fusione bifasicoin presenza del suo ligando coenzima A (CoA). (A) WcbI, in assenza di ligando (blu), mostra un semplice schema di fusione monofasica. Ad alte concentrazioni di ligando (1 mm linea verde), un modello simile è stato osservato. Tuttavia, a concentrazioni di ligando intermedie. (60 mM; linea rossa), due picchi di fusione distinti, corrispondenti agli stati libera-ligando e ligando-bound si osservano (B) Il passaggio tra le due serie di picchi è dose-dipendente per tutta la gamma di concentrazioni. (C) Modellazione della fusione bifasica come somma di una parte del ligando risultati legante libero e alta dà una buona misura per i dati (linea tratteggiata viola, rispetto a linea rossa). Questo fit è migliorata estrapolando il risultato osservato per la concentrazione alta ligando (dove il modello suggerisce ~ 95% di occupazione) alla piena occupazione (linea tratteggiata blu). (D) I dati ottenuti per la proporzione di WcbI legato al CoA sho ws una misura eccellente per un modello di legame semplice, con una Kd di 58 ± 2 mM (questi dati rappresentano i dati raccolti in due giorni separati, con leggermente diverse concentrazioni di ligando scelto per il secondo giorno basato sulla prima serie di risultati). Pannelli. (A - C) sono stati preparati utilizzando Excel, e il pannello (D) utilizzando GraphPad Cliccate qui per vedere una versione più grande di questa figura.
{kind=link}
Tabella 1 Ricetta per esperimenti iniziali.
| Reagente | Volume in mix (ml) |
| Proteina | Per concentrazione finale di 0,11 mg / ml |
| 0.3 | |
| 0,5 M HEPES pH 7,0 | 3.7 |
| 5 M NaCl | 5.6 |
| Acqua | Per 180 ml |
Questo descrive il "master mix" di proteine, reagente di rilevamento e buffer per un esperimento scouting iniziale per fornire una stima di K d, come descritto nella sezione protocollo 1 Questa miscela buffer è adatto per le proteine generiche. Qualora i risultati precedenti suggeriscono altri buffer devono essere utilizzati, questi dovrebbero essere sostituiti. Se la proteina magazzino è ad una concentrazione bassa (cioè, meno di 0,3 mg / ml), può essere necessario ridurre la quantità di buffer aggiuntivo aggiunto per compensare tampone già presente nel campione proteico.
Tabella 2 Ricetta per la determinazione di K d.| Reagente | Volume in mix (ml) |
| Proteina | Per concentrazione finale di 0,11 mg / ml |
| 5,000X SYPRO Arancione | 1.78 |
| 0,5 M HEPES pH 7,0 | 22.2 |
| 5 M NaCl | 33.3 |
| Acqua | Per 180 ml |
Questo descrive il "master mix" di proteine, reagente di rilevamento, e bUffer per una determinazione piena di K d per un campione di proteine, come descritto nella sezione del protocollo 2 Questa miscela buffer è adatto per le proteine generiche. Qualora i risultati precedenti suggeriscono altri buffer devono essere utilizzati, questi dovrebbero essere sostituiti. Se la proteina magazzino è ad una concentrazione bassa (cioè, meno di 0,3 mg / ml), può essere necessario ridurre la quantità di buffer aggiuntivo aggiunto per compensare tampone già presente nel campione proteico.
Tabella 3. equazioni e parametri per l'analisi dei dati.
d| Passo a protocollo sperimentale | Equazione richiesta | Parametri obbligatori | Descrizione di variabili e parametri |
| 3.3 | |||
| Sito unico legame ligando | Y = Basso + ((Alto-Basso) * (1 - ((PK d -X + sqrt (((P + X + K d) ^ 2) - (4 * P * X))) / (2 * P )))) | P: concentrazione di proteine. Kd: costante di dissociazione. P e Kd sono riportati nelle stesse unità che sono stati utilizzati per le concentrazioni di ligando. Superiore, Inferiore: temperature di fusione a concentrazione del ligando infinita e nessuna concentrazione ligando, rispettivamente. | |
| 3.4 | In basso = * YMIN | YMIN: Valore minimo di Y (più basso di proteine sperimentale Tm, in questo caso) | |
| Top = * YMAX | YMAX: Valore massimo di Y (più alto proteina sperimentale Tm) | ||
| K d = * X YMID | YMID: valore di Y che corrisponde alla media di YMIN e YMAX. X è il valore corrispondente di X (qui, la concentrazione di ligando pertinente) | ||
| P = (Valore iniziale, per essere in forma) | |||
| 4.1 | |||
| Semplice modello cooperativo | Y = Basso + ((Alto-Basso) * (((X / Kd) ^ n) / (1 + ((X / Kd) ^ n)))) | n: HCoefficiente di ill. Questo descrive la cooperatività, o altre proprietà biochimiche, della proteina, e non è necessariamente una stima del numero di siti di legame del ligando della proteina. Un coefficiente di Hill rappresenta uno non cooperatività; valori inferiori indicano una cooperatività negativa, e valori maggiori di uno cooperatività positiva. | |
| In basso = * YMIN | |||
| Top = * YMAX | |||
| K d = * X YMID | |||
| P = (Valore iniziale, per essere in forma ) | |||
| n = (valore iniziale, per essere in forma) | |||
| Sequenziale legame di due ligandi | Y = Basso + ((Alto-Basso) * ((X ^ 2) / (K d * K2)) / (1 + (X / K d) + ((X ^ 2) / (K d * K2))) ) | K2: costante di dissociazione per il secondo evento vincolante. | |
| In basso = * YMIN | |||
| Top = * YMAX | |||
| K2 = * X YMID | |||
| P = (Valore iniziale, per essere in forma) | |||
| Indipendente legame di due ligandi | Y = Basso + ((Alto-Basso) * ((X ^ 2) / (K d * K2)) / (1 + (2 * X / K d) + ((X ^ 2) / (K d K2 *) ))) | ||
| In basso = * YMIN | |||
| K d = * X YMID | |||
| K2 = * X YMID | |||
| P = (Valore iniziale, per essere in forma) | |||
| 5.5 | |||
| Analisi degli spostamenti binari in temperatura di fusione | Y = 1 - ((PK d -X + sqrt (((P + X + K d) ^ 2) - (4 * P * X))) / (2 * P)) | ||
| In basso = * YMIN | |||
| Top = * YMAX | |||
| K d = * X YMID | |||
| P = (Valore iniziale, per essere in forma) | |||
| 5.8 | |||
| L'estrapolazione di concentrazione ligando infinita | (C2 - ((1-$ R $ 2) * B2)) / $ R $ 2 | B2: cella contenente il risultato senza ligando. C2: cella contenente il risultato con la massima ligando. $ R $ 2: cella contenente la percentuale legata alla massima concentrazione del ligando. |
I passaggi 3, 4 e 5 richiedono l'aggiunta di equazioni dettagliate nel software di analisi, e precisa definizione dei parametri per l'analisi dei dati di partenza. Le equazioni per ciascun passaggio rilevante sono mostrate, con le corrette selezioni dei parametri. Una spiegazione del significato delle variabili e dei parametri è fornita per riferimento.
Tabella 4 Concentrazioni per lo screening di interazione di esochinasi con glucosio.
| Punto di campionamento | Ligando (glucose) concentrazione (mM) |
| 1 | 0 |
| 2 | 0.001 |
| 3 | 0.005 |
| 4 | 0.01 |
| 5 | 0.03 |
| 6 | 0.1 |
| 7 | 0.3 |
| 8 | 0.4 |
| 9 | 0.7 |
| 10 | 1.1 |
| 11 | 2.1 |
| 12 | 3.7 |
| 13 | 5.3 |
| 14 | 7 |
| 15 | 9 |
| 16 | 11 |
Esochinasi dal lievito Saccharomyces cerevisiae in erba è stato aggiunto al mix master come descritto nel protocollo, supplementato con 10 mM MgCl 2 come il magnesio è un cofattore nota. La stima iniziale del K d era tra 0,5 e 2 mm. Gli esperimenti sono stati istituiti per fornire le concentrazioni finali indicate di glucosio.
Tabella 5 Concentrazioni per lo screening di interazione di WCBM con il PIL.
| Punto di campionamento | Ligand (GTP) concentrazione (mM) |
| 1 | 0 |
| 2 | 0.5 |
| 3 | 1 |
| 4 | 5 |
| 5 | 10 |
| 6 | 25 |
| 7 | 50 |
| 8 | 100 |
| 9 | 250 |
| 10 | |
| 11 | 1.000 |
| 12 | 2.500 |
| 13 | 5.000 |
| 14 | 7.500 |
| 15 | 10.000 |
| 16 | 20.000 |
WCBM da Burkholderia pseudomallei è stato aggiunto al mix master come descritto nel protocollo. La stima iniziale del K D è stato di circa 100 micron. Gli esperimenti sono stati istituiti per fornire le concentrazioni finali indicate di GTP, con l'obiettivo di coprire almeno due ordini di grandezza al di sopra e al di sotto K d.
Discussione
Fluorimetria differenziale a scansione ha dimostrato il suo potere come un metodo robusto e versatile per la caratterizzazione delle proteine, e identificare potenziali ligandi proteici. I successi ben documentati in expediting stabilizzazione proteica, la scoperta di farmaci (in particolare nei laboratori meno ben finanziati) e cristallizzazione 10,23-25 hanno reso un metodo interessante per lo screening iniziale dei composti. Composti aggiunti alle proteine mostrano una dose di chiara dipendente aumento della temperatura di fusione apparente 7,9. Tuttavia, ci sono stati alcuni tentativi di utilizzare i risultati di questi esperimenti per determinare le costanti di legame apparente per aiutare nella classifica composti per la loro affinità. Qui, presentiamo un metodo per determinare sistematicamente una costante di dissociazione apparente di proteine in presenza di un ligando.
I risultati qui presentati dimostrano che DSF può rapidamente e robusta fornire stime della costante di dissociazione peruna combinazione di proteina-ligando. I dati osservati possono essere manipolati con gli strumenti disponibili in commercio di fornire una rapida determinazione di K d, senza la necessità di effettuare assunzioni riguardanti il valore probabile di parametri. Il metodo ha un vantaggio significativo su alcuni metodi comparabili di essere parsimoniosi in proteine e tempo necessario. L'esperimento descritto qui consumerà 0,13 mg di proteine per dell'esperimento (circa 0,4 mg per esperimenti ripetuti in triplice copia). Questo confronto con Calorimetria isotermica di titolazione (ITC), in cui un singolo esperimento con una media di 40 kDa consumerà un importo simile. La serie completa di esperimenti necessari per questo protocollo sarebbe consumare circa 4 ore, compresa la preparazione, per un singolo set di esperimenti. Ancora una volta, questo è probabile che sia molto più veloce di metodi come ITC o risonanza plasmonica di superficie, che pur potenti spesso richiedono notevole ottimizzazione per ottenere migliori dati.
I nostri risultati dimostrano che permane l'obbligo di esaminare attentamente i dati grezzi, la misura di questi dati per determinare la temperatura di fusione, e la misura dei dati di temperatura di fusione per determinare la costante di dissociazione. Un primo problema è la forma dei dati grezzi prodotti nella proteina di fusione. In alcuni casi, la forma non può approssimare a quello osservato nella Figura 1A. Problemi comuni includono bassi turni di temperatura sulla ligando vincolanti, sfondo di alta fluorescenza, e inusuali più transizioni di temperatura. A bassa temperatura turni sono visti sul legame un numero di ligandi. Per questo metodo, il parametro più critico è l'errore nella misurazione T m, rispetto al cambiamento di temperatura. I dati di solito può essere montato abbastanza bene quando la deviazione standard delle misurazioni triplice copia non supera il 10% del turno di temperatura di fusione tra proteine non legato e completamente legato. La nostra esperienza è che quando tale temperaturaturni lette- sono solo 2 ° C, questo può essere sufficiente per il montaggio dei dati, se i singoli punti di dati sono molto accurate. Un secondo problema è insolitamente a forma di curve. Questi spesso differiscono tra proteina libera e forme ligando legato, come il legame ligando influisce modalità della proteina dispiegarsi. In questi casi, l'utente deve esaminare se i dati possono essere utilizzati con idonei considerazione dei modelli da utilizzare per la determinazione della temperatura di fusione e la costante di dissociazione. Un altro problema comune è che l'aggiunta di un cofattore per la proteina (ad esempio, MgCl 2 nel nostro esempio con esochinasi) è necessario per ottenere i dati più affidabili. La nostra esperienza è stata che un attento esame di tutti i fattori che possono nell'esperimento in fase di prendere dei valori iniziali è essenziale per ottenere i migliori risultati. Inoltre, i trattamenti teorici alternativi possono rivelare le caratteristiche di questi dati 15,17. Infine, non è raro che alcune proteine che contain nativamente esposto regioni idrofobiche di mostrare sfondo ad alta fluorescenza. Ci sono una serie di soluzioni a questi problemi, che sono stati ampiamente recensito altrove 6,9.
In particolare, l'utente deve valutare se usare la Boltzmann o modelli derivati (ad esempio, figura 4), e nel caso di utilizzo di derivati, se più fusi devono essere modellati. I due metodi di modellazione del dispiegamento termico differiscono dal fatto che il metodo di Boltzmann adatta ai dati sperimentali di Boltzmann, assumendo una forma sigmoidale regolare la curva di dispiegamento. Al contrario, il metodo derivato prende la derivata prima dei dati sperimentali in ogni punto (pannello inferiore nella Figura 1A), e considera la temperatura di fusione di essere il punto di massima derivata prima. Il metodo derivato restituisce generalmente una temperatura di fusione più elevata di circa 2 - 3 ° C. La maggior parte delle proteine torneranno una più coerenterisultato (ovvero, l'errore standard della temperatura di fusione per esperimenti in triplo è inferiore) per uno dei due metodi. Questo è di solito intimamente legata alla forma precisa della proteina dispiegarsi curva, ed è necessario determinare empiricamente il metodo migliore in ciascun caso. Quando viene utilizzato il modello derivato, è anche importante considerare più eventi di fusione. Alcuni dati mostrano chiaramente le prove per più transizioni, e in questi casi i risultati sono suscettibili di essere più facile da interpretare, se questi eventi di fusione più sono modellati. Nel contesto di questo protocollo, è spesso il caso che l'aggiunta del ligando può causare una proteina di passare da avere più transizioni di fusione di una singola transizione (ad esempio, stabilizzando il sottodominio più fragile termicamente), o viceversa. Vorremmo quindi sostenere che i dati grezzi vengono esaminati insieme prima di considerare quale approccio sarà meglio usare.
Seguendo il modelling delle singole temperature di fusione, ulteriori problemi possono sorgere in raccordo questi per i modelli presentati nella sezione del protocollo. E 'indispensabile esaminare attentamente la misura di dissociazione equazione costante utilizzando una scala logaritmica, in quanto questa analisi evidenzia spesso discrepanze tra i dati osservati e il modello (es., Figura 3). Sebbene i risultati ottenuti sono generalmente robusti, cura nella interpretazione offre la possibilità di estrarre i risultati migliori, e più significato, dai dati.
Un particolare problema sollevato da questi dati è l'interpretazione che deve essere posizionato su proteine che mostrano cooperatività, o più eventi vincolanti, in DSF. Abbiamo, ad oggi, solo osservato questo fenomeno in proteine che sono tenuti ad avere più eventi di legame specifici (ad esempio, WCBM, una proteina il cui omologo migliore è un Multimer 26, e che agisce come un Multimer sulla dimensione cromatografia di esclusione [dati nonmostrato]). Non è affatto chiaro che la cooperatività negativa osservata in DSF denaturazione indica che l'enzima mostrerà infine cooperatività negativa: anzi, questo può essere un indizio del legame complesso che deve essere esplorato più a fondo utilizzando una vasta gamma di metodi. Ciò suggerisce a noi, però, che più studi approfonditi di tali proteine sono suscettibili di individuare gli effetti interessanti.
I valori indicati per la costante di dissociazione con questo metodo sono generalmente dello stesso ordine di quelli forniti da altri metodi, come ad esempio Calorimetria isotermica di titolazione e risonanza plasmonica di superficie. Tuttavia, i valori assoluti osservati sono spesso superiori a quelli osservati con questi metodi. Questo è almeno in parte una conseguenza del fatto che la costante di dissociazione si osserva alla temperatura di fusione della proteina con ligando. Questo Kd è generalmente superiore a quella a temperature fisiologiche. Il dissociation costante è correlata alla temperatura della reazione dalle equazioni:
 [1]
[1]
 [2]
[2]
(Dove c θ è la concentrazione di riferimento standard, Δ r G è la variazione di energia libera di Gibbs della reazione, R è la costante dei gas molare, Δ H è la variazione di entalpia nella reazione, e Δ S è la variazione di entropia nella reazione .)
Reazioni con costanti di dissociazione nell'intervallo misurabile di questo metodo generalmente hanno un negativo Δ r G, e quindi l'effetto di un aumento della temperatura sulla equazione [1] saranno aumentare la costante di dissociazione. Entrambi i termini Δ H e Δ S che costituiscono l'energia libera di Gibbs (equazione [2]) sono temperature 27 dipendenti, e l'effetto sulla costante di dissociazione dipenderanno dalla grandezza e segno di queste dipendenze di temperatura, e saranno necessariamente interazione dipendente. Di conseguenza, non è inaspettato che le costanti di dissociazione determinate con questo metodo sono a volte superiori a quelli determinati con metodi che operano a RT. Dipendenza dalla temperatura è, naturalmente, anche un avvertimento di molti altri metodi, che tendono a fornire la costante di dissociazione a temperature inferiori alla temperatura fisiologica.
Un altro avvertimento del metodo DSF è che è un metodo marcato, a differenza ITC. L'etichetta fluorescente utilizzato (SYPRO arancione) è idrofobo, e così in alcuni casi può competere con il legame di ligandi idrofobici alle proteine. Di conseguenza, è probabile che in alcuni casi, la costante di dissociazione ottenuto sarà artificialmente elevato a causa della competizione con l'etichetta. Tuttavia, per il confronto di diversi ligandi, (l'uso primario diDSF), le differenze è improbabile che siano sufficientemente significativo ai fini della graduatoria di composti per affinità.
Un potenziale svantaggio di questo metodo è il limite di rilevamento che può essere raggiunto. In linea di principio, non dovrebbe essere possibile misurare con precisione un valore per K d, che è inferiore al 50% della concentrazione di proteine, e anche i valori di questa gamma sono suscettibili di essere di dubbia precisione. Mentre il limite di rivelazione in questo fine dell'intervallo può essere esteso un po riducendo le concentrazioni di proteine e colorante, la sensibilità dello strumento impedirà un'ulteriore riduzione della concentrazione proteica. Analogamente, l'estremità superiore della sensibilità sarà determinata dalla solubilità del ligando. Per ottenere una stima matematicamente robusto per K d, è molto importante per ottenere dati con il 90% delle proteine presenti nella forma ligando-bound, che richiede concentrazioni di ligando ad essere approssimativaly dieci volte K d (supponendo che non cooperatività). Il limite di rivelazione sarà quindi necessariamente essere un decimo della solubilità del ligando nel buffer pertinente. Ciò significa che i limiti di rivelazione del metodo in genere compresi tra 1 mM e tra 1 e 100 mm, a seconda della proteina e ligando.
In conclusione, a scansione differenziale fluorimetria è una tecnica versatile applicabile ad una vasta gamma di proteine. Usando i metodi qui presentati, è possibile determinare rapidamente ed economico l'affinità di una proteina per diversi ligandi. Questo ha un grande potenziale per l'applicazione nella purificazione della proteina e la stabilizzazione, chiarendo la funzione o la specificità degli enzimi provenienti da metagenomi, e nella scoperta della droga, soprattutto nei piccoli laboratori.
Divulgazioni
The authors declare that they have nothing to disclose.
Riconoscimenti
This work was funded by grant from the BBSRC (grant number BB/H019685/1 and BB/E527663/1) to the University of Exeter.
Materiali
| Name | Company | Catalog Number | Comments |
| StepOne real time PCR instrument | Life Technologies | 4376357 | DSF can be performed with many other instruments. The StepOne instrument has very convenient software for data analysis. |
| Protein thermal shift software v1.0 | Life Technologies | 4466037 | |
| MicroAmp Fast optical 48-well plates | Life Technologies | 4375816 | |
| Optical sealing tape | Life Technologies | 4375323 | Bio-rad part no. 223-9444 is an alternative supplier |
| U-bottomed 96-well plates | Fisher | 11521943 | |
| SYPRO Orange | Life Technologies | S6650 | For a smaller volume supplier, use Sigma part no. S5692 |
| SPSS statistics version 20 | IBM | N/A | Other statistics packages will provide similar functionality |
| GraphPad Prism 6.02 | GraphPad | N/A | Other statistics packages will provide similar functionality |
| Hand applicator (PA1) | 3M | 75-3454-4264-6 | |
| Hexokinase from Saccharomyces cerevisiae | Sigma-Aldrich | H5000 | |
| Glucose | Fisher scientific | 10141520 |
Riferimenti
- Freyer, M. W., Lewis, E. A. Isothermal titration calorimetry: experimental design, data analysis, and probing macromolecule/ligand binding and kinetic interactions. Methods Cell Biol. 84, 79-113 (2008).
- Ladbury, J. E. Calorimetry as a tool for understanding biomolecular interactions and an aid to drug design. Biochem Soc Trans. 38, 888-893 (2010).
- Abdiche, Y., Malashock, D., Pinkerton, A., Pons, J. Determining kinetics and affinities of protein interactions using a parallel real-time label-free biosensor, the Octet. Anal Biochem. 377, 209-217 (2008).
- Pantoliano, M. W., et al. High-density miniaturized thermal shift assays as a general strategy for drug discovery. J Biomol Screen. 6, 429-440 (2001).
- Senisterra, G., Chau, I., Vedadi, M. Thermal denaturation assays in chemical biology. Assay Drug Dev Technol. 10, 128-136 (2012).
- Ericsson, U. B., Hallberg, B. M., Detitta, G. T., Dekker, N., Nordlund, P. Thermofluor-based high-throughput stability optimization of proteins for structural studies. Anal Biochem. 357, 289-298 (2006).
- Lo, M. C., et al. Evaluation of fluorescence-based thermal shift assays for hit identification in drug discovery. Anal Biochem. 332, 153-159 (2004).
- Nettleship, J. E., Brown, J., Groves, M. R., Geerlof, A. Methods for protein characterization by mass spectrometry, thermal shift (ThermoFluor) assay, and multiangle or static light scattering. Methods Mol Biol. 426, 299-318 (2008).
- Niesen, F. H., Berglund, H., Vedadi, M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc. 2, 2212-2221 (2007).
- Geders, T. W., Gustafson, K., Finzel, B. C. Use of differential scanning fluorimetry to optimize the purification and crystallization of PLP-dependent enzymes. Acta Crystallogr Sect F Struct Biol Cryst Commun. 68, 596-600 (2012).
- Vedadi, M., et al. Chemical screening methods to identify ligands that promote protein stability, protein crystallization, and structure determination. Proc Natl Acad Sci USA. 103, 15835-15840 (2006).
- Davis, B. J., Erlanson, D. A. Learning from our mistakes: the 'unknown knowns' in fragment screening. Bioorg Med Chem Lett. 23, 2844-2852 (2013).
- Larsson, A., Jansson, A., Aberg, A., Nordlund, P. Efficiency of hit generation and structural characterization in fragment-based ligand discovery. Curr Opin Chem Biol. 15, 482-488 (2011).
- Scott, D. E., et al. Using a fragment-based approach to target protein-protein interactions. Chembiochem. 14, 332-342 (2013).
- Cimmperman, P., et al. A quantitative model of thermal stabilization and destabilization of proteins by ligands. Biophys. J. 95, 3222-3231 (2008).
- Matulis, D., Kranz, J. K., Salemme, F. R., Todd, M. J. Thermodynamic stability of carbonic anhydrase: measurements of binding affinity and stoichiometry using ThermoFluor. Biochemistry. 44, 5258-5266 (2005).
- Zubriene, A., et al. Measurement of nanomolar dissociation constants by titration calorimetry and thermal shift assay - radicicol binding to Hsp90 and ethoxzolamide binding to CAII. Int J Mol Sci. 10, 2662-2680 (2009).
- Weiss, J. N. The Hill equation revisited: uses and misuses. FASEB J. 11, 835-841 (1997).
- Cuccui, J., et al. Characterization of the Burkholderia pseudomallei K96243 capsular polysaccharide I coding region. Infect Immun. 80, 1209-1221 (2012).
- DeShazer, D., Waag, D. M., Fritz, D. L., Woods, D. E. Identification of a Burkholderia mallei polysaccharide gene cluster by subtractive hybridization and demonstration that the encoded capsule is an essential virulence determinant. Microb Pathog. 30, 253-269 (2001).
- Berman, H., Henrick, K., Nakamura, H. Announcing the worldwide Protein Data Bank. Nat Struct Biol. 10, 980 (2003).
- Vivoli, M., Ayres, E., Beaumont, E., Isupov, M., Harmer, N. Structural insights into WcbI, a novel polysaccharide biosynthesis enzyme. IUCr Journal. 1 (1), 28-38 (2014).
- Sorrell, F. J., Greenwood, G. K., Birchall, K., Chen, B. Development of a differential scanning fluorimetry based high throughput screening assay for the discovery of affinity binders against an anthrax protein. J Pharm Biomed Anal. 52, 802-808 (2010).
- Uniewicz, K. A., et al. Differential scanning fluorimetry measurement of protein stability changes upon binding to glycosaminoglycans: a screening test for binding specificity. Anal Chem. 82, 3796-3802 (2010).
- Wan, K. F., et al. Differential scanning fluorimetry as secondary screening platform for small molecule inhibitors of Bcl-XL. Cell Cycle. 8, 3943-3952 (2009).
- Koropatkin, N. M., Holden, H. M. Molecular structure of alpha-D-glucose-1-phosphate cytidylyltransferase from Salmonella typhi. J Biol Chem. 279, 44023-44029 (2004).
- Paleskava, A., Konevega, A. L., Rodnina, M. V. Thermodynamics of the GTP-GDP-operated conformational switch of selenocysteine-specific translation factor SelB. J Biol Chem. 287, 27906-27912 (2012).
Ristampe e Autorizzazioni
Richiedi autorizzazione per utilizzare il testo o le figure di questo articolo JoVE
Richiedi AutorizzazioneThis article has been published
Video Coming Soon
Personale delle biblioteche
Copyright © 2025 MyJoVE Corporation. Tutti i diritti riservati