
Method Article
A Novel In Vitro Model of Blast Traumatic Brain Injury
In This Article
Summary
This paper describes a novel model of primary blast traumatic brain injury. A compressed-air driven shock tube is used to expose in vitro mouse hippocampal slice cultures to a single shock wave. This is a simple and rapid protocol generating a reproducible brain tissue injury with a high throughput.
Abstract
Traumatic brain injury is a leading cause of death and disability in military and civilian populations. Blast traumatic brain injury results from the detonation of explosive devices, however, the mechanisms that underlie the brain damage resulting from blast overpressure exposure are not entirely understood and are believed to be unique to this type of brain injury. Preclinical models are crucial tools that contribute to better understand blast-induced brain injury. A novel in vitro blast TBI model was developed using an open-ended shock tube to simulate real-life open-field blast waves modelled by the Friedlander waveform. C57BL/6N mouse organotypic hippocampal slice cultures were exposed to single shock waves and the development of injury was characterized up to 72 h using propidium iodide, a well-established fluorescent marker of cell damage that only penetrates cells with compromised cellular membranes. Propidium iodide fluorescence was significantly higher in the slices exposed to a blast wave when compared with sham slices throughout the duration of the protocol. The brain tissue injury is very reproducible and proportional to the peak overpressure of the shock wave applied.
Introduction
Blast traumatic brain injury (TBI) is a complex type of brain injury that results from the detonation of explosive devices1,2. Blast TBI has emerged as a major health issue in the last 15 years with the recent military conflicts in Iraq and Afghanistan2,3. Overall, it is estimated that between 4.4% and 22.8% of soldiers returning from Iraq and Afghanistan have suffered mild TBI, a large proportion of these being blast-related, with a higher reported rate of blast TBI in the US forces compared with the UK forces4,5.
The use of improvised explosive devices has been responsible for most of the blast-associated trauma, including blast TBI, endured by the military forces6. The detonation of an explosive charge results in a very rapid — but transient — increase in pressure, occurring in milliseconds. The resulting overpressure wave from a real-life free-field explosion is modelled by the Friedlander function, with a sudden rise to the peak overpressure followed by an exponential decay7,8. The range of extreme forces and their rapid time course seen in a blast event are usually not experienced in non-blast traumas1,9. The peak overpressure, which is the maximum pressure of the waveform, and the duration of the positive wave are believed to be important contributors to blast brain injury and these depend on the explosive charge and the distance from the detonation10,11.
The trauma that results from an explosive blast is classified as four discrete components, designated as primary, secondary, tertiary and quaternary blast injury10,12,13,14. Each of these components is associated with specific mechanisms of injury. Primary blast injury results from the direct action of the overpressure wave on organs and tissues2,13. Secondary blast injury results from the impact of projectile fragments, causing penetrating and non-penetrating wounds2,15. Tertiary blast injury occurs when the victim's body is displaced against the ground or surrounding objects and is associated with acceleration/deceleration forces1,10,13. Quaternary blast injury describes a heterogeneous group of injuries directly related to the explosion not covered by the first three injury mechanisms described12,13. It includes (but is not limited to) thermal injury, smoke inhalation, radiation, electromagnetic waves, and adverse psychological effects13,15. Most blast-associated TBI results directly from the first three mechanisms of injury, while the quaternary mechanisms of blast injury are usually associated with systemic injury13. The effects of acceleration/deceleration forces (e.g., whiplash), blunt and penetrating traumatic brain injury have been extensively studied in relation to other types of TBI (e.g., motor vehicle crashes, falls, ballistic injury). However, the primary blast overpressure wave is unique to blast injury and its effects on brain tissue are much less well understood16. The primary blast injury mechanisms, associated with an overpressure wave, are the first of the mechanical forces to interact with the brain.
Numerous preclinical TBI models have been developed over the last decades that have been invaluable to understand blast TBI mechanisms of injury and pathophysiology and investigate potential new treatments, which would otherwise be impossible to do exclusively in the clinical setting17,18,19. Although no single preclinical model can reproduce the complexity of clinical blast brain trauma, typically different preclinical TBI models replicate distinct aspects of human TBI. The damaging action of the forces associated with a blast explosion can be studied in isolation or in combination in both in vitro and in vivo blast TBI models. In vitro models have the advantage of allowing a tight control of the experimental environment (tissue physiologic conditions and injury biomechanics), which reduces biological variability and improves reproducibility, permitting the study of specific molecular cascades without the confounders present in animal models20. Our goal was to develop an in vitro model to investigate the effects of primary blast on brain tissue. We aimed to develop a model with a supersonic shockwave with a Friedlander waveform representative of a free-field explosion such as that produced by an improvised explosive device (IED).
Protocol
The experiments described in this manuscript were done in compliance with the United Kingdom Animals (Scientific procedures) Act of 1986 and have been approved by the Animal Welfare & Ethical Review Body of Imperial College London. Animal care was in compliance with the institutional guidelines of Imperial College London.
1. Hippocampal Organotypic Slice Preparation and Culture
NOTE: This protocol allows the production of organotypic hippocampal slices according to the interface method described by Stoppini and colleagues with minor modifications21,22,23. Ideally, no more than three animals should be euthanized and dissected in one session to ensure each step is done swiftly and to avoid compromising the quality of the slices. Use aseptic technique throughout.
- Ensure the following steps are taken before starting the dissection protocol.
- Prepare and filter sterilize all solutions in advance using a 0.22 µm filter.
- Keep the solutions at 4 °C during the storage and throughout the brain dissection and hippocampal slice preparation by keeping the storage containers and Petri dishes on a metal heat sink sitting on wet ice at all times.
- Autoclave all metal instruments, rings and paper tissues.
- Ensure all instruments and material to be used for each step of the protocol are ready to use, laid out in advance and sprayed with 70% ethanol. Ensure that they are allowed to cool down and dry.
- Euthanize a 5–7 day-old C57BL/6N mouse pup by cervical dislocation and briefly wipe the whole skin surface with sterile paper tissue soaked in 70% ethanol. Pat the skin dry and decapitate the pup using Mayo scissors.
- Cut the scalp skin with iris scissors along the midline of the head starting in the occipital region and ending near the snout and retract it laterally.
- Insert the tip of Vannas scissors in the foramen magnum and make two small lateral cuts on the bone along the transverse sinuses, then cut the skull along the midline up to the olfactory bulb and make two small cuts perpendicularly to the midline in this region.
- Using fine point curved forceps, retract the flaps of bone laterally away from the midline, carefully remove the brain with a small spatula and transfer it to a 90 mm silicone elastomer-coated Petri dish containing "dissection medium".
NOTE: Dissection medium is Gey's balanced salt solution (5 mg/mL D-glucose, 1% antibiotic-antimycotic solution, with 10,000 units/mL penicillin, 10 mg/mL streptomycin and 25 µg/mL amphotericin B). - Remove the cerebellum and separate the cerebral hemispheres along the midline using a razor blade. Use a spatula to transfer the cerebral hemispheres to a new 90 mm silicone elastomer-coated Petri dish filled with fresh ice-cold dissection medium. If more than one animal is to be used in one session, repeat Steps 1.2–1.6.
- Under a stereomicroscope, cut the olfactory bulb and the tip of the frontal cortex with a razor blade and separate the cerebral cortex from the rest of the cerebral tissue using fine tip forceps. This step leaves the hippocampus exposed on the medial surface of the cortical tissue. From this step onward, use a laminar flow tissue culture hood (ultraviolet sterilized and cleaned with 70% ethanol solution).
- Apply ice-cold dissection medium to a flat plastic chopping disk and, using a spatula, position the brain tissue on the disk such that the medial surface of the cortex is facing up and the axis of the hippocampus is perpendicular to the axis of the chopping blade.
- Remove as much as possible of the dissection medium from the chopping disk using a fine tip Pasteur pipette.
- Cut the brain into 400 µm slices using a tissue chopper at a 50% chopping speed and force.
NOTE: It is very important that this step is done as quickly as possible as the brain tissue is not submerged in dissection medium. - Replace the dissection medium in the silicone elastomer-coated Petri dish with fresh ice-cold medium.
- Once the tissue chopper is finished, carefully submerge the brain tissue in fresh dissection medium, and transfer the cut tissue back to the 90 mm silicone elastomer-coated Petri dish using a straight blade scalpel.
- Under a stereomicroscope, carefully separate the cortical slices using fine tip forceps. For each slice, inspect the hippocampus for morphology and potential tissue damage resulting from the dissection or slicing.
- Separate the hippocampi from the entorhinal cortex and from the fimbria using fine tip forceps and small Vannas scissors. Typically, around 6 to 8 hippocampal slices are generated per hemisphere.
- Transfer up to 6 hippocampal slices to a tissue culture insert using a cut Pasteur pipette and place it inside a 35 mm Petri dish. Ensure that the slices are spread a few millimeters apart (this will ensure each slice can be imaged individually).
- Immediately add ice-cold "growth medium" to the bottom of each Petri dish, under the tissue culture insert, just below the top of the tissue culture insert rim.
NOTE: Growth medium contains 50% Minimum essential medium Eagle, 25% Hanks' balanced salt solution, 25% horse serum, 5 mg/mL D-glucose, 2 mmol/L L-glutamine, 1% antibiotic-antimycotic solution, and 10 mmol/L HEPES, titrated to pH 7.2 with sodium hydroxide. - Change the growth medium the day after the dissection and then every 2 to 3 days thereafter (use growth medium at 37 °C). Ensure that the growth medium is added in sufficient amount, but not so much that it overflows over the tissue culture insert membrane, which could compromise the viability of the tissue slices.
- Keep the tissue slices in a humidified incubator at 37 °C with 5% carbon dioxide in air for 12 to 14 days prior to being used in experiments.
2. Preparation of the Hippocampal Organotypic Slices for the Experimental Blast TBI Protocol
NOTE: All the steps of this section, except imaging, take place in a laminar flow tissue culture hood.
- Insert the custom-made stainless-steel rings into a 6 well plate (one per well).
NOTES: The rings have a rim with a notch (which should sit in the 12 o'clock position) and fit snugly inside the wells, while the tissue culture inserts fit easily in this rim. Ensure that the rings are washed with a bactericidal disinfectant, thoroughly rinsed with purified water, autoclaved and allowed to cool down in advance. - Fill the well of the 6-well plate with pre-warmed (37 °C) serum-free "experimental medium" with propidium iodide.
NOTE: Experimental medium with propidium iodide is: 75% Minimum essential medium Eagle, 25% Hanks' balanced salt solution, 5 mg/mL D-glucose, 2 mmol/L L-glutamine, 1% antibiotic-antimycotic solution, 10 mmol/L HEPES and 4.5 µmol/L propidium iodide, titrated pH to 7.2 with sodium hydroxide. - Ensure that the level of the medium does not reach above the notch of the ring. Transfer the 6-well plate with the rings back to the incubator for 1 h to ensure that the medium is at 37 °C immediately before the tissue culture inserts are transferred.
- Transfer the tissue culture inserts with the organotypic slices from the 35 mm Petri dishes into the 6-well plate with the rings and the experimental medium with forceps.
- Make a dot on the insert rim in the 3 o'clock position with a permanent marker pen.
NOTE: The inserts are going to be removed from the 6-well plate during the experiment and this step facilitates returning the insert to its original position after the shock wave exposure and to easily keep track of each slice throughout the protocol. - Label each 6-well plate with a unique name & date and make a map of the wells of each plate, naming each well (e.g., A, B, C, etc.) and each slice in each well (e.g., 1, 2, 3, etc.), so that each slice has a unique identifier (e.g., A1, A2, A3, etc.).
- Transfer the 6-well plate to the incubator for 1 h to ensure the slices are at 37 °C immediately before imaging.
NOTE: Avoid overflow of medium or any bubbles of air underneath the tissue culture insert membrane, which could compromise the viability of the slices. - 1 h after transferring to experimental medium, image each slice individually using a fluorescence microscope (2X objective, NA 0.06) fitted with an appropriate excitation (BP 535/50 nm) and emission (LP 610 nm) filter to assess slice health before the injury protocol is carried out.
NOTES: The slices that exhibit areas of dense red staining at this stage should be considered to present compromised viability and should be excluded from further analysis (these typically represent less than 10% of the total number of slices generated). Ensure that imaging is performed in a sequential manner and as swiftly as possible to minimize the time that the slices are outside the incubator (typically 6 wells should take just under 30 min to image). - Keep the lid of the 6-well plate on at all times. Some condensation may build up on the inside of the lid. If this happens, briefly use a hairdryer on the low setting.
- Ensure that all imaging conditions are identical on different days and between experiments.
NOTE: The aim of imaging is to quantify the tissue fluorescence, hence this step is important to ensure the reproducibility of the results and allow the comparison of the data obtained.
3. Submersion and Transport of the Tissue Culture Inserts with the Hippocampal Organotypic Slices
- Immediately after imaging, in a laminar flow hood, take one tissue culture insert out of the 6-well plate using forceps.
- Carefully transfer the insert to a sterile polyethylene bag (3" x 5") pre-filled with 20 mL of warm (37 °C) experimental medium freshly bubbled with 95% oxygen and 5% carbon dioxide.
NOTE: Ensure that the oxygen and carbon dioxide enriched experimental medium was bubbled for at least 40 min with 95% oxygen and 5% carbon dioxide using a scintered-glass bubbler inside a Dreschel bottle and transferred into the sterile polyethylene bags inside a laminar flow tissue culture hood using a 20 mL syringe with a bacterial filter and a sterile filling tube (127 mm) attached. Seal the bags immediately and transfer them to a 37 °C incubator for at least 1 h before the tissue culture insert transfer. - Ensure that each sterile bag is correctly labelled (with the plate and well identification). Repeat this step for each tissue culture insert. Exclude the air bubbles carefully upon sealing the sterile bags (done securely by twisting the top of the bags and by applying a plastic clamp).
- Return the sterile bags with the tissue culture inserts and the 6-well plates with experimental medium into the 37 °C incubator.
- After 1 h, carefully pack the sterile bags with the tissue culture inserts in plastic boxes inside a thermo-regulated box filled with de-ionized water at 37 °C in order to keep the organotypic slices at physiologic temperature throughout the shock wave exposure protocol.
4. Preparation of the Shock Tube and Hippocampal Organotypic Slice Shock Wave Exposure
- Wear steel-toe protective boots, a laboratory coat and gloves during the preparation of the shock tube and the shock wave exposure.
- Bolt the sterile bag holder frame to the shock tube distal flange, ensuring that the central hole is aligned with the shock tube outlet (using a blanking rod).
- Mount two pressure transducers radially: sensor 1, in the middle part of the driven section and sensor 2 in the distal flange of the shock tube (Figure 1A). Connect the pressure transducers to an oscilloscope through a current source power unit.
- Ensure that all shock tube release valves and flow controls are closed.
- Open the external compressed air line and charge the solenoid valve to 2.5 bar.
- Open the compressed air cylinder safety valve and slowly open the pressure regulator to increase the pressure to approximately 5 bar.
NOTE: The pressure set for this regulator should be slightly above the highest diaphragm bursting pressure. - Prepare the diaphragms by cutting 23 µm-thick polyester sheets into 10 x 10 cm2 squares. Prepare the handles using autoclave tape and sticking them to the top and bottom of each diaphragm.
- Position one diaphragm (single slot of the double breech - single diaphragm configuration) or two diaphragms (both slots of the double breech - double diaphragm configuration) in the breech (Figure 1B).
- Center the diaphragms, and clamp them using four M24 bolts and nuts, fastening them sequentially in a diagonally symmetric way, and ensuring that the diaphragms are wrinkle-free.
- Clamp each sterile bag individually in vertical position on the holder frame, ensuring that the surface of the tissue culture inserts with the organotypic hippocampal slices is facing the shock tube outlet and the tissue culture insert is centered inside the sterile bag (Figure 1C). Make sure that the sterile bag is securely clamped all around to ensure firm and even immobilization.
- Wear ear defenders and safety spectacles when pressuring the shock tube. Switch on the current source power unit and oscilloscope to acquire the shock wave data (acquisition rate of 50 mega-samples/s, record length 20 ms, 1 million points) and close solenoid valve.
- Using the flow control knob on the shock tube control panel, slowly pressurize the driver volume section of the shock tube for single diaphragm configuration or both the driver volume section and the double breech section of the shock tube for double diaphragm configuration.
NOTE: For single diaphragm configuration, the burst pressure will depend only on the diaphragm material and thickness and the diaphragm will rupture spontaneously once the material bursting pressure is reached. For double diaphragm configuration, the bursting pressure will also depend on the gas pressure differential in both the driver and the double breech chambers and, for the diaphragms to burst in a controlled way, the double breech safety valve is opened manually once the target pressures are reached. - As soon as the diaphragm ruptures (producing a loud noise), quickly close the compressed air flow using the flow knob and open the solenoid valve.
NOTE: The total volume of the driver section can be modified by the insertion of blanking segments, allowing a wider range of shock wave peak overpressure and durations to be obtained. The ideal combination of shock wave parameters should be enough to cause tissue injury but not so high that it causes tissue culture insert or sterile bag distortion or rupture. - Expose each sterile bag with a tissue culture insert to a single shock tube wave and return it immediately to the thermo-regulated box before a new sterile bag is taken from the box and clamped on the holder frame. Ensure that Steps 4.10–4.14 are performed as smoothly and swiftly as possible (within a few minutes) to prevent the experimental medium cooling down, as temperatures below 37 °C may interfere with injury development.
- Once all tissue culture inserts have been exposed to a shock wave (or sham protocol), return the tissue culture inserts to the original 6-well plate and to their respective well (inside a laminar flow tissue culture hood) and return to the incubator.
- Keep the 6-well plate in the incubator with 5% carbon dioxide in air at 37 °C until further imaging.
- Include sham controls for each experiment, together with the slice shock wave exposure.
NOTE: Sham slices are treated identically to the slices exposed to a shock wave (sealed in the sterile bags with experimental medium, transported to the shock tube laboratory in the same thermo-regulated box and suspended on the metal frame for an equivalent period of time) but the shock tube is not fired.
5. Hippocampal Organotypic Slice Injury Quantification
- At 24 h, 48 h and 72 h, image the slices as described in Steps 2.8 and 2.9.
- After imaging at 72 h post shock wave exposure, discard the tissue following local biological waste protocols and disinfect and autoclave the metal rings.
Results
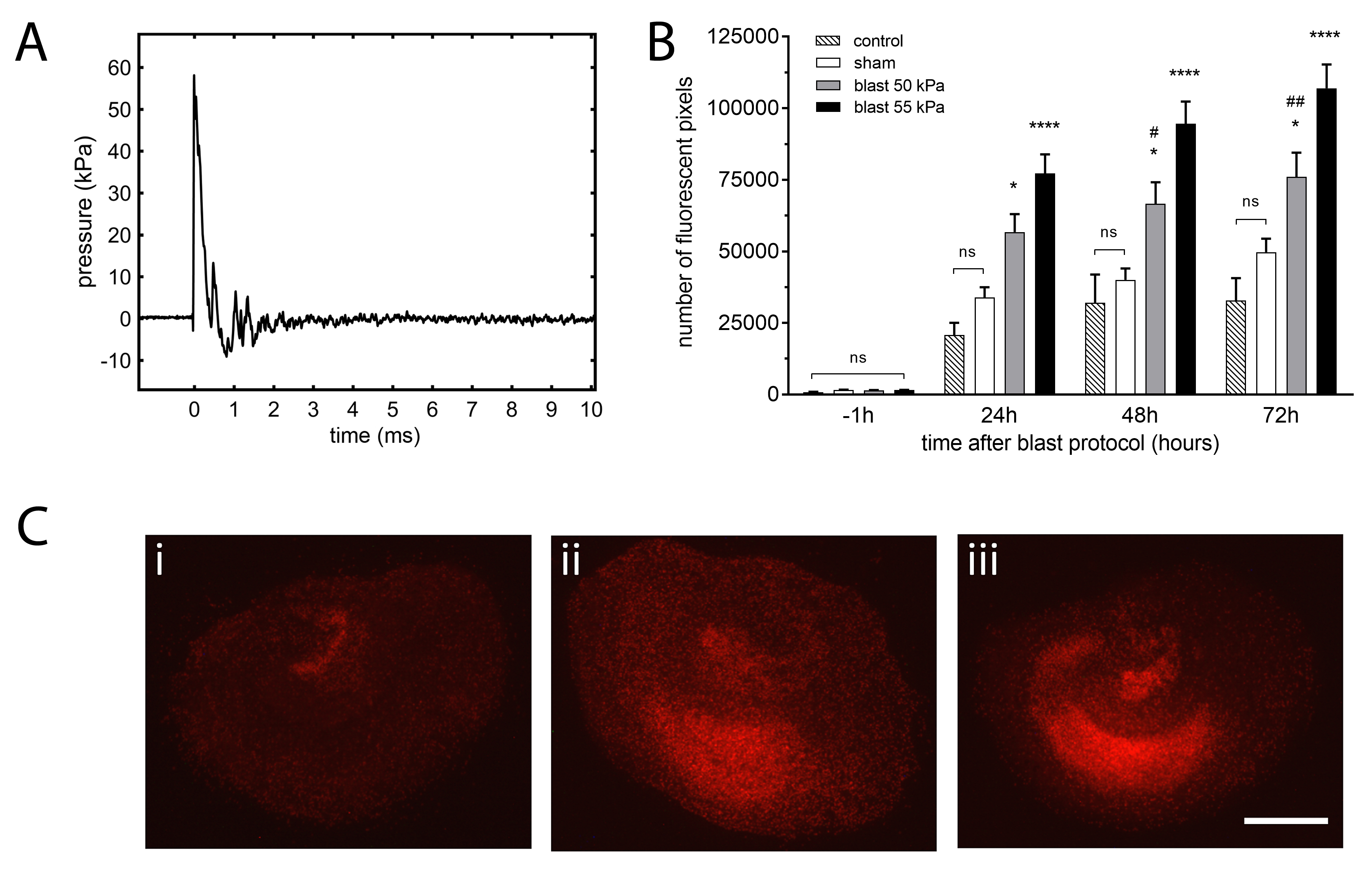
The shock tube used in this method allows the generation of overpressure transients that simulate real-life open field explosions modelled by the Friedlander function7,8. Supersonic shockwaves with a velocity of 440 m/s (Mach 1.3) were obtained (Figure 2A). The waveform data reported is from sensor 2, positioned radially at the end of the driven section of the shock tube.
Using the protocol described above, organotypic hippocampal slice cultures exposed to a single shock wave (Figure 2A) develop significant injury quantified using propidium iodide, a highly polar fluorescent dye that only penetrates the cells with compromised cellular membranes24,25 (Figure 2B, C).
Even under optimal conditions, and consistent to other OHSC published models21,22, there is a low level of background propidium iodide fluorescence due, in part, to minor damage resulting from the inherent tissue manipulations (such as media changes during the culture period or removal from the incubator for imaging). This blast TBI protocol involves substantial manipulation that includes the submersion of the slices in medium inside sterile bags and a considerable degree of handling during the shock wave exposure protocol (e.g., clamping the sterile bags to the holder frame). However, if all steps are performed carefully, this additional manipulation does not have an impact on the underlying health of the OHSC as no significant differences were seen between a control group of slices kept in the 6-well plates at all times (i.e., the inserts were not submerged or handled) and the sham group, which included slices that were submerged inside sterile bags clamped to the shock tube (Figure 2B).
The two shock waves chosen, at 50 kPa and 55 kPa peak overpressure, produced significant (p <0.05 and p <0.0001, respectively) and reproducible injury when compared to the uninjured sham slices at all time-points after the blast exposure protocol (Figure 2B) without causing any damage to the tissue culture inserts or the sterile bags. In order to determine the sensitivity of the model to small differences in peak-overpressure, we decided to select values that were different by ~10 %. These results also show that, as expected, the injury resulting from 55 kPa is higher than that after a 50 kPa shock wave.
The data are expressed as mean ± standard error of the mean. Significance was assessed using a 2-way repeated measures analysis of variance using Holm-Sidak post hoc test. Factor 1 was group (control, sham, blast) and factor 2 was time after the injury (-1 h, 24 h, 48 h and 72 h), where factor 1 was the repeated factor. The adjustment of p value for multiple comparisons was used. P values of less than 0.05 were taken to indicate a significant difference between groups. Statistical tests were implemented using a graphing and statistics software package.

Figure 1: Schematic of the shock tube device with the sterile bag holder frame. (A). The shock tube is a 3.8 m long stainless-steel tube, made of three 1.22 m long sections, connected by gaskets and flanges, with an internal diameter of 59 mm. (B) Inset shows the double breech assembly. One or two Mylar diaphragms can be clamped in the assembly with seal provided by rubber o-rings. (C) Sterile bag holder frame. The body of the frame consists of two metal plates with a centered circular hole (59 mm diameter) that aligns with the shock tube outlet. Two thin (4 mm) sheets of silicone elastomer are fitted between the two metal plates. The purpose of these sheets is to provide an even and non-slippery surface to clamp the sterile bags. The distance between the bag and the outlet of the shock tube is 7 cm. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Typical shockwave and resulting injury in organotypic hippocampal slice cultures. (A) Representative example of shock wave obtained using 23 µm-thick polyester film, 2.16 bar burst pressure, 55 kPa peak overpressure, 0.4 ms positive wave duration, 10.1 kPa·ms impulse. Waveform data were obtained from sensor 2 radially mounted on the distal flange of the shock tube driven section. The shock wave velocity was 440 m/s (Mach 1.3). (B) The development of injury is proportional to the intensity of the shock wave. Both 50 kPa and 55 kPa peak overpressure shock waves caused significant injury that developed throughout the 72 h protocol when compared with the sham group. The injury resulting from a 55 kPa peak overpressure wave exposure was significantly higher than after 50 kPa at 48 h and 72 h. Sham slices were treated identically to blast slices but shock tube was not fired. Control slices were maintained in 6 well plates in an incubator without any manipulation. Bars represent mean values and error bars are standard errors (n = 7, controls; n = 48, sham; n = 30, blast 50 kPa; n = 51, blast 55 kPa; n = number of slices, from 6 separate experiments). *p <0.05, ****p <0.0001 compared with sham. #p <0.05, ##p <0.01 compared with blast 55 kPa. (C) Representative propidium iodide fluorescence images of organotypic slices from (i) sham, (ii) blast 50 kPa and (iii) blast 55 kPa groups at 72 h after injury. The sham slice shows low levels of fluorescence, i.e., injury, and the blast exposed slices show high levels of diffuse injury, more pronounced on the 55 kPa peak overpressure exposed slice (Scale bar = 500 µm). Please click here to view a larger version of this figure.
{kind=link}
Discussion
Among all the mechanisms of injury associated with blast TBI (primary, secondary and tertiary blast injury mechanisms), primary blast injury is unique to blast trauma and it is the least understood of the blast-associated mechanisms1,2. The novel protocol described here was developed to study primary blast TBI using an open-ended shock tube to expose in vitro mouse hippocampal slice cultures to a single shock wave using a simple and rapid protocol that allows the creation of a reproducible primary blast TBI with a high throughput.
The first in vitro primary blast TBI models applied hydrostatic pressure waves to cells26,27. However, the pressure output did not model the Friedlander function as the duration of a hydrostatic pressure pulse was much longer than airborne blast overpressure waves13. The characteristic Friedlander function can be easily modelled in the laboratory using a shock tube1,8. The shock tube can produce shock waves that simulate real-life open field explosions in a conventional laboratory environment, while allowing the precise control of wave parameters, such as peak overpressure, positive wave duration and impulse, by varying the diaphragm material and thickness, and the driver volume8,28,29.
Simple in vitro models such as cell cultures usually lack the heterogeneity of cell types and synaptic connectivity30. Recently, the effect of blast on in vitro brain cell 'spheroids' incorporating different cell types has been investigated31. Further investigation of these interesting preparations is merited; however, it is not clear how their cellular organization and connectivity mirrors the intact brain. OHSC are a well-established in vitro experimental model23,32, are easy to culture and their three-dimensional tissue cytoarchitecture, cell differentiation and synaptic connectivity are well preserved and very similar to that in vivo33,34,35,36. OHSCs represent an intermediate level of complexity between cell culture and an in vivo model23,32. OHSCs have been demonstrated to reproduce in vitro pathological neurodegenerative cascades seen in in vivo models and have been very useful in the screening of potential neuroprotective drugs and in understanding their mechanisms of action17,21,22,37,38. Finally, the anatomic area studied, the hippocampus, is highly relevant in translational TBI studies, as this region is frequently damaged in TBI patients39,40,41.OHSC have been used to model blast TBI28,42,43,44, however, our model is relatively simple and can be adapted to existing shock-tubes in either horizontal or vertical configurations without complex adaptations.
OHSC can be kept in culture for many days, which facilitates the investigation of biological processes over time34. In this model, the tissue injury that resulted from shock wave exposure was measured daily over three days, following the blast exposure using propidium iodide, a well-established marker of cell damage. Propidium iodide is a nontoxic highly polar dye that penetrates the cells with compromised cellular membranes, where it binds to nucleic acids and exhibits a characteristic bright red fluorescence24,25,45. The fluorescence measured with propidium iodide has been shown to have a good correlation with injured cell count using Nissl staining46,47.
Given that the injury produced in this model was diffuse (Figure 2C), the fluorescence of the whole slice was measured when performing the analysis, similar to previously published work in other brain injury paradigms21,22, instead of using specific regions, as has been done in other in vitro blast TBI models28,43,44,48. The global approach used in the model described in this article also eliminates the potential variability that is introduced when outlining defined regions of interest and provides a more comprehensive picture of the blast-related injury. Both shock wave peak overpressures, 50 kPa and 55 kPa, produced significant (p <0.05 and p <0.0001, respectively) injury when compared to sham slices (Figure 2B). As anticipated, the shock wave with the highest peak overpressure, 55 kPa, produced more injury than the 50 kPa wave. In an in vitro model with isolated brain tissue directly exposed to a shockwave, how to accurately scale to the whole organism or a human being is not straightforward. Nevertheless, the shockwaves we used are within the range of peak overpressures observed in the field, typically 50–1,000 kPa8,49.
In order to maintain the OHSC exposed to physiologic temperature and levels of oxygen and carbon dioxide, while ensuring that they were free from contamination throughout the shock wave exposure protocol, the tissue culture inserts were sealed into sterile polyethylene bags following an aseptic technique, submerged in experimental medium warmed to 37 °C and freshly bubbled with 95% oxygen and 5% carbon dioxide, similarly to previously published work28,43,44,48. Contrary to these models where complex devices were used to hold the sterile bags during shock wave exposure, in this protocol, a simple and rapid method was used to suspend the OHSC tissue culture inserts in front of the shock tube outlet (Figure 1A, C). The model described in this paper allows rapid processing and high throughput, while minimizing the risk of hypothermia. These aspects are particularly relevant for neuroprotection studies given that some therapeutic interventions may have a very limited time window of potential application after TBI. This novel shock wave exposure protocol allows 6 to 9 tissue culture inserts (typically 36 to 54 hippocampal organotypic tissue slices) to be exposed to a shock wave in a short interval of time (approximately 1 h).
The OHSCs require good aseptic technique throughout. It is important to use an aseptic laminar flow hood throughout the culturing and when transferring to the sterile bags for blast. In order to carry out the slice imaging under aseptic conditions with the lids of the 6-well plates in place, we use custom-made metal rings to raise the cell culture inserts to the focal plane of the microscope. An important part of our protocol is that we include uninjured sham slices in every experiment. Sham slices are treated identically to blast slices with the exception that the shock-tube is not fired; another important step is that all slices are imaged 1 h before injury or sham treatment, to ensure that the health of the population of slices used are identical (Figure 2B).
In addition to quantifying cell injury in the slices over time, the tissue can be fixed at the end of the experiment for conventional immunohistochemistry50. We developed and evaluated the method using mouse hippocampal slices. However, our technique could be easily adapted to use other tissue that can be grown in culture, such as spinal cord, retina, lung or epithelial tissue. In this paper and our previous work with the model, we investigated only the effect of exposure to a single blast. However, the model would be well suited to investigate the effects of repeated low-level blasts on brain or other tissue. OHSCs can be kept in culture for many weeks or even months, allowing chronic effects to be investigated.
In vitro models, being simpler than in vivo models, have a higher throughput, are less expensive and experiments can usually be completed on a shorter time scale17. However, the results obtained using in vitro models need to be validated in animal models as in vitro cultured tissues are kept in an artificial environment and may respond to injury differently from what they would in vivo17. Nonetheless, in vitro models have been extremely valuable in increasing our understanding of brain injury cascades and in screening neuroprotective drugs before the use of more complex in vivo models17,22,51,52. Despite the many advantages offered by this model, it is important to note that in vitro models lack the key features of TBI present in animals and in vivo models, such as the effects on vascular system, increased intracranial pressure, systemic immune response and functional behavioral impairment, which highlights the need to validate the results found in in vitro models in the whole animal. Nevertheless, in vitro models such as the model described in this paper are extremely useful translationally relevant scientific tools.
In conclusion, this work describes a simple and straightforward novel method where mouse organotypic hippocampal tissue cultures are exposed to tightly controlled and reproducible real-life relevant shock waves using a laboratory shock tube. The resulting global injury, which was quantified using propidium iodide, a well-established marker of cell damage, is very reproducible and is proportional to the peak overpressure of the shock waves applied.
Disclosures
The authors have no competing financial interests.
Acknowledgements
Supported by: Royal Centre for Defence Medicine, Birmingham, United Kingdom, Royal British Legion Centre for Blast Injury Studies, Imperial College London, United Kingdom. Medical Research Council, London, United Kingdom (MC_PC_13064; MR/N027736/1). The Gas Safety Trust, London, United Kingdom. Rita Campos-Pires was the recipient of a doctoral training award from the Fundação para a Ciência e a Tecnologia, Lisbon, Portugal. Katie Harris was the recipient of a PhD studentship from the Westminster Medical School Research Trust, London, United Kingdom.
This model was developed with the support of the Royal British Legion Centre for Blast Injury Studies (RBLCBIS) at Imperial College. We would like to acknowledge the financial support of the Royal British Legion. Researchers interested in collaborations or further detail may contact the authors or RBLCBIS.
We thank Dr Amarjit Samra, Director of Research, Royal Centre for Defence Medicine, Birmingham, United Kingdom, for supporting this work, Scott Armstrong, Department of Surgery & Cancer, Imperial College London, for assistance with preliminary experiments, Theofano Eftaxiopolou, Hari Arora & Luz Ngoc Nguyen, Department of Bioengineering Imperial College London, & William Proud, Department of Physics Imperial College London, for advice on the shock-tube, Raquel Yustos, research technician, Department of Life Sciences, Imperial College London, for technical support, Paul Brown MBE, workshop manager and Steve Nelson, workshop technician, Department of Physics, Imperial College London, for making the metal rings, Neal Powell of the Department of Physics, Imperial College London, for artwork.
Materials
| Name | Company | Catalog Number | Comments |
| Geys balanced salt solution | Sigma UK | G9779 | |
| D-glucose | Sigma UK | G8270 | |
| Antibiotic/antimycotic | Sigma UK | A5955 | |
| Minimum essential medium Eagle | Sigma UK | M4655 | |
| Hanks balanced salt solution | Sigma UK | H9269 | |
| Horse serum | Sigma UK | H1138 | |
| L-glutamine | Sigma UK | G7513 | |
| HEPES | VWR Prolabo, Belgium | 441476L | |
| Sodium hydroxide | Sigma UK | S-0945 | |
| Tissue culture inserts | Millicell CM 30 mm low height Millipore | PICM ORG 50 | |
| 6-well plates | NUNC, Denmark | 140675 | |
| Propidium iodide | Sigma UK | P4864 | |
| Sterile polyethylene bags - Twirl'em sterile sample bags | Fisherbrand | 01-002-30 | |
| Portex Avon Kwill Filling Tube 5" (127 mm) | Smiths Medical Supplies | E910 | |
| Epifluorescence microscope | NIKON Eclipse 80i, UK | ||
| Microscope objective | Nikon Plan UW magn. 2x, NA 0.06, WC 7.5 mm | ||
| Microscope filter | Nikon G-2B (longpass emission) | ||
| Mylar electrical insulating film, 304 mm x 200 mm x 0.023 mm | RS Components UK | 785-0782 | |
| Pressure transducer | Dytran Instruments Inc. | 2300V1 | |
| Tissue chopper | Mickle Laboratory Engineering Co., Guildford, Surrey, United Kingdom. | Mcllwain tissue chopper | |
| Silicone elastomer | Dow Corning, USA | Sylgard 184 | |
| Graphing & statistics software | GraphPad Software, USA | Prism 7.0 |
References
- Risling, M., Davidsson, J. Experimental animal models for studies on the mechanisms of blast-induced neurotrauma. Frontiers in Neurology. 3, 30(2012).
- Nakagawa, A., et al. Mechanisms of primary blast-induced traumatic brain injury: insights from shock-wave research. Journal of Neurotrauma. 28 (6), 1101-1119 (2011).
- Goldstein, L. E., McKee, A. C., Stanton, P. K. Considerations for animal models of blast-related traumatic brain injury and chronic traumatic encephalopathy. Alzheimer's Research & Therapy. 6 (5), 64(2014).
- Rona, R. J., et al. Mild traumatic brain injury in UK military personnel returning from Afghanistan and Iraq: cohort and cross-sectional analyses. Journal of Head Trauma and Rehabilitation. 27 (1), 33-44 (2012).
- Terrio, H., et al. Traumatic brain injury screening: preliminary findings in a US Army Brigade Combat Team. Journal of Head Trauma and Rehabilitation. 24 (1), 14-23 (2009).
- Elder, G. A., Stone, J. R., Ahlers, S. T. Effects of low-level blast exposure on the nervous system: is there really a controversy. Frontiers in Neurology. 5, 269(2014).
- Ling, G., Bandak, F., Armonda, R., Grant, G., Ecklund, J. Explosive blast neurotrauma. Journal of Neurotrauma. 26 (6), 815-825 (2009).
- Bass, C. R., et al. Brain injuries from blast. Annals of Biomedical Engineering. 40 (1), 185-202 (2012).
- Young, L. A., Rule, G. T., Bocchieri, R. T., Burns, J. M. Biophysical mechanisms of traumatic brain injuries. Seminars in Neurology. 35 (1), 5-11 (2015).
- Wolf, S. J., Bebarta, V. S., Bonnett, C. J., Pons, P. T., Cantrill, S. V. Blast injuries. Lancet. 374 (9687), 405-415 (2009).
- Kluger, Y., Nimrod, A., Biderman, P., Mayo, A., Sorkin, P. The quinary pattern of blast injury. American Journal of Disaster Medicine. 2 (1), 21-25 (2007).
- Champion, H. R., Holcomb, J. B., Young, L. A. Injuries from explosions: physics, biophysics, pathology, and required research focus. Journal of Trauma: Injury, Infection, and Critical. 66 (5), 1468-1477 (2009).
- Chen, Y. C., Smith, D. H., Meaney, D. F. In-vitro approaches for studying blast-induced traumatic brain injury. Journal of Neurotrauma. 26 (6), 861-876 (2009).
- Edwards, D. S., Clasper, J. Blast Injury Science and Engineering: A Guide for Clinicians and Researchers. Bull, A. M. J., Clasper, J., Mahoney, P. F. , Springer International Publishing. 87-104 (2016).
- Kirkman, E., Watts, S., Cooper, G. Blast injury research models. Philosophical Translations of the Royal Society B: Biological Sciences. 366 (1562), 144-159 (2011).
- Hicks, R. R., Fertig, S. J., Desrocher, R. E., Koroshetz, W. J., Pancrazio, J. J. Neurological effects of blast injury. J Trauma. 68 (5), 1257-1263 (2010).
- Morrison, B., Elkin, B. S. 3rd, Dolle, J. P., Yarmush, M. L. In vitro models of traumatic brain injury. Annual Reviews of Biomedical Engineering. 13, 91-126 (2011).
- Johnson, V. E., Meaney, D. F., Cullen, D. K., Smith, D. H. Animal models of traumatic brain injury. Handbook of Clinical Neurology. , 115-128 (2015).
- Xiong, Y., Mahmood, A., Chopp, M. Animal models of traumatic brain injury. Nature Reviews Neuroscience. 14 (2), 128-142 (2013).
- Morganti-Kossmann, M. C., Yan, E., Bye, N. Animal models of traumatic brain injury: is there an optimal model to reproduce human brain injury in the laboratory? Injury. 41, Suppl 1. S10-S13 (2010).
- Banks, P., Franks, N. P., Dickinson, R. Competitive inhibition at the glycine site of the N-methyl-D-aspartate receptor mediates xenon neuroprotection against hypoxia-ischemia. Anesthesiology. 112 (3), 614-622 (2010).
- Harris, K., et al. Neuroprotection against traumatic brain injury by xenon, but not argon, is mediated by inhibition at the N-methyl-D-aspartate receptor glycine site. Anesthesiology. 119 (5), 1137-1148 (2013).
- Stoppini, L., Buchs, P. A., Muller, D. A simple method for organotypic cultures of nervous tissue. Journal of Neuroscience Methods. 37 (2), 173-182 (1991).
- Noraberg, J., Kristensen, B. W., Zimmer, J. Markers for neuronal degeneration in organotypic slice cultures. Brain Research Protocols. 3 (3), 278-290 (1999).
- Macklis, J. D., Madison, R. D. Progressive incorporation of propidium iodide in cultured mouse neurons correlates with declining electrophysiological status: a fluorescence scale of membrane integrity. Journal of Neuroscience Methods. 31 (1), 43-46 (1990).
- Salvador-Silva, M., et al. Responses and signaling pathways in human optic nerve head astrocytes exposed to hydrostatic pressure in vitro. Glia. 45 (4), 364-377 (2004).
- Howard, D., Sturtevant, B. In vitro study of the mechanical effects of shock-wave lithotripsy. Ultrasound Medical Biology. 23 (7), 1107-1122 (1997).
- Effgen, G. B., et al. A Multiscale Approach to Blast Neurotrauma Modeling: Part II: Methodology for Inducing Blast Injury to in vitro Models. Front Neurol. 3, 23(2012).
- Nguyen, T. T. The characterisation of a shock tube system for blast injury studies. , Imperial College London. PhD Thesis (2016).
- Noraberg, J., et al. Organotypic hippocampal slice cultures for studies of brain damage, neuroprotection and neurorepair. Current Drug Targets CNS and Neurological Disorders. 4 (4), 435-452 (2005).
- Sawyer, T. W., et al. Investigations of primary blast-induced traumatic brain injury. Shock Waves. 28 (1), 85-99 (2017).
- Gahwiler, B. H. Organotypic monolayer cultures of nervous tissue. Journal of Neuroscience Methods. 4 (4), 329-342 (1981).
- Gahwiler, B. H., Capogna, M., Debanne, D., McKinney, R. A., Thompson, S. M. Organotypic slice cultures: a technique has come of age. Trends in Neuroscience. 20 (10), 471-477 (1997).
- De Simoni, A., Yu, L. M. Preparation of organotypic hippocampal slice cultures: interface method. Nature Protocols. 1 (3), 1439-1445 (2006).
- Gogolla, N., Galimberti, I., DePaola, V., Caroni, P. Long-term live imaging of neuronal circuits in organotypic hippocampal slice cultures. Nature Protocols. 1 (3), 1223-1226 (2006).
- De Simoni, A., Griesinger, C. B., Edwards, F. A. Development of rat CA1 neurones in acute versus organotypic slices: role of experience in synaptic morphology and activity. Journal of Physiology. 550 (Pt 1), 135-147 (2003).
- Sundstrom, L., Morrison, B., Bradley, M. 3rd, Pringle, A. Organotypic cultures as tools for functional screening in the CNS). Drug Discovery Today. 10 (14), 993-1000 (2005).
- Cater, H. L., et al. Stretch-induced injury in organotypic hippocampal slice cultures reproduces in vivo post-traumatic neurodegeneration: role of glutamate receptors and voltage-dependent calcium channels. Journal of Neurochemistry. 101 (2), 434-447 (2007).
- Atkins, C. M. Decoding hippocampal signaling deficits after traumatic brain injury. Translational Stroke Research. 2 (4), 546-555 (2011).
- Bigler, E. D., et al. Hippocampal volume in normal aging and traumatic brain injury. American Journal of Neuroradiology. 18 (1), 11-23 (1997).
- Umile, E. M., Sandel, M. E., Alavi, A., Terry, C. M., Plotkin, R. C. Dynamic imaging in mild traumatic brain injury: support for the theory of medial temporal vulnerability. Archives of Physical Medicine and Rehabilitation. 83 (11), 1506-1513 (2002).
- Effgen, G. B., et al. Primary Blast Exposure Increases Hippocampal Vulnerability to Subsequent Exposure: Reducing Long-Term Potentiation. Journal of Neurotrauma. 33 (20), 1901-1912 (2016).
- Effgen, G. B., et al. Isolated primary blast alters neuronal function with minimal cell death in organotypic hippocampal slice cultures. Journal of Neurotrauma. 31 (13), 1202-1210 (2014).
- Vogel Iii, E. W., et al. Isolated Primary Blast Inhibits Long-Term Potentiation in Organotypic Hippocampal Slice Cultures. Journal of Neurotrauma. , (2015).
- Vornov, J. J., Tasker, R. C., Coyle, J. T. Direct observation of the agonist-specific regional vulnerability to glutamate, NMDA, and kainate neurotoxicity in organotypic hippocampal cultures. Experimental Neurology. 114 (1), 11-22 (1991).
- Cho, S., et al. Spatiotemporal evidence of apoptosis-mediated ischemic injury in organotypic hippocampal slice cultures. Neurochemistry International. 45 (1), 117-127 (2004).
- Newell, D. W., Barth, A., Papermaster, V., Malouf, A. T. Glutamate and non-glutamate receptor mediated toxicity caused by oxygen and glucose deprivation in organotypic hippocampal cultures. The Journal of Neuroscience. 15 (11), 7702-7711 (1995).
- Miller, A. P., et al. Effects of blast overpressure on neurons and glial cells in rat organotypic hippocampal slice cultures. Frontiers in Neurology. 6, 20(2015).
- Panzer, M. B., Wood, G. W., Bass, C. R. Scaling in neurotrauma: how do we apply animal experiments to people. Experimental Neurology. , 120-126 (2014).
- Campos-Pires, R., et al. Xenon Protects against Blast-Induced Traumatic Brain Injury in an In Vitro Model. Journal of Neurotrauma. 35 (8), 1037-1044 (2018).
- Coburn, M., Maze, M., Franks, N. P. The neuroprotective effects of xenon and helium in an in vitro model of traumatic brain injury. Critical Care Medicine. 36 (2), 588-595 (2008).
- Campos-Pires, R., et al. Xenon improves neurologic outcome and reduces secondary injury following trauma in an in vivo model of traumatic brain injury. Critical Care Medicine. 43 (1), 149-158 (2015).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved