
Method Article
RNA-based Reprogramming of Human Primary Fibroblasts into Induced Pluripotent Stem Cells
In This Article
Summary
Here we describe a clinically relevant, high-efficiency, feeder-free method to reprogram human primary fibroblasts into induced pluripotent stem cells using modified mRNAs encoding reprogramming factors and mature microRNA-367/302 mimics. Also included are methods to assess reprogramming efficiency, expand clonal iPSC colonies, and confirm expression of the pluripotency marker TRA-1-60.
Abstract
Induced pluripotent stem cells (iPSCs) have proven to be a valuable tool to study human development and disease. Further advancing iPSCs as a regenerative therapeutic requires a safe, robust, and expedient reprogramming protocol. Here, we present a clinically relevant, step-by-step protocol for the extremely high-efficiency reprogramming of human dermal fibroblasts into iPSCs using a non-integrating approach. The core of the protocol consists of expressing pluripotency factors (SOX2, KLF4, cMYC, LIN28A, NANOG, OCT4-MyoD fusion) from in vitro transcribed messenger RNAs synthesized with modified nucleotides (modified mRNAs). The reprogramming modified mRNAs are transfected into primary fibroblasts every 48 h together with mature embryonic stem cell-specific microRNA-367/302 mimics for two weeks. The resulting iPSC colonies can then be isolated and directly expanded in feeder-free conditions. To maximize efficiency and consistency of our reprogramming protocol across fibroblast samples, we have optimized various parameters including the RNA transfection regimen, timing of transfections, culture conditions, and seeding densities. Importantly, our method generates high-quality iPSCs from most fibroblast sources, including difficult-to-reprogram diseased, aged, and/or senescent samples.
Introduction
Reprogramming somatic cells into induced pluripotent stem cells (iPSCs) requires extended expression of a core set of transcription factors that are important in maintaining pluripotency1,2. When producing iPSCs for clinical applications, it is essential that the mutational burden in input cells is minimized during processing and the efficiency of iPSC generation is maintained at a relatively high level across patient samples. However, the majority of reprogramming methods, including integration-free protocols, suffers from very low reprogramming efficiencies, which limit clinical usefulness of these approaches3. The low reprogramming efficiency may also promote selective reprogramming of cells carrying preexisting mutations, increasing mutational burden in resulting iPSCs. In addition, all DNA-based reprogramming methods, such as lentivirus- and episomal-based approaches, suffer from the safety concern that DNA may randomly integrate into the genome and create the opportunity for harmful insertional mutagenesis and unwanted (potentially oncogenic) expression of pluripotency genes in downstream tissue derivatives4.
A promising approach to achieve efficient induction of pluripotency in somatic cells and reduce mutational burden in resulting iPSCs is to use synthetic capped messenger RNAs containing modified nucleobases (modified mRNAs) for reprogramming5. The efficiency of modified mRNA-based reprogramming approaches can be further enhanced by adding embryonic stem cell (ESC)-specific microRNAs (miRNAs)-367/302s3, which have been shown to reprogram somatic cells with increased efficiency6,7. However, even with the addition of miRNAs-367/302s, the modified mRNA-based reprogramming approach often fails during application to freshly isolated patient cells3. To address inconsistencies of this modified mRNA-based approach, we have recently reported an optimized, integration-free strategy that induces pluripotency in human primary fibroblasts with a high success rate and utilizes both modified mRNAs encoding reprogramming factors and mature miRNA-367/302 mimics8. In our method, the reprogramming modified mRNA cocktail includes a modified version of OCT4 fused with the MyoD transactivation domain (called M3O)9 and five other reprogramming factors (SOX2, KLF4, cMYC, LIN28A, and NANOG). Combining the modified mRNA encoding the pluripotency factors with the miRNA mimics appeared to have a synergistic effect on reprogramming efficiencies in this protocol. Additional optimizations of the RNA transfection regimen, cell seeding, and culturing conditions were also necessary to increase reprogramming efficiency of the approach to an ultra-high level8.
Unlike many other protocols, our reprogramming approach typically requires only a few thousand input fibroblasts. In addition, many common non-integrating strategies using episomal plasmids, Sendai virus, or self-replicating RNA involve extensive passaging to dilute the reprogramming vector in generated iPSCs. Conversely, modified mRNA and mature miRNA mimics have a short half-life and are rapidly eliminated from cells. Taken together, the amount of cumulative cell culture time between collecting patient samples and the generation of usable iPSCs is minimal in this approach, effectively limiting mutation accumulation in resulting iPSCs and improving cost-effectiveness.
Here, we present the detailed step-by-step protocol to achieve high-efficiency reprogramming of adult human fibroblasts into iPSCs using our combinatorial modified mRNA/miRNA-based approach8. This RNA-based reprogramming protocol provides a straightforward, cost-effective, and robust method for generating integration-free iPSCs for research and potential clinical applications. Furthermore, it is applicable to the reprogramming of a variety of fibroblast lines including difficult-to-reprogram, disease-associated, aged, and senescent fibroblasts. A schematic of the protocol for reprogramming human fibroblasts is shown in Figure 1. The protocol specifically describes a method for reprogramming three wells of human adult primary fibroblasts in a 6-well format plate. Two wells typically yield a sufficient number of high-quality iPSC colonies. In many cases, only one well is necessary, and the third well can be used for analysis of reprogramming efficiency. If required, the number of wells can be scaled up.
Protocol
Work under RNase-free conditions and use aseptic techniques when possible. Perform all cell culture-related manipulations in a biological safety cabinet using aseptic techniques. Follow institutional biosafety standards for work with human cells.
1. Reagents and Equipment for Preparation of Reprogramming Initiation
- Prepare the modified-mRNA cocktail encoding reprogramming factors.
- Follow the previously published protocol10 to perform in vitro transcription, capping, and dephosphorylation procedures for each modified mRNA encoding individual reprogramming factor. After the final purification step, elute the modified mRNA with nuclease-free water, supplement the purified modified mRNA solution with 1 U/µL RNase inhibitor, quantitate the modified mRNAs using a spectrophotometer, and store at -80 °C for up to 6 months. Prepare multiple aliquots of each modified mRNA to minimize the number of freeze-thaw cycles.
NOTE: Similar protocols for modified mRNA production have been reported elsewhere5,8,11. Templates for in vitro transcription can be generated by PCR amplification from corresponding plasmid templates using primers listed in the Table of Materials according to the previously published protocol10. Plasmid templates for each reprogramming factor (M3O9, SOX2, KLF4, cMYC, NANOG, LIN28A) and mWasabi (control for transfection) are available from Addgene, a non-profit plasmid repository. - Mix together all the modified mRNAs at a molar ratio of 3:1:1:1:1:1 (M3O : SOX2 : KLF4 : cMYC : NANOG : LIN28A) and include 10% mWasabi modified mRNA as a control for the transfection efficiency. Adjust the concentration of the complete modified mRNA reprogramming mix to a final concentration of 100 ng/µL by adding nuclease-free water supplemented with 1 U/µL RNase inhibitor. Prepare seven 33 µL aliquots of the complete modified mRNA cocktail. Store the mixed reprogramming aliquots at -80 °C.
NOTE: For each transfection, 1,000 ng of the modified mRNA cocktail is added per well (i.e., a total of 3,000 ng for 3 wells). Each 33 µL aliquot is sized to transfect 3 wells of a 6-well format plate and includes 3 µL of excess volume to account for pipetting errors. Preparing seven 33 µL aliquots is sufficient to complete a full fibroblast reprogramming of 3 wells in a 6-well format plate.
- Follow the previously published protocol10 to perform in vitro transcription, capping, and dephosphorylation procedures for each modified mRNA encoding individual reprogramming factor. After the final purification step, elute the modified mRNA with nuclease-free water, supplement the purified modified mRNA solution with 1 U/µL RNase inhibitor, quantitate the modified mRNAs using a spectrophotometer, and store at -80 °C for up to 6 months. Prepare multiple aliquots of each modified mRNA to minimize the number of freeze-thaw cycles.
- Prepare the cocktail of reprogramming miRNA mimics.
- Dissolve lyophilized miRNA mimics (Syn-hsa-miR-302a-3p, Syn-hsa-miR-302b-3p, Syn-hsa-miR-302c-3p, Syn-hsa-miR-302d-3p, Syn-hsa-miR-367-3p) to a 5 pmol/µL (5 µM) final concentration in nuclease-free water supplemented with 1 U/µL of RNase inhibitor. Prepare multiple aliquots of each miRNA mimic and store at -80 °C for long term storage.
- Mix all miRNA mimics in a 1:1:1:1:1 molar ratio to a final concentration of 5 pmol/µL (5 µM). Prepare seven 14 µL aliquots of the miRNA mimics mix. Store the mixed aliquots at -80 °C.
NOTE: For each transfection, 20 pmol of the miRNA mimics mix is added per well (i.e., a total of 60 pmol for 3 wells). Each 14 µL aliquot is sized to transfect 3 wells of a 6-well format plate and includes 2 µL of excess volume to account for pipetting errors. Preparing seven 14 µL aliquots is sufficient to complete a full fibroblast reprogramming of 3 wells in a 6-well format plate.
- Prepare the transfection buffer.
- Pre-warm one 500 mL bottle and one 100 mL bottle of fresh reduced-serum medium to room temperature (RT) for approximately 2 h. Do not use a water bath.
- Transfer a pH meter to a biosafety cabinet. Wash the meter's glass electrode with nuclease-free water. Calibrate the pH meter according to the manufacture's instructions. Wash the electrode again with nuclease-free water.
- Transfer both bottles of reduced-serum medium into the biosafety cabinet. Use the 500 mL RT bottle to adjust the pH.
- Measure the base pH of the reduced-serum medium by inserting the pH meter's glass electrode into the buffer. Wait up to 1 min before reading the pH on the meter.
- Add 3–4 mL of 1 M NaOH into 500 mL of reduced-serum medium, close the bottle, and mix well. Wait up to 5 min before opening the bottle for pH measurement. Insert the pH meter's electrode into the buffer and wait until the reading on the pH meter stabilizes.
- Continue adding small volumes of 1 M NaOH until the pH of reduced-serum medium reaches 8.15–8.17. Calibrate the pH meter several times during the process. If at any point the pH becomes higher than 8.18, reduce it by adding fresh reduced-serum medium from the pre-warmed 100 mL bottle (step 1.3.1).
NOTE: The pH of the reduced-serum medium-based transfection buffer is critical. Reprogramming will be successful if the pH of the transfection buffer is about 8.2 ± 0.05 but may fail if the pH is higher than 8.25. Therefore, it is advised to stop adding NaOH once the buffer's pH reaches 8.15–8.17. This will ensure that the final pH of the transfection buffer is approximately 8.2–8.22 after sterilization. - Filter sterilize the transfection buffer using a 0.22 µm vacuum filtration system. Aliquot the sterilized buffer into 5 or 15 mL tubes with minimal air space.
- Store aliquots of the transfection buffer for up to 3 months at 4 °C. Measure the pH of the aliquots periodically. Discard the aliquots if the pH is higher than 8.25. Limit aliquot usage to 2 transfections only since exposure of the transfection buffer to atmospheric air increases the pH of the buffer.
- Prepare the fibroblast expansion medium (FEM): minimum essential medium supplemented with 10% fetal bovine serum (FBS), 1x non-essential amino acids, 1x glutamine supplement, 55 µM 2-mercaptoethanol, 1x pen/strep/fungizone. Store the FEM at 4 °C.
- Prepare the reprogramming medium: DMEM/F12 (no HEPES) supplemented with 20% knockout serum replacement (KOSR), 0.5x non-essential amino acids, 0.5x glutamine supplement, 55 µM 2-mercaptoethanol, 50 µg/mL ascorbic acid, 1x pen/strep/fungizone, 100 ng/mL bFGF, and 200 ng/mL B18R. Prepare the medium at the initiation of reprogramming without bFGF and B18R, and store it at 4 °C. Do not use it beyond 1 month following preparation. Add bFGF and B18R immediately before each use to an aliquot sized for that day's use.
- Prepare the plating medium by adding 5% heat-inactivated (HI) FBS into the reprogramming medium containing 20% KOSR without bFGF and B18R from step 1.5. Prepare the medium fresh on the day of intended use. Add bFGF and B18R immediately before use on day 0 of reprogramming.
- Calibrate the tissue culture incubators before initiating reprogramming according to the manufacturer's instructions using a handheld digital CO2 analyzer. Use a low-O2 incubator for the reprogramming steps to ensure successful iPSC generation.
2. Culturing Fibroblasts for Reprogramming
- Prepare fibroblasts prior to the initiation of reprogramming (day -2).
- Coat a new 10 cm tissue culture plate with 5 mL of 0.1% gelatin. Tap or swirl the plate to ensure that the entire surface is coated. Incubate for 15 min at 37 °C. Aspirate gelatin and add 10 mL of FEM. Set aside. Do not allow the coated surface of the plate to dry before adding FEM.
- Carefully aspirate the spent medium from the fibroblasts. Rinse the cells once with 5 mL of DPBS to remove the residual serum. Add 3 mL of trypsin-EDTA. Gently rock the plate to ensure complete coverage over the cells.
- Aspirate excess fluid, leaving ~500 µL of trypsin. Do not over-aspirate, since this can dry and kill the fibroblasts. Incubate the fibroblasts with trypsin-EDTA for 3 min at 37 °C.
- Remove the plate from the incubator and firmly but gently tap the side of the plate to dislodge the cells. Check the cells under the microscope. If the cells are detached and floating, proceed. If the cells are still attached, incubate for another 3 min.
- Quickly rinse/collect the detached cells using 5 mL of FEM to neutralize the trypsin-EDTA. Move the fibroblast suspension into a 15 mL conical tube. Ensure that the cells are well-mixed.
- Gently mix the cell suspension to break large clumps of cells, then count them on a hemocytometer. Transfer 2.5 x 105 cells into the gelatin-coated 10-cm dish prepared in step 2.1.1. Incubate the cells overnight in the 37 °C, 5% CO2 humidified regular tissue culture incubator.
NOTE: The cell density is critical to achieve high reprogramming efficiency, as it is important that the fibroblasts are healthy and rapidly dividing. Plating 2.5 x 105 cells yields a 40-60% confluency 2 days later for most fibroblast samples. Adjust the amount accordingly for diseased or senescent cells to attain the desired 40-60% confluency 48 h later. - Replace the medium with 10 mL of fresh FEM the following day (day -1).
- Plate the fibroblasts to initiate reprogramming (day 0).
- Verify that the fibroblasts to be reprogrammed are at 40–60% confluency (Figure 2, day 0). If the cells are over- or under-confluent, passage the fibroblasts as described in step 2.1 and adjust the plating density accordingly. Culture for another 2 days.
- Transfer 4 mL of DPBS into a 15 mL conical tube. Add 100 µL of recombinant human (rh) laminin-521. Pipette up and down to mix thoroughly.
- Add 1 ml per well of diluted rhLaminin-521 into 3 wells of a 6-well plate. Incubate the coated plate at 37 °C for 2 h.
- Warm 6 mL of FEM and 4 mL of plating medium to 37 °C. Supplement the plating medium with bFGF to a final concentration of 100 ng/mL and B18R to a final concentration of 200 ng/mL.
- Carefully aspirate the spent medium from fibroblasts (step 2.2.1). Rinse the cells with 5 mL of DPBS and add 3 mL of trypsin-EDTA. Gently rock the plate to cover the cells with trypsin-EDTA.
- Aspirate excess fluid, leaving ~500 µL of trypsin, as done in step 2.1.3. Incubate the fibroblasts with trypsin-EDTA for 3 min at 37 °C. Remove the plate from the incubator and firmly but gently tap the side of the plate to dislodge cells. Check the cells under the microscope. If the cells are detached and floating, proceed. If the cells are still attached, incubate for another 3 min.
- Rinse/collect the detached cells using 5 mL of FEM to neutralize the trypsin-EDTA. Move the fibroblast suspension into a 15 mL conical tube. Ensure that the cells are well-mixed and count them on a hemocytometer. Pipette 12,000 cells into 4 mL of prewarmed plating medium from step 2.2.4.
NOTE: Do not centrifuge the cells at any point. The number of plated cells can be adjusted up or down as required for slow- or fast-growing lines, respectively. - Remove the coated plate from the incubator and aspirate diluted rhLaminin-521 from the coated wells. Do not allow the surface of the wells to dry. Gently resuspend cells in the plating medium. Pipette 1 mL of cell suspension into each coated well (i.e., 3,000 cells per well).
- Place the plated cells into a tri-gas tissue culture incubator with O2 set to 5% (low-O2). Once the plate is set down, gently but thoroughly disperse cells by alternating between an up/down then left/right motion. Repeat the motions 2 more times. Incubate the cells overnight. Do not swirl the plate to mix.
- Pipette 4 mL of reprogramming medium to a 15 mL conical tube and place it in the low-O2 incubator with a loosened cap to equilibrate overnight. Do not add bFGF and B18R until the following day.
3. Initiation of Reprogramming (day 1)
NOTE: Once the reprogramming is initiated, daily maintenance is required for approximately 1 month. Be sure to plan accordingly. All subsequent cell incubations must be performed under low-O2 conditions in the 37 °C, 5% CO2, 5% O2 humidified tri-gas tissue culture incubator.
- Replace the plating medium at least 1 h prior to transfection.
- Remove the equilibrated reprogramming medium from the low-O2 incubator. Add bFGF to a final concentration of 100 ng/mL and B18R to a final concentration of 200 ng/mL. Mix well.
- Proceeding with 1 well at a time, use a 1 mL pipette to remove the spent medium and replace it with 1 mL of reprogramming medium supplemented with bFGF and B18R. Repeat this for each well. Do not use vacuum aspiration, which excessively dries the cells, causes stress, and reduces reprogramming efficiency. Move the plate with cells back into the low-O2 incubator.
- Transfect fibroblasts with 5 µL of RNAiMax transfection reagent per 1 µg of modified mRNA, and 1 µL of the transfection reagent per 6 pmol of miRNA mimics per transfection.
NOTE: Optimal results are obtained when transfections proceed for 16–20 h. The transfection reagent is diluted 10x; the 100 ng/µL modified mRNA cocktail is diluted 5x; and the 5 pmol/µL miRNA mimics mix is diluted 8.33x with the transfection buffer prior to complexation. See Table 1 for a summary of the transfection setup.- While preparing the transfection mixes, minimize potential exposure of reagents to RNase by following standard laboratory practice.
- Equilibrate an aliquot of the transfection buffer (step 1.3) for approximately 1 h at RT. Do not use a 37 °C water bath or incubator to warm up the transfection buffer.
- Remove a single aliquot of modified mRNAs (33 µL) and miRNA mimics (14 µL) from -80 °C and warm them to RT for approximately 3-5 min, until thawed. Do not thaw the aliquots at 37 °C. Spin them down briefly in a microfuge.
- Warm the transfection reagent to RT for approximately 3-5 min. Do not use a 37 °C water bath or incubator. Invert the closed tube 2–3 times to mix the reagent. Spin it down briefly in a microfuge.
- Transfer 279 µL of the RT transfection buffer into an RNase-free microcentrifuge tube. Add 31 µL of the transfection reagent to achieve a final volume of 310 µL. Mix thoroughly by pipetting. Do not vortex. Incubate the tube at RT for 1 min.
NOTE: This volume of the diluted transfection reagent is sufficient to complex both the modified mRNA and miRNA mimics aliquots from step 3.2.3. - Add 132 µL of the RT transfection buffer to the 33 µL aliquot of modified mRNA. Gently pipette to mix: final volume is 165 µL.
- Add 102.6 µL of the RT transfection buffer to the 14 µL aliquot of miRNA mimics. Gently pipette to mix: final volume is 116.6 µL.
- Add 165 µL of the diluted transfection reagent from step 3.2.5 to the diluted modified mRNA from step 3.2.6. Pipette to mix: final volume is 330 µL.
- Add 116.6 µL of the diluted transfection reagent from step 3.2.5 to the diluted miRNA mimics mix from step 3.2.7. Mix well (final volume is 233.2 µL). Incubate for 15 min at RT to allow the transfection buffer to complex with the modified mRNAs and miRNA mimics.
- Remove the plate with cells from the low-O2 incubator. Add 100 µL (1 µg) of the complexed modified mRNA transfection mix to each well, dropwise across the well. Disperse the transfection complexes by gently but thoroughly agitating the plate with an up/down then left/right motion. Do not swirl the plate to mix.
- Add 66.7 µL (20 pmol) of the complexed miRNA mimics transfection mix to each well, dropwise across the well. Disperse transfection complexes by gently but thoroughly agitating the plate with an up/down then left/right motion. Do not swirl the plate to mix.
- Place the transfected cells into the tri-gas incubator with O2 set to 5% (low-O2). Once the plate is set down, disperse the transfection complexes again by gently but thoroughly agitating the plate with an up/down then left/right motion.
- Pipette 4 mL of reprogramming medium into a 15 mL conical tube and place it in the low-O2 incubator with loosened cap to equilibrate overnight. Do not add bFGF and B18R until the following day.
| Tube 1 - RNAiMax dilution (1st mix) | ||
| Reagent | Concentration | Volume |
| Transfection Buffer | 279 µL | |
| RNAiMax (add 2nd) | 10x | 31 µL |
| Total: 310 µL | ||
| (incubate 1 min at room temperature) | ||
| Tube 2 - modified mRNA mix (2nd mix) | ||
| Reagent | Concentration | Volume |
| modified mRNA mix | 100 ng/µL (5x) | 33 µL |
| Transfection Buffer | 132 µL | |
| Total: 165 µL | ||
| (add equal volume of diluted RNAiMax from Tube 1) | ||
| Tube 3 - miRNA mimics mix (3rd mix) | ||
| Reagent | Concentration | Volume |
| miRNA mimics mix | 5 pmol/µL (8.33x) | 14 µL |
| Transfection Buffer | 102.6 µL | |
| Total: 116.6 µL | ||
| (add equal volume of diluted RNAiMax from Tube 1) | ||
Table 1: Preparation of transfection mix.
4. Replacing Reprogramming Medium between Transfections (days 2, 4, 6, 8, 10, and 12)
- Replace the medium 16–20 h post-transfection as described in 3.1.1–3.1.2. Add bFGF to a final concentration of 100 ng/mL and B18R to a final concentration of 200 ng/mL into reprogramming medium aliquots.
- Monitor mWasabi expression daily to confirm transfection quality using a microscope configured to visualize EGFP.
NOTE: mWasabi expression should be minimally apparent on day 2 and increase in brightness with each additional transfection.
5. Every-Other-Day Transfections (days 3, 5, 7, 9, 11, and 13)
- Perform the transfection as described in step 3.2. Do not change the medium on days of transfection.
- Prepare a 4 mL aliquot of reprogramming medium and place it in the low-O2 incubator to equilibrate for the medium change on the following day. Do not add bFGF and B18R until the following day.
6. Procedures to be Performed after Final Transfection
- Perform daily medium changes from day 14 through approximately day 17. Warm 7 mL of reprogramming medium to 37 °C. Add bFGF to a final concentration of 100 ng/mL.
NOTE: B18R is no longer necessary after the final transfection. Beyond day 14, it is no longer necessary to equilibrate the medium overnight in the low-O2 incubator. - Remove the medium from all wells using a serological pipette. Continue to avoid using aspiration. Add 2 mL of reprogramming medium supplemented with bFGF per well.
- On days 17 and 18, analyze the reprogrammed wells under an inverted or dissecting microscope. If colonies are formed in close proximity to each other due to high efficiency of reprogramming and cannot be manually isolated, separate the colonies by performing an optional passaging of reprogrammed wells described in step 7 below. If colonies are sparse and well-separated, manually pick the clones directly from the reprogrammed well as described in step 8 and subculture iPSCs according to standard protocols.
7. Optional Procedure: Passaging Cells from Reprogrammed Wells using EDTA
- Prepare 0.5 mM EDTA in DPBS (EDTA) by diluting the 0.5 M EDTA stock solution. Filter sterilize EDTA using a 0.22 µm vacuum filtration system.
- Prepare feeder-free pluripotent stem cell (PSC) medium (e.g., mTeSR1) according to the manufacturer's instructions. Pre-warm 32 mL of PSC medium to 37 °C.
- Coat all wells of two 6-well format plates with hESC-qualified extracellular matrix (ECM) following the manufacturer's instructions. Seal plates with paraffin film and incubate them for 1 h at RT.
- Aspirate the ECM solution from the pre-warmed plates and replace it with 2 mL of PSC medium per well. Do not allow the surface of the wells to dry. Set them aside.
- Aspirate the reprogramming medium from 2 reprogrammed wells on a day when the colonies are large and clearly formed (usually day 18). Rinse once with 1 mL of EDTA and aspirate.
- Add 1 mL of EDTA per well. Incubate for 4 min at 37 °C.
- Gently remove the plate from the incubator and place it in the biosafety cabinet.
NOTE: At this point, cells may be very loosely adhered and easily dislodged. - Carefully aspirate EDTA from both wells. Add 3 mL of pre-warmed PSC medium to each well treated with EDTA. Use a cell-scraper to gently but thoroughly dislodge cells from both wells.
- Use a serological pipette to gently pipette the cell suspension and break up large clumps. Do not pipette until all the cell clumps are gone. Preserve iPSC clusters.
NOTE: Plating large clumps will not affect iPSC colony outgrowth. If there are excessively large cell clumps, simply avoid transferring them into the next dish. - Using a serological pipette, evenly distribute cells from each EDTA-treated well by pipetting 0.5 mL into each well of the ECM-coated 6-well plate.
NOTE: Do not combine cells from the reprogrammed wells (i.e., reprogrammed well 1 should be evenly distributed into the first 6-well plate, and reprogrammed well 2 should be evenly distributed into the second 6-well plate). - Move the plated cells back into the low-O2 incubator. To evenly distribute cells, shake each plate back-and-forth and side-to-side. Do not swirl. Replace the PSC medium daily.
8. Picking iPSC Colonies
- Pre-warm 15 mL of PSC medium to 37 °C. Coat all wells of a single 6-well plate with hESC-qualified ECM following the manufacturer's instructions. Seal the plates with paraffin film and incubate them for 1 h at RT.
- Aspirate the culture medium from wells being used to collect iPSCs. Rinse once with 1 mL of EDTA and aspirate. Add 1 mL of EDTA per well. Incubate for 4 min at 37°C. While the cells are incubating, aspirate the ECM solution from the pre-warmed plates and replace with 2 mL of PSC medium per well. Set the plates aside.
- Gently remove the plate with iPSCs to be picked from the incubator and place it in the biosafety cabinet.
NOTE: At this point, cells may be very loosely adhered and easily dislodged. - Carefully aspirate EDTA. Very gently add 3 mL of pre-warmed PSC medium, taking care not to dislodge iPSC colonies.
- Move the plate to an inverted or dissecting scope to better visualize colonies. Prepare a 1 mL pipette with a sterile tip. Depress the plunger fully, then use the pipette tip to gently scrape a colony while slowly drawing liquid into the tip to collect the colony. Draw as little medium as possible while picking the iPSC colony.
- To transfer the colony, pipette up and down 3–4 times in a single well of the ECM-coated plate from step 8.2. Repeat until 6 colonies have been picked and transferred into individual wells. Pick no more than 2 colonies from any single well if an optional passaging described in step 7 has been performed.
- Use a standard human stem cell protocol to freeze down all remaining wells. Thaw and re-plate frozen stocks if more colonies must be picked later.
9. Characterization of iPSCs
- Perform TRA-1-60 staining for analysis of reprogramming efficiency (day 18).
NOTE: 2 out of 3 reprogrammed wells are passaged for future colony picking. The remaining well can be used for TRA-1-60 staining. This procedure can be performed on the laboratory bench top.- Wash the reprogrammed well with 1 mL of PBS. Fix cells with ice-cold methanol at -20 °C for 5 min.
- Aspirate methanol. Dry the well for approximately 2 min. Be careful not to over-dry. The cells have dried enough when they become a translucent/matte color.
- Wash the well 3 times with 1 mL of PBS for 5 min with gentle shaking. During the washes, prepare 3 mL of 3% hydrogen peroxide in PBS.
- Aspirate the PBS. Add 2 mL of diluted peroxide solution. Incubate for 15 min at RT with gentle shaking. Prepare 4 mL of the blocking solution: 10% bovine-serum albumin (BSA) in PBS.
- Aspirate the peroxide solution. Wash the well 2 times with 1 mL of PBS for 5 min.
- Aspirate PBS and add 2 mL of blocking solution into the well. Incubate for 1 h at RT.
- Wash the well 3 times with 1 mL of PBS for 5 min with gentle shaking.
- Dilute primary anti-TRA-1-60 antibody at 1:100 in the blocking solution supplemented with 0.05% sodium azide. Prepare 1 mL of antibody dilution for 1 well of a 6-well format dish. Add the antibody dilution to the well and wrap the plate with parafilm to avoid evaporation. Incubate overnight at 4 °C with gentle shaking.
NOTE: If needed, the incubation with the primary antibody can be done for 1 h at RT with gentle shaking. The primary antibody dilution can be re-used up to 5 times. - After incubation with the primary antibody, wash the well 3 times with 1 mL of PBS for 5 min with gentle shaking.
- Dilute the secondary anti-mouse HRP-conjugated antibody at 1:200 in the blocking solution. Incubate the well with the secondary antibody dilution for 2 h at RT with gentle shaking.
- Aspirate the secondary antibody dilution and wash the well 3 times with 1 mL of PBS for 5 min with gentle shaking.
- During the third wash, prepare the substrate solution following the manufacturer's instruction. After the final wash, aspirate PBS. Add 1 mL of the substrate solution and incubate until desired color develops (approximately 10 min).
- Aspirate the substrate solution. Rinse the well with water for 5 min while gently shaking.
- Count the colonies, if desired. Reprogramming efficiency = (number of colonies) / (number of plated fibroblasts) x 100%.
- For long-term storage, aspirate water from the stained plate and air-dry overnight at RT. Seal the dried plate with parafilm and store at 4 °C for up to 2 years to assess reprogramming efficiency later.
Results
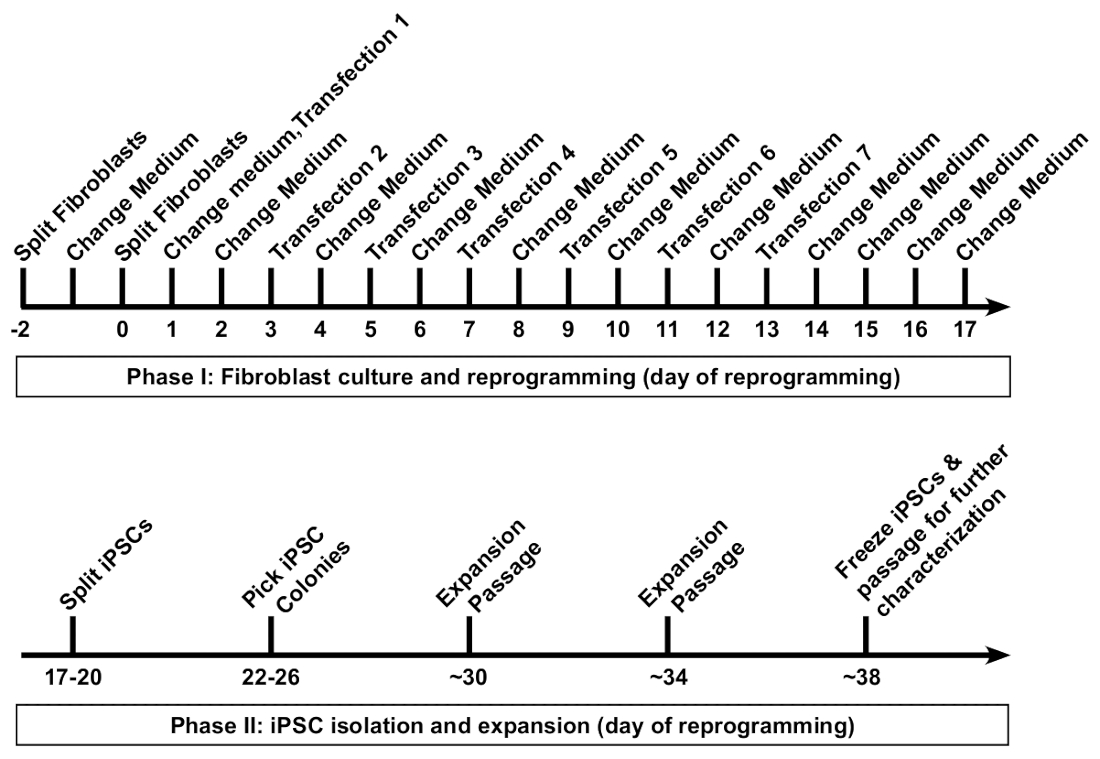
It typically takes approximately 5–6 weeks from the initiation of fibroblast reprogramming to freezing of the first vials of iPSCs (Figure 1). The reprogramming protocol can be generally categorized into two phases. Phase 1 includes the fibroblast culture and seven transfections, with the reprogramming RNA cocktail performed every 48 h. Phase 2 includes isolation, expansion, and characterization of the iPSC colonies.
Before initiating the protocol, it should be ensured that the fibroblasts to be reprogrammed are of good quality. Healthy fibroblasts should appear spindle-shaped, bipolar, and refractile with a doubling time of approximately 24 h. By day 0, 250,000 cells plated into a 10 cm dish on day -2 should grow to 40–60% confluency (Figure 1, day 0) and yield approximately 6–10 x 105 cells. Cells proliferating at a slower rate can be compensated for by plating at a higher density on day -2 and on day 0 for reprogramming.
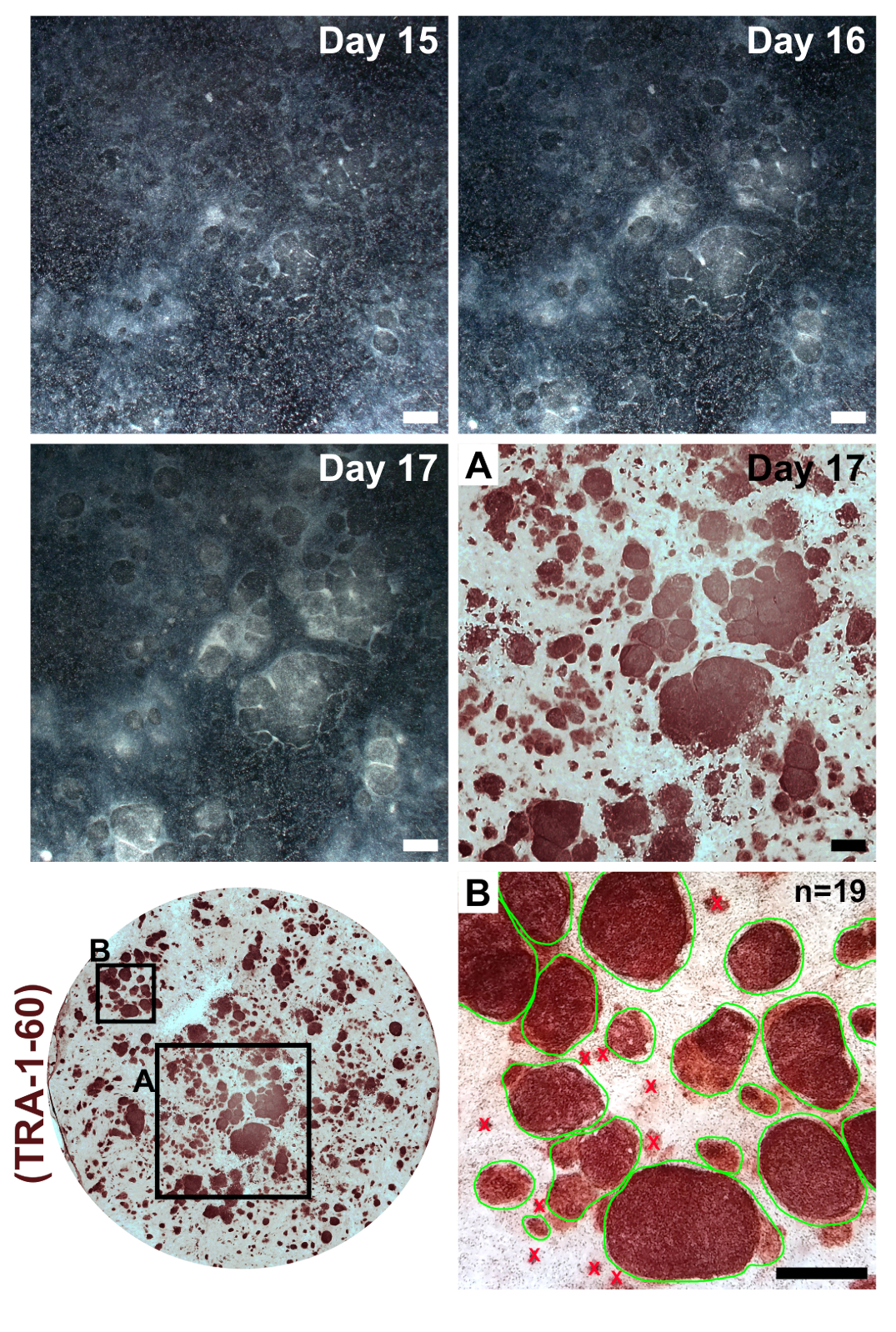
Fibroblasts should appear very sparse following plating into a well of a 6-well format dish for reprogramming (Figure 2, day 1). Twenty-four hours following the first transfection, fibroblasts will lose their spindle shape and adopt a more rounded morphology (Figure 2, day 2), which is maintained through the remainder of reprogramming. Green fluorescence from mWasabi mRNA should be minimally observable on day 2 and steadily increase in brightness to be clearly visible by day 4. The ability to detect mWasabi fluorescence can depend on scope sensitivity and setup. Cell density will gradually and consistently increase throughout the first three transfections (days 1-6), with an apparent burst in proliferation occurring between days 7 and 8. The cells should appear largely confluent by day 10 (Figure 2, day 10). The first iPSC colonies can appear as early as day 11 (Figure 2, day 11); however, colonies may not be observable until as late as day 18. Generally, by days 15–18, there will be large and obvious iPSC colonies that are clearly distinct from surrounding, incompletely reprogrammed fibroblasts (Figure 2, day 15 and Figure 3, day 17). Immunostaining for the pluripotency marker TRA-1-60 can be performed to assess reprogramming efficiency (Figure 3, day 17, TRA-1-60). In our experience, most fibroblast lines yield hundreds of colonies per reprogrammed well (Figure 3, inset B).
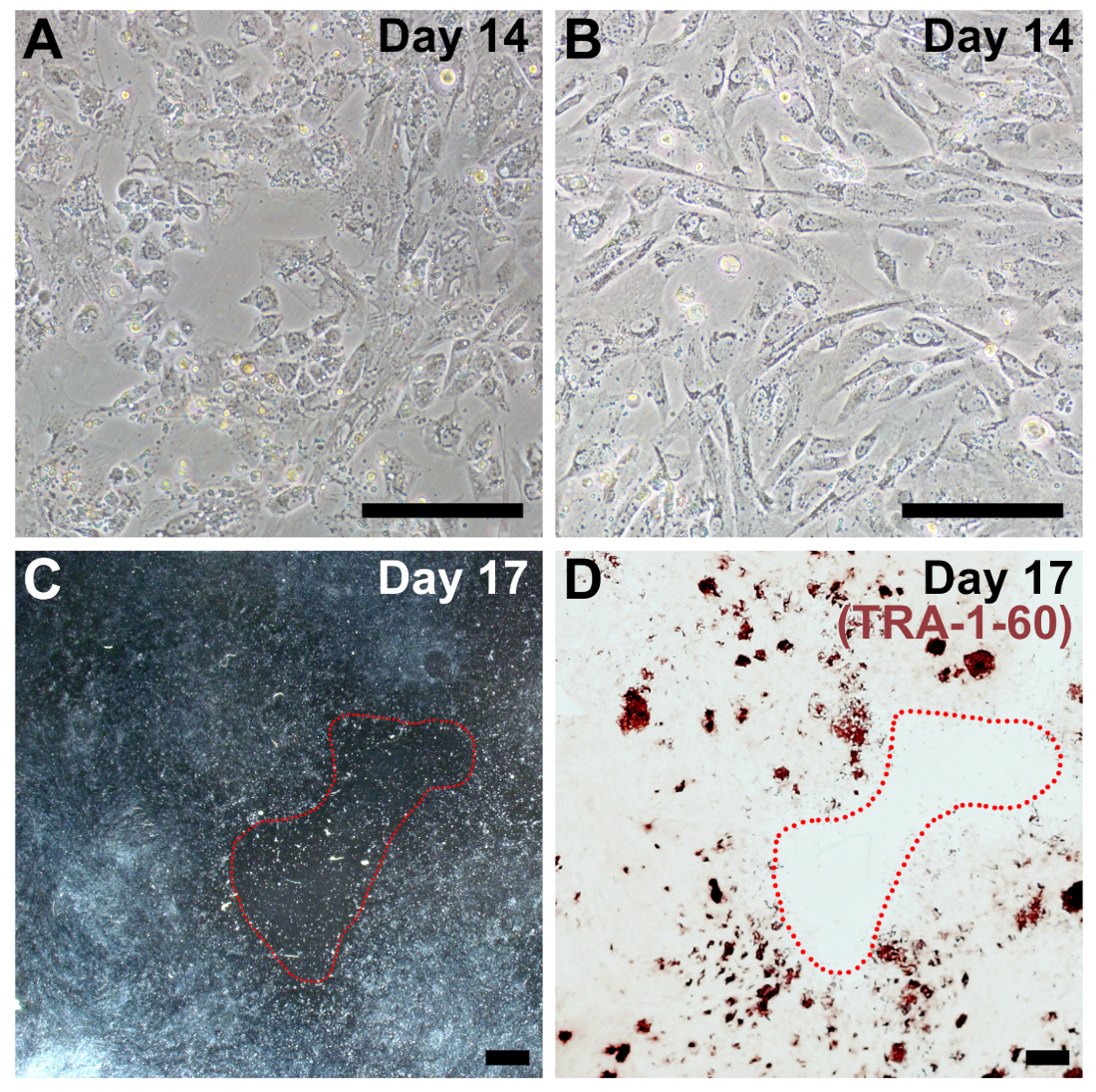
Suboptimal plating density is the most common reason for reduced efficiency of reprogramming in our protocol and is frequently associated with fibroblasts that are diseased, senescent, and/or high-passage. If plating density is too low, there will be large acellular barren patches at the end of reprogramming (Figure 4C), and iPSC colonies may not form (Figure 4D). Reprogrammed cells should be very confluent by day 14 (compare Figure 4A and 4B to Figure 2, day 14). Similarly, if cells are plated too densely or proliferate too quickly, reprogramming efficiency is dramatically reduced.
To maintain homogeneity of patient-derived iPSCs, it is important to expand cell lines from a single colony. Since reprogramming efficiency is very high in our protocol, neighboring iPSC colonies may form in close proximity to and grow into each another (Figure 3, days 15–17). This sometimes makes it difficult to mechanically separate a single colony for clonal expansion. We have found that it is helpful to first passage a reprogrammed well and dilute it across a larger culture area. A good passage ratio consists of evenly splitting a reprogrammed 6-well-format plate well across an entire 6-well plate.
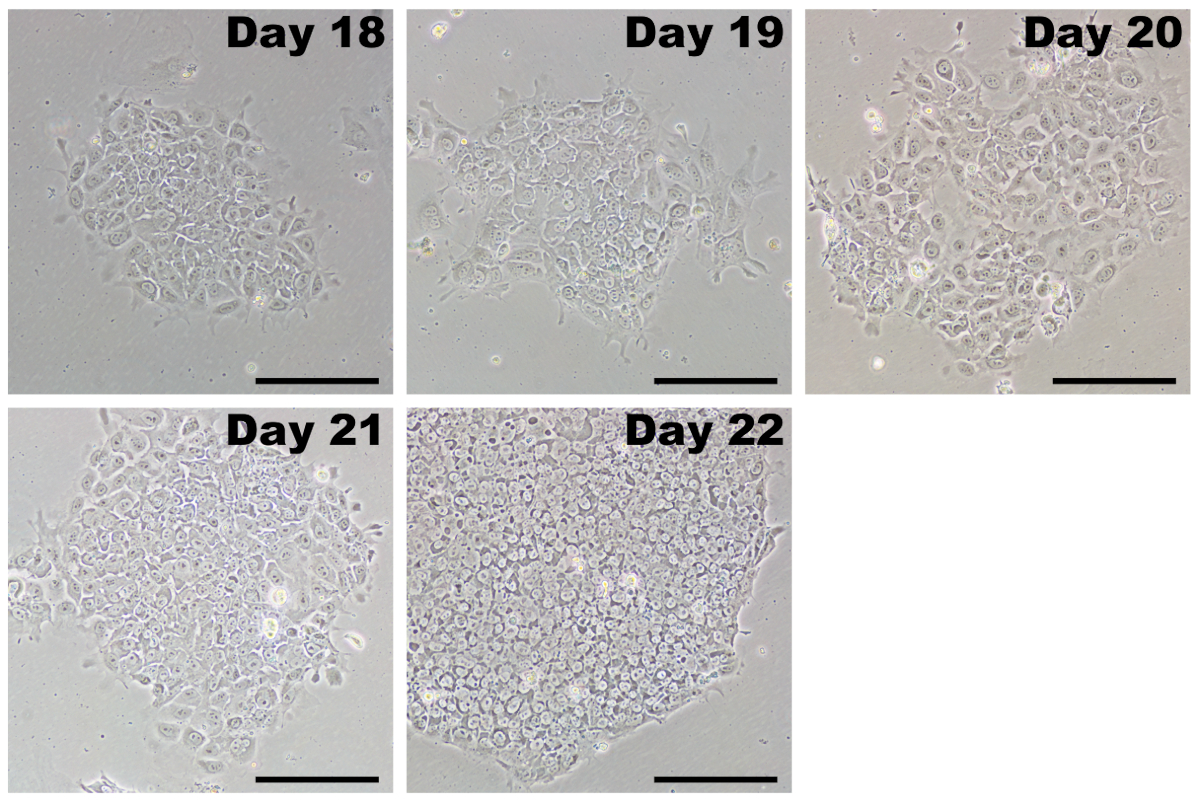
Following the dilution passage, iPSCs grow as colonies and are easily distinguishable from fibroblasts (Figure 5, day 18). Initially, iPSC colonies may be loosely packed, and individual cells have a relatively large cytoplasmic area. Over the course of 4–7 days, the iPSCs proliferate and form a characteristic tightly-packed colony with defined edges. Individual cells within the colony have a large nuclear fraction with prominent nucleoli (Figure 5, day 22). There should be many colonies that form in each well, and only those with classic iPSC morphology should be picked for expansion.

Figure 1: Reprogramming of human fibroblasts into induced pluripotent stem cells (iPSCs). A schematic protocol for the reprogramming of human fibroblasts is presented. Fibroblasts are first passaged at a low density into a well of a 6-well format dish, followed by seven transfections performed at 48 h intervals. Medium is replaced 16–20 h after each transfection. Reprogrammed iPSCs are first passaged at approximately day 18, and clonal colonies are picked by day 26. Typically, fibroblast-derived iPSC lines can be frozen for long-term storage by day 38. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Representative daily images during each day of reprogramming. Fibroblasts should be approximately 40–60% confluent at the time of passage to initiate reprogramming (day 0, cells are plated in a 10 cm dish). The first transfection (T1) occurs on day 1, and the cells should appear very sparse at this point. The following day (day 2), a more rounded morphology should become apparent. Cells will continue to increase in density throughout the protocol with iPSC clusters beginning to appear as early as day 11 (circled in red). By day 15, iPSC colonies will be large with discreet boundaries. Scale bar = 200 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Colony formation following transfections with reprogramming modified mRNAs and miRNA mimics. Low-magnification images were taken of a representative reprogramming on days 15–17. Following the final transfection, reprogrammed iPSCs will form clear colonies with defined boundaries that expand in size and condense to become clearly distinct from incompletely reprogrammed surrounding fibroblasts. Immunostaining for the pluripotency marker TRA-1-60 indicates the presence of iPSCs (inset A) and can be used for calculation of reprogramming efficiency by counting all colonies within a single well (inset B, examples of countable colonies circled in green). Scale bar = 1 mm. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Representative images of sub-optimal plating density for reprogramming. (A, B) Examples of fibroblasts that are too sparse by day 14 of reprogramming (compare to Figure 2, day 14). (C) Low magnification image on day 17 of reprogramming with a large, barren patch circled in red. (D) The same well was fixed and stained for TRA-1-60 to confirm overall poor reprogramming efficiency due to low cell density. Scale bars = 200 µm (A, B), and 1 mm (C, D). Please click here to view a larger version of this figure.
{kind=link}

Figure 5: Representative images of iPSCs after initial passage. After completing transfections with reprogramming modified mRNAs and miRNA mimics, reprogrammed cells are passaged by days 17–20. iPSCs have a growth advantage in PSC medium and rapidly overtake any fibroblasts that were incompletely reprogrammed. Initially, iPSCs will form colonies that may appear loose with poorly defined borders. Within several days, the cells rapidly proliferate and take on the characteristic morphology of tightly packed cells with a high nucleus-to-cytoplasm ratio, tightly clustering into colonies with distinct borders. Scale bar = 200 µm. Please click here to view a larger version of this figure.
{kind=link}
Discussion
This protocol describes a clinically-relevant, non-integrating, RNA-based method that allows for the reprogramming of normal and disease-associated human fibroblast lines into iPSCs at an ultra-high efficiency. To date, every human fibroblast line we have attempted to reprogram with the described protocol has yielded a satisfactory number of high-quality iPSCs for downstream applications. Resulting iPSCs can immediately be transferred and expanded in feeder-free culture conditions.
Quality of fibroblasts for reprogramming:
Reprogramming success is heavily dependent on the quality of starting fibroblasts. Ideally, reprogramming should be initiated with the lowest passage fibroblasts available to achieve the highest efficiency. Reprogramming efficiency is best with fibroblasts of passage 2–4. Reprogramming can still work with high-passage (passage 5–8), even senescent fibroblasts, albeit with a reduced efficiency. Sometimes low-passage fibroblasts are unavailable or patient samples have a genetic mutation that prevents healthy growth. In this case, optimization of initial plating density may be required. The reprogramming of compromised fibroblast lines is usually associated with increased cell death during RNA transfections. As a result, the cells in the reprogramming well will appear to be sparse by days 10–14 of reprogramming. Large acellular areas will also be visible in the well. If this is the case, the reprogramming protocol will need to be re-initiated with a higher initial starting number of fibroblasts. Plating 3,000 input cells per well of a 6-well format dish works consistently for most adult fibroblast lines. However, increasing the plating number to 5,000–10,000 (50,000 for senescent lines) may help to improve reprogramming of disease-associated samples, as has been reported in our previous publication8. Conversely, cells reaching confluency too early can also be resistant to reprogramming. If cells undergoing transfections with reprogramming RNAs proliferate too rapidly (as is sometimes the case with primary neonatal fibroblasts), initiate reprogramming with 500 fibroblasts per well of a 6-well format dish8.
Handling of the transfection buffer:
The pH of the transfection buffer (reduced-serum medium adjusted to the pH of 8.2) is paramount to achieving the optimal transfection efficiencies required for this reprogramming protocol. For this reason, several precautions regarding the handling of the transfection buffer are recommended. We have found that even short exposure of the transfection buffer to atmospheric air affects the pH of the buffer. Therefore, the transfection buffer should be aliquoted into a screw cap container with minimal air space (we use either 5 or 15 mL screw cap conical tubes). To further minimize air exposure, use each transfection buffer aliquot for a maximum of two transfections. Lastly, since temperature impacts pH, it is critical that the transfection buffer is equilibrated to RT for assembly of the transfection complexes. It is advised to not warm the transfection buffer to 37 °C.
Passaging of iPSCs:
While many previously published protocols recommend picking individual iPSC colonies at the end of reprogramming, this may be difficult to achieve when the efficiency of reprogramming is very high or if the colonies cluster together, as is often the case in our protocol. Therefore, if iPSC colonies are in close proximity to each other, we recommend first passaging the cells to spread out reprogrammed iPSCs before manually picking colonies. There are several advantages to performing this early passage step. Spreading out the colonies gives them more room to grow, yielding much larger colonies for picking than could otherwise be achieved in the original well. This greatly improves the success rate in establishing an iPSC line from a picked clone. We also find that the additional culture time with fibroblasts, albeit at a diluted ratio, appears to improve the average quality of picked iPSC colonies. The incompletely reprogrammed fibroblasts may provide supporting paracrine factors, which continue to help establish the iPSCs and ease the direct transition into feeder-free cell culture conditions. Fibroblasts fortuitously have a selective growth disadvantage compared to iPSCs when cultured in mTeSR1. Therefore, the contaminating fibroblast cell population is quickly diluted to negligible quantities within 3–4 passages.
ROCK inhibitors such as Y-27632 are frequently used for routine culture of human iPSCs. We have found that frequent and/or extended culture of some iPSC lines with Y-27632 can have deleterious effects on overall quality. When using a clump passaging method, such as with EDTA, Y-27632 is not necessary to maintain iPSC viability after splitting. We have completely eliminated supplementing media with Y-27632 for all iPSC isolation, expansion, or routine culture.
Protocol limitations:
One limitation to the described RNA-based reprogramming approach is the initial cost and complexity associated with the preparation of the reprogramming reagents. Although preparatory procedures to generate mRNA reagents are all routine and have been previously described (PCR, in vitro transcription, DNase treatment, capping, dephosphorylation, purification), cumulatively the production of mRNA reagents is a relatively lengthy and non-trivial process. The other major challenge to this protocol is the need to transfect cells every 48 h, increasing the labor intensity of the reprogramming protocol. These considerations may be prohibitive if reprogramming of only a few patient samples is desired. However, if the primary consideration is the generation of clinically relevant iPSCs or achieving very high reprogramming efficiency, the described RNA-based reprogramming approach is ideal.
In summary, the described high-efficiency RNA-based reprogramming method hinges on optimized transfection efficiency of the somatic cell type to be reprogrammed as described in our previous publication8. The RNA transfection protocol presented in this study is highly tuned for human primary fibroblasts but can potentially be tailored to other cell types to improve reprogramming efficiency of various somatic cells.
Disclosures
I.K. and G.B. are co-inventors on a patent application entitled "Methods and Compositions for Reprogramming Cells", PCT Application No. PCT/US2016/063258.
Acknowledgements
We are grateful for funding support from the National Institutes of Health (T32AR007411-33) and the University of Colorado Skin Diseases Research Core Center (P30AR057212). We also thank the Epidermolysis Bullosa (EB) Research Partnership, EB Medical Research Foundation, Cure EB Charity, Dystrophic Epidermolysis Bullosa Research Association (DEBRA) International, King Baudouin Foundation's Vlinderkindje Fund, and Gates Frontiers Fund.
Materials
| Name | Company | Catalog Number | Comments |
| Plasmid templates for PCR | |||
| pcDNA3.3_KLF4 | Addgene | 26815 | |
| pcDNA3.3_SOX2 | Addgene | 26817 | |
| pcDNA3.3_c-MYC | Addgene | 26818 | |
| pcDNA3.3_LIN28A | Addgene | 26819 | |
| pCR-Blunt_hM3O | Addgene | 112638 | |
| pCR-Blunt_hNANOG | Addgene | 112639 | |
| pCR-Blunt_mWasabi | Addgene | 112640 | |
| Modified mRNA in vitro transcription and miRNA mimics | |||
| Forward Primer | Integrated DNA Technologies | TTGGACCCTCGTACAGAAGC TAATACG | |
| Reverse Primer (Ordered as ultramer, 4nmol scale) | Integrated DNA Technologies | TTTTTTTTTTTTTTTTTTTTTTTT TTTTTTTTTTTTTTTTTTTTTTTT TTTTTTTTTTTTTTTTTTTTTTTT TTTTTTTTTTTTTTTTTTTTTTTT TTTTTTTTTTTTTTTTTTTTTTTT CTTCCTACTCAGGCTTTATTCA AAGACCA | |
| (ARCA Cap) 3´-0-Me-m7G(5')ppp(5')G | New England Biolabs | S1411S | |
| Pfu Ultra II Hotstart 2x Master Mix | Agilent | 600850-51 | |
| 5-Methylcytidine-5'-Triphosphate | Trilink Biotechnologies | N-1014 | |
| Antarctic Phosphatase | New England Biolabs | M0289L | |
| DNase I | NEB | M0303S | |
| MEGAscript T7 Transcription Kit | ThermoFisher Scientific | AM1334 | |
| Pseudouridine-5'-Triphosphate | Trilink Biotechnologies | N-1019 | |
| Riboguard RNase Inhibitor | Lucigen | RG90910K | |
| RNA Clean & Concentrator | ZymoResearch | R1019 | |
| Syn-hsa-miR-302a-3p miScript miRNA Mimic | Qiagen | MSY0000684 | |
| Syn-hsa-miR-302b-3p miScript miRNA Mimic | Qiagen | MSY0000715 | |
| Syn-hsa-miR-302c-3p miScript miRNA Mimic | Qiagen | MSY0000717 | |
| Syn-hsa-miR-302d-3p miScript miRNA Mimic | Qiagen | MSY0000718 | |
| Syn-hsa-miR-367-3p miScript miRNA Mimic | Qiagen | MSY0000719 | |
| Water (Nuclease free, Not DEPC-treated) | Fisher | AM9937 | Use to dilute modified mRNAs and miRNA mimics |
| Fibroblast culture and reprogramming | |||
| 0.1% Gelatin in H2O | StemCell technologies | #07903 | |
| Stericup-GV Sterile Vacuum Filtration System | EMD Millipore | SCGVU05RE | Use to sterilize the transfection buffer and 0.5 mM EDTA in DPBS |
| 2-Mercaptoethanol | ThermoFisher Scientific | 21985023 | |
| 6-well plates (tissue culture treated) | Corning | 3516 | |
| DMEM/F12 | ThermoFisher Scientific | 11320033 | |
| Fetal Bovine Serum | Fisher | 26-140-079 | |
| FGF Basic | ThermoFisher Scientific | phg0263 | |
| GlutaMax Supplement | ThermoFisher Scientific | 35050061 | Glutamine supplement used for the prepration of media |
| Heat Inactivated FBS | Gibco Technologies | 10438026 | |
| KnockOut Serum Replacement | ThermoFisher Scientific | 10828010 | |
| Lipofectamine RNAiMAX Transfection Reagent | ThermoFisher Scientific | 13778500 | The protocol is optimized for the Lipofectamine RNAiMax transfection reagent |
| MEM | ThermoFisher Scientific | 11095080 | |
| MEM Non-essential amino acids | ThermoFisher Scientific | 11140050 | |
| Opti-MEM I Reduced Serum Medium | ThermoFisher Scientific | 31985070 | Reduced-serum medium, use to make the transfection buffer by adjusting the pH to 8.2, 500 mL |
| Opti-MEM I Reduced Serum Medium | ThermoFisher Scientific | 31985062 | Reduced-serum medium, use to make the transfection buffer by adjusting the pH to 8.2, 100 mL |
| 10 M NaOH | Sigma-Aldrich | 72068 | Make a 1 M solution by diluting in nuclease free water and use for pH adjustment |
| Water (Nuclease free, Not DEPC-treated) | Fisher | AM9932 | Use to dilute NaOH and wash a pH meter |
| Pen/Strep/Fungizone | GE Healthcare | SV30079.01 | |
| rhLaminin-521 | ThermoFisher Scientific | A29248 | Supplied at a concentration of 100 µg/mL, use as a matrix for the reprogramming procedure |
| Dulbecco's phosphate-buffered saline (DPBS) | Life Technologies | 14190144 | |
| Trypsin-EDTA 0.25% Phenol Red | Life Technologies | 2520056 | |
| Vaccinia Virus B18R (CF) | ThermoFisher Scientific | 34-8185-86 | |
| iPSC culture | |||
| Corning Matrigel hESC-Qualified Matrix | Corning | 354277 | Extracellular matrix (ECM) for culturing iPSCs |
| EDTA, 0.5 M stock solution | K&D Medical | RGF-3130 | Dilute to 0.5 mM in DPBS, filter sterilize and use for iPSC passaging |
| mTeSR1 | StemCell Technologies | 85850 | Pluripotent stem cell (PSC) medium, provides growth advantage to iPSCs over fibroblasts |
| Antibodies and Detection | |||
| Rabbit anti Mouse (HRP conjugated) | Abcam | ab97046 | |
| Tra-1-60 (mouse anti human) | Stemgent | 09-0010 | |
| Hydrogen Peroxide (30%) | LabChem | LC154301 | Dilute to 3% with PBS |
| Bovine Serum Albumin (BSA) | Fisher | BP9703100 | |
| Phosphate-buffered salin (PBS) | Hyclone | SH30258.02 | Supplied as 10x, dilute to 1x |
| VECTOR NovaRED Peroxidase Substrate Kit | Vector Laboratories | SK-4800 | |
| Special Equipment | |||
| Description | Notes | ||
| Biological safety cabinet | |||
| Regular humidified tissue culture incubator | Calibrate CO2 using digital meter, fyrite, or equivalent. Equilibrate incubator to 5% CO2, 37 °C. | ||
| Tri-gas humidified tissue culture incubator | Calibrate CO2 using digital meter, fyrite, or equivalent. Equilibrate incubator to 5% CO2, 5% O2, 37 °C. Use for the reprogramming procedure. | ||
| pH Meter | Must have resolution to two decimal places. Designate to RNA work if possible. | ||
| Inverted microscope | Microscope configured to visualize EGFP for monitoring transfection efficiency. |
References
- Takahashi, K., Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell. 126 (4), 663-676 (2006).
- Takahashi, K., et al. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell. 131 (5), 861-872 (2007).
- Schlaeger, T. M., et al. A comparison of non-integrating reprogramming methods. Nature Biotechnology. 33 (1), 58-63 (2015).
- Hacein-Bey-Abina, S., et al. LMO2-Associated Clonal T Cell Proliferation in Two Patients after Gene Therapy for SCID-X1. Science. 302 (5644), 415-419 (2003).
- Warren, L., et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell. 7 (5), 618-630 (2010).
- Judson, R. L., Babiarz, J. E., Venere, M., Blelloch, R. Embryonic stem cell-specific microRNAs promote induced pluripotency. Nature Biotechnology. 27 (5), 459-461 (2009).
- Anokye-Danso, F., et al. Highly efficient miRNA-mediated reprogramming of mouse and human somatic cells to pluripotency. Cell Stem Cell. 8 (4), 376-388 (2011).
- Kogut, I., et al. High-efficiency RNA-based reprogramming of human primary fibroblasts. Nature Communications. 9 (1), (2018).
- Hirai, H., et al. Radical acceleration of nuclear reprogramming by chromatin remodeling with the transactivation domain of MyoD. Stem Cells. 29 (9), 1349-1361 (2011).
- Mandal, P. K., Rossi, D. J. Reprogramming human fibroblasts to pluripotency using modified mRNA. Nature Protocols. 8 (3), 568-582 (2013).
- Avci-Adali, M., et al. In vitro synthesis of modified mRNA for induction of protein expression in human cells. Journal of Visualized Experiments. , e51943 (2014).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved