
Method Article
Combined Nucleotide and Protein Extractions in Caenorhabditis elegans
In This Article
Summary
Here, we present a protocol for the isolation of RNA, DNA, and protein from the same sample, in an effort to reduce variation, improve reproducibility, and facilitate interpretations.
Abstract
A single biological sample holds a plethora of information, and it is now common practice to simultaneously investigate several macromolecules to capture a full picture of the multiple levels of molecular processing and changes between different conditions. This protocol presents the method of isolating DNA, RNA, and protein from the same sample of the nematode Caenorhabditis elegans to remove the variation introduced when these biomolecules are isolated from replicates of similarly treated but ultimately different samples. Nucleic acids and protein are extracted from the nematode using the acid guanidinium thiocyanate-phenol-chloroform extraction method, with subsequent precipitation, washing, and solubilization of each. We show the successful isolation of RNA, DNA, and protein from a single sample from three strains of nematode and HeLa cells, with better protein isolation results in adult animals. Additionally, guanidinium thiocyanate-phenol-chloroform-extracted protein from nematodes improves the resolution of larger proteins, with enhanced detectable levels as observed by immunoblotting, compared to the traditional RIPA extraction of protein.
The method presented here is useful when investigating samples using a multiomic approach, specifically for the exploration of the proteome and transcriptome. Techniques that simultaneously assess multiomics are appealing because molecular signaling underlying complex biological phenomena is thought to occur at complementary levels; however, it has become increasingly common to see that changes in mRNA levels do not always reflect the same change in protein levels and that the time of collection is relevant in the context of circadian regulations. This method removes any intersample variation when assaying different contents within the same sample (intrasample.)
Introduction
Multiomics, the analytical approach that uses a combination of omics, such as genome, proteome, transcriptome, epigenome, microbiome, or lipidome, has become increasingly popular when processing large data sets for disease characterization1,2. Mounting evidence has shown that limiting approaches to a single "ome" provides an incomplete molecular analysis (reviewed by Rotroff and Motsinger-Reif1). Large data sets are generated, particularly when performing screens using high-throughput techniques, but in order to paint a complete picture or to identify the most relevant targets, multiomic approaches are preferable. With the use of multiomics approaches, however, there is the frequent observation of discrepancies between mRNA and protein levels3,4,5,6. Notably, mRNA and protein used for side-by-side transcriptomic and proteomic analyses with RNA sequencing (RNAseq) and liquid chromatography-tandem mass spectrometry (LC-MS/MS), respectively, are often obtained from similarly treated samples from different replicates, potentially introducing variation between the same conditions3,4,5,6. Harvald et al. performed an elegant C. elegans starvation time-course study that compared the transcriptome and proteome of wild-type (WT) worms to that of hlh-30 mutant worms that lack an important transcription factor in longevity7. Of note, the RNA and protein were harvested from the same condition replicates, so not from the same sample. Their findings show a low correlation between the mRNA levels and protein levels at each time point (r = 0.559 to 0.628). In fact, their heatmap formed four clusters: Cluster I had a large decrease in mRNA levels but little or no decrease in corresponding protein levels, Cluster II had little or no increase in mRNA levels but an increase in protein levels, Cluster III had an increase in mRNA levels but a decrease in protein levels, and Cluster IV had an increase in mRNA levels but only a subtle change in protein levels4. In addition, this intersample variation may be introduced in cases where the samples of the same condition are not collected at the exact same time. For example, mRNA and proteins regulated by the circadian cycle fluctuate depending on the time of day8,9, or, more specifically, the exposure of C. elegans to light9; expression of these circadian proteins may be delayed up to 8 h after gene expression induction10. Nevertheless, the prevalence of this observation does not necessarily mean it is wrong; in fact, this may prove to be informative. Protein and mRNA are in a constant dynamic state between formation and degradation. Moreover, proteins are often posttranslationally modified to increase stability or to induce their degradation11. For instance, their ubiquitination status can lead to either activation or targeting to the proteasome or lysosome for degradation12. Additionally, noncoding RNAs play an important role in regulating gene expression at the transcriptional and posttranscriptional stages13. Thus, the question is how to limit the variables to confirm that the discrepancies we observe in these nematode studies are real.
Here, we propose a method that removes the intersample variable by allowing analyses of different macromolecules from the same sample. The goal of this protocol is to offer a method to consistently isolate DNA, RNA, and protein from a single sample of C. elegans (also referred to as worms) in an effort to reduce variation, improve reproducibility, and facilitate interpretations. Additional benefits of using the same sample include the saving of time and resources during sample collection, facilitating the cross-sectional analysis of valuable and limited samples, including strains that are difficult to grow and maintain, and gaining insights into the differential regulation of macromolecules based on intrasample variations in mRNA and protein levels.
This method is suitable for assessing gene expressions and protein levels from a single sample of worms, allowing for a more comprehensive evaluation of multiple levels of molecular processing. Guanidinium thiocyanate-phenol-chloroform (GTCp) reagent14, a commonly used chemical to isolate RNA, is used for the extraction of nucleic acids and protein from the worms, with subsequent precipitation, washing, and solubilization of each. This protocol is a compilation of various protocols15,16 with minor modifications, designed with a focus on C. elegans, but we have also successfully isolated RNA, protein, and DNA from a pellet of HeLa cells following the same steps. Although not tested here, this protocol is likely to work on tissues as well17,18.
Protocol
NOTE: Each macromolecule precipitation step is performed sequentially, followed by washes done concurrently; however, it is recommended to complete the RNA isolation first as it is intrinsically unstable.
1. Sample Collection
- Seed 1,000 worm eggs per 10 cm plate with appropriate growth conditions19. Incubate at 20 °C for 72 h.
NOTE: Bleach egg-bearing adults in order to collect eggs as previously described19. - Wash the plate with around 5 mL of M9 buffer and collect 1,000 adult worms into a tube.
NOTE: M9 buffer is composed of 35 mM sodium phosphate dibasic, 102 mM sodium chloride, 22 mM potassium phosphate monobasic, and 1 mM magnesium sulfate in sterile water19. - Centrifuge the worms at 1,000 x g for 1 min, discard the supernatant, and transfer the pelleted worms with 1 mL of M9 buffer to a 1.5 mL microcentrifuge tube.
- Centrifuge again at 845 x g for 1 min and discard most of the supernatant. Store the pelleted worms at -80 °C for a minimum of 4 h.
NOTE: Using a frozen pellet produces a higher yield than a fresh pellet. A series of freeze/thaw cycles using liquid nitrogen or 95% ethanol on dry ice to crack the worm cuticle is recommended if using a fresh pellet. Leaving a small amount of M9 on the pellet will help break the cuticle when freezing.
2. Nucleotide and Protein Isolation
- Remove any supernatant from the thawed pellet and add 1 mL of cold GTCp reagent. Mix well by pipetting up and down. Place the sample on ice for 10 min and mix it occasionally by turning it upside down.
CAUTION: Phenol is corrosive, neurotoxic, and highly toxic and may cause chemical burns and blindness.
NOTE: We used up to 5,000 adult worms as the starting material with the reported volumes; each volume is based on the use of 1 mL of GTCp reagent. When using a larger sample, it may be necessary to scale up. - To the solution of worms and GTCp, add 200 µL of cold chloroform. Hold the tube between fingers and shake vigorously for 15 s. Leave it at room temperature (RT) for 3 min.
CAUTION: Chloroform is a toxic irritant, may cause skin sores and other organ-targeted harm, and is a possible carcinogen. - Centrifuge the tube at 13,500 x g for 15 min at 4 °C.
NOTE: Three layers are formed after this spin: the top, clear aqueous phase, the bottom, pink organic phase, and the small, cloudy interphase that contains lipids and DNA. - Using a micropipette, move the clear top layer (aqueous phase) to a new RNase-free 1.5 mL microcentrifuge tube (tube A) to isolate RNA via alcohol precipitation (as detailed in step 2.5) and move the pink layer (organic phase) from the remaining pellet to a new tube (tube B) and place it on ice.
NOTE: The pink organic phase can be frozen at -80 °C until the isolation of DNA and protein from this sample. Larger samples will produce a thick white layer between the aqueous and organic phase. This fraction contains DNA. If this layer can be removed without disturbing the other phases, do so and put it in a separate tube (tube B2). - Isolate RNA as follows.
- From the clear aqueous phase in tube A, precipitate the RNA with 500 µL of 100% isopropanol. Incubate for 10 min at RT. Then, centrifuge tube A at 13,500 x g for 10 min at 4 °C.
NOTE: A small white pellet of RNA should be visible at the bottom of the tube. - Decant the majority of the supernatant. Remove the rest with a 1 mL syringe with a needle and discard the supernatant.
NOTE: The size of the needle is not important; however, a larger needle gauge will offer more control so as not to disturb the pellet. A micropipette can be used if needles are not available. - Add 1 mL of 75% ethanol to tube A to wash the pellet. Spin the tube at 5,300 x g for 5 min at 4 °C.
- Remove the supernatant from the pellet by decanting and using a 1 mL syringe with a needle, as described in step 2.5.2, and discard it.
- Let the pellet air-dry for 5–10 min, but do not let it overdry. Use 50 µL of RNase-free water to reconstitute the pellet of RNA before it becomes completely transparent.
NOTE: This is an adequate starting volume for 1,000–3,000 worms, but it may need to be adjusted depending on how much starting material was used. - Incubate the pellet at 55–60 °C for 10 min to dissolve it.
- Measure the concentration and the purity using a spectrophotometer. Record absorbances at 260 nm for RNA concentration and at 230 and 280 nm to identify any impurities.
- Clean the RNA to remove any contaminants, such as phenol residue or DNA, either with column purification or with subsequent ethanol washes and DNase treatment.
NOTE: At this point, the RNA is ready to be sent for RNAseq analysis or can be used to make cDNA to use for RT-qPCR (see section 3.1 for details). RNA can be stored at -80 °C until further use.
- From the clear aqueous phase in tube A, precipitate the RNA with 500 µL of 100% isopropanol. Incubate for 10 min at RT. Then, centrifuge tube A at 13,500 x g for 10 min at 4 °C.
- Isolate DNA as follows.
- From the pink organic phase in tube B and the sample in tube B2, if any, precipitate the DNA by adding 300 µL of 100% ethanol and mix by inversion. Leave the tube(s) at RT for 2–3 min.
- Centrifuge tubes B and B2 at 375 x g for 5 min at 4 °C to pellet DNA.
- Move the supernatant from tubes B and B2 by pouring it into a new 2 mL tube (tube C) and leave it on ice for subsequent protein isolation. Wash the DNA pellet in tube B or B2 with 1 mL of 0.1 M sodium citrate in 10% ethanol for 30 min. Centrifuge tubes B and B2 at 375 x g for 5 min at 4 °C.
NOTE: Sodium binds to the DNA backbone, making DNA precipitate more readily in sodium citrate and ethanol, allowing impurities to be removed by low-speed centrifugation. - Repeat the wash step as described in step 2.6.3. Resuspend the pellet in 1.5 mL of 75% ethanol and leave it at RT for 20 min, with occasional mixing by upending.
- Centrifuge tube B and B2 at 375 x g for 5 min at 4 °C.
- Discard the supernatant and allow it to dry for 5–10 min.
- Dissolve the pellet in 150 µL of 8 mM sodium hydroxide. Adjust to the desired pH with HEPES if necessary. Spin the sample at 375 x g for 5 min at 4 °C.
- Using a micropipette, move the supernatant (DNA) to a new tube. Measure the concentration and determine the purity using a spectrophotometer. Record absorbances at 260 nm for DNA concentration and at 230 and 280 nm to identify any impurities.
- Isolate protein as follows.
- To precipitate the protein, add up to 1.5 mL of 100% isopropanol to the pink supernatant in tube C, mix by inverting several times, and incubate at RT for 10 min.
- Centrifuge tube C at 13,500 x g for 10 min at 4 °C.
- Discard the supernatant and wash the pellet with 2 mL of 0.3 M guanidine hydrochloride in 95% ethanol for 20 min at RT. Centrifuge tube C at 5,300 x g for 5 min at 4 °C.
- Repeat the wash step as described in step 2.7.3 2x.
- Move the protein pellet to a new 1.5 mL tube (tube C2) and add up to 1.5 mL of 95% ethanol. Vortex and let it sit at RT for 20 min.
- Centrifuge tube C2 at 5,300 x g for 5 min at 4 °C. Discard the supernatant and let the pellet dry for 10 min at RT. Dissolve the pellet in 300 µL of 5% SDS at 50 °C for 60 min.
NOTE: A longer incubation time may be required to fully dissolve the protein pellet. In the past, the pellet has been incubated for up to 6 h without sacrificing quality. - Centrifuge tube C2 at 13,500 x g for 10 min at 17 °C. Move the supernatant to a new tube.
- Measure the concentration using a preferred protein quantification assay that is compatible with detergent.
NOTE: The protein is ready to be used in SDS-PAGE and western blotting. It can be stored at -20 °C for future use. For other techniques, such as LC-MS/MS, the detergent will need to be dialyzed off.
3. Evaluating mRNA and Protein Levels
- RT-qPCR
- Prepare cDNA by reverse transcription with 1 ng of RNA, using the following thermocycler protocol: priming (5 min at 25 °C), reverse transcription (20 min at 46 °C), and RT inactivation (1 min at 95 °C).
- Make a stock plate of cDNA by adding 100 µL of 1:100 dilutions of each cDNA sample to individual wells of a 96-well plate. Include appropriate controls, such as no template/water-only wells, and serially diluted samples from 1:25 to 1:400 (originated with pooled cDNA from all of the samples analyzed) to allow for a standard curve to establish primer efficiency for each gene analyzed.
NOTE: This format can be used with the 96-well microdispenser, which allows for the transfer of all samples in a plate to an RT-qPCR plate and for the mitigation of well-to-well pipetting variations. The appropriate master mix (e.g., SYBR + primers) can be added to each well thereafter, with a multichannel pipette. Overall, this approach reduces errors introduced when hand-pipetting each sample, thus further reducing variability. - Add 3 µL of cDNA from the stock plate to the wells of the RT-qPCR plate.
- Make a master mix for each set of primers that includes 1x supermix containing a DNA-intercalating cyanine dye, 5 µM each of forward and reverse primers, and water up to 7 µL per sample. Add 7 µL to the appropriate wells of an RT-qPCR plate and agitate lightly to mix.
NOTE: Include samples to measure housekeeping genes to use as a reference to normalize the results20. - Run the plate using an RT-qPCR protocol that is suitable for the primers being used.
- Western blot
NOTE: For detailed information about western blots, please refer to Mahmood and Yang21.- Prepare samples for SDS-PAGE by combining 20 µg of protein with an equal volume of 2x Laemmli Sample Buffer and boil at 95 °C for 10 min.
- Load the samples onto a 4%–15% Tris-glycine gel and run them at 150 V for 1 h, or until the dye front reaches the bottom of the gel.
- Transfer proteins from the gel to a nitrocellulose membrane at 25 V for 30 min in a semidry transfer apparatus.
- Confirm the proper transfer by staining the membrane with Ponceau S stain.
- Thoroughly wash the stain from the membrane with Tris-buffered saline (TBS) with 0.01% polysorbate 20 (TBS-T).
- Block the membrane with 5% nonfat milk in TBS-T for 1 h at RT.
- Incubate the membrane in the primary antibody at the appropriate dilution according to the supplier’s recommendations. Leave it on the rocker at 4 °C overnight.
- Wash the membrane 3x, for 5 min each, with TBS-T.
- Incubate the membrane in the appropriate secondary antibody at the appropriate dilution according to the supplier’s recommendations. Leave it on the rocker for 1 h at RT.
- Wash the membrane 3x, for 5 min each, with TBS-T.
- Image and quantify the western blot, using preferred methods.
Results
The RNA, DNA, and protein samples were analyzed, and representative results using the RNA and protein isolates are presented here. Additionally, we compare the protein sample harvested using the GTCp method to the common RIPA lysis method22. For our experiment, we used four independent replicates of each WT (N2) worms and two long-lived mutant worms, eat-2 and rsks-123,24.
RNA and DNA quantity and quality
The concentration of RNA when resuspended in 50 µL of water ranged from 0.2–2 µg/µL, using around 3,000 adult worms as starting material. The absorbance ratios, which indicate purity, ranged from 2.0 to 2.2 for A260/A280 and 1.9 to 2.5 for A260/A230 (Table 1), indicating the successful extraction of RNA without contamination with GTCp components, such as phenol or guanidine hydrochloride.
The DNA extraction was successful, but the quality was variable. The DNA concentration when resuspended in 150 µL of NaOH ranged from 0.04 to 1.1 µg/µL. The absorbance ratios ranged from 1.8 to 2.1 for A260/A280 and 0.9 to 1.6 for A260/A230 (Table 1), indicating the successful extraction of DNA; however, some of the samples were contaminated with GTCp components, like phenol or guanidine hydrochloride. If DNA isolation is necessary, care must be taken when separating the middle layer from the GTCp phase separation, and more washes may be necessary.
RT-qPCR of selected targets
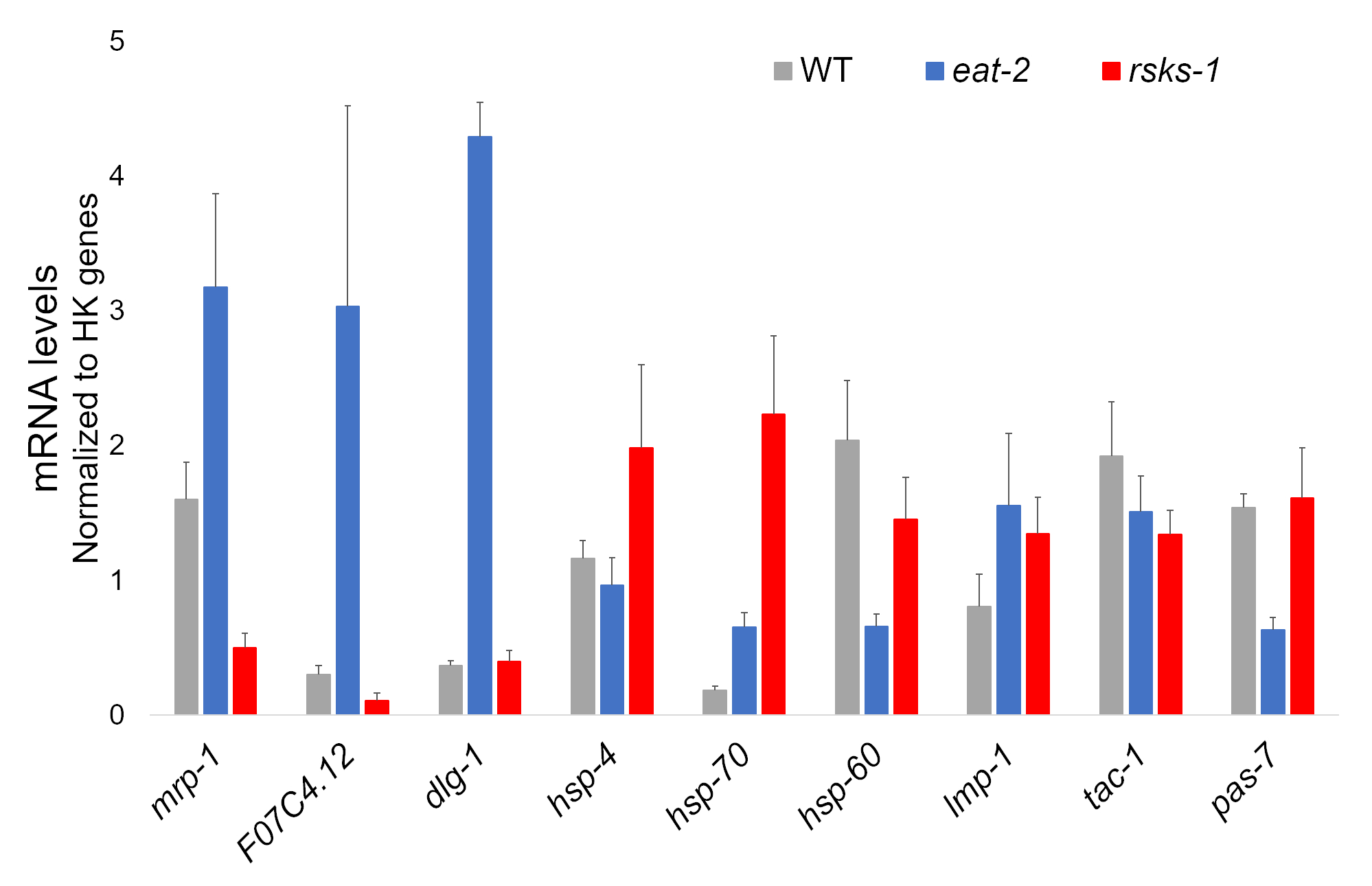
The RNA isolation from four independent samples of each worm strain was successful and was sent for RNAseq analysis, with subsequent RT-qPCR performed using a supermix containing a cyanine dye. Targets analyzed by RT-qPCR were selected from a large RNAseq dataset based on the most differentially regulated genes common in both mutants as compared to the WT worms. F07C4.1225, a homolog to human neuroligin 3, isoform b, was the selected shared upregulated target. We also included a differentially regulated gene between both mutants, mrp-126, for additional analysis, which was upregulated in eat-2 worms but downregulated in rsks-1 worms. Expression changes of mrp-1 were confirmed via RT-qPCR; however, the F07C4.12 upregulation in rsks-1 worms predicted by the RNAseq analysis was not seen by RT-qPCR (Figure 1).
Additional targets with antibodies validated for worms were investigated as well. These included several organelle markers characterized in Hadwiger et al.27. A number of these markers were differentially expressed at the mRNA level in the mutant worms. As seen in Figure 1, lmp-128 and dlg-129 were upregulated in eat-2 worms; hsp-430, hsp-7030, and lmp-1 were upregulated in rsks-1 worms; hsp-6031 and pas-732 were downregulated in eat-2 worms; hsp-60 and tac-133 were downregulated in rsks-1 worms.
Simultaneous vs. RIPA protein preparation
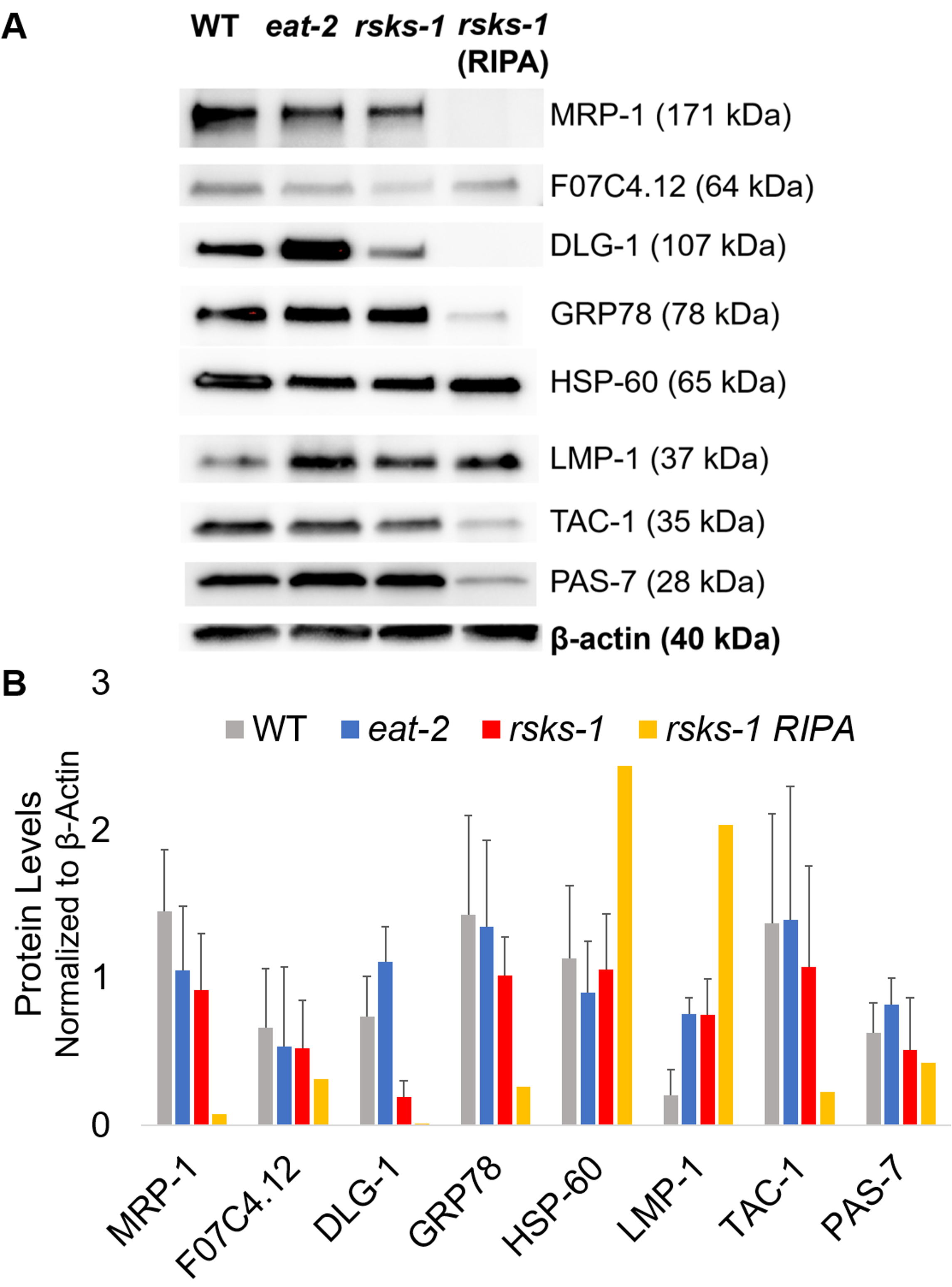
To affirm the efficiency and quality of the protein with the GTCp method as described above, we compared it to protein collected using the more traditional method of RIPA lysis22. Using the same amount of starting material, namely either adults only (about 10,000 worms) or a mixed population of adults, larvae, and eggs (about 130,000 worms), the overall quantity collected with GTCp was less when resuspended in equal end volumes (Table 1); however, the extraction from an adult-only population was more successful than from a mixed population. Additionally, the quality of the proteins was similar, albeit the GTCp-extracted protein showed a better resolution of larger proteins upon equal loading of total protein, as shown by Coomassie stain in Figure 2. Indeed, targets evaluated by western blot in Figure 3 showed similar levels in most cases; however, the proteins larger than 75 kDa showed lower levels in the RIPA-extracted protein compared to the GTCp-extracted protein (Figure 3A, right lane; Figure 3B, yellow bar).
The extraction method presented here was also evaluated in the mammalian cell line HeLa. For reference, the amount of protein extracted with RIPA from a compact pellet of six million HeLa cells (an ~25 µL pellet) was comparable to that extracted from a 130,000 mixed population of worms (an ~75 µL compact pellet), or about half of what is extracted from 10,000 adult worms (an ~100 µL gravity-settled pellet), as seen in Table 1. We show that the efficiency of protein extraction (Table 1) and the resolution of larger proteins (Figure 2) was decreased in GTCp compared to RIPA-extracted protein in HeLa cells, suggesting this technique works better in an adult population of worms. The incomplete solubilization of the protein pellet from GTCp-extracted HeLa protein may be a limitation of this method in mammalian cells.
Immunoblotting targets analyzed by RT-qPCR
Next, we investigated the protein levels of the gene products tested by RT-qPCR to determine if the mRNA levels correlated with the protein levels. As seen in Figure 3, the average expression changes of protein correlated with the average mRNA expressions changed for many targets; however, some protein levels did not reflect the changes in mRNA levels. Often, the mRNA levels in eat-2 worms were higher than those in the other strains, but the protein levels were either the same or lower than the other strains of the same target. The most outstanding observation was the very high levels of dlg-1 mRNA in eat-2 worms, which did not translate to more protein; in fact, there were lower dlg-1 protein levels when compared to the other strains (Figure 3).
Finally, the levels in the GTCp-extracted protein and the RIPA-extracted protein showed a striking difference. The RNA was extracted in the same manner for both; however, the similar but separate sample collected by RIPA lysis showed markedly reduced protein levels, particularly for those larger than 75 kDa (Figure 3A, right lane; Figure 3B, yellow bar).
Intrasample comparison of mRNA and protein levels
One of the goals of this protocol was to determine if the variation we saw at the mRNA and protein level was real, or if it could be an artifact of intersample variation. In Figure 4, a subset of targets was compared within the single samples. Each individual sample is shown as the same shade and color of the dot, with sample 1 being the darkest shade and sample 4 as the lightest shade of gray (WT), blue (eat-2), or red (rsks-1). Within most samples, there was a low variability of the mRNA levels, with greater variability at the protein level, a potential flaw of the semiquantitative analysis of western blots. When looking at the position of each of the colored dots between the mRNA and protein pairs, the order of highest-to-lowest often did not match; for example, in WT, the hsp-60 mRNA order was sample 4, 3, 2, 1, but the protein levels were 1, 2, 3, 4. Thus, differences between mRNA and protein levels within a sample certainly exist, but the presented method allows users to remove the time of collection as a possible source of the observed difference.

Table 1: Concentrations and absorption ratios. RNA and DNA concentrations and purity ratios were measured on a spectrophotometer. Protein concentrations were determined using a colorimetric protein quantification assay. The gray, blue, and red highlight the samples used for RT-qPCR and western blot. The yellow indicates samples used to compare GTCp to RIPA protein extraction or worm protein to HeLa protein. Please click here to download this file.

Figure 1: Gene expression by RT-qPCR. RT-qPCR analysis for the mRNA obtained by this method, confirming targets identified from RNAseq data, and of organelle marker genes. The error bars represent standard deviation; n = 4. The concentration of mRNA was defined against a standard curve for each set of primers. All mRNA levels were normalized to the average of a set of six housekeeping genes used as reference genes, which include act-1, cdc-42, ama-1, nhr-23, pmp-3, and cyn-1. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Comparison of GTCp- versus RIPA-extracted protein in worms and HeLa cells. Total protein from wild-type (WT) worms or HeLa cells harvested via GTCp or RIPA lysis method, separated by SDS-PAGE, and stained with Coomassie blue. Each lane contains 25 µg of total protein. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: Protein levels in worms. (A) Western blot of targets investigated by RT-qPCR and (B) densitometry analysis of signal intensity. Each lane contains 20 µg of total GTCp-extracted protein from wild-type (WT), eat-2, or rsks-1 mutant worms. The image shown is a representation of four independent replicates per worm strain. β-actin (bottom) was used to control for equal loading for each target; only one representative set is shown. The right lane contains 20 µg of total RIPA-extracted protein from rsks-1 mutant worms. (B) Corresponding signal intensities were quantified using ImageJ. The error bars represent standard deviation; n = 4. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: Intrasample mRNA and protein level comparison. The mRNA and protein levels from individual samples from a subset of targets for each strain are aligned. The color is the representative of the Table 1. The gray/black is WT, the blue is eat-2, and the red is rsks-1. mRNA and protein of the same target are paired next to each other and separated by nematode strain. Please click here to view a larger version of this figure.
{kind=link}
Discussion
Methods for biomolecule isolations, such as DNA, RNA, and proteins, are often optimized without technical overlap or combinations. This is particularly disadvantageous when samples are difficult to obtain, which could lead to harvesting samples under the same conditions at different times. Depending on the cellular pathways, replicates collected at different times may generate variation. This manuscript offers a procedure to circumvent this hurdle by enabling the concurrent isolation and purification of each biomolecule from the same sample of worms, reducing variations introduced by different isolation techniques, the timing of sample harvest, or unequal harvesting. Controlling these variables not only saves time and resources, but also facilitates reproducibility. Here, we demonstrate a combinatorial approach that avoids compromising RNA and protein quality, albeit with variable results with DNA. Preparations can be further optimized using DNA cleaning procedures. We demonstrated the approach using material from the nematode C. elegans and HeLa cells.
Previous work exploring the transcriptome and proteome of N2 WT animals and eat-2 and rsks-1 mutants have offered insight into various pathways, including mechanisms that extend lifespan4,34,35,36. In an effort to investigate the mechanism of caloric restriction in extending lifespan, a Stable Isotope Labeling by/with Amino acids in Cell culture (SILAC) analysis found that eat-2 worms have an overall downregulation of global protein synthesis 36. The data presented here is consistent with this finding, even as the mRNA levels of the same targets are greatly increased. Another group aimed to identify effectors of S6K-mediated longevity and, thus, performed a proteomic screen of rsks-1 worms34. From the RNAseq data found with the current study, we identified at least three genes that corroborate with proteins identified from this screen; MRP-1 and homologs of CPA and neuroligin (F07C4.12) were discovered as being differentially expressed in rsks-1 worms as compared to WT N234.
The data generated using this method is consistent with previous multiomic investigations. The mRNA levels of nine targets were used to predict the protein levels in each sample. Of these targets, many had predictable protein levels based on the mRNA levels. Nevertheless, there were notable discrepancies between the mRNA and protein levels. Importantly, the protocol presented here allows scientists to confidently evaluate and interpret these differences by removing intersample variability by collecting mRNA and protein from the same sample. Furthermore, we compared the mRNA levels to the protein levels collected from the same sample or collected from a similar but different sample harvested with RIPA. We showed that for a number of the targets, there were much lower levels of protein in the RIPA-extracted sample. Without controlling for the intersample variation, it would be impossible to know if this difference was due to the differential regulation of mRNA and protein.
It is important to keep in mind that there are protocols optimized specifically for these different macromolecules, so if a cross-sectional analysis is not the final goal of the experiment, then it would be pertinent to employ those methods instead. Using GTCp to isolate DNA and protein causes them to become less soluble, requiring the reconstitution of DNA in a weak base, such as NaOH, and solubilizing the protein in a buffer with a high concentration of detergent with heating, at the risk of incomplete solubilization. Additionally, GTCp contains guanidinium thiocyanate and acidic phenol, which inactivates enzymes such as proteases, but will slowly degrade protein over time unless frozen. It will be to the discretion of the researcher to decide if the circumvention of these limitations will be worthwhile.
Importantly, when working with RNA, a sterile technique, keeping the sample on ice unless otherwise noted, and the use of commercially available decontaminating reagent is recommended to keep the RNA intact. Notably, larger samples will need more GTCp to start with, in which each extraction solvent will also need to be scaled up. This protocol does not require any manual homogenization of the worm or cell samples when the correct amount of GTCp is used. In the context of DNA isolation, the yield is highly dependent on the proficiency to recover the organic (pink) layer. Finally, to improve the solubilization of proteins during isolation, increasing the volume of resolubilization buffer or adding other detergents besides SDS may be necessary. Indeed, proteins from an adult-only population of worms instead of a mixture of adults, larvae, and eggs are much easier to resolubilize.
Overall, using this protocol provides an integrated approach to biomolecule isolations and facilitates the interpretation of correlations, or lack thereof, between mRNA and protein levels that can arise from separately harvesting biomolecules from different samples. Using this method can help scientists to identify correctly cases where the translation of mRNA to protein is not correlative and can lead to the deeper investigation of posttranscriptional and posttranslational regulatory mechanisms under various conditions.
Disclosures
The authors have nothing to disclose.
Acknowledgements
L.R.L. was funded by grants from the NIH/NIA (R00 AG042494 and R01 AG051810), a Glenn Foundation for Medical Research Award for Research in Biological Mechanisms of Aging and a Junior Faculty Grant from the American Federation for Aging Research. The authors would like to thank Anita Kumar and Shi Quan Wong for their useful feedback in the writing of this manuscript.
Materials
| Name | Company | Catalog Number | Comments |
| 4–15% Mini-PROTEAN® TGX Stain-Free™ Protein Gels | BIORAD | 4568084 | |
| Antibodies: | |||
| CePAS7-s | DHSB- U of Iowa | ||
| CeTAC1-s | DHSB- U of Iowa | ||
| DLG1-s | DHSB- U of Iowa | ||
| GRP78 | Novus Bio. | NBp1-06274 | |
| HRP Goat Anti-Mouse | Li-Cor | 926-80010 | |
| HRP Goat Anti-Rabbit | Li-Cor | 92680011 | |
| HSP60-s | DHSB- U of Iowa | ||
| LMP1-s | DHSB- U of Iowa | ||
| MRP-1 | Abcam | ab24102 | |
| Neuroligin 3 | Abcam | ab172798 | |
| β-actin | Millipore | MAB1501R | |
| ChemiDoc MP Imaging System | BIORAD | ||
| Chloroform | Fisher | C298-500 | Health hazard, Irritant, Toxic |
| Coomassie Brilliant Blue | ThermoSci | 20279 | |
| DC Protein assay | BIORAD | 500-0116 | |
| Epoch 2 microplate reader | BioTek | ||
| Ethanol (200 proof) | Fisher | 04-355-223 | Flammable, health hazard |
| HeLa cells | ATCC | CCL-2 | BSL2 |
| iScript Reverse Transcription Supermix | BIORAD | 1708840 | |
| Hydra microdispenser: Matrix Hydra | Robbins/ThermoFisher | ||
| Isopropanol | Fisher | A516-4 | Flammable, health hazard |
| M9 buffer: 3 g KH2PO4 6 g Na2HPO4 5 g NaCl Add H2O to 1 liter. Sterilize by autoclaving. After solution cools down, add 1 mL sterile 1 M MgSO4 | |||
| Laemmli Sample Buffer (2x) | BIORAD | 161-0737 | |
| NanoDrop One | ThermoSci | ||
| PAGE apparatus | BIORAD | ||
| Ponceau S | Alfa Aesar | J60744 | |
| Primers | IDT | 25 nmole DNA Oligo with Standard Desalting | |
| dlg-1: F (5'-GGTCCTACCA GGCAGTTGAG-3') R (5'-CACGTCCGTT AACCTCTCCC-3') | |||
| hsp-4: F (5'-AGAGGGCTTT GTCAACCCAG-3') R (5'-TCGTCAGGGT TGATTCCACG-3') | |||
| hsp-70: F (5'-CGGCATGTGA ACGTGCTAAG-3') R (5'-GAGCAGTTGA GGTCCTTCCC-3') | |||
| hsp-60: F (5'-ATTGAGCAAT CGACGAGCGA-3') R (5'-CAACACCTCC TCCTGGAACG-3') | |||
| lmp-1: F (5'-ACAACAACAC CGGACTCACG-3') R (5'-ATCGAGCTCC CACTCTTTGG-3') | |||
| tac-1: F (5'-AGTGGCAGGC AAAGTTCCTC-3') R (5'-TGAGCACCTT GATCTCGTCG-3') | |||
| pas-7: F (5'-GTACGCTCAA AAGGCTGTCG-3') R (5'-CTGAATCGGC ATTGGCTCAC-3') | |||
| mrp-1: F (5'-TTTGCCTTGC GCTTGTTCTG-3') R (5'-AGTTCCAGTG CGGAGCATAC-3') | |||
| F07C4.12: F (5'-TGCTGAGCAT GAAGGACTGT-3') R (5'-TGGCAATAGC TCCTCCGTTG-3') | |||
| HK Actin: F (5'-CTACGAACTT CCTGACGGACAAG-3') R (5'-CCGGCGGACT CCATACC-3') | |||
| HK cyn-1: F (5'-GTGTCACCAT GGAGTTGTTC-3') R (5'-TCCGTAGATT GATTCACCAC-3') | |||
| HK nhr-23: F (5'-CAGAAACACT GAAGAACGCG-3') R (5'-CGATCTGCAG TGAATAGCTC-3') | |||
| HK ama-1: F (5'-TGGAACTCTG GAGTCACACC-3') R (5'-CATCCTCCTT CATTGAACGG-3') | |||
| HK cdc-42: F (5'-CTGCTGGACA GGAAGATTACG-3') R (5'-CTCGGACATT CTCGAATGAAG-3') | |||
| HK pmp-3: F (5'-GTTCCCGTGT TCATCACTCAT-3') R (5'-ACACCGTCGA GAAGCTGTAGA-3') | |||
| LightCycler 96 qPCR machine | Roche | ||
| RIPA buffer: 10 mM Tris-Cl (pH 8.0) 1 mM EDTA 1% Triton X-100 0.2% SDS 140 mM NaCl 1 tablet of Roche protease inhibitor per 20 mL | |||
| SsoAdvanced Universal SYBR Supermix | BIORAD | 1725274 | |
| SuperSignal West Femto Max Sens Substrate | ThermoSci | 34095 | |
| Trans-Blot Transfer apparatus | BIORAD | ||
| Trans-Blot Turbo Transfer Pack | BIORAD | 170-4159 | |
| TRIzol reagent | Invitrogen | 15596026 | Health hazard (skin, eyes) |
| Worm strains: | Caenorhabditis Genetics Center (CGC) | ||
| N2 (wild type) | Caenorhabditis Genetics Center (CGC) | ||
| eat-2 (MAH95) | Caenorhabditis Genetics Center (CGC) | ||
| rsks-1 (VB633) | Caenorhabditis Genetics Center (CGC) |
References
- Rotroff, D. M., Motsinger-Reif, A. A. Embracing Integrative Multiomics Approaches. International Journal of Genomics. 2016, 1715985 (2016).
- Chen, R., Snyder, M. Promise of personalized omics to precision medicine. Wiley Interdisciplinary Reviews: System Biology and Medicine. 5 (1), 73-82 (2013).
- Anderson, L., Seilhamer, J. A comparison of selected mRNA and protein abundances in human liver. Electrophoresis. 18 (3-4), 533-537 (1997).
- Harvald, E. B., et al. Multi-omics Analyses of Starvation Responses Reveal a Central Role for Lipoprotein Metabolism in Acute Starvation Survival in C. elegans. Cell Systems. 5 (1), (2017).
- Griffin, T. J., et al. Complementary profiling of gene expression at the transcriptome and proteome levels in Saccharomyces cerevisiae. Molecular and Cell Proteomics. 1 (4), 323-333 (2002).
- Greenbaum, D., Colangelo, C., Williams, K., Gerstein, M. Comparing protein abundance and mRNA expression levels on a genomic scale. Genome Biology. 4 (9), 117 (2003).
- Lapierre, L. R., et al. The TFEB orthologue HLH-30 regulates autophagy and modulates longevity in Caenorhabditis elegans. Nature Communications. 4, 2267 (2013).
- Doherty, C. J., Kay, S. A. Circadian control of global gene expression patterns. Annual Reviews Genetics. 44, 419-444 (2010).
- Goya, M. E., Romanowski, A., Caldart, C. S., Benard, C. Y., Golombek, D. A. Circadian rhythms identified in Caenorhabditis elegans by in vivo long-term monitoring of a bioluminescent reporter. Proceedings of the National Academy of Sciences USA. 113 (48), E7837-E7845 (2016).
- Dalvin, L. A., Fautsch, M. P. Analysis of Circadian Rhythm Gene Expression With Reference to Diurnal Pattern of Intraocular Pressure in Mice. Investigative Ophthalmology and Visual Science. 56 (4), 2657-2663 (2015).
- Narayanan, A., Jacobson, M. P. Computational studies of protein regulation by post-translational phosphorylation. Current Opinion in Structural Biology. 19 (2), 156-163 (2009).
- Swatek, K. N., Komander, D. Ubiquitin modifications. Cell Research. 26 (4), 399-422 (2016).
- Cech, T. R., Steitz, J. A. The noncoding RNA revolution-trashing old rules to forge new ones. Cell. 157 (1), 77-94 (2014).
- Chomczynski, P., Sacchi, N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Analytical Biochemistry. 162 (1), 156-159 (1987).
- Triant, D. A., Whitehead, A. Simultaneous Extraction of High-Quality RNA and DNA from Small Tissue Samples. Journal of Heredity. 100 (2), 246-250 (2009).
- Liu, X., Harada, S. RNA Isolation from Mammalian Samples. Current Protocols in Molecular Biology. 103 (1), 11-14 (2013).
- Hummon, A. B., Lim, S. R., Difilippantonio, M. J., Ried, T. Isolation and solubilization of proteins after TRIzol extraction of RNA and DNA from patient material following prolonged storage. Biotechniques. 42 (4), 467-470 (2007).
- Kopec, A. M., Rivera, P. D., Lacagnina, M. J., Hanamsagar, R., Bilbo, S. D. Optimized solubilization of TRIzol-precipitated protein permits Western blotting analysis to maximize data available from brain tissue. Journal of Neuroscience Methods. 280, 64-76 (2017).
- Stiernagle, T. Maintenance of C. elegans. WormBook. , (2006).
- Hoogewijs, D., Houthoofd, K., Matthijssens, F., Vandesompele, J., Vanfleteren, J. R. Selection and validation of a set of reliable reference genes for quantitative sod gene expression analysis in C. elegans. BMC Molecular Biology. 9 (1), 9 (2008).
- Mahmood, T., Yang, P. -. C. Western blot: technique, theory, and trouble shooting. North American Journal of Medical Sciences. 4 (9), 429-434 (2012).
- Peach, M., Marsh, N., MacPhee, D. J., Kurien, B. T., Scofield, R. H. Protein solubilization: Attend to the choice of lysis buffer. Protein Electrophoresis: Methods and Protocols. , 37-47 (2012).
- Lakowski, B., Hekimi, S. The genetics of caloric restriction in Caenorhabditis elegans. Proceedings of the National Academy of Science USA. 95 (22), 13091-13096 (1998).
- Pan, K. Z., et al. Inhibition of mRNA translation extends lifespan in Caenorhabditis elegans. Aging Cell. 6 (1), 111-119 (2007).
- Gaudet, P., Livstone, M. S., Lewis, S. E., Thomas, P. D. Phylogenetic-based propagation of functional annotations within the Gene Ontology consortium. Briefings in Bioinformatics. 12 (5), 449-462 (2011).
- Broeks, A., Gerrard, B., Allikmets, R., Dean, M., Plasterk, R. H. Homologues of the human multidrug resistance genes MRP and MDR contribute to heavy metal resistance in the soil nematode Caenorhabditis elegans. The EMBO Journal. 15 (22), 6132-6143 (1996).
- Hadwiger, G., Dour, S., Arur, S., Fox, P., Nonet, M. L. A Monoclonal Antibody Toolkit for C. elegans. PLoS One. 5 (4), e10161 (2010).
- Kostich, M., Fire, A., Fambrough, D. M. Identification and molecular-genetic characterization of a LAMP/CD68-like protein from Caenorhabditis elegans. Journal of Cell Science. 113 (14), 2595 (2000).
- Firestein, B. L., Rongo, C. DLG-1 is a MAGUK similar to SAP97 and is required for adherens junction formation. Molecular Biology of the Cell. 12 (11), 3465-3475 (2001).
- Heschl, M. F., Baillie, D. L. Characterization of the hsp70 multigene family of Caenorhabditis elegans. DNA. 8 (4), 233-243 (1989).
- Yoneda, T., et al. Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. Journal of Cell Science. 117 (18), 4055 (2004).
- Takahashi, M., Iwasaki, H., Inoue, H., Takahashi, K. Reverse Genetic Analysis of the Caenorhabditis elegans 26S Proteasome Subunits by RNA Interference. Biological Chemistry. 383, 1263 (2002).
- Le Bot, N., Tsai, M. C., Andrews, R. K., Ahringer, J. TAC-1, a regulator of microtubule length in the C. elegans embryo. Current Biology. 13 (17), 1499-1505 (2003).
- McQuary, P. R., et al. C. elegans S6K Mutants Require a Creatine-Kinase-like Effector for Lifespan Extension. Cell Reports. 14 (9), 2059-2067 (2016).
- Larance, M., et al. Global Proteomics Analysis of the Response to Starvation in C. elegans . Molecular and Cell Proteomics. 14 (7), 1989-2001 (2015).
- Yuan, Y., et al. Enhanced energy metabolism contributes to the extended life span of calorie-restricted Caenorhabditis elegans. Journal of Biological Chemistry. 287 (37), 31414-31426 (2012).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved