
Method Article
Targeted Studies Using Serial Block Face and Focused Ion Beam Scan Electron Microscopy
In This Article
Summary
Here, we present a protocol for efficiently combining serial block face and focused ion beam scanning electron microscopy to target an area of interest. This allows for efficient searching, in three dimensions, and locating rare events in a large field of view.
Abstract
This protocol allows for the efficient and effective imaging of cell or tissue samples in three dimensions at the resolution level of electron microscopy. For many years electron microscopy (EM) has remained an inherently two-dimensional technique. With the advent of serial scanning electron microscope imaging techniques (volume EM), using either an integrated microtome or focused ion beam to slice then view embedded tissues, the third dimension becomes easily accessible. Serial block face scanning electron microscopy (SBF-SEM) uses an ultramicrotome enclosed in the SEM chamber. It has the capability to handle large specimens (1,000 µm x 1,000 µm) and image large fields of view at small X,Y pixel size, but is limited in the Z dimension by the diamond knife. Focused ion beam SEM (FIB-SEM) is not limited in 3D resolution, (isotropic voxels of ≤5 nm are achievable), but the field of view is much more limited. This protocol demonstrates a workflow for combining the two techniques to allow for finding individual regions of interest (ROIs) in a large field and then imaging the subsequent targeted volume at high isotropic voxel resolution. Preparing fixed cells or tissues is more demanding for volume EM techniques due to the extra contrasting needed for efficient signal generation in SEM imaging. Such protocols are time consuming and labor intensive. This protocol also incorporates microwave assisted tissue processing facilitating the penetration of reagents, which reduces the time needed for the processing protocol from days to hours.
Introduction
This protocol describes a workflow for the efficient targeting of high-resolution, three-dimensional electron microscopy (EM) to a specific region of interest (ROI). Since its beginnings in the 1930s, EM has been an essentially two-dimensional technique. The first published images were of whole tissues or cells but that soon gave way to sections that were cut by hand using an ultramicrotome and imaged using a transmission electron microscope (TEM). TEM produces very high-resolution micrographs where even the smallest of cellular structures are clearly discernable. However, the thinness of section necessary for the tissue to be imaged by the electron beam made information in the Z dimension minimal. Since cells are three-dimensional structures, interactions between cell structures and cell surfaces had to be inferred from limited data. This raised the potential for misinterpretation, especially in structures that were complex. Some microscopists managed to obtain more accurate 3D structures by serial-sectioning cells and tissues and then painstakingly reconstructing them from individual TEM images1. This was a very labor-intensive process and before the advent of digital imaging and computer rendering the results were also difficult to visualize. In recent years two techniques have been introduced which have become collectively known as volume electron microscopy (volume EM)2 that have made EM in three dimensions accessible to more laboratories.
The idea of obtaining a stack of images from an embedded block inside an electron microscope can be traced back to 1981 when Steve Leighton and Alan Kuzirian built a miniature microtome and placed it in the chamber of a scanning electron microscope3 (SBF-SEM). This prototype was eventually copied and improved 23 years later by Denk and Horstmann4 and subsequently commercialized. At roughly the same time biological scientists became aware of another technology used primarily in materials science, the focused ion beam. This technique uses an ion beam of some kind (Gallium, plasma) to remove a very small amount of surface material from a sample (FIB-SEM)5. Both techniques employ sectioning followed by imaging providing a series of images which can be combined into an X, Y, Z, stack. Both techniques provide 3D information but at different resolution scales. SBF-SEM is limited by the physical properties of the diamond knife to slices no thinner than 50 nm for long serial imaging runs; however the sample block size that can be sectioned is large, up to 1 mm x 1 mm x 1 mm. Due to the large digital acquisition format of the back scattered electron detector (32k x 32k pixels) that receives a signal from the block face, image pixel sizes can be as small as 1 nm. This results in non-isotropic voxels where the X,Y dimension is frequently smaller than the Z. Because of the precision of the ion beam, FIB-SEM has the ability to collect images with isotropic voxels ≤5 nm. However, the total area that can be imaged is quite small. A summary table of various samples and volumes imaged with the two techniques has been published previously3.
Tissue preparation for volume EM is more difficult than for standard TEM or SEM because samples must be stained to provide adequate signal generation in the SEM. Frequently, stainings need to be optimized not only for the particular tissue type but also for adding contrast to certain cellular structures to make identification and reconstruction easier. The protocol used here is based on the NCMIR standard6. Additional staining usually means additional protocol steps. Thus for volume EM, standard protocols need to be extended to ensure sufficient time for reagents to penetrate the sample. Microwave assisted processing can reduce the time needed for staining from hours to minutes and makes volume EM sample preparation more efficient7. This method is applicable to all cell and tissue types8 and to research questions where the inhomogeneity of the tissue makes sampling of a specific area essential9.
Once a data stack is obtained it can be aligned and the structures of interest segmented from the rest of the data and modeled in 3D. Although the automation of imaging many slices of tissue has made image acquisition relatively straightforward, the process of digitally reconstructing and visualizing the data is a time-consuming task. Software for this purpose is not yet integrated nor fully automated. Since much of the early work using volume EM was directed towards neuroscience, the techniques for staining and digitally segmenting structures such as axons is fairly far advanced compared to other cells and organelles. While the literature on other non-neuronal tissues is growing quickly, nonlinear or irregular structures require more manual input.
Using both SBF-SEM and FIB-SEM is a useful approach for targeting and imaging specific, nonhomogeneous, tissue structures at high resolution in 3D. Combining that with microwave assisted tissue processing that vastly decreases the time needed for sample preparation. Together this workflow will make generating high-resolution isotropic voxel image datasets of fine structures an efficient and more rapid process.
Protocol
1. Sample fixation and processing for electron microscopy
- Fix seedlings of Arabidopsis thaliana grown on agar plates in 0.5% paraformaldehyde, 2.5% glutaraldehyde in 0.1 M phosphate buffer (PB) pH 6.8 for 2 h at room temperature (RT).
CAUTION: Aldehydes are irritants and corrosive and have carcinogenic, mutagenic and teratogenic potential. All solutions must be handled with appropriate protective equipment and in a fume hood. - Cut root tips of the plant grown in step 1.1 and put 2-3 tips in 0.5 mL tubes containing the same fixative overnight at 4 °C.
NOTE: The volume of this and any solutions in the remaining steps is determined by the sample volume; a minimum ratio of sample to solution is 10:1. Samples larger than 1 mm in any dimension will be difficult to stain, so working with larger tissue blocks is more difficult. Not all tissues have the same characteristics; for example, plant leaves and stems can be difficult to stain. If larger samples or difficult tissue types are desired, optimization of sample processing must be performed before moving on to data collection. - Prepare the thiocarbohydrazide (TCH) solution, needed fresh and available before step 1.7. Add 0.1 g of thiocarbohydrazide to 10 mL of double distilled water (ddH2O) and dissolve by heating to 60 °C in the oven for 1 h. Before use, filter TCH solution using a 0.22 µm syringe filter.
- Remove fixative from the tubes and replace with 0.1 M PB pH 6.8. Put the tubes on an orbital shaking table at 100 rpm and wash for 10 min. Repeat the washing using fresh PBS 5 times.
- Post fix the root tips by replacing PB with 2% osmium tetroxide (OsO4) and 0.2% ruthenium red in 0.1 M PB pH 6.8. Put the tubes in the microwave with the lids open and start program 9 (Table 1).
CAUTION: Osmium is extremely hazardous in case of ingestion, very hazardous in case of inhalation, and hazardous in case of skin contact. Always handle using appropriate protective equipment and in a fume hood.
NOTE: Throughout the protocol, the lids of the tubes are always open during the microwave steps. - Wash the root tips twice with ddH2O for 5 min each on the benchtop. For the third and fourth ddH2O wash use program 15 on the microwave (Table 1). After the first 40 second ddH2O wash, take samples out of the microwave and replace the buffer with fresh ddH2O. Put samples back in the microwave and continue the program.
NOTE: The microwave will sound an alarm when buffer needs to be refreshed. Make sure the lid for the vacuum chamber is replaced correctly every time. - Incubate samples in previously prepared TCH solution at RT for 2 min on the bench and for further incubation use the microwave program 8 (Table 1). Do not change solution between bench and microwave.
- Wash samples as described in step 1.6.
- Place samples in 1% OsO4 in ddH2O for microwave program 9 (Table 1).
- Wash samples as described in step 1.6.
- Incubate samples in 1% uranyl acetate in ddH2O using microwave program 16 (Table 1).
CAUTION: Uranyl acetate is toxic, an irritant and has carcinogenic, mutagenic and teratogenic potential. Always handle using appropriate protective equipment. - Wash samples as described in step 1.6.
- Prepare Walton’s lead solution for use in step 1.14. First make a stock solution of L-aspartic acid by adding 0.998 g of L-aspartic acid to 250 mL of ddH2O and adjusting pH to 3.8 with 1 M KOH. Next, dissolve 0.066 g of lead nitrate in 10 mL of L-aspartic acid stock solution and adjust pH to 5.5. Leave the solution in the oven at 60 °C for 30 min.
NOTE: No precipitates should form. - Incubate samples in Walton’s lead solution for 30 min in the oven at 60 °C.
- Wash samples as described in step 1.6.
- Dehydrate samples in EtOH in graded steps of 50%, 70%, 90% in ddH2O, and then 2x in 100% EtOH. Use microwave program 10 (Table 1) and the microwave will prompt users every 40 s to replace solution with the next EtOH step. This is the last step done in the microwave.
- Further dehydrate in 100% propylene oxide 2x for 10 min each at RT on the bench, replacing the solution between steps.
NOTE: Propylene oxide can dissolve some plastics such as polystyrenes; either use glass vials for this step or pre-test plastics for resistance.
CAUTION: Propylene oxide is highly flammable. Always handle using appropriate protective equipment and in a fume hood. - Start infiltration of root tips by incubating in 50% Spurr’s resin in propylene oxide (min 2 h).
CAUTION: Spurr’s resin components are irritants. Always handle using appropriate protective equipment and in a fume hood. - Replace solution with 100% Spurr’s and leave overnight at RT.
- Change to fresh 100% Spurr’s resin 2 times (min 2 h incubations).
- Place samples in an embedding mold, again containing fresh 100% Spurr’s resin and polymerize in an oven at 65 °C for 36−48 h.
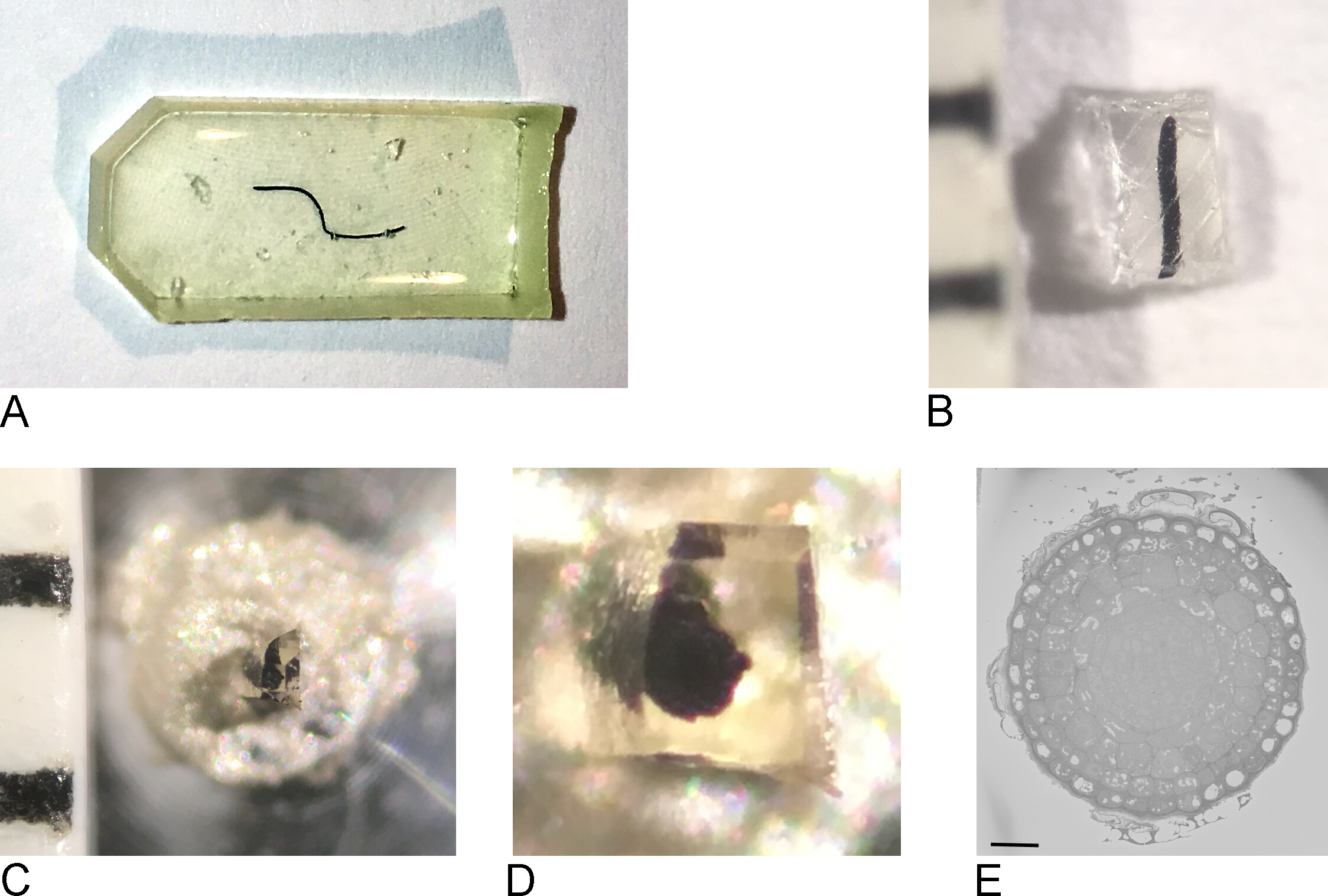
NOTE: The embedding mold used depends on the type of tissue and the approach used for imaging. Here, a flat silicone embedding mold was used (Figure 1A).
2. Prepare embedded samples for imaging
- Remove samples from the oven, remove resin from the embedding mold (Figure 1B).
- Using a razor blade, roughly trim the sample to a block of maximum 0.5 mm x 0.5 mm x 0.5 mm (Figure 1C).
NOTE: To prevent charging in SBF-SEM, it is important to trim away as much bare resin as possible and make the sample as flat/thin as possible. Ideally all sides of the block contain tissue already, but most importantly the side which will be attached to the metal pin (see step 2.3) has to contain exposed tissue so that the tissue is in direct contact with the conductive metal. - Remove the sample from the extraneous resin and attach it to a metal pin (Figure 1D) with conductive epoxy resin, making sure that part of the tissue is touching the metal pin. Let the epoxy cure overnight in the oven at 65 °C.
NOTE: Make sure the sample is positioned in the center of the pin, because the movement of the stage in the SB-SEM is limited. Removing the very small resin encased sample from extra resin can be difficult as the small sample will have a tendency to fly away when detached. A simple and effective solution to this is to cover the sample with a sheet of paraffin film as shown in the supplementary movie of reference1. - Fasten the carrier in the holder for the ultramicrotome. Use a razor blade to remove any excess epoxy and use the ultramicrotome and a diamond knife to smooth the face and sides of the block, forming a pyramid. Make sure that at least some of the tissue is already exposed on the block-face.
NOTE: The extra step of using a diamond knife is optional but it makes the resulting block easier to approach in the SBF-SEM because the shadow of the knife on the block face is clearer thus making estimating the distance between knife and block face easier to determine. - Place the trimmed sample block in the sputter coater and coat the sample with a thin layer (2−5 nm) of platinum (Pt).
NOTE: The platinum on the block-face will be cut away during the approach in the SBF-SEM (see below), but the platinum on the sides of the pyramid will provide additional conductivity. In this example, the sample was coated with platinum, but gold, or gold/ palladium is also effective; however, coating with gold resulted in increased debris on the block-face during an imaging run.
3. Imaging in the SBF-SEM
- Insert the carrier in the SBF-SEM microscope and bring the knife close to the sample surface. Using the diamond knife trim off the upper portion of the sample so that the Pt layer has already been removed and at least part of the tissue is exposed.
NOTE: Since this process is different for each SBF-SEM microscope, not every step is specified here. As long as the sample surface is free from Pt and ready for imaging, the next steps will be possible. - Start imaging at low resolution and short dwell times to get an overview of the sample and locate a region of interest (Figure 1E).
NOTE: Here, 512 x 512 pixels and 1 µs dwell time were used for quick scanning and positioning of the stage and 2,000 x 2,000 pixels with 1 µs dwell time were used for optimizing the imaging window and adjusting focus. - Using an accelerating voltage of 1.5-2.0 kV at a current of 80−100 pA, capture an image of the tissue.
NOTE: The example shown here was imaged on a high vacuum system where the beam current needs to be adjusted to minimize charging, which is very sample dependent. Typically, the electron beam is set to 1.5−2.0 kV but this will be sample dependent. At higher magnifications (usually > 10,000x) the resin is too much affected by the beam to guarantee smooth sectioning, so typically pixel-size is set to 8−20 nm at an image size of 8,000−10,000 x 8,000−10,000 pixels with a corresponding magnifications of 430 – 1,400x and field sizes of 64 X 64 μm and 200 x 200 μm respectively. - Determine a region of interest and decide how many sections are needed to cover the volume of interest and start the imaging run, using the back scattered electron detector.
NOTE: In the example presented here, 500 sections of 80 nm were imaged at 10 nm pixels and 10,000 x 10,000 pixel images (dwell time 1 µs). The microscope was set to 1.6 kV and 100 pA. In general, the number of sections is dependent on sample and size of the ROI and can vary from 100 s to 1,000 s of consecutive sections. The resulting dataset consists of single images of every section. These images need to be converted to a 3D stack.
4. SBF-SEM data processing
- Using Fiji, select file-import-image sequence and locate the image stack to load the images. Depending on the size of the dataset, check the ‘use virtual stack’ box.
NOTE: If the dataset is really big, first convert it to 8 bit (if collected at 16 bit) and if necessary bin the data until it has a workable size. - Using the play button at the bottom of the image scroll through the dataset to see if the imaging run was successful. Check for typical imaging artefacts for SBF-SEM, such as sections falling off the knife on the block-face, charging in areas of bare resin, cutting artefacts of the knife (horizontal lines on the image).
- Using the command image-properties adjust the pixel size and voxel depth (i.e., section-thickness) used during the run. If the data is already binned, take this into account.
- Using the command plugins-registration-linear stack alignment with SIFT to register the data.
NOTE: Registration of SBF-SEM data is needed because there might be slight sample movement during the imaging due to charging or drift of the sample. As this is only a minimal movement in XY, only translation is needed. - Check the alignment by scrolling through the dataset and if OK use the save command under the file menu to save the aligned dataset as a 3D-tif file.
- Analyze the dataset carefully to see if ROI is included and contains the information that is needed for the biological question. On the last image of the stack (= the current block-face), select a new ROI for FIB-SEM imaging.
NOTE: If there is no good region for FIB-SEM imaging present on the current block-face, more sections can be cut from the block (which is still in the SBF-SEM) until a ROI appears. There is a limit to the volume that can be imaged with the FIB. The ROI on the SBF-SEM image can be maximum in X,Y of 30-40 µm x 15-20 µm.
5. Imaging in the FIB-SEM
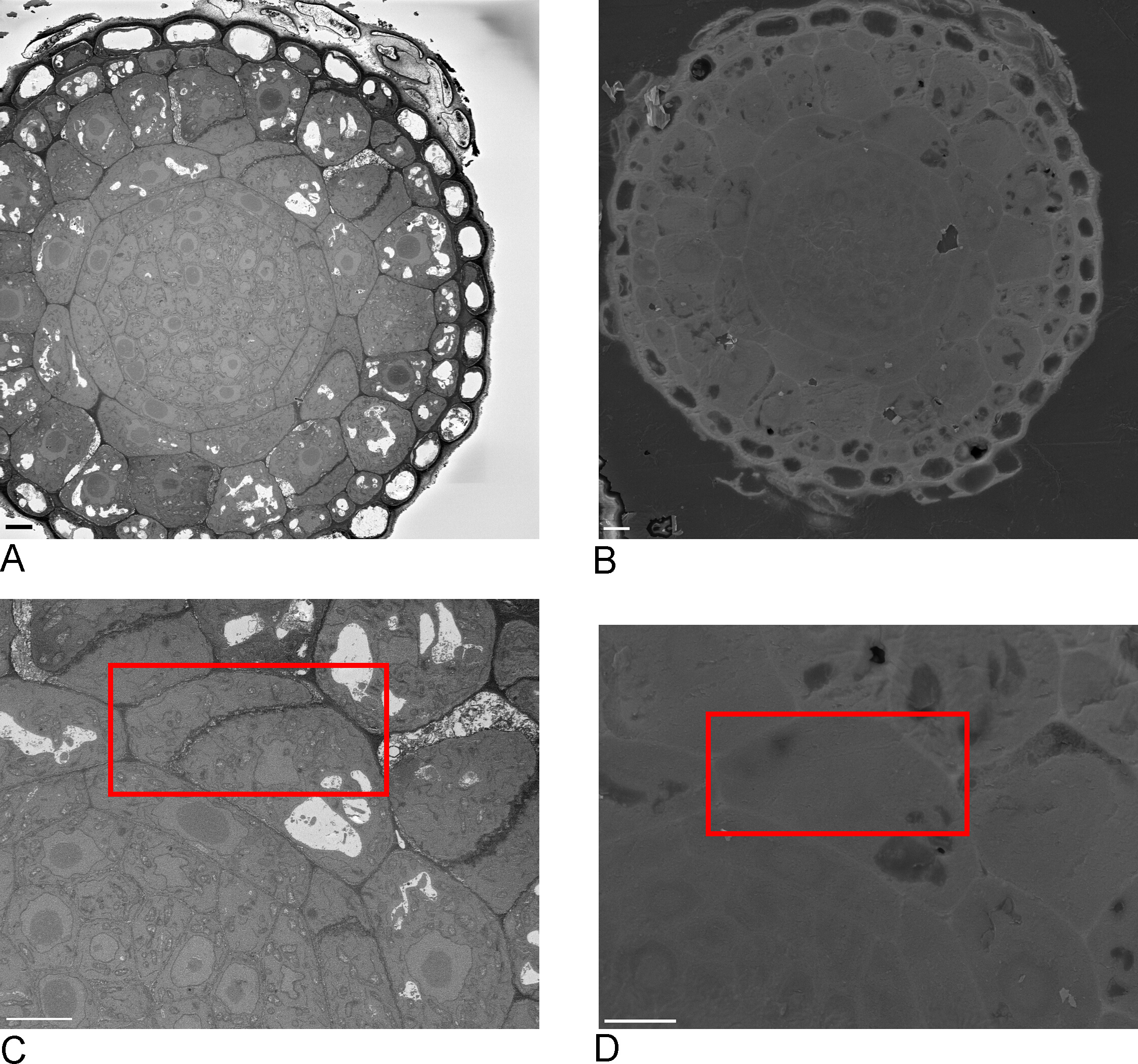
- Take overview images of the sample in the SBF-SEM, ideally including one or more edges of the sample that are then recognizable in the FIB-SEM (Figure 2A,C).
NOTE: In this example the SBF-SEM overview images were taken at 10 nm pixel size, 8,000 x 8,000 pixels at 1 µs dwell time. The microscope was still set at 1.6 kV and 100 pA. - Remove the sample from the SBF-SEM and place in the sputter coater. Coat the sample with ≥20 nm platinum for FIB-SEM imaging.
- Load sample into FIB-SEM and using secondary electron detector at 15 kV, 1 nA locate the ROI identified in the SBF-SEM on the block-face (Figure 2B,D).
NOTE: Imaging at 15 kV is necessary to see through the platinum coating. - Bring the ROI on the sample into the coincidence point of the FIB and SEM beams, by moving and tilting the stage (Figure 3A).
NOTE: The FIB column is usually mounted under an angle (Figure 3A). This means that any sample needs to be tilted in such a way that the surface to be imaged and sectioned is positioned parallel to the FIB beam. The surface to be imaged is now tilted with respect to the SEM beam and a trench of tissue needs to be removed before the SEM is able to image the ROI (Figure 3E) - Using the FIB beam and gas injection system, deposit a 1 μm protective layer of platinum on the surface above the ROI (Figure 3C). Next, using a low milling current (50-100 pA), mill fine lines into the platinum deposition for auto-focusing and 3D tracking during the imaging run (Figure 3D). Using the FIB column and carbon gas injector, cover these lines with carbon deposition.
NOTE: The size of the ROI here corresponds to the size of the ROI on the SBF-SEM image and the maximum size can thus be 30-40 µm x 15-20 µm. In this example a ROI of 17 µm x 8 µm was imaged. Carbon deposition is needed for protection of the lines and to create a black-white contrast between the carbon and platinum which is ideal for auto-focusing. The milling currents used for each step can be found in Table 2. - Using a high milling current, mill a trench of 30 µm in front of the ROI, creating the imaging surface for the SEM beam (Figure 3E).
NOTE: The FIB beam is inherently destructive, even more so at high currents. Make sure to keep imaging at high currents to a minimum and image at low magnification and fast scan speeds to avoid melting the resin at the ROI. - Smooth the imaging surface with a milling current closer to the current used during the imaging run. Stop polishing when all auto-focus and 3D tracking marks are clearly visible on the imaging surface (Figure 3F). The progress of the FIB can be followed by imaging the surface with the EM (using a back scattered electron detector) while polishing.
- Determine the area to be imaged on the newly created surface and set the imaging parameters. Make sure the electron beam is focused on the surface, set brightness and contrast and set the pixel-size and section thickness. Keep the imaging time below 1 minute by adjusting dwell time and line average.
NOTE: It is important to use a low voltage for imaging with the electron beam to ensure that only the surface of the block is imaged (i.e., that no electrons from deeper into the sample are imaged). This is done by keeping the voltage below 2 kV and using a back scattered electron detector with a grid voltage, allowing only high energy electrons to be imaged. In this example, the electron beam was set at 1.5 kV and 1 nA with a grid voltage of 1.2 kV on the back scattered electron detector. Also here, a pixel-size of 5 nm was used with 5nm sections to result in a dataset with isotropic voxels. An area of 17 µm x 10 µm was imaged at 6.5 µs dwell time and a line average of 1.0. - Set the windows for auto-tuning and 3D tracking, using the same pixel size, dwell time and line average as used for the imaging.
NOTE: This process will differ for different systems, therefore only the step is mentioned without specifying the different actions. - Start the imaging run and monitor the stability of the process during the first 50−100 sections. Once the system is running smoothly, leave the room and ensure that there is as little disturbance to the room as possible.
NOTE: The duration of the run and the number of sections will depend on the size of the ROI and the section thickness. In FIB-SEM the Z axis is actually the height of the ROI box on the SBF-SEM image (max 15−20 µm; Figure 3E). - Register the FIB-SEM data in the same way as described above for the SBF-SEM data.
Results
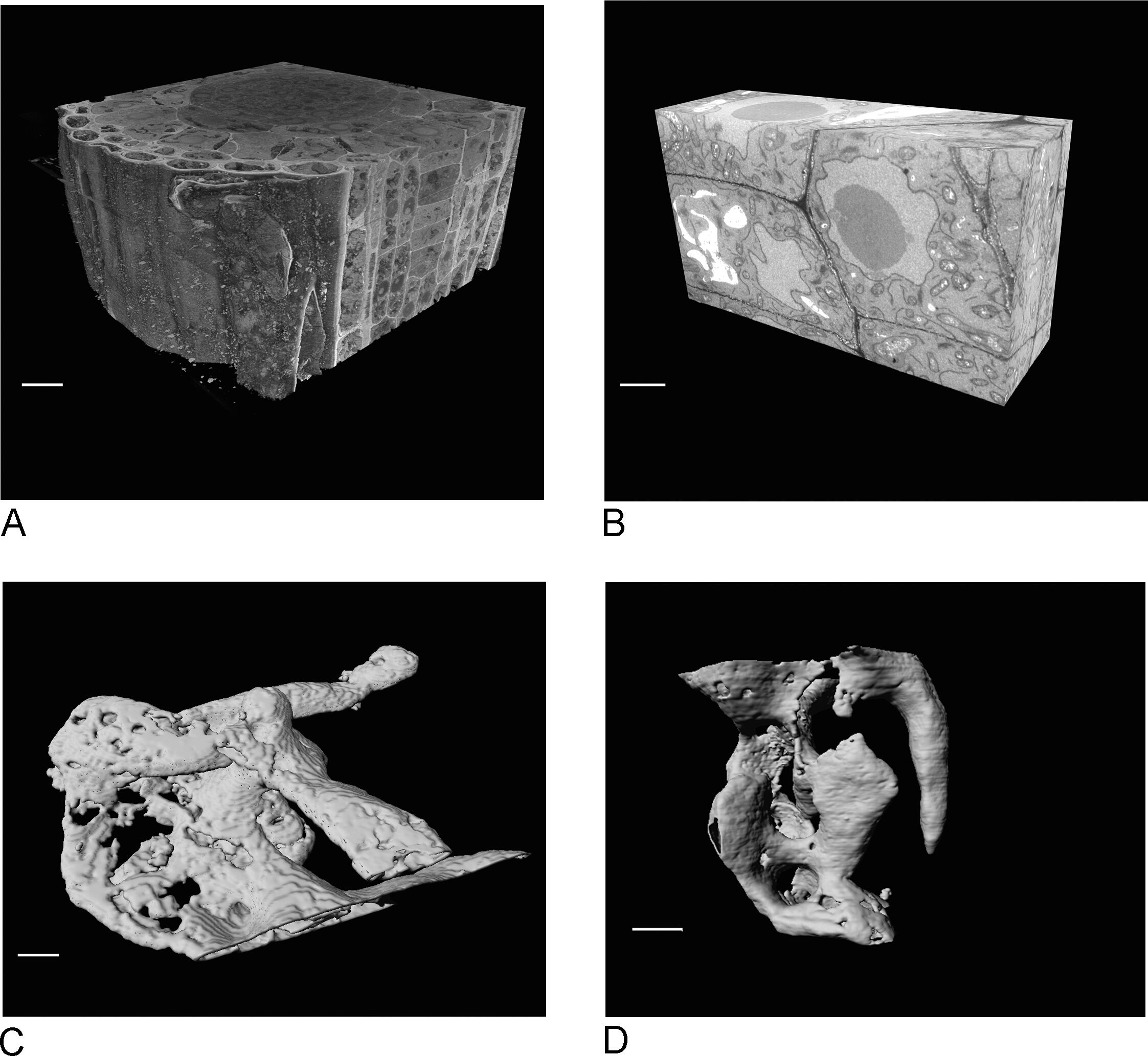
Images from the SBF-SEM provide an overview of the tissue, giving insight into the spatial orientation of cells and intercellular connections (Figure 4A). The subsequent FIB-SEM imaging on a new region, which is usually a region of interest determined after inspection of the SBF-SEM run, adds high-resolution detail of specific cells and/or structures (Figure 4B).
Figure 4C,D show the difference in rendering of the non-isotropic voxels of the SBF-SEM data (Figure 4C) and the isotropic voxel FIB-SEM data (Figure 4D). The z thickness used in SBF-SEM means that the rendering clearly shows the sections, resulting in a 'staircase' effect in on the surface. In the FIB-SEM data the 5 nm sections ensure that the rendering appears much smoother and individual sections blend into the surface completely.

Figure 1: Creation of the block face from a resin embedded sample. (A) A root-tip embedded in resin. (B) Using a razor blade the excess resin is trimmed away until a block of 0.5 mm2 remains. (C,D) The trimmed block is glued onto a metal pin and after a night in the oven, the sides of the block are trimmed and the surface smoothened with a diamond knife using an ultramicrotome. (E) Inside the SBF-SEM, the sample is oriented so that the blockface and ROI can be recognized, scale bar = 20 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 2: Correlation between SBF-SEM and FIB-SEM. Overview images of the block-face using the SBF-SEM (A) and the FIB-SEM (B), scale bar = 5 µm. (C,D) Zoom on the ROI. Red box delineates the region to be imaged with FIB-SEM, scale bar = 5 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 3: FIB-SEM scheme and preparation steps. (A) Scheme showing the orientation of the FIB beam, SEM beam and sample. The sample needs to be positioned to the coincidence point of the FIB and SEM beams to be able to mill and image on the same region. (B) Schematic drawing of the trench needed for SEM imaging of the sections removed by the FIB. (C) Image taken with the FIB beam showing Platinum deposition on the ROI, scale bar 5 µm. (D) Image taken with the FIB beam showing the lines used for auto-focus and 3D tracking. The lines in the middle are used for auto-focus and the outside lines provide 3D tracking. Carbon deposition on top of the lines provides the needed contrast (platinum vs carbon) to perform these tasks, scalebar 5 µm. (E) Image taken with the FIB beam after milling of the trench, scalebar 5 µm. (F) Image taken with the SEM beam showing the region of interest imaged during the FIB-SEM run, scale bar 2 µm. Please click here to view a larger version of this figure.
{kind=link}

Figure 4: SBF-SEM and FIB-SEM results before and after segmentation. (A) 3D view of SBF-SEM dataset (100 x 100 x 40 µm, scale bar = 10 µm), (B) 3D view of FIB-SEM dataset (17 x 10 x 5.4 µm, scale bar = 2 µm), (C) Rendered vacuoles segmented from SBF-SEM data by thresholding, scale bar = 2 µm D. Rendered granules segmented from FIB-SEM data by thresholding, scale bar = 2 µm. Please click here to view a larger version of this figure.
{kind=link}
| Protocol for Microwave Processing | |||||||||
| Program # | Description | User Prompt (on/off) | Time (hr:min:sec) | Power (Watts) | Temp (°C) | Load Cooler (off/auto/on) | Vacuum/Bubbler Pump (off/bubb/vac cycle/vac on/vap) | Steady Temp | |
| Pump (on/off) | Temp (°C) | ||||||||
| 8 | TCH | OFF | 0:01:00 | 150 | 50 | OFF | VACUUM CYCLE | ON | 30 |
| OFF | 0:01:00 | 0 | 50 | OFF | VACUUM CYCLE | ON | 30 | ||
| OFF | 0:01:00 | 150 | 50 | OFF | VACUUM CYCLE | ON | 30 | ||
| 9 | OSMIUM | OFF | 0:02:00 | 100 | 50 | OFF | VACUUM CYCLE | ON | 30 |
| OFF | 0:02:00 | 0 | 50 | OFF | VACUUM CYCLE | ON | 30 | ||
| OFF | 0:02:00 | 100 | 50 | OFF | VACUUM CYCLE | ON | 30 | ||
| OFF | 0:02:00 | 0 | 50 | OFF | VACUUM CYCLE | ON | 30 | ||
| OFF | 0:02:00 | 100 | 50 | OFF | VACUUM CYCLE | ON | 30 | ||
| 10 | 50% EtOH | ON | 0:00:40 | 150 | 50 | OFF | OFF | ON | 30 |
| 70% EtOH | ON | 0:00:40 | 150 | 50 | OFF | OFF | ON | 30 | |
| 90% EtOH | ON | 0:00:40 | 150 | 50 | OFF | OFF | ON | 30 | |
| 100% EtOH | ON | 0:00:40 | 150 | 50 | OFF | OFF | ON | 30 | |
| 100% EtOH | ON | 0:00:40 | 150 | 50 | OFF | OFF | ON | 30 | |
| 15 | 0.1M CACODYLATE | ON | 0:00:40 | 250 | 50 | OFF | VACUUM CYCLE | ON | 30 |
| 0.1M CACODYLATE | ON | 0:00:40 | 250 | 50 | OFF | VACUUM CYCLE | ON | 30 | |
| 15 | ddH2O | ON | 0:00:40 | 250 | 50 | OFF | VACUUM CYCLE | ON | 30 |
| ddH2O | ON | 0:00:40 | 250 | 50 | OFF | VACUUM CYCLE | ON | 30 | |
| 16 | Uranyl Acetate | OFF | 0:01:00 | 150 | 50 | OFF | VACUUM CYCLE | ON | 30 |
| OFF | 0:01:00 | 0 | 50 | OFF | VACUUM CYCLE | ON | 30 | ||
| OFF | 0:01:00 | 150 | 50 | OFF | VACUUM CYCLE | ON | 30 | ||
| OFF | 0:01:00 | 0 | 50 | OFF | VACUUM CYCLE | ON | 30 | ||
| OFF | 0:01:00 | 150 | 50 | OFF | VACUUM CYCLE | ON | 30 | ||
| OFF | 0:01:00 | 0 | 50 | OFF | VACUUM CYCLE | ON | 30 | ||
| OFF | 0:01:00 | 150 | 50 | OFF | VACUUM CYCLE | ON | 30 | ||
Table 1. Detailed protocol for microwave processing.
| Step | Current | Estimated Time |
| Deposition Platinum | 3n A | 10-15 minutes |
| Milling Autotune and Tracking Marks | 50-100 pA | 4-6 minutes |
| Deposition Carbon | 3 nA | 5-10 minutes |
| Milling Coarse Trench | 15-30 nA | 30-50 minutes |
| Polishing Surface | 1.5-3 nA | 15-20 minutes |
| Imaging Run | 700 pA-1.5 nA | Hours-days |
Table 2. FIB milling currents used for sample preparation and imaging run
Discussion
Volume electron microscopy is more challenging and time consuming than conventional SEM or TEM. Because of the need to stain tissues or cells en bloc, processing steps must be long enough to ensure penetration of reagents throughout the sample. Using microwave energy to facilitate penetration makes for shorter, more efficient processing and improves staining. Because preparation for EM is much more stringent than for light microscopy all solutions and reagents must be quality controlled strictly. Changes in pH, tonicity, the use of impure reagents, and introduction of contaminants due to poor technique can all have deleterious effects on the final image.
Volume EM also requires individually tailored protocols for each different sample type. Mammalian tissues of different types: plants, single cells such as yeast, trypanosomes, C. elegans, etc., all need their own variations to achieve optimal results. Fixation and staining must be designed so as to preserve structural integrity and keep the sample as close to its in vivo morphology as possible. Fixation of tissues at physiological temperature, pH and tonicity is critical to making the sample as life-like as it can be. High-pressure freezing (HPF) of samples may help to preserve a more life-like situation, (or perhaps just yield different artifacts), but for other than cells and very thin tissues HPF will fail as vitreous ice can only be generated in small volumes. Therefore for many questions chemical fixation is the only option. No matter if the fixation is HPF or chemical, in any EM experiment the structural results need to be carefully compared to similar results from live cell or tissue imaging to see if they are consistent. Staining must also be optimized while considering the specific question that needs to be answered and the protocol that will be used for visualization of the digital images.
Having both an SBF-SEM and FIB system in close proximity is a great advantage in many experiments. The large field of view and high X,Y resolution of SBF-SEM makes finding individual structures/cells/events straightforward and provides an overall spatial orientation of cells in tissues. In addition, its ability to allow imaging through a sample in Z is very powerful; however, reconstructions that require fine geometric detail can fail or produce artifacts using this technique due to the non-isotropic voxels it generates. The FIB is limited by the physics of the process to a smaller imaging field but its 3D resolution is sufficient for very accurate reconstructions. Combining the two techniques is straightforward as samples can move from SBF-SEM to FIB without further processing or preparation. We acknowledge that using the SBF-SEM for searching through a sample to find a particular area is a very expensive use of a much more capable tool. However, the ability to immediately see the new blockface and determine whether the ROI has been reached is a great advantage. Additionally, the alternatives of using serial semi-thin (0.5 µm) LM sections may remove small structures before they are detected, and inspecting a block using single TEM sections which have to be cut, put on a grid and then viewed in an equally expensive TEM is not as efficient as the method presented.
Because many programs exist to segment and render the data, and the needs of a given structure may not be best served by a single application, no standard workflow can be proposed. Some simple structures may be segmented with a thresholding algorithm if they fall within very narrow grey scale values. Neuronal structures can be semi-automatically segmented using a program such as Ilastik11 but it will be less useful on more random or complex shaped organelles such as ER. Microscopy Image Browser is a very flexible program that can align, segment, and render volume EM data, but requires significant user interaction12. As a general rule the amount of time needed to digitally visualize the results will greatly exceed the time for preparing the sample and imaging.
Volume EM techniques have opened up the third dimension to ultrastructural analysis. Other methods of obtaining 3D EM have limitations in their volume (TEM tomography), or their efficiency (serial section TEM). Although for the most part volume EM techniques are too complex and costly to be implemented in individual laboratories, the number of shared core facilities offering them has been growing and the number of sample types successfully imaged has increased rapidly. For those with a specific question and a particular tissue it is likely someone will be able to offer advice and instructions on its preparation and imaging. Volume EM equipment can be improved to include the capacity to handle larger samples in the SBF-SEM and the capability of milling larger ROIs with the FIB. Software which is able to segment out structures of interest in a more automated way will vastly simplify the process of analyzing the data and improvements in computing speed will reduce the time needed to do so. Despite its current limitations, volume EM is still a useful tool and combining SBF-SEM and FIB-SEM provides an efficient workflow for identifying rare events and imaging them at high resolution.
Disclosures
The authors have nothing to disclose.
Acknowledgements
The equipment for volume EM was provided by a generous grant from the Government of Flanders.
Materials
| Name | Company | Catalog Number | Comments |
| 3View 2XP | Gatan | NA | In chamber ultramicrotome for SBFI |
| Cacodylate buffer 0.2M solution | EM Sciences | 11652 | |
| Conductive epoxy resin (circuit works) | RS components | 496-265 | |
| Diatome Histo 4.0mm diamond knife | EM Sciences | 40-HIS | |
| Digitizing tablet | Wacom | DTV-1200W | No longer available |
| Eppendorf tubes | Eppendorf | 0030 120.094 | |
| Flat Embedding Mold | EM Sciences | 70900 | |
| Gluteraldehyde 25% solution | EM Sciences | 16220 | |
| High MW Weight Tannic Acid | EM Sciences | 21700 | |
| Lead Citrate | Sigma-Aldrich | 22861 | |
| NaCl | Sigma-Aldrich | 746398 | |
| Osmium Tetroxide 4% solution | EM Sciences | 19170 | |
| Paraformaldehyde | Sigma-Aldrich | P6148 | |
| Pelco Biowave Pro + | Ted Pella | 36700 | |
| Potassium Ferrocyanide | Sigma-Aldrich | P3289 | |
| Quorum Q150T ES sputter coater | Quorum Technologies | 27645 | |
| Reichert-Jung Ultracut ultramicrotome | NA | NA | No longer available |
| Sodium Cacodylate 0.2M | EM Sciences | 11653 | |
| Spurrs Resin kit | EM Sciences | 14300 | |
| Uranyl Acetate | EM Sciences | 22400 |
References
- Linberg, K. A., Fisher, S. K. An ultrastructural study of interplexiform cell synapses in the human retina. Journal of Comparative Neurology. 243, 561-576 (1986).
- Peddie, C. J., Collinson, L. M. Exploring the third dimension: volume electron microscopy comes of age. Micron. 61, 9-19 (2014).
- Leighton, S. SEM images of block faces, cut by a miniature microtome within the SEM – A technical note. Scanning Electron Microscopy. (pt 2), 71-76 (1981).
- Denk, W., Horstmann, H. Serial block-face scanning electron microscopy to reconstruct three dimensional tissue nanostructure. PLoS Biology. 11, 329 (2004).
- Heymann, J. A., Hayles, M., Gestmann, I., Giannuzzi, L. A., Lich, B., Subramaniam, S. Site specific 3D imaging of cells and tissues with a dual beam microscope. Journal of Structural Biology. 155 (1), 63-73 (2006).
- . NCMIR Methods for 3D EM: A new protocol for preparation of biological specimens for serial block face scanning electron microscopy. V7_01_10 Available from: https://ncmir.ucsd.edu/sbem-protocol (2019)
- Giberson, R. T., Austin, R. L., Charlesworth, J., Adamson, G., Herrera, G. A. Microwave and digital imaging technology reduce turnaround times for diagnostic electron microscopy. Ultrastructural Pathology. 27 (3), 187-196 (2003).
- Kremer, A., et al. Developing 3D EM in a broad biological context. Journal of Microscopy. 259 (2), 80-96 (2015).
- Vanslembrouck, B., Kremer, A., Pavie, B., van Roy, F., Lippens, S., van Hengel, J. Three-dimensional reconstruction of the intercalated disc including the intercellular junctions by applying volume scanning electron microscopy. Journal of Histochemistry and Cell Biology. 149, 479-490 (2018).
- Russel, M. R., et al. 3D correlative light and electron microscopy of cultured cells using serial blockface scanning electron microscopy. Journal of Cell Science. 130 (1), 278-291 (2017).
- Sommer, C., Strähle, C., Köthe, U., Hamprecht, F. A. ilastik: Interactive Learning and Segmentation Toolkit. Eighth IEEE International Symposium on Biomedical Imaging (ISBI). Proceedings. , 230-233 (2011).
- Belevich, I., Joensuu, M., Kumar, D., Vihinen, H., Jokitalo, E. Microscopy Image Browser: A Platform for Segmentation and Analysis of Multidimensional Datasets. PLoS Biology. 14 (1), e1002340 (2016).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request PermissionThis article has been published
Video Coming Soon
Copyright © 2025 MyJoVE Corporation. All rights reserved